
Cu/M:ZnO (M = Mg, Al, Cu) colloidal nanocatalysts for the solution hydrogenation of carbon dioxide to methanol†
Alice H. M.
Leung
a,
Andrés
García-Trenco
a,
Andreas
Phanopoulos
 a,
Anna
Regoutz
b,
Manfred E.
Schuster
c,
Sebastian D.
Pike
a,
Milo S. P.
Shaffer
*de and
Charlotte K.
Williams
*a
a,
Anna
Regoutz
b,
Manfred E.
Schuster
c,
Sebastian D.
Pike
a,
Milo S. P.
Shaffer
*de and
Charlotte K.
Williams
*a
aChemistry Research Laboratory, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: charlotte.williams@chem.ox.ac.uk
bDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK
cJohnson Matthey Technology Centre, Blount's Court, Sonning Common, RG4 9NH, UK
dDepartment of Chemistry, Imperial College London, London, SW7 2AZ, UK. E-mail: m.shaffer@imperial.ac.uk
eDepartment of Materials, Imperial College London, London, SW7 2AZ, UK
First published on 27th May 2020
Abstract
Doped-ZnO nanoparticles, capped with dioctylphosphinate ligands, are synthesised by the controlled hydrolysis of a mixture of organometallic precursors. Substitutional doping of the wurtzite ZnO nanoparticles with 5 mol% Mg(II), Al(III) and Cu(I) is achieved by the addition of sub-stoichiometric amounts of the appropriate dopant [(n-butyl)(sec-butyl)magnesium, triethylaluminium or mesitylcopper] to diethylzinc in the precursor mixture. After hydrolysis, the resulting colloidal nanoparticles (sizes of 2–3 nm) are characterised by powder X-ray crystallography, transmission electron microscopy, inductively-coupled plasma optical emission spectrometry and X-ray photoelectron spectroscopy. A solution of the doped-ZnO nanoparticles and colloidal Cu(0) nanoparticles [M:ZnO : Cu = 1 : 1] are applied as catalysts for the hydrogenation of CO2 to methanol in a liquid-phase continuous flow stirred tank reactor [210 °C, 50 bar, CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2 = 1:3, 150 mL min−1, mesitylene, 20 h]. All the catalyst systems display higher rates of methanol production and better stability than a benchmark heterogeneous catalyst, Cu–ZnO–Al2O3 [480 μmol mmolmetal−1 h−1], with approximately twice the activity for the Al(III)-doped nanocatalyst. Despite outperforming the benchmark catalyst, Mg(II) doping is detrimental towards methanol production in comparison to undoped ZnO. X-Ray photoelectron spectroscopy and transmission electron microscopy analysis of the most active post-catalysis samples implicate the migration of Al(III) to the catalyst surface, and this surface enrichment is proposed to facilitate stabilisation of the catalytic ZnO/Cu interfaces.
:H2 = 1:3, 150 mL min−1, mesitylene, 20 h]. All the catalyst systems display higher rates of methanol production and better stability than a benchmark heterogeneous catalyst, Cu–ZnO–Al2O3 [480 μmol mmolmetal−1 h−1], with approximately twice the activity for the Al(III)-doped nanocatalyst. Despite outperforming the benchmark catalyst, Mg(II) doping is detrimental towards methanol production in comparison to undoped ZnO. X-Ray photoelectron spectroscopy and transmission electron microscopy analysis of the most active post-catalysis samples implicate the migration of Al(III) to the catalyst surface, and this surface enrichment is proposed to facilitate stabilisation of the catalytic ZnO/Cu interfaces.
Introduction
The doping of semiconductors is critical for manipulating carrier density and band-gap in (opto)electronics, and in catalysis to control activity, selectivity and lifetime.1,2 Zinc oxide, a wide band-gap (3.3 eV), n-type semiconductor, is an important material in both contexts, providing useful properties such as transparency and accessible redox chemistry, while using only abundant elements.3–7 Consequently, ZnO nanoparticles (NPs) have found use as solution-processible electronic inks, and as high surface area catalysts.8–10 Relevant to this work are elegant examples of property enhancement through heterovalent substitutional doping of ZnO.3,11–14 Depending on the dopant either shallow energy level donor (n-type) or acceptor (p-type) states are incorporated, adjusting the properties.3 To synthesise these materials, a judicious choice of reactants and conditions must be employed to balance the reactivity of the host and dopant i.e. to prevent phase separation.1,4 n-Type ZnO doping is more easily achieved and is most often accomplished through incorporation of Group 13 elements at interstitial sites.3 For instance, aluminium doping enhanced the optical and electronic properties of ZnO, affording a cheaper and less toxic competitor to the ubiquitous transparent conducting oxide, indium tin oxide (ITO).11 Alternatively, doping with Mg(II) afforded colloidal solutions that were better reductants than ZnO, as demonstrated by the rapid and spontaneous electron transfer from Zn0.75Mg0.25O to ZnO.14 On the other hand, p-type doping often suffers from compensating mechanisms during synthesis, such as the presence of low-energy native defects (interstitial zinc atoms or oxygen vacancies) or background impurities.3 Nevertheless, ZnO doping with Group 1 elements15 or Cu(I)16 has been achieved. For instance, the thermal treatment of heterometallic cubane ([Me3Zn3M(THF)OtBu4], M = Li, Na, K), at 750 °C for 3 h, produced p-type ZnO nanoparticles (20–70 nm) with dopant levels up to 10 mol%.15,17 Ultimately, the successful and reproducible, large-scale fabrication of both n- and p-type doped-ZnO could allow for production of ZnO p–n junctions for use in thin film, large area, potential transparent semiconducting electronic devices.18–20Beyond semiconducting applications, ZnO is also one component of the industrial heterogeneous three-component catalyst Cu–ZnO–Al2O3, which is used to produce methanol on a 50 million-tonne scale in conjunction with a syn-gas feedstock.21,22 Methanol is a clean burning liquid fuel that can be blended with petrol providing an attractive ‘drop-in’ substitute with existing fuels, and moreover can be generated from renewable raw materials (i.e. CO2/H2).23–26 Doping the ZnO component can enhance the catalytic performance using either syn-gas or CO2/H2 gas mixtures.27–29 The range of dopants includes alkaline metals (e.g. Cs(I) or Mg(II)),30 transition (e.g. Zr(II) or Mn(II)/(III))27,31 or main group metals (e.g. Al(III) or Ga(III)).32,33 These dopants are proposed to increase activity by acting as electronic and/or structural promoters, i.e. by altering the electronic structure through addition of energy levels to reduce activation energy, or by modifying the catalyst nanostructure to increase the number of active sites, respectively.28,34 In particular, there is an enhancement in activity when n-type dopants, such as Al(III) and Ga(III) are incorporated into Cu/ZnO catalysts.28,29 It is proposed that dopants facilitate formation of Cu/ZnO interfaces by labilising the ZnOx (x < 1) component.35 These interfaces, often observed at step-defects in the Cu phase,36 are the putative catalytic active sites.29,37 Conversely, by lowering the density of donor states, e.g. by doping Cu/ZnO with Mg(II), an increase in optical band-gap and reduction potential were observed.28 The lower efficiency for methanol production was attributed to the converse effects limiting the formation of critical Cu/ZnOx interfaces.28,29
Most methanol production occurs using syn-gas in fixed bed reactor configurations but slurry phase methods, where the catalyst is suspended in solution, are attractive alternatives. This slurry process can improve reaction kinetics and may increase the intrinsic activity. As long as high enough pressures and dilute conditions are implemented, the mass transport effects can be managed, though for productivity reasons, reactors may, in fact, be operated near the mass transport limit.38 Additionally, liquid phase synthesis may mitigate local hot spots, accelerate catalyst activation and facilitate continuous product removal. These benefits have led to the development of a commercial liquid-phase methanol synthesis process (LPMeOH™).39 One method to optimize liquid phase processes is to develop colloidal (quasi-homogeneous) catalysts. Our group,8,9,40–43 and others,44–46 have previously outlined some of the advantages of such species including easy control over catalyst composition (Cu:Zn ratio), access to small, well-defined, high surface area nanoparticles, and control over surface chemistry. These colloidal catalysts have been applied either to the synthesis of methanol from syn-gas21,47 or by carbon dioxide hydrogenation (CO2/H2).9,40,41 They can show high activity and selectivity over the reverse water–gas shift (rWGS) reaction.28
To make these colloidal catalysts, we have developed a synthesis using the hydrolysis of organozinc reagents. The method generates small (2–4 nm) and monodispersed ZnO nanoparticles, which are soluble in a range of organic solvents.48–50 The hydrolysis of organozinc compounds (e.g. ZnPh2, ZnCy2, ZnEt2), in the presence of an aqueous stable, anionic coordinating ligand (e.g. carboxylate or phosphinate), readily generates soluble ZnO nanoparticles,48,50–57 with a high density of surface defects, proposed to be beneficial for catalysis.9,44,58 Hydrolytic syntheses provide straightforward control of ligand coverage and kinetic control over the growth of particles, allowing access to small ZnO nanoparticles (2–4 nm).4,50,54,57 The methodology is potentially attractive for dopant incorporation since a second organometallic reagent can simply be added to the one-pot hydrolysis reaction. The synthesis of doped-ZnO nanoparticles via co-precipitation methods has allowed doping levels >10% (e.g. 18% Mg-,14 13% Al-29 or 12% Cu-doping59) with a range of metals (e.g. Group 1,15,17 Group 2,14 Group 13,28,29 and Group 1116,59−61); however, there are only a few examples of successful doping using organometallic routes.4,62,63 Chaudret and co-workers demonstrated that the addition of Li-amides during the hydrolysis of ZnCy2, in the presence of octylamine, led to the formation of spherical Li-doped-ZnO nanoparticles (2.4–4.3 nm, Li content 1–10 mol%). The nanoparticle growth was controlled by the localisation of LiOH on the surface of ZnO.62 Alternatively, our group prepared Mg-doped ZnO nanoparticles by mixing ZnEt2 with MgBu2 before hydrolysis, in the presence of dioctylphosphinic acid (DOPA–H, (C8H17)2PO2H). The resultant particles showed up to 10% Mg incorporation into the wurtzite ZnO structure.63
This work aims to investigate the preparation of other doped ZnO materials, e.g. with Al(III), Mg(II) and Cu(I). These colloidal nanoparticles will be tested as components in liquid phase catalysts for the reduction of CO2 to methanol.
Experimental
Materials and methods
The syntheses of all the ZnO nanoparticles were carried out in a dry nitrogen-filled glovebox and using Schlenk line techniques. All solvents were purchased from VWR, UK; all chemicals from Sigma Aldrich and were used as supplied, unless otherwise stated. Toluene was dried using an MBraun SPS-800 solvent purification system and degassed by sparging with N2 for 1 h to remove oxygen. Diethyl zinc, triethyl aluminium and (n-butyl)(sec-butyl)magnesium are highly pyrophoric and volatile: extreme caution must be taken during usage. HPLC-grade water was used for the controlled hydrolysis experiments.Elemental analyses were performed by Mr Stephen Boyer at London Metropolitan University and inductively-coupled plasma optical emission spectrometry (ICP-OES) was performed by Mr Alan Dickerson at the University of Cambridge.
Powder X-ray diffraction (XRD) experiments were performed using a PANalytical X'pert PRO diffractometer in the theta/theta reflection mode, fitted with a nickel filter, 0.04 rad Soller slit, 10 mm mask, 1/4° fixed divergence slit, and 1/2° fixed antiscatter slit. An operating voltage of 40 kV was used with a step size of 0.033° 2θ, scan step time of 70 s and scan range of 10–80° 2θ. Air sensitive samples were prepared in a glovebox and sealed with polyimide tape. The diffraction patterns were analysed using Fityk (version 0.9.0; Marcin Wojdyr, 2010). The peaks were fitted to a SplitPearson7 function, and the crystallite size was calculated from the full-width half-maximum of the fitted curve, using the Scherrer equation, to the most intense and not overlapped reflections; Cu: 43.5° (hkl = 111), ZnO: 47.5° (hkl = 102), 57.2° (hkl = 110). For the detailed XRD study of the (101) ZnO nanoparticle peak, the diffraction scans were performed using the same diffractometer with a step size of 0.0167° 2θ, scan step time of 120 s and scan range of 32–38° 2θ. The expansion/contraction of the ZnO lattice was determined, using Bragg's equation, from the position of the (101) peak which was determined by Gaussian fit of the diffraction peak. UV-Vis absorption spectra of ZnO nanoparticles were recorded using a PerkinElmer Lambda 950 spectrometer, in transmission mode, from 300–500 nm with a scan speed of 1 nm s−1. Fourier transform infrared (FT-IR) spectra were recorded using a PerkinElmer Spectrum 100 spectrometer fitted with an ATR attachment and each spectrum was collected with 32 scans, at a resolution of 4 cm−1. Thermogravimetric analysis (TGA) was carried out using a Mettler Toledo TGA/DSC 1, under a flow of dry N2 at 60 mL min−1, from 100–700 °C, at a heating rate of 5 °C min−1.
Bright field (BF) TEM and HAADF-STEM images were acquired with Johnson Matthey's probe corrected ARM200F, at the ePSIC facility (Diamond Light Source), with an acceleration voltage of 200 keV. Measurement conditions were a CL aperture of 30 μm, convergence semiangle of 24.3 mrad, beam current of 12 pA, and scattering angles of 0–10 and 35–110 mrad, for BF and HAADF-STEM respectively.
XPS was performed on a Thermo Scientific Kα+ X-ray photoelectron spectrometer system operating at 2 × 10−9 mbar base pressure. This system incorporates a monochromated, microfocused Al Kα X-ray source (hν = 1486.6 eV) and a 180° double focusing hemispherical analyser with a 2D detector. The X-ray source was operated at 6 mA emission current and 12 kV anode bias and a flood gun was used to minimize sample charging. Samples were mounted using conductive carbon tape and transferred to the spectrometer, using a special glovebox module, which ensured that samples were transferred without exposure to air. Data were collected at 20 eV pass energy for core levels and Auger lines. Data were collected over six measurement positions using an X-ray spot size of 400 μm to avoid beam damage affecting the results. Data were then averaged across the different measurement positions. All data were analysed using the Avantage software package.
:ZnO nanoparticles were added directly to the reactor vessel together with mesitylene (89 mL). The solution was stirred and degassed by a flow of N2 (600 mL min−1) for 30 min. The air-sensitive Cu@DOPA colloid was introduced into the reactor using a syringe under a flow of N2, giving a total reactor volume of 100 mL. A Zn:Cu ratio of 1:1 (0.4 mmol each) was added in all cases. The reactor was then pressurised to 50 bar using a gas mixture of 96 vol% of H2:CO2 = 3:1 and 4 vol% Ar (used as internal standard for GC analysis), and heated to 210 °C. The experiments were performed at a flow rate of 150 mL min−1 for 20 h. The stability study with Al:ZnO@DOPA was performed at a flow rate of 150 mL min−1 for 40 h.
The commercial CuO–ZnO–Al2O3 catalyst was obtained from Alfa Aesar (45776, pellet size 5.4 × 3.6 mm, mass composition CuO, 63.5%; ZnO, 25.1%; Al2O3, 10.1%; MgO, 1.3%). It was ground to a fine powder and tested as a reference material. The choice of catalyst loading was dictated by the normalisation considerations, making sure that the total molar concentration of Cu and Zn was the same as in the Cu/ZnO colloids. The catalyst is pre-activated using a diluted H2 stream (5 vol% H2/N2) at 4.5 bar and 240 °C (ramp 2 °C min−1) for 3 h, according to a standard activation protocol for the ternary Cu–ZnO–Al2O3 methanol synthesis catalyst in a slurry reactor.65 The catalytic runs were performed under the conditions stated above.
The reaction products and unreacted material were monitored by an online gas chromatograph (Bruker 450 GC), equipped with a thermal conductivity detector (TCD) and a flame ionisation detector (FID). TCD was used to quantify CO, CO2 and Ar, whilst FID was used to quantify methanol and other oxygenates or hydrocarbons. The lines from the reactor to the GC were heated to 180 °C to avoid the condensation of any products. All experiments were conducted under differential conditions, with an overall CO2 conversion <2%, avoiding mass transport limitations (see ESI† for details). Selectivity values are given on a carbon basis, where the only two products were methanol and CO.
Results and discussion
Synthesis and characterisation of M:ZnO@DOPA
Small (2–3 nm) colloidal ZnO nanoparticles (ZnO@DOPA) were previously prepared by the controlled hydrolysis of diethylzinc and sub-stoichiometric amounts (vs. ZnEt2) of DOPA–H, in a molar ratio of Zn:DOPA–H = 5:1.9,40 The [DOPA]− ligand stabilises the nanoparticles and results in high solubility in organic solvents.9,40,57 Here, this general hydrolysis procedure was applied to doped-ZnO nanoparticles by the reaction of ZnEt2 with the desired dopant organometallic precursors (MRx) prior to hydrolysis. Three dopants were targeted as they either showed precedent for acting as structural and/or electronic promoters (Mg(II) and Al(III))28,29,32,66,67 or are required for the highly investigated Cu–ZnO synergy (Cu(I)),24,28,29,36,37,68–75 which is proposed to be critical in methanol synthesis.
In each case, a pre-hydrolysis mixture was obtained by reacting 0.2 equiv. of DOPA–H with commercially available diethylzinc (0.95 equiv.) and 0.05 equiv. of commercially available (n-butyl)(sec-butyl)magnesium, triethylaluminium or mesitylcopper (CuMes) in toluene. These precursors were selected not only due to their availability, but also high solubility in organic solvents and rapid reactivity with water to deter phase separation between zinc and dopant. The overall metal:ligand ratio was maintained at 5:1 as these conditions yielded particles fully saturated with ligand. Upon reaction with water, the organometallic reagents produce only inert hydrocarbons as byproducts (i.e. ethane, butane and/or mesitylene, respectively) which are easily removed from the final product in vacuo. The post-hydrolysis mixture, consisting of a colloidal suspension of ZnO nanoparticles, was purified by reducing the solvent volume and via centrifugation, yielding Mg:ZnO@DOPA and Al:ZnO@DOPA as white powders and Cu:ZnO@DOPA as a dark green powder.
Analysis of the as-synthesised particles by X-ray diffraction (XRD) showed the formation of small crystalline particles (2–3 nm) with sizing determined via peak analysis using the Scherrer equation (Table 1). In each case, only the wurtzite ZnO phase was observed (Fig. 1a–d), suggesting incorporation of any dopants into the crystalline structure rather than co-crystallisation of other phases. High-angle angular dark-field scanning tunnelling electron microscopy (HAADF-STEM) images (Fig. 1e–g) revealed spherical nanoparticles with narrow size distributions (2.6–2.8 nm, Fig. S1†) and no obvious amorphous phases were observed by electron microscopy.
| M:ZnO samples |
Size (nm) | (101) reflectionc | |||
|---|---|---|---|---|---|
| XRDa | TEMb | 2θ° | d Å−1 | Δd Å−1 | |
| a Average particle size determined by application of the Scherrer equation to XRD (102) and (110) reflections (Fig. 1a–d). b Average particle size ± standard error (standard deviation) determined by size analysis of TEM images (Fig. 1e–f). The standard error of the mean is defined as standard deviation/(no. of measurements). c The positions of (101) peaks were determined using Gaussian fits to the XRD reflection in the range of 30–35° 2θ (Fig. S2). | |||||
| ZnO@DOPA | 2.3 | 2.8 ± 0.02 (0.6) | 34.62 | 2.59 | — |
| Mg:ZnO@DOPA | 2.0 | 2.7 ± 0.04 (0.5) | 34.31 | 2.61 | +0.02 |
| Al:ZnO@DOPA | 2.3 | 2.7 ± 0.06 (0.9) | 35.75 | 2.51 | −0.08 |
| Cu:ZnO@DOPA | 2.2 | 2.6 ± 0.03 (0.4) | 34.82 | 2.58 | −0.01 |
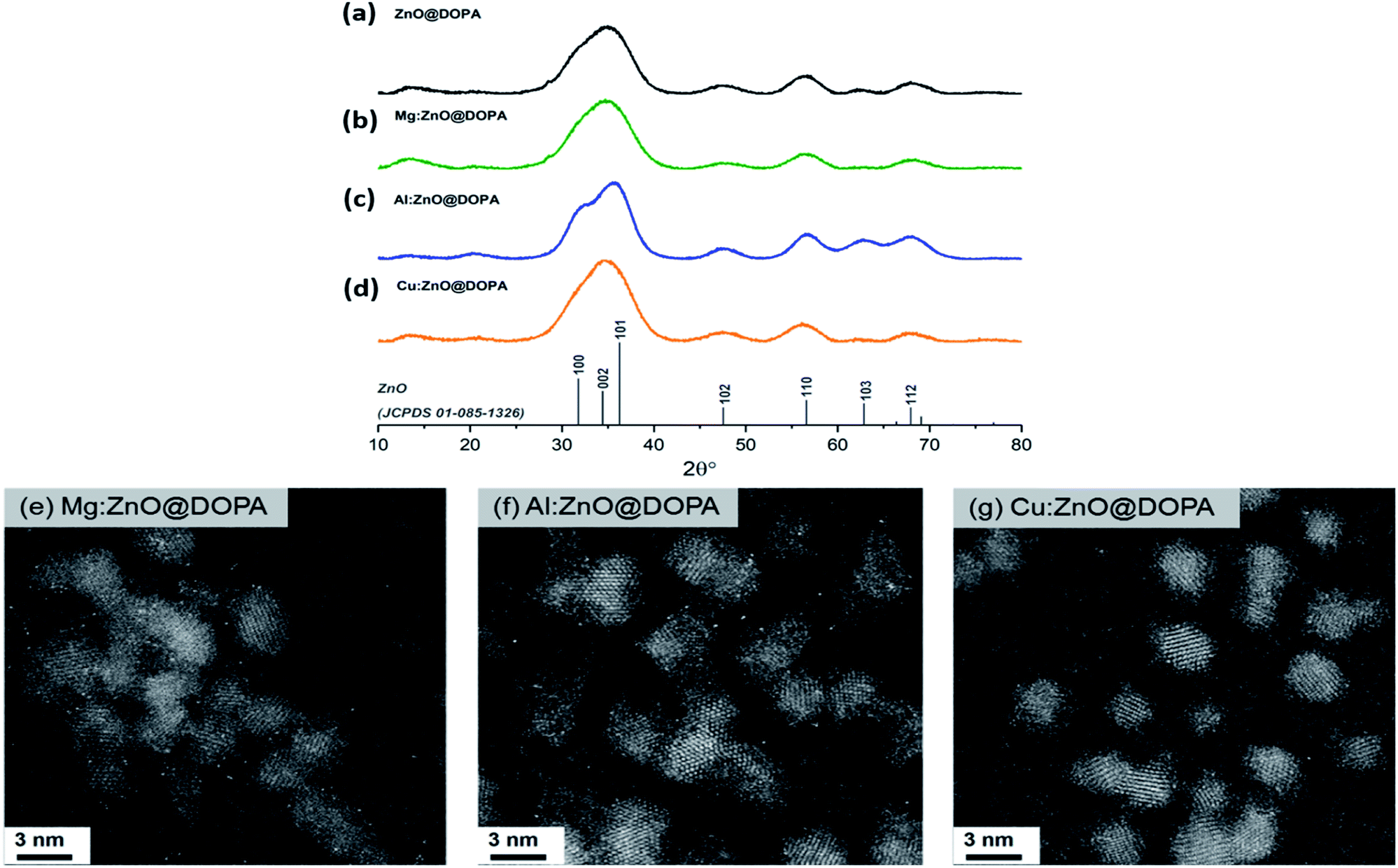 | ||
| Fig. 1 Powder XRD patterns and HAADF-STEM images of (a) ZnO@DOPA, (b) & (e) Mg:ZnO@DOPA, (c) & (f) Al:ZnO@DOPA and (d) & (g) Cu:ZnO@DOPA. Patterns are indexed against ZnO as grey vertical bars (JCPDS 01-085-1326). All scale bars in the STEM images are 3 nm. | ||
Incorporation of the second metal was assessed further, by considering changes to the lattice parameters, evidenced in the strongest (101) reflection (Table 1, Fig. S2†). A significant contraction was observed in Al:ZnO@DOPA, indicative of successful substitution of tetrahedral Zn(II) sites (rion[Zn(II)] = 0.74 Å) with Al(III) (rion[Al(III)] = 0.53 Å).76,77 No clear lattice changes were observed for Mg:ZnO@DOPA and Cu:ZnO@DOPA due to the similar ionic radii of the metals (rion[Mg(II)] = 0.71 Å and rion[Cu(I)] = 0.74 Å) and the peak breadth resulting from the small particle sizes.14,78
Bulk compositional analysis by ICP-OES confirmed and quantified the presence of the dopants. For Al:ZnO@DOPA and Cu:ZnO@DOPA, the heteroatom concentration of 5 mol% is in line with starting reagent stoichiometry, but the value for Mg:ZnO@DOPA is slightly lower than expected (Table 2). The solid-state27 Al-MAS-NMR spectrum of Al:ZnO@DOPA (Fig. S3†) confirms the replacement of tetrahedral Zn(II) with Al(III), indicated by the sharp signal at δ 55 ppm which corresponds to tetrahedral Al(III) environments. In addition, pentahedral (δAl: 47 ppm) and octahedral (δAl: −17 and 16 ppm) Al environments are also present and are tentatively assigned as surface sites.28,66,67
| M:ZnO@DOPA | Dopant quantity (rel. at%) | Optical band-gap (eV) | |
|---|---|---|---|
| ICP-OES | XPS | UV-Visc | |
| a Derived from comparison of the peak area from peak fit analysis of Zn 2p3/2 with Cu 2p3/2 and Mg 1s (Fig. 2). Error of ±0.5%. b Derived from comparison of the peak area from peak fit analysis of Zn 3s with Al 2p (Fig. 2). Error of ±0.5%. c Error of ±0.04 eV, derived from uncertainty of linear fits (coefficient of determination, R2 > 0.99) (Fig. S4).28 | |||
| ZnO@DOPA | — | — | 3.71 |
| Mg:ZnO@DOPA | 3.77 ± 0.05 | 4.5a | 3.84 |
| Al:ZnO@DOPA | 4.96 ± 0.07 | 5.0b | 3.65 |
| Cu:ZnO@DOPA | 5.05 ± 0.07 | 3.3a | 3.67 |
The chemical environments and oxidation states for all samples were analysed using XPS (Fig. 2). All samples show a binding energy (BE) for Zn 2p3/2 at 1021.7 eV, however from the Zn core level alone, it is not possible to determine its oxidation state, as the BEs of Zn metal and ZnO are identical.79 However, the Zn L3M4,5M4,5 Auger lines clearly show the typical structure of ZnO, with two features at 987.9 eV and 991.1 eV kinetic energy (KE). The P 2p and C 1s lines show BEs expected for the DOPA ligand. The O 1s core level has two contributions, one from ZnO at 530.4 eV and one at 531.5 eV from DOPA.80 The Mg 1s and Al 2p core level appear at BEs typical for Mg(II) and Al(III), 1304.2 eV and 73.9 eV, respectively. The Cu 2p3/2 core level is at a BE of 932.3 eV, which can either stem from Cu(0) or Cu(I).81 Usually, the Cu LMM Auger lines can be used to distinguish between these two oxidation states, but in these samples, the lines overlap with the much stronger Zn LMM lines, preventing further analysis. The core level also exhibits a small shoulder towards higher BE, at 933.7 eV, indicating a small amount of Cu(II). From peak fit analysis of the Zn 2p3/2 and the metal core levels, relative atomic ratios were determined (Table 2). Comparing the dopant quantities obtained by XPS and ICP-OES highlights the likely surface speciation of Mg, compared to Al which appears to be distributed throughout the particles.
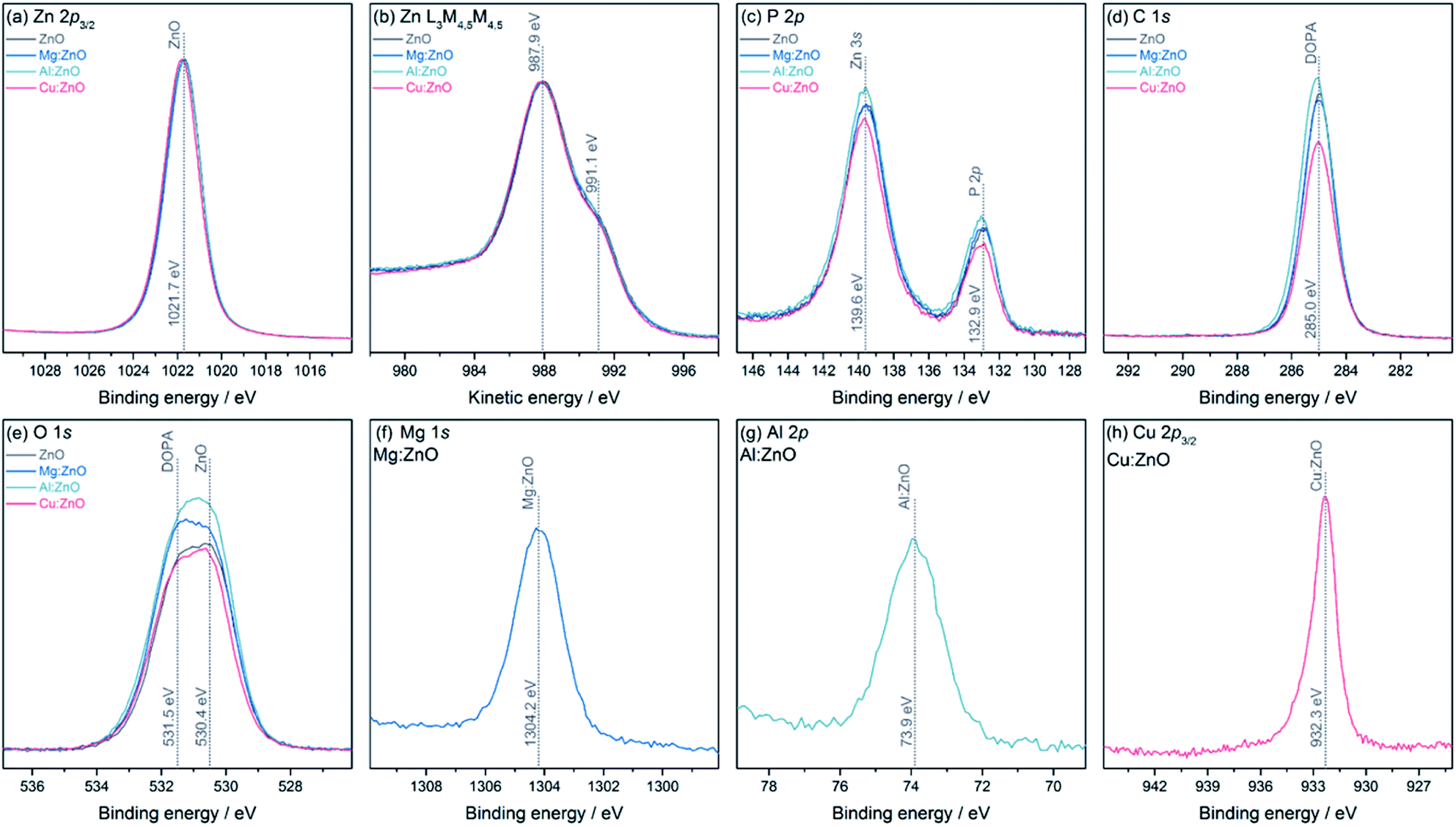 | ||
| Fig. 2 XPS core levels and Auger spectra for ZnO@DOPA and M:ZnO@DOPA samples, including (a) Zn 2p3/2, (b) Zn L3M4,5M4,5 (c) P 2p/Zn 3s, (d) C 1s, (e) O 1s, and (f) Mg 1s of Mg:ZnO@DOPA, (g) Al 2p of Al:ZnO@DOPA, and (h) Cu 2p3/2 of Cu:ZnO@DOPA. | ||
The optical band-gaps were determined via Tauc plots from the on-set absorption in solid-state UV-Vis spectra (Table 2, Fig. S4†). The Al-doped sample showed a smaller band-gap than ZnO, presumably due to the introduction of Al(III) additional donor levels.11,82–84 In contrast, Mg(II) doping widens the band-gap, as observed by others.14,28,78,85–87 A small decrease in the band-gap of Cu:ZnO@DOPA (3.67 eV) suggests partial incorporation of Cu(I)/Cu(II) into the lattice, where the dopant provides additional shallow acceptor levels,88,89 as a larger decrease would be expected for 5% doping.90 It is possible that some of the CuMes is also reduced to Cu(0), rather than doping the ZnO. It was previously reported that CuMes reacts with diethylzinc to produce Cu(0) nanoparticles.41 In this work, however, there was no direct evidence for such small Cu(0) nanoparticles by TEM or XRD.
Analysis of all the new nanoparticles by TGA showed a mass loss of 25–30% between 150–500 °C for all M:ZnO@DOPA, assigned to the thermal decomposition of the [DOPA]− ligand (Table S2, Fig. S5†). Along with elemental analysis (Table S2†), ratios close to M:ZnO : [DOPA]− = 6 : 1 were found for M:ZnO@DOPA. The ratio differs slightly to the starting reagent stoichiometry and it is proposed that low quantities of soluble molecular clusters e.g. [Zn4O(DOPA)6], are removed during nanoparticle purification and account for the lower ligand loading.57
From IR spectroscopy (Fig. S6†), the presence of C–H stretches (2800–3000 cm−1), along with phosphinate, PO2−, stretches (∼1047 cm−1, asymm; ∼1126 cm−1, symm) associated with the ligand were detected. The separation of 79–81 cm−1 between the asymmetric and symmetric phosphinate stretches indicates κ2-ligand coordination (see Fig. S6† inset). The presence of O–H stretches centred at 3380 cm−1, is indicative of hydroxyl and water coordinated to the nanoparticle surface. Overall, all characterisation data strongly suggest the successful incorporation of Mg(II) and Al(III) into the lattice of ZnO nanoparticles, whilst Cu(I) appears to have been partially incorporated.
Catalytic CO2 hydrogenation to methanol
The M:ZnO@DOPA nanoparticles were combined with colloidal Cu@DOPA nanoparticles and tested as catalysts for the hydrogenation of CO2 to methanol (Fig. 3), under differential conditions (CO2 conversion <2%). A short residence time allowed accurate calculation of reaction rates, avoiding complications due to competing side reactions such as the rWGS reaction. Cu@DOPA nanoparticles were synthesised by the hydrogenation of CuMes in the presence of DOPA–H and detailed characterisation data are consistent with previous reports (Cu : DOPA–H = 10 : 1).64,91 For each catalytic experiment, the colloidal catalysts (0.4 mmol Cu and 0.4 mmol M:Zn@DOPA) were introduced into the continuous flow stirred tank reactor with mesitylene as solvent (total of 100 mL), under a dynamic flow of nitrogen. Mesitylene was selected on the basis of its non-coordinating nature and high boiling point (165 °C), while being sufficiently volatile to allow for its complete removal under vacuum for analysis of spent catalysts. The reactor was pressurised to 50 bar with a gas feed of a CO2:H2 mixture (molar ratio of 1:3), at 210 °C, following previous protocols.39,70 Established colloidal Cu/ZnO@DOPA and the commercial heterogeneous catalyst Cu–ZnO–Al2O3 were also tested as benchmarks for this study. The activation of heterogeneous Cu–ZnO–Al2O3 was conducted following a standard procedure.65
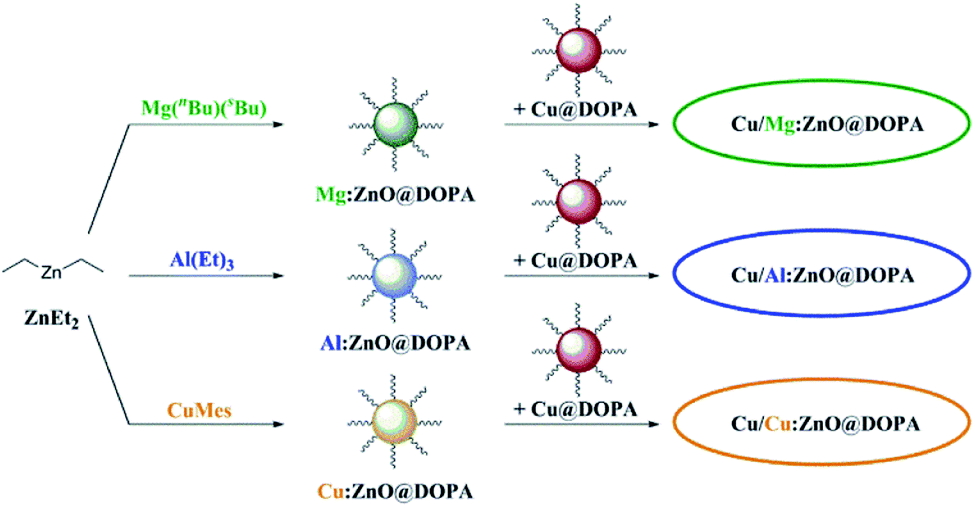 | ||
| Fig. 3 Overview of M:ZnO@DOPA syntheses and the subsequent combination with Cu@DOPA for the formation of colloidal Cu/ZnO nanocatalysts in hydrogenation of CO2 to methanol. | ||
All catalysts reached peak methanol production rate after 3–4 h time-on-stream and remained stable for over 15 h (Fig. S7 and S8†). High methanol selectivity was observed in all cases and CO was the only by-product (from the rWGS reaction). Both doped and undoped colloidal catalysts displayed higher activities than the commercial heterogeneous catalyst (Fig. 4). Mg-doping was somewhat detrimental to activity in comparison with undoped ZnO@DOPA; Cu/Mg:ZnO@DOPA displays 30–40% lower activity than the other three colloidal catalysts. Cu/Al:ZnO@DOPA showed a small but noticeable improvement when compared to Cu/ZnO@DOPA (+10%), but no clear difference was observed for the Cu-doped sample.
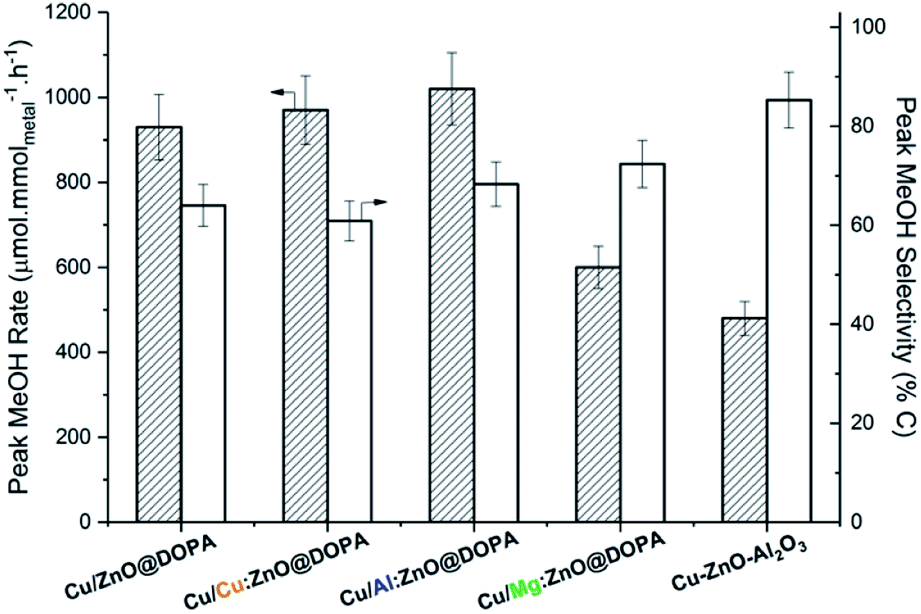 | ||
| Fig. 4 Peak methanol rates and selectivity for the colloidal Cu/ZnO@DOPA, Cu/M:ZnO@DOPA catalysts and reference catalyst Cu–ZnO–Al2O3 in the hydrogenation of CO2 to methanol. Reaction conditions: 210 °C, 50 bar, CO2 : H2 = 1 : 3, 150 mL min−1. | ||
All post-catalysis colloids were characterised using air-sensitive techniques to prevent the oxidation of Cu(0). By XRD, all samples displayed typical diffraction peaks attributed to crystalline ZnO and metallic Cu(0), with no additional crystalline phases observed (Fig. S9–S11†). Some ripening of the original M:ZnO@DOPA and Cu(0) nanoparticles was indicated by XRD and TEM (Table 3).
| Catalyst | ZnO (nm) | Cuf (nm) | ||||
|---|---|---|---|---|---|---|
| Pre-catalysis | Post-catalysis | Post-catalysis | ||||
| XRDa | TEMb | XRDa | TEMb | XRDa | TEMb | |
| a Average particle size determined by application of the Scherrer equation to XRD (102) and (110) reflections (Fig. 1a–d and S9–S11). b Average particle size ± standard error (standard deviation) determined by size analysis of TEM images (Fig. 1e–f and S14, S15). The standard error of the mean is defined as standard deviation/(no. of measurements). c The sizing measurement is taken from previously published system.40 d Measurement taken from 40 h time-on-stream study. e Crystallite size of catalyst after 20 h time-on-stream. f For pre-catalysis Cu@DOPA, sizes of ∼2 nm were reported using TEM.64 | ||||||
| Cu/ZnO@DOPAc | 2.3 | 2.8 ± 0.02 (0.6) | 3.2 | 4.9 ± 0.1 (1.1) | 6.4 | 7.2 ± 0.2 (1.9) |
| Cu/Mg:ZnO@DOPA | 2.0 | 2.7 ± 0.04 (0.5) | 2.3 | 3.5 ± 0.04 (0.4) | 4.4 | 4.3 ± 0.06 (0.6) |
| Cu/Al:ZnO@DOPAd | 2.3 | 2.7 ± 0.06 (0.9) | 3.6 (3.1)e | 4.5 ± 0.07 (1.3) | 3.0 (2.4)e | 4.6 ± 0.02 (1.1) |
| Cu/Cu:ZnO@DOPA | 2.2 | 2.6 ± 0.03 (0.4) | 3.3 | — | 2.0 | — |
The difference in activity between the highest (Al-doped) and lowest (Mg-doped) samples is tentatively attributed to the quantity of Cu/ZnO interfaces. These interfaces, formed under the reductive catalytic conditions, are commonly suggested to be the active site for methanol synthesis. They can be identified using electron microscopy, through analysis of the degree of contact between different identifiable crystalline phases.36,37,46,68,69 To this end, the nanoscale structures of post-catalysis samples of Cu/Mg:ZnO@DOPA and Cu/Al:ZnO@DOPA were investigated using BF-TEM, and were found to differ significantly.
Cu/Al:ZnO@DOPA showed contact between Cu and Al:ZnO nanoparticles (Fig. 5a & S13†). The nanocrystalline particles were additionally surrounded by an amorphous network that is rich in Al(III) (identified by EDX) and likely to be alumina as suggested by the emergence of a second, higher BE species (76.8 eV) in the Al 2p core line by XPS (Fig. S12 & S13†). The formation of surface alumina under reducing conditions has also been found in the heterogeneous Cu–ZnO–Al2O3 catalyst. In this case it appears the Al(III) that remains within the lattice acts as an electronic promoter, while the in situ formed alumina matrix acts as a structural promoter.28,29,67 It is proposed that Al-doping of ZnO increases the density of donor states (n-type) and contributes to the formation of oxygen vacancies which accelerates migration of Zn onto Cu(0) and hence increases the formation of Cu/ZnO interfaces.71–73
 | ||
| Fig. 5 BF-TEM images of the post-catalysis sample of (a) Cu/Al:ZnO@DOPA and (b) Cu/Mg:ZnO@DOPA. | ||
More isolated nanoparticles and fewer Cu/ZnO interfaces were observed for Cu/Mg:ZnO@DOPA (Fig. 5b & S14†). Additionally, the Mg-rich surface of Mg:ZnO@DOPA (as suggested by XPS) might hinder ZnO and Cu(0) interface formation. EDX of the post-catalysis sample shows that some Mg is distributed in between Cu(0) and ZnO nanoparticles (Fig. S14†). It appears Mg(II) is detrimental to activity as both a structural and electronic dopant. This is in line with a previous study of Mg(II) doping into the heterogeneous Cu–ZnO–Al2O3 methanol catalysts from syn-gas (230 °C, 30 bar, CO:CO2:H2: inert gas = 6:8:59:27).28
Stability of the colloidal catalyst
During the 20 h time-on-stream (210 °C, 50 bar, mesitylene), the colloidal catalysts all displayed good short-term stability. In contrast, the commercial heterogeneous catalyst Cu–ZnO–Al2O3 was significantly deactivated, by ∼15%, over this period. This reduction in activity may be attributed to sintering and agglomeration of Cu(0) under the operating conditions.92,93 In this sense, CO2/H2 is a considerably more demanding feed-gas than syn-gas. In order to explore colloidal catalyst stability over a longer period, the Cu/Al:ZnO@DOPA sample was tested against the heterogeneous catalyst over 40 h (210 °C, 50 bar, 150 mL min−1). Only a small reduction in activity was observed for Cu/Al:ZnO@DOPA (∼7%), whereas the Cu–ZnO–Al2O3 catalyst experienced ∼30% deactivation (Fig. 6 & S15†). With both catalysts, methanol selectivity remained high throughout (Fig. S16†). The better stability of the colloidal catalyst is tentatively attributed to the high particle stability, as post catalysis analysis revealed that agglomeration was significantly prevented. Specifically, the Cu(0) nanoparticles sizes only increase from 2.4 to 3.0 nm and the ZnO from 3.1 to 3.6 nm (Table 3 & Fig. S10†). Whilst the solvated, phosphinate ligand stabilised particles ripen more slowly than the heterogeneous catalyst, the presence of the alumina network may help to decrease the sintering further through structural promotion. CO2 conversion could likely be increased with longer residence times (lower supplied gas space velocities), but at the expense of rate and methanol selectivity.38,43 A detailed process study would be needed to optimise any one specific catalyst.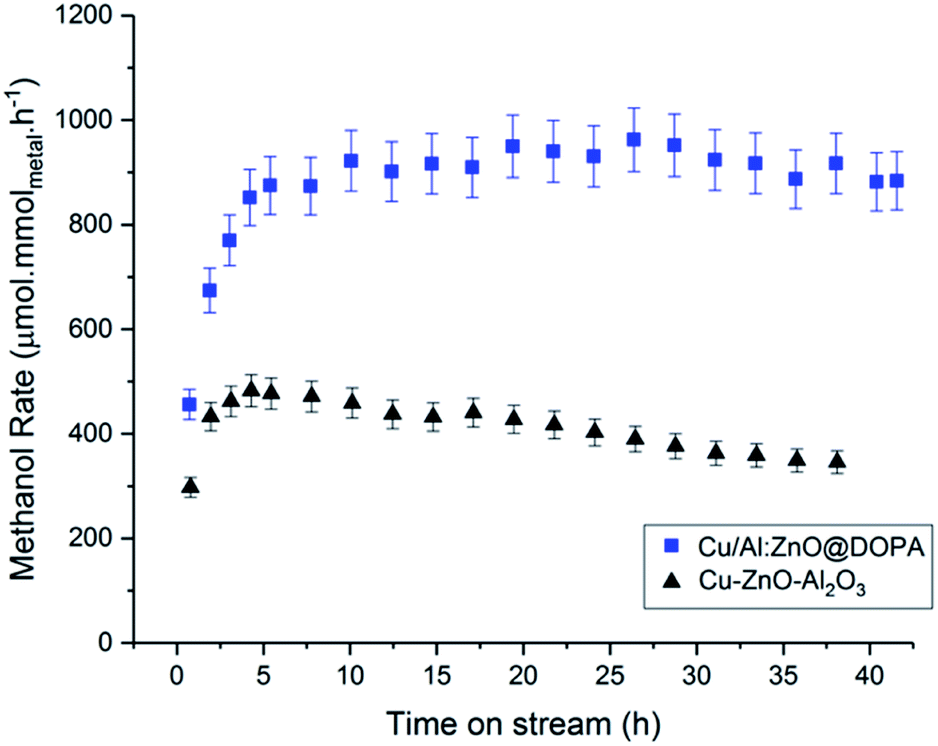 | ||
| Fig. 6 Methanol rates of Cu/Al:ZnO@DOPA and commercial heterogeneous Cu–ZnO–Al2O3, over 40 h time-on-stream. | ||
Conclusions
Doped-ZnO nanoparticles (2–3 nm), with ∼5 mol% dopant (Mg(II), Al(III) and Cu(I)), and phosphinate capping ligands, were synthesised by the room temperature hydrolysis of a mixture of organometallic reagents. The particle analysis suggests the localisation of Mg(II) on the surface of the ZnO nanoparticles, whilst Al(III) is distributed throughout the structure. For Cu-doped ZnO, incomplete incorporation of Cu(I) is suggested by the optical band-gap and XPS, probably due to partial Cu(0) formation. UV-Vis spectroscopy showed a smaller band-gap for Al-doping but a larger band-gap for Mg-doped particles compared to ZnO alone.The doped ZnO nanoparticles were combined with Cu(0) nanoparticles (<2 nm), capped by phosphinate, to form a series of active colloidal nanocatalysts for the hydrogenation of CO2 to methanol in the liquid phase (210 °C, 50 bar, H2:CO2 molar ratio of 3:1, 150 mL min−1). All catalysts showed methanol production rates higher than a benchmark heterogeneous catalyst Cu–ZnO–Al2O3, although the Mg-doped catalyst showed ∼40% lower activity than the other colloidal catalysts. It is proposed that the Mg rich surface layer prevents formation of catalytically important Cu/ZnO interfaces. In contrast, Al-doping improved activity compared to ZnO nanoparticles. An amorphous surface alumina network is indicated in these post-catalysis samples and may hinder nanoparticle ripening and improve catalyst stability over a 40 h time-on-stream.
The organometallic synthesis of doped colloidal ZnO nanoparticles is a useful means to tune properties and appears to be somewhat generalizable. Future studies should target mixed metal oxides and dopant levels. In particular, transition metal and Group 13 n-type ZnO dopants should be explored, as these materials show promising performances in syn-gas liquid phase methanol synthesis.26
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Engineering and Physical Sciences Research Council are acknowledged for research funding (EP/H046380/1, EP/K035274/1; DTA studentships to AL). AR acknowledges the support from Imperial College London for her Imperial College Research Fellowship and from the Analytical Chemistry Trust Fund for her CAMS-UK Fellowship. Johnson Matthey PLC is acknowledged for using their aberration corrected JEOL 200F microscope located within the ePSIC facility (Diamond Light Source).Notes and references
- R. Buonsanti and D. J. Milliron, Chem. Mater., 2013, 25, 1305–1317 CrossRef CAS.
- D. Mocatta, G. Cohen, J. Schattner, O. Millo, E. Rabani and U. Banin, Science, 2011, 332, 77–81 CrossRef CAS PubMed.
- Ü. Özgür, Y. I. Alivov, C. Liu, A. Teke, M. A. Reshchikov, S. Doğan, V. Avrutin, S.-J. Cho and H. Morkoç, J. Appl. Phys., 2005, 98, 041301 CrossRef.
- J. A. Garden and S. D. Pike, Dalton Trans., 2018, 47, 3638–3662 RSC.
- A. M. Schimpf, C. E. Gunthardt, J. D. Rinehart, J. M. Mayer and D. R. Gamelin, J. Am. Chem. Soc., 2013, 135, 16569–16577 CrossRef CAS.
- C. N. Valdez, M. Braten, A. Soria, D. R. Gamelin and J. M. Mayer, J. Am. Chem. Soc., 2013, 135, 8492–8495 CrossRef CAS.
- J. N. Schrauben, R. Hayoun, C. N. Valdez, M. Braten, L. Fridley and J. M. Mayer, Science, 2012, 336, 1298–1301 CrossRef CAS.
- A. García-Trenco, E. R. White, M. S. P. Shaffer and C. K. Williams, Catal. Sci. Technol., 2016, 6, 4389–4397 RSC.
- N. J. Brown, J. Weiner, K. Hellgardt, M. S. P. Shaffer and C. K. Williams, Chem. Commun., 2013, 49, 11074–11076 RSC.
- S. Sharma, S. S. Pande and P. Swaminathan, RSC Adv., 2017, 7, 39411–39419 RSC.
- R. Buonsanti, A. Llordes, S. Aloni, B. A. Helms and D. J. Milliron, Nano Lett., 2011, 11, 4706–4710 CrossRef CAS PubMed.
- Z. Sun, J. He, A. Kumbhar and J. Fang, Langmuir, 2010, 26, 4246–4250 CrossRef CAS PubMed.
- S. T. Ochsenbein and D. R. Gamelin, Nat. Nanotechnol., 2011, 6, 112–115 CrossRef CAS PubMed.
- A. W. Cohn, K. R. Kittilstved and D. R. Gamelin, J. Am. Chem. Soc., 2012, 134, 7937–7943 CrossRef CAS PubMed.
- S. Polarz, A. Orlov, A. Hoffmann, M. R. Wagner, C. Rauch, R. Kirste, W. Gehlhoff, Y. Aksu, M. Driess, M. W. E. van den Berg and M. Lehmann, Chem. Mater., 2009, 21, 3889–3897 CrossRef CAS.
- Y. Kanai, Jpn. J. Appl. Phys., 1991, 30, 703–707 CrossRef CAS.
- C. Rauch, W. Gehlhoff, M. R. Wagner, E. Malguth, G. Callsen, R. Kirste, B. Salameh, A. Hoffmann, S. Polarz, Y. Aksu and M. Driess, J. Appl. Phys., 2010, 107, 024311 CrossRef.
- S. B. Zhang, S.-H. Wei and A. Zunger, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 1–7 Search PubMed.
- K. Mohanta, S. K. Batabyal and A. J. Pal, Chem. Mater., 2007, 19, 3662–3666 CrossRef CAS.
- R. Frisenda, A. J. Molina-Mendoza, T. Mueller, A. Castellanos-Gomez and H. van der Zant, Chem. Soc. Rev., 2018, 47, 3339–3358 RSC.
- C. Baltes, S. Vukojević and F. Schüth, J. Catal., 2008, 258, 334–344 CrossRef CAS.
- A. Galadima and O. Muraza, J. Nat. Gas Sci. Eng., 2015, 25, 303–316 CrossRef CAS.
- L. C. Grabow and M. Mavrikakis, ACS Catal., 2011, 1, 365–384 CrossRef CAS.
- S. Natesakhawat, J. W. Lekse, J. P. Baltrus, P. R. Ohodnicki Jr, B. H. Howard, X. Deng and C. Matranga, ACS Catal., 2012, 2, 1667–1676 CrossRef CAS.
- M. Saito and K. Murata, Catal. Surv. Asia, 2004, 8, 285–294 CrossRef CAS.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- H. Y. Chen, J. Lin, K. L. Tan and J. Li, Appl. Surf. Sci., 1998, 126, 323–331 CrossRef CAS.
- J. Schumann, M. Eichelbaum, T. Lunkenbein, N. Thomas, M. C. Á. Galván, R. Schlögl and M. Behrens, ACS Catal., 2015, 5, 3260–3270 CrossRef CAS.
- M. Behrens, S. Zander, P. Kurr, N. Jacobsen, J. Senker, G. Koch, T. Ressler, R. W. Fischer and R. Schlögl, J. Am. Chem. Soc., 2013, 135, 6061–6068 CrossRef CAS PubMed.
- G. A. Vedage, P. B. Himelfarb, G. W. Simmons and K. Klier, in Solid State Chemistry in Catalysis, American Chemical Society, 1985, ch. 18, vol. 279, pp. 295–312 Search PubMed.
- C. Li, X. Yuan and K. Fujimoto, Appl. Catal., A, 2014, 469, 306–311 CrossRef CAS.
- J. G. Nunan, P. B. Himelfarb, R. G. Herman, K. Klier, C. E. Bogdan and G. W. Simmons, Inorg. Chem., 1989, 28, 3868–3874 CrossRef CAS.
- M. M.-J. Li, Z. Zeng, F. Liao, X. Hong and S. C. E. Tsang, J. Catal., 2016, 343, 157–167 CrossRef CAS.
- P. L. Hansen, J. B. Wagner, S. Helveg, J. R. Rostrup-Nielsen, B. S. Clausen and H. Topsøe, Science, 2002, 295, 2053–2055 CrossRef CAS PubMed.
- M. B. Fichtl, J. Schumann, I. Kasatkin, N. Jacobsen, M. Behrens, R. Schlögl, M. Muhler and O. Hinrichsen, Angew. Chem., Int. Ed., 2014, 53, 7043–7047 CrossRef CAS PubMed.
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B.-L. Kniep, M. Tovar, R. W. Fischer, J. K. Nørskov and R. Schlögl, Science, 2012, 336, 893–897 CrossRef CAS PubMed.
- J. C. Frost, Nature, 1988, 334, 577–580 CrossRef CAS.
- M. Sahibzada, I. S. Metcalfe and D. Chadwick, J. Catal., 1998, 174, 111–118 CrossRef CAS.
- S. Lee and A. Sardesai, Top. Catal., 2005, 32, 197–207 CrossRef CAS.
- S. D. Pike, A. García-Trenco, E. R. White, A. H. M. Leung, J. Weiner, M. S. P. Shaffer and C. K. Williams, Catal. Sci. Technol., 2017, 7, 3842–3850 RSC.
- N. J. Brown, A. García-Trenco, J. Weiner, E. R. White, M. Allinson, Y. Chen, P. P. Wells, E. K. Gibson, K. Hellgardt, M. S. P. Shaffer and C. K. Williams, ACS Catal., 2015, 5, 2895–2902 CrossRef CAS.
- A. García-Trenco, E. R. White, A. Regoutz, D. J. Payne, M. S. P. Shaffer and C. K. Williams, ACS Catal., 2017, 7, 1186–1196 CrossRef.
- A. García-Trenco, A. Regoutz, E. R. White, D. J. Payne, M. S. P. Shaffer and C. K. Williams, Appl. Catal., B, 2018, 220, 9–18 CrossRef.
- M. K. Schröter, L. Khodeir, M. W. E. van den Berg, T. Hikov, M. Cokoja, S. Miao, W. Grünert, M. Muhler and R. A. Fischer, Chem. Commun., 2006, 2498–2500 RSC.
- S. Schimpf, A. Rittermeier, X. Zhang, Z.-A. Li, M. Spasova, M. W. E. van den Berg, M. Farle, Y. Wang, R. A. Fischer and M. Muhler, ChemCatChem, 2010, 2, 214–222 CrossRef CAS.
- M. A. Sliem, S. Turner, D. Heeskens, S. B. Kalidindi, G. V. Tendeloo, M. Muhler and R. A. Fischer, Phys. Chem. Chem. Phys., 2012, 14, 8170–8178 RSC.
- C. Kiener, M. Kurtz, H. Wilmer, C. Hoffmann, H.-W. Schmidt, J.-D. Grunwaldt, M. Muhler and F. Schüth, J. Catal., 2003, 216, 110–119 CrossRef CAS.
- M. Monge, M. L. Kahn, A. Maisonnat and B. Chaudret, Angew. Chem., Int. Ed., 2003, 42, 5321–5324 CrossRef CAS PubMed.
- D. Prochowicz, K. Sokołowski and J. Lewiński, Coord. Chem. Rev., 2014, 270–271, 112–126 CrossRef CAS.
- K. L. Orchard, M. S. P. Shaffer and C. K. Williams, Chem. Mater., 2012, 24, 2443–2448 CrossRef CAS.
- D. Lee, M. Wolska-Pietkiewicz, S. Badoni, A. Grala, J. Lewiński and G. De Paëpe, Angew. Chem., Int. Ed., 2019, 131, 17323–17328 CrossRef.
- J. Paczesny, M. Wolska-Pietkiewicz, I. Binkiewicz, M. Wadowska, Z. Wróbel, K. Matuła, W. Nogala, J. Lewiński and R. Hołyst, ACS Appl. Mater. Interfaces, 2016, 8, 13532–13541 CrossRef CAS.
- J. Paczesny, M. Wolska-Pietkiewicz, I. Binkiewicz, Z. Wróbel, M. Wadowska, K. Matuła, I. Dzięcielewski, D. Pociecha, J. Smalc-Koziorowska, J. Lewiński and R. Hołyst, Chem.–Eur. J., 2015, 21, 16941–16947 CrossRef CAS.
- M. Wolska-Pietkiewicz, A. Grala, I. Justyniak, D. Hryciuk, M. Jędrzejewska, J. Grzonka, K. J. Kurzydłowski and J. Lewiński, Chem.–Eur. J., 2017, 23, 11856–11865 CrossRef CAS PubMed.
- M. Wolska-Pietkiewicz, K. Tokarska, A. Grala, A. Wojewódzka, E. Chwojnowska, J. Grzonka, P. J. Cywiński, K. Kruczała, Z. Sojka, M. Chudy and J. Lewiński, Chem.–Eur. J., 2018, 24, 4033–4042 CrossRef CAS PubMed.
- M. L. Kahn, M. Monge, V. Collière, F. Senocq, A. Maisonnat and B. Chaudret, Adv. Funct. Mater., 2005, 15, 458–468 CrossRef CAS.
- S. D. Pike, E. R. White, M. S. P. Shaffer and C. K. Williams, Nat. Commun., 2016, 7, 1–11 Search PubMed.
- T. Chen and V. O. Rodionov, ACS Catal., 2016, 6, 4025–4033 CrossRef CAS.
- D. L. Hou, X. J. Ye, H. J. Meng, H. J. Zhou, X. L. Li, C. M. Zhen and G. D. Tang, Appl. Phys. Lett., 2007, 90, 142502 CrossRef.
- Y. Kanai, Jpn. J. Appl. Phys., 1991, 30, 2021–2022 CrossRef CAS.
- C. H. Park, S. B. Zhang and S.-H. Wei, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 073202 CrossRef.
- A. Glaria, M. L. Kahn, T. Cardinal, F. Senocq, V. Jubera and B. Chaudret, New J. Chem., 2007, 32, 662–669 RSC.
- S. K. Sehmi, S. Noimark, S. D. Pike, J. C. Bear, W. J. Peveler, C. K. Williams, M. S. P. Shaffer, E. Allan, I. P. Parkin and A. J. MacRobert, ACS Omega, 2016, 1, 334–343 CrossRef CAS PubMed.
- S. D. Pike, E. R. White, A. Regoutz, N. Sammy, D. J. Payne, C. K. Williams and M. S. P. Shaffer, ACS Nano, 2017, 11, 2714–2723 CrossRef CAS PubMed.
- A. Sawant, M. K. Ko, V. Parameswaran, S. Lee and C. J. Kulik, Fuel Sci. Technol. Int., 1987, 5, 77–88 CrossRef CAS.
- S. Miao, R. N. d'Alnoncourt, T. Reinecke, I. Kasatkin, M. Behrens, R. Schlögl and M. Muhler, Eur. J. Inorg. Chem., 2009, 910–921 CrossRef CAS.
- M. Behrens, G. Lolli, M. Muratova, I. Kasatkin, M. Hävecker, R. N. d'Alnoncourt, O. Storcheva, K. Köhler, M. Muhler and R. Schlögl, Phys. Chem. Chem. Phys., 2013, 15, 1374–1381 RSC.
- F. Liao, Y. Huang, J. Ge, W. Zheng, K. Tedsree, P. Collier, X. Hong and S. C. E. Tsang, Angew. Chem., Int. Ed., 2011, 50, 2162–2165 CrossRef CAS PubMed.
- F. Liao, Z. Zeng, C. Eley, Q. Lu, X. Hong and S. C. E. Tsang, Angew. Chem., Int. Ed., 2012, 51, 5832–5836 CrossRef CAS PubMed.
- X.-M. Liu, G. Q. Lu, Z.-F. Yan and J. Beltramini, Ind. Eng. Chem. Res., 2003, 42, 6518–6530 CrossRef CAS.
- A. le Valant, C. Comminges, C. Tisseraud, C. Canaff, L. Pinard and Y. Pouilloux, J. Catal., 2015, 2015, 41–49 CrossRef.
- C. Tisseraud, C. Comminges, T. Belin, H. Ahouari, A. Soualah, Y. Pouilloux and A. le Valant, J. Catal., 2015, 330, 533–544 CrossRef CAS.
- C. Tisseraud, C. Comminges, S. Pronier, Y. Pouilloux and A. le Valant, J. Catal., 2016, 343, 106–114 CrossRef CAS.
- Y. Choi, K. Futagami, T. Fujitani and J. Nakamura, Appl. Catal., A, 2001, 208, 163–167 CrossRef CAS.
- J. Nakamura, Y. Choi and T. Fujitani, Top. Catal., 2003, 22, 277–285 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- S. Mehra, A. Bergerud, D. J. Milliron, E. M. Chan and A. Salleo, Chem. Mater., 2016, 28, 3454–3461 CrossRef CAS.
- S. Heitz, Y. Aksu, C. Merschjann and M. Driess, Chem.–Eur. J., 2011, 17, 3904–3910 CrossRef CAS PubMed.
- G. Schön, J. Electron Spectrosc. Relat. Phenom., 1973, 2, 75–86 CrossRef.
- R. Al-Gaashani, S. Radiman, A. R. Daud, N. Tabet and Y. Al-Douri, Ceram. Int., 2013, 39, 2283–2292 CrossRef CAS.
- T. Futagami, Y. Aoki, O. Yoda and S. Nagai, Nucl. Instrum. Methods Phys. Res., Sect. B, 1994, 88, 261–266 CrossRef CAS.
- K. J. Chen, T. H. Fang, F. Y. Hung, L. W. Ji, S. J. Chang, S. J. Young and Y. J. Hsiao, Appl. Surf. Sci., 2008, 254, 5791–5795 CrossRef CAS.
- B. E. Sernelius, K.-F. Berggren, Z.-C. Jin, I. Hamberg and C. G. Granqvist, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 10244–10248 CrossRef CAS PubMed.
- T. Ratana, P. Amornpitoksuk, T. Ratana and S. Suwanboon, J. Alloys Compd., 2009, 470, 408–412 CrossRef CAS.
- V. Etacheri, R. Roshan and V. Kumar, ACS Appl. Mater. Interfaces, 2012, 4, 2717–2725 CrossRef CAS.
- S. Heitz, Y. Aksu, C. Merschjann and M. Driess, Chem. Mater., 2010, 22, 1376–1385 CrossRef CAS.
- A. S. H. Hameed, C. Karthikeyan, S. Sasikumar, V. S. Kumar, S. Kumaresan and G. Ravi, J. Mater. Chem. B, 2013, 1, 5950–5962 RSC.
- X. Peng, J. Xu, H. Zang, B. Wang and Z. Wang, J. Lumin., 2008, 128, 297–300 CrossRef CAS.
- M. Suja, S. B. Bashar, M. M. Morshed and J. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 8894–8899 CrossRef CAS PubMed.
- C. Boukaous, B. Benhaoua, A. Telia and S. Ghanem, Mater. Res. Express, 2017, 4, 105024 CrossRef.
- C. Barrière, G. Alcaraz, O. Margeat, P. Fau, J. B. Quoirin, C. Anceau and B. Chaudret, J. Mater. Chem., 2008, 18, 3084–3086 RSC.
- M. V. Twigg and M. S. Spencer, Top. Catal., 2003, 22, 191–203 CrossRef CAS.
- H. H. Kung, Catal. Today, 1992, 11, 443–453 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Tables outlining amount of dopant precursor used and mass analysis by EA and TGA. Additional figures showing nanocatalyst characterisation, methanol production rates and selectivity as well as post-catalysts characterisation. Rationale for catalytic reaction conditions used. See DOI: 10.1039/d0ta00509f |
| This journal is © The Royal Society of Chemistry 2020 |