
The impact of cation structure upon the acidity of triazolium salts in dimethyl sulfoxide†
Nicholas
Konstandaras
 ,
Michelle H.
Dunn
,
Max S.
Guerry
,
Christopher D.
Barnett
,
Marcus L.
Cole
and
Jason B.
Harper
*
,
Michelle H.
Dunn
,
Max S.
Guerry
,
Christopher D.
Barnett
,
Marcus L.
Cole
and
Jason B.
Harper
*
School of Chemistry, University of New South Wales, Sydney, NSW 2052, Australia. E-mail: j.harper@unsw.edu.au; Fax: +612 9385 6141; Tel: +612 9385 4692
First published on 14th November 2019
Abstract
A series of triazolium salts, selected for their varying electronic and steric properties, were prepared and their pKa values were determined in DMSO at 25 °C using the bracketing indicator method. The effect of each systematic structural variation upon the acidity of the triazolium cation has been considered, in particular examining the effects of systematically altering electronic properties, quantified through the use of Hammett σ parameters. The first pKa value for an azolium salt that generates a mesionic carbene is also reported. These new data allow for the selection of appropriate bases for the deprotonation of such triazolium salts and the potential to correlate the pKa values determined herein with the nucleophilicity of the corresponding carbenes.
Introduction
N-Heterocyclic Carbenes (NHCs) have attracted significant interest as organocatalysts,1–7 being able to catalyse a wide range of reactions including the widely described benzoin condensations8–10 and Stetter reactions.11–13 Triazolylidenes are of particular interest as the introduction of chiral centres on such ring substituents introduces the potential for these species to act as enantioselective catalysts.14,15 The various properties of NHCs have been investigated over the years;16,17 particularly notable are the studies towards understanding the catalytic ability of these systems performed by Berkessel et al.,18–22 Rovis et al.,23–25 O'Donoghue et al.26,27 and others.28–31 In terms of the mechanism of reaction, both Rovis and O'Donoghue have shown qualitatively that reaction rates increased with the electron withdrawing nature of the substituents on the triazolylidene catalyst used.When used as organocatalysts, NHCs are most commonly generated in situ by deprotonation of the parent azolium salt. As a result, the pKa values of the corresponding azolium salts are of interest as they allow both for an understanding of the extent of deprotonation of the azolium salt with any given base but also an indication of the nucleophilicity of the carbene (through the Brønsted relationship32).31 The latter feature is key, given the ubiquity of carbenes acting as nucleophiles in organocatalysis. The acidities of a number of azolium salts have been reported in the gas phase,31,33,34 in THF,35 in aqueous solutions36–41 and in DMSO.38,42–46 In terms of triazolium salts, O'Donoghue et al.39,40 have determined the pKa values of a large number triazolium cations in aqueous solutions using deuterium exchange methodologies, particularly examining the effects of structural variations and varying the electronic nature of substituents. Cheng et al.44 have also considered the pKa values of triazolium salts; these studies were carried out in DMSO using the bracketing indicator method and focussed particularly on the effect of changing the electronic nature of the triazolium through variation of the electronic character of an N-aryl substituent. In both of these cases, there was significant interest in triazolium salts incorporating a fused bicyclic structure, as these are prevalent in the literature, particularly as precursors for chiral organocatalysts.14,29,47–49
This study focuses on extending the available data, examining the effects of common structural variations of triazolium salts on their acidity in DMSO. A series of triaryl substituted species are used to examine electronic effects on pKa values and a 1,2,3-triazolium salt is examined in the first example of considering the acidity of a species leading to an ‘abnormal’ carbene.
Results and discussion
The pKa values for the triazolium salts presented herein were determined using the bracketing indicator method43,50via titration with the deprotonated form of an appropriate fluorene indicator (Fig. 1) in DMSO at 25 °C in a manner analogous to that reported previously by our group.46 Each titration followed the decrease in the absorbance due to the fluorenide anion in the UV-Vis region upon addition of each triazolium salt.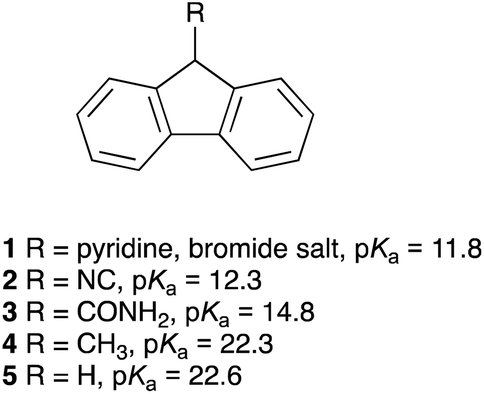 | ||
| Fig. 1 The fluorene indicators 1–5 used in this study and their corresponding pKa values in DMSO.51–54 | ||
In the determination of the equilibrium constant in each case, the absorbance of both the triazolium salt, and the NHC were also considered, though the absorbance due to the fluorenide dominated in all cases.
It is important that the pKa values of the indicators used are within two of the pKa value of the triazolium salt as beyond this range changes in the equilibrium constant are too difficult to determine reliably.43 Each pKa value reported herein results from at least four titrations with at least two fluorene indicators with known pKa values, where each equilibrium constant was determined using all available data points for the titration. The uncertainties reported herein are based on replicate experiments; these uncertainties are typically greater than the uncertainties in the regression analysis.
It is worth noting that there is the potential for ion pairing even in a dissociating solvent such as DMSO.55,56 However, it has been demonstrated that, at the concentrations used in this work, pKa values in DMSO are not altered by ion pairing effects of the anion.43,44,46,56 All but one of the salts examined herein possess tetrafluoroborate anions for the sake of consistency.
Utilising the methodology outlined above, the pKa values of a range of triazolium salts were determined in order to examine the effects of altering the structure of triazolium salts upon their acidity. Initially, the salts 6–10 were considered (Fig. 2); these salts allow evaluation of several key features that affect the acidities of these species. These features include the nature of the substituent at the 3-position, the potential for linking the 3- and 4-substituents, along with the electronic nature of the aryl group at the 1-position.
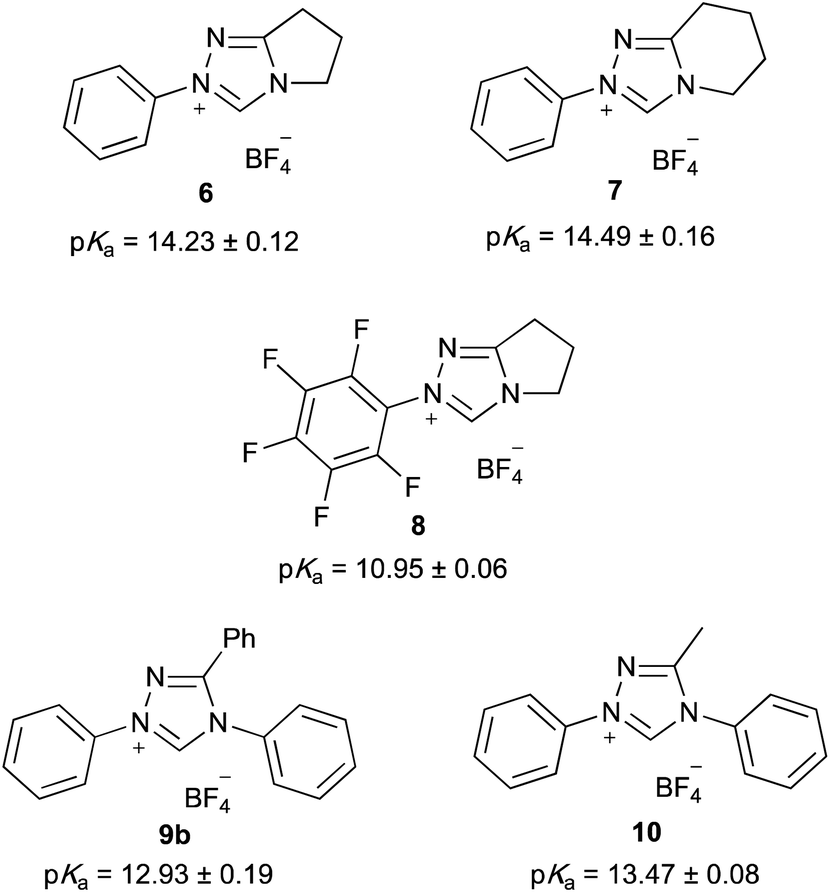 | ||
| Fig. 2 The triazolium salts 6–10 and their pKa values determined in DMSO at 25 °C using the bracketing indicator method. Uncertainties are reported as the standard deviation multiplied by the Student t-value for at least four measurements. | ||
The pentafluoro derivative 8 is commonly used in literature as the precursor to a carbene catalyst26 and is hence readily available while the triphenyl derivative 9b‡ has been widely reported.57,58 The systems 6 and 7 containing a bicyclic triazolium cation were prepared from the appropriate lactam in three steps and overall moderate yield (Scheme 1). The 3-methyltriazolium salt 10 was prepared through the thione intermediate 11 as outlined in Scheme 2, once again in moderate overall yield. The pKa values for the five triazolium salts 6–10 were determined and are summarised in Fig. 2.
 | ||
| Scheme 1 Synthesis of the 1,2,4-triazolium salts 6 and 7; (a) (CH3)3O(BF4), DCM, 18 h, r.t.; (b) PhNHNH2, DCM, 2 days, r.t.; (c) CH(OEt)3, MeOH, 18 h, 100 °C. | ||
 | ||
| Scheme 2 Synthesis of the 1,2,4-triazolium salt 10; (a) CH3COOH, 1.5 h, 80 °C. (b) KOH, EtOH, PhNCS, 45 min, r.t. (c) CH3COOH, HBF4, H2O2, 1 h, r.t. | ||
Initially, it is worthwhile noting that the pKa values obtained in this work were broadly similar to those obtained by Cheng et al.44 for the related systems 12 and 13 (Fig. 3). Further, the pKa values of the triazolium salts 6–10 in DMSO reported herein (ca. 11.0–14.4) are significantly lower than the pKa values determined in aqueous media; that is, the triazolium salts are more acidic in DMSO. Reports by O'Donoghue et al. using the kinetic deuteration method indicate that the pKa values of triazolium salts 6, 8 and 9b in water are 17.5, 16.5 and 16.8, respectively.39 This effect has been observed previously for triazolium salts44 and also has been noted in studies of the pKa values of imidazolium43 and thiazolium59 salts. These changes have been attributed to the fact that, while the neutral carbene will be relatively insensitive to solvent stabilisation, the azolium cation will likely be stabilised by hydrogen bonding in water to a greater extent than in DMSO.43,60,61 This stabilisation of the protonated form will result in a larger pKa value (a less acidic species).
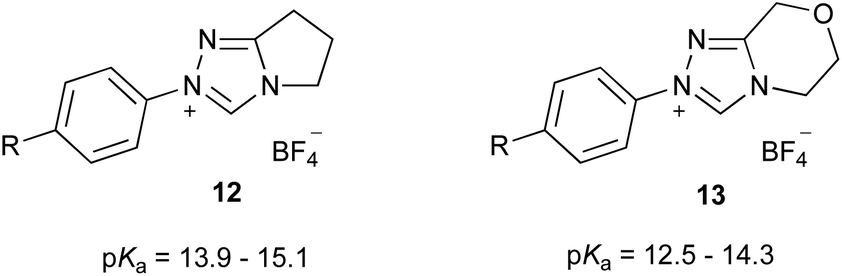 | ||
| Fig. 3 The triazolium salt series 12 and 13 considered previously and their pKa values in DMSO at 25 °C.44 R = OMe, Me, H, F, Cl, Br, CN. | ||
Similarly it is of interest to note that the pKa values of the triazolium salts reported in Fig. 2 are significantly lower (that is, the triazolium salts 6–10 are more acidic) than the pKa values of a similar series of imidazolium salts reported previously in DMSO under identical conditions (ca. 18.0–23.8).46 The significant difference in the pKa values of triazolium and imidazolium salts is likely due to the presence of the additional electronegative nitrogen atom in the triazolium ring, which will destabilise the azolium salt relative to the carbene, thereby increasing its acidity.
By examining the triazolium salts 6–10, it can be seen that other structural changes can also have significant and varied effects upon their pKa values. For example, the pKa values of salts 6 and 7 are the same within uncertainty. This similarity indicates that moving from a five- to a six-membered ring has no significant impact upon the electronic properties of the triazolium core. Such similarity is reasonable as the addition or removal of a single methylene group would be expected to minimally alter the electronic nature of the cycloalkyl ring.§
In contrast, the pKa value of triazolium salt 8 is markedly lower (by ca. 3) than that of triazolium salt 6. This difference can be attributed to the electron withdrawing nature of fluorine atoms on the phenyl ring,62,63 which destabilise the triazolium cation relative to the NHC, causing the triazolium salt 8 to be markedly more acidic than the salt 6. Such a decrease in pKa value with the addition of increasingly electron withdrawing substituents is consistent with previous observations of triazolium salts39 and imidazolium salts.43,46 The effects of such phenyl ring substitution shall be discussed further below.
It is also of interest to compare the pKa values of triazolium salts 6, 7, 9b and 10 in order to examine the effects that the addition of phenyl rings about the triazolium core have upon their acidity. As a starting point, consider salts 6 and 7, where the triazolium cores are substituted with one phenyl ring and the equivalent of two alkyl substituents. Upon comparison with salt 10, which has two phenyl rings and one alkyl substituent, it can be seen that substituting the equivalent of one alkyl substituent with a phenyl ring in this case causes the pKa value to decrease by ca. 0.8–1.0. This change can be rationalised by noting that phenyl rings are notably more electron withdrawing than alkyl substituents (σp(Ph) = 0.05, σp(Me) = −0.1462,63![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ¶); as discussed above, decreasing the electron density about the triazolium ring will destabilise the cation relative to the carbene, thereby increasing its acidity.
¶); as discussed above, decreasing the electron density about the triazolium ring will destabilise the cation relative to the carbene, thereby increasing its acidity.
Similarly, upon comparing the diphenyl substituted salt 10 with the triphenyl substituted triazolium salt 9b, it can be seen that replacing the methyl group with a phenyl ring causes a further decrease in pKa of ca. 0.55. This change is consistent with the increased electron withdrawing character of the phenyl ring destabilising the cation relative to the carbene, thereby decreasing the pKa value of salt 9b compared to salt 10.
Systematic electronic variation in triazolium salts
The variation in the pKa values of triazolium salts 6–10 demonstrates that the acidity of triazolium salts are susceptible to structural variations about the triazolium ring, such as the addition of phenyl and cycloalkyl rings commonly seen among triazolium salts. Though such changes in pKa values are generally consistent with the trend of greater electron donation to the triazolium core leading to stabilisation of the azolium cation and therefore a less acidic salt, it is of interest to examine the effects in a more quantitative fashion. To this end the triazolium salts 9a–e were synthesised (using modifications of the literature preparation of the parent salt 9b) from the corresponding anilines through the appropriate benzamides 14a–e and intermediates 15a–e (Scheme 3) in fair to moderate yields, noting that no effort was made to optimise these yields and that the intermediates in the process are susceptible to hydrolysis. The pKa values of the salts 9a–e (Table 1) were determined as described above in order to examine the effects of systematically altering electron donation and withdrawal to the triazolium core through substitution at the para-aryl position.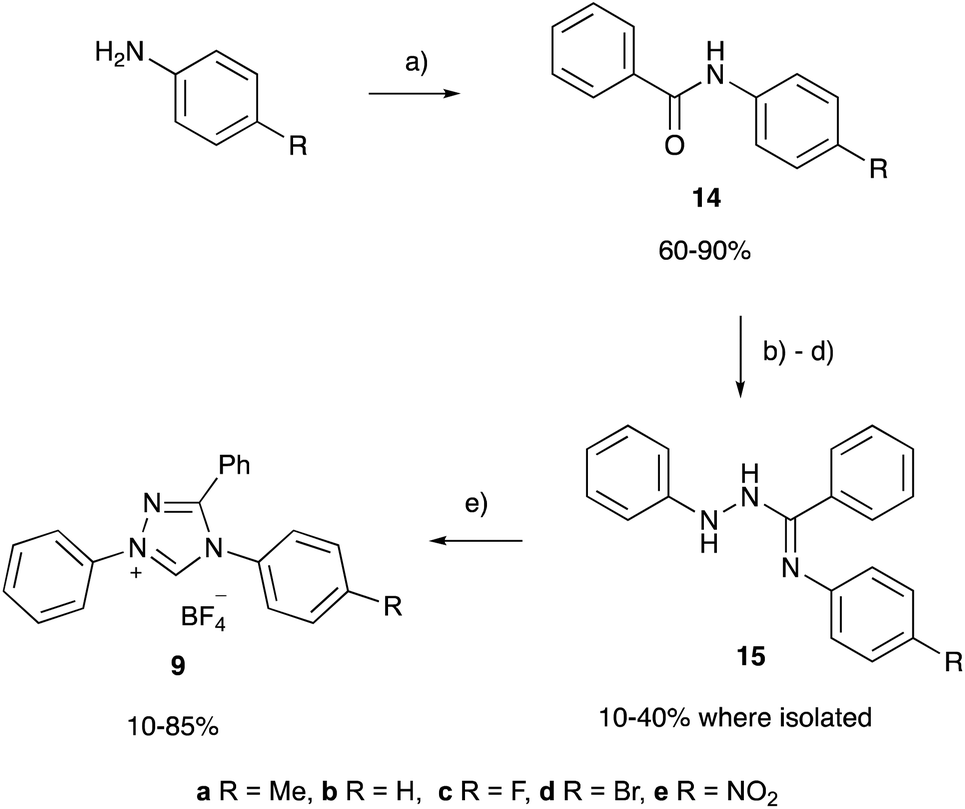 | ||
| Scheme 3 Synthesis of the 1,2,4-triazolium salts 9a–e; (a) PhCOCl, DCM, 20 h, r.t.; (b) SOCl2, 16 h, reflux; (c) PhNHNH2, NEt3, 48 h, r.t.; (d) AcOH, 15 min, r.t.; (e) NH4BF4, CH(OEt)3, 16–48 h, reflux. | ||
The pKa values shown in Table 1 demonstrate the general trend that triazolium salts become more acidic as the electron withdrawing nature of the para-aryl substituents (quantified by the Hammett σ value) increases.63 This observation is consistent with similar observations made for imidazolium salts37,46 along with other triazolium salts,39,44 and is consistent with greater destabilisation of the triazolium cation, relative to the carbene, with increased electron withdrawal from substituents (as discussed above). Such effects can be quantified by considering a plot of the pKa values of triazolium salts 9a–evs. the Hammett σ value of the para-aryl substituent (Fig. 4), which shows a clear linear trend. Though such a plot is not a Hammett plot, as no normalisation to the value of the unsubstituted case has been performed, it is worth noting that the slope of such a plot corresponds to the negative of the Hammett susceptibility constant (ρ), and therefore allows comparison with literature. As a result, the slope shown in Fig. 4 (−1.73 ± 0.10) is a quantitative measure of the magnitude of the impact of altering the electronic properties of the para-aryl substituent upon the pKa values of triazolium salts 9a–e. The magnitude of the slope of the correlation shown in Fig. 4 demonstrates that acidity of triazolium salts 9a–e is more greatly affected by substitution than the acidity of substituted benzoic acids (ρ = 1).64
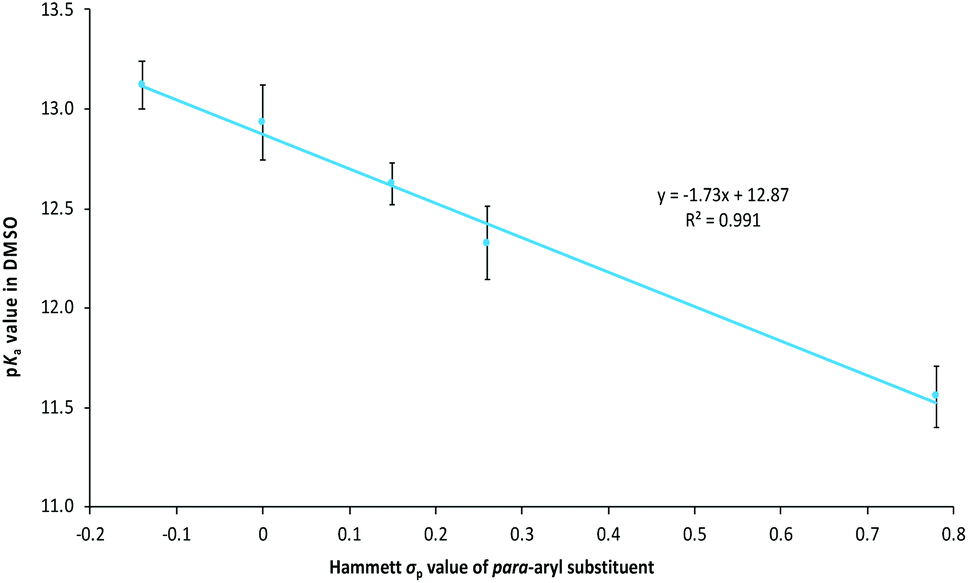 | ||
| Fig. 4 The correlation between the pKa values of the triazolium salts 9a–e in DMSO at 25 °C and the Hammett σp value of the 4′-aryl substituent.63 | ||
It is useful to compare these data to similar series reported in the literature. The equivalent slopes determined by Cheng et al. for the plots of the pKa values of the triazolium salt series 12 and 13 in DMSO (Fig. 3) vs. the Hammett σ value of the para-aryl were determined to be −2.18 and −2.00,|| respectively.44 These slopes are very similar to, if slightly larger in magnitude than, that reported for the triazolium salts 9a–e presented here (−1.73 ± 0.10). This minor change might be due to a slightly lesser degree of charge localisation upon the nitrogen atom of triazolium salts 9a–e compared to in the series 12 and 13, due to delocalisation of the charge onto the additional phenyl rings.
Whilst Hammett analysis was not carried out, the data presented by O'Donoghue et al.39 for the series of triazolium salts 12 (the same series studied by Cheng et al.44) and 16 (a closely related series to that presented by Cheng et al.44) allows determination of the effect of the electronic nature of the N-aryl substituent on pKa values in water (Fig. 5). Analysis (see ESI† for details) shows that the slopes of the equivalent plots in these cases are (−0.93 ± 0.05) and (−0.19 ± 0.17) for series 12 and 16, respectively. It should be noted that there are fewer variants for each series in water than in either the case presented herein or by Cheng et al.;44 that, along with the smaller range of acidities measured in the salt series 16, is the probable cause of the larger uncertainties in the slope in that case. Irrespective, these data in comparison with that presented here and by Cheng,44 shows that the electronic effects of N-aryl substituents on pKa values of triazolium salts are smaller in water than in DMSO, likely because of the stabilising effect of water (as discussed above). It is also worth noting that the significant decrease in electronic susceptibility seen in the series 16 is not replicated in DMSO for the related series 13 (cf. series 12); this suggests that a key hydrogen bond donating effect of water, stabilisation by the additional phenyl ring in series 16, or a combination of both potential effects minimises any electronic impacts.**
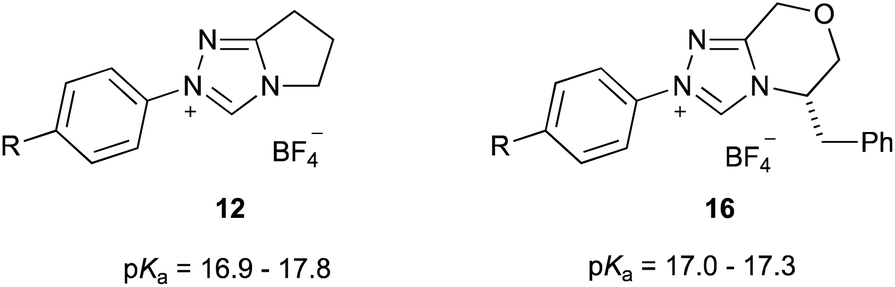 | ||
| Fig. 5 The triazolium salt series 12 and 16 that been considered previously to examine electronic effects on pKa values of azolium salts in water at 25 °C.39 R = OMe, H, F, CN. | ||
It is also of interest to compare these data with the equivalent slopes for plots of pKa values vs. the Hammett σ value of the para-aryl substituents for other families of azolium salts. To our knowledge, at present, the only such plots considered involve the two series of imidazolium salts 17 (slope −1.25 ± 0.09) and 18 (slope −1.23 ± 0.10) (Fig. 6††) considered previously by our group using equivalent analyses.46 Comparison of that data with the equivalent for protonated anilines suggested delocalisation of the charge equally across the two nitrogen centres.46 The larger magnitude of the slope in the cases of series 9, 12 and 13 reported here in DMSO indicates that changing the substituent on the aryl group has a greater effect than in the imidazolium case. The origin of this effect is not immediately clear, though it is important to note that it is consistent for both 1-aryl (12 and 13) and 4-aryl (9) series. One explanation might be that the substituents on both nitrogen centres change in the case of series 17 and 18, whilst for the triazolium series 9, 12 and 13 only one nitrogen substituent is varied; the result is that the extent of charge localisation in the triazolium systems may vary with the electronic nature of the nitrogen substituent and result in a greater effect of the substituted aryl group on the acidity of these species cf. imidazolium salts.
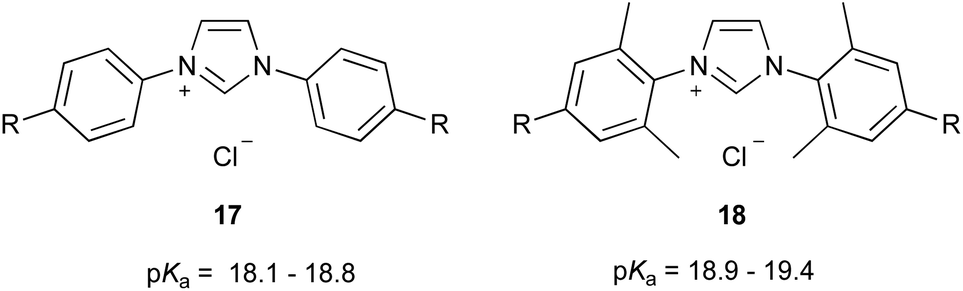 | ||
| Fig. 6 The imidazolium salt series 17 and 18 that have been considered previously to examine electronic effects on pKa values of azolium salts in DMSO.46 R = OMe, Me, H, F, Cl, Br. | ||
Precursors to abnormal carbenes
“Abnormal carbenes” is the term given to mesoionic NHCs, which are NHCs that do not possess a resonance structure without charge separation;65 these species are of interest as they are stronger σ electron donors than normal NHCs.65 In order to examine such species, the 1,2,3-triazolium salt 19 was synthesised, through copper-catalysed azide alkyne cycloaddition of substituted mesityl precursors,66 followed by methylation.45,67 This salt 19‡‡ has a pKa value of 22.26 ± 0.09 and deprotonates to give the mesoionic 1,2,3-triazol-5-ylidene 20 (Scheme 4), one of the most common types of abnormal carbene.66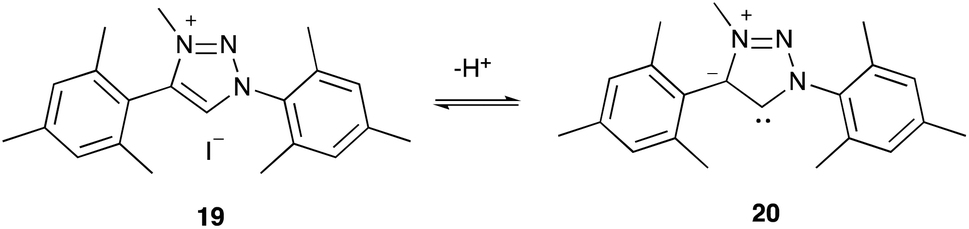 | ||
| Scheme 4 The deprotonation of the 1,2,3-triazolium salt 19 to give the abnormal carbene 20. | ||
It is worth comparing this pKa value with that of triazolium salt 10. The pKa value of salt 19 is significantly higher (the cation is less acidic) by 8.79 ± 0.12. It must first be noted that this increase in the pKa value is in part due to the addition of methyl groups upon the phenyl rings, which would stabilise the cation through inductive electron donation. Similarly, the increased steric hindrance between the mesityl groups and the N-methyl substituent would prevent the mesityl and triazolium rings from lying in the same plane, thereby reducing the impact of the electron donating phenyl substituents.46 A crude estimate of these effects can be made by comparing the pKa values of the imidazolium salts 21 and 22 previously reported46 (Fig. 7) suggesting that the addition of such methyl groups would account for an increase in pKa value of ca. 4.5.§§ This estimate suggests that the high pKa value of salt 19 cannot be solely due to the effects of these methyl groups alone.
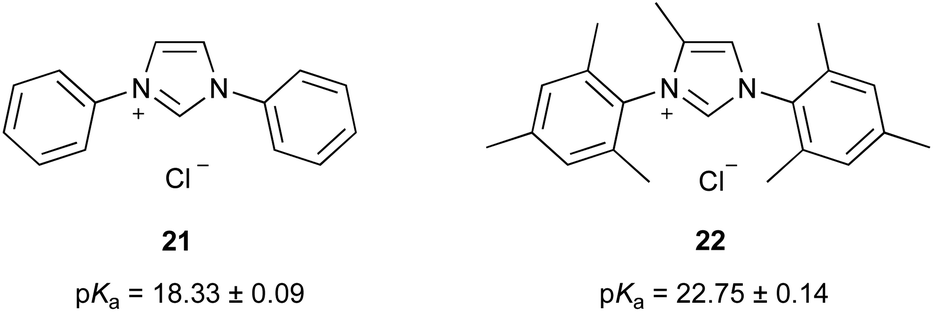 | ||
| Fig. 7 The pKa values of imidazolium salts 21 and 22 in DMSO at 25 °C reported previously.46 | ||
Further contributing to the large difference in the pKa values of triazolium salts 10 and 19 are the differences in azolium ring. For example, salt 19 bears a methyl group directly upon the partially positively charged nitrogen atom of the triazolium cation, which would lead to a greater stabilisation of the cation than the presence of the aryl ring,63 and therefore increase the pKa value. However, likely more significant is the fact that the charge separation in the carbene 20 decreases its stability (relative to non-charge separated cases).
With the above in mind, it is useful to compare the pKa value of the triazolium salt 19 with that of the imidazolium salt 22. It might initially be expected that the charge separated form of the abnormal carbene 20 would be sufficiently unstable, compared to a standard neutral carbene, to cause the salt 19 to be markedly less acidic than salt 22. The fact that salt 19 is instead more acidic (by approximately 0.5 pKa) than salt 22 instead suggests that the abnormal carbene 20 may not be as significantly different in energy from the normal carbene as implied by the charge separation. Regardless, the pKa value of salt 19 is the highest currently reported pKa value for any triazolium salt in DMSO, demonstrating how altering the structure may be useful in producing triazolium salts to generate carbenes with markedly increased basicity and therefore nucleophilicity.
Conclusions
This work has demonstrated how the pKa values of triazolium salts in DMSO are affected by commonly seen structural moieties, noting a general decrease in pKa with the addition of phenyl rings and no significant change in pKa between five- and six-membered cycloalkyl rings. Altering the electronics of the phenyl rings has been shown to significantly alter pKa values with the effects quantified through a Hammett analysis of triphenyl triazolium salts. This work also reports the first pKa value of a salt that forms an abnormal NHC upon depronotation, demonstrating how such structures may be of use to produce carbenes with markedly increased nucleophilicities, noting that higher basicity may not need to a greater nucleophilicity for systems with adjacent sterically inflexible units.68 Such information allows for the selection of appropriate bases to allow for the full deprotonation of triazolium salts when used as precatalysts along with allowing for the prediction of the pKa values of triazolium salts and correlation with the nucleophilicity of the corresponding carbenes.Experimental
The fluorenes 1–4 have been previously reported and were prepared according to modified literature procedures51,69–75 from commercially available starting materials (see ESI† for full details). Fluorene 5 (>99%) was commercially available and was used as received.The fluorinated imidazolium salt 8 was commercially available and was dried in vacuo prior to use. The bicyclic triazolium salt 6 was prepared from N-methylpyrrolidinone76 and the thione 11 from hydrazine77,78 (see ESI† for full details). The abnormal carbene precursor 19 was prepared in a modification of literature procedures from mesityl azide and mesityl acetylene,66 followed by methylation,45,67 with full experimental given in the ESI.†
6,7,8,9-Tetrahydro-2-phenyl-5H-1,2,4-triazolo[4,3-a]azepinium tetrafluoroborate 7
In a method based on that reported by Alexakis,76 to a solution of 2-piperidone (1.07 g, 10.8 mmol) in dry dichloromethane (50 mL), trimethyloxonium tetrafluoroborate (1.75 g, 11.8 mmol) was added in one portion. The resulting colourless mixture was stirred at room temperature for 18 hours. To this solution phenylhydrazine (1.15 mL, 1.26 g, 11.7 mmol) was added which resulted in a bright orange solution that was stirred at room temperature overnight. The mixture was concentrated under reduced pressure resulting in a yellow oil. To this diethyl ether (30 mL) was added, the solid that was formed was triturated with ethyl acetate and then collected using filtration. This solid was dried under reduced pressure and then dried under reduced pressure to give the hydrazone intermediate an offwhite solid. Dry toluene (15 mL) and triethyl orthoformate (4.80 mL, 4.28 g, 28.9 mmol) were added under argon and the mixture was heated at reflux for 48 hours. The mixture was then cooled to room temperature and concentrated under reduced pressure to give a dark orange oil. To this diethyl ether (30 mL) was added, the solid that formed was triturated with ethyl acetate and then collected using filtration. The resulting off-white solid was recrystallized from ethanol to give the title compound 7 as off-white crystalline solid (1.52 g, 5.30 mmol, 49%). M.p. 155–159 °C. 1H NMR (500 MHz, dimethyl sulfoxide-d6) δ 10.73 (s, 1H, C5–H), 7.93–7.84 (m, 2H, Ar–H), 7.74–7.59 (m, 3H, Ar–H), 4.31 (t, J = 5.9 Hz, NCH2), 3.09 (t, J = 5.9 Hz, CCH2), 2.09–1.92 (m, 4H, CH2CH2). 13C NMR (125 MHz, dimethyl sulfoxide-d6) δ 153.3 (C3–CH2), 140.7 (C5), 135.0 (C1′), 130.4 (C4′), 130.3 (C2′/3′), 120.4 (C2′/3′), 45.4 (NCH2), 20.7 (CCH2), 20.6 (NCH2CH2), 18.3 (CCH2CH2). IR (Solid): vmax 3140 (w), 2961 (w), 1582 (m), 1497 (w), 1425 (w), 1329 (w), 1024 (s), 760 (s) cm−1. Found HR-MS (ESI) m/z: 200.1181 (M+, 100%). C12H14N3+ requires m/z: 200.1182 (M+, 100%).3-Methyl-1,4-diphenyl-4H-1,2,4-triazolium tetrafluoroborate 10
To a solution of the thione 11 (1.32 g, 4.94 mmol) in acetic acid (4 mL) and tetrafluoroboric acid (48% w/w in water, 4.0 mL, 1.2 g, 14 mmol), aqueous hydrogen peroxide (30% w/w in water, 4.0 mL, 1.2 g, 35 mmol) was added dropwise over 30 minutes. During this time the solution turned yellow and a white precipitate formed. This mixture was stirred for an hour, before being filtered and a white solid collected. Recrystrallisation from methanol gave the triazolium salt 10 as a white solid (1.13 g, 3.51 mmol, 71%). M.p. 183–186 °C. 1H NMR (600 MHz, dimethyl sulfoxide-d6) δ 11.13 (s, 1H, C5–H), 8.01–7.97 (m, 2H, Ar–H), 7.86–7.79 (m, 2H, Ar–H), 7.78–7.70 (m, 5H, Ar–H), 7.70–7.62 (m, 1H, Ar–H), 2.60 (s, 3H, CH3). 13C NMR (125 MHz, dimethyl sulfoxide-d6) δ 153.3 (C3), 142.1 (C5), 134.8 (Ar–C), 131.5 (Ar–C), 131.5 (Ar–C), 131.4 (Ar–C), 130.6 (Ar–C), 130.3 (Ar–C), 126.0 (Ar–C), 120.4 (Ar–C), 10.5 (CH3). 19F NMR (282 MHz, dimethyl sulfoxide-d6): δ −148.32 (s, 4F, BF4), −148.27 (s, 1F, BF4). IR (solid): vmax 3108 (w), 1582 (m), 1486 (w), 1223 (m), 1018 (s), 764 (m), 678 (m) cm−1. Found HR-MS (ESI) m/z: 236.1180 (M+, 100%). C15H14N3+ requires m/z: 236.1182 (M+, 100%).General procedure for the synthesis of the triazolium salts 9a–e
The benzamides 14a–e were prepared according to literature procedures79 (see ESI† for full details). The appropriate benzamide (1 equiv.) was then dissolved in boiling thionyl chloride (ca. 1 mL mmol−1 benzamide) and stirred at reflux overnight. The thionyl chloride was removed through distillation at reduced pressure initially affording an oil that crystallised on standing; the corresponding imidoyl chloride, which was used promptly to minimise hydrolysis. This solid and dry triethylamine (1.5 equiv.) were dissolved in dry THF (ca. 2 mL mmol−1 benzamide), the solution was cooled to 0 °C and phenylhydrazine (1 equiv.) was added dropwise over 10 minutes with stirring. A drying tube was affixed, the ice bath was removed and the solution was stirred at room temperature for 48 hours; the volatiles were then removed under reduced pressure affording a viscous brown oil. This oil was triturated with 2% aqueous acetic acid solution (2 mL mmol−1 benzamide) and the mixture was scratched until a beige powder formed. Where practical, the residue was collected through filtration and purified using flash column chromatography affording intermediate 15, which was used immediately. The appropriate intermediate 15 (1 equiv.) was dissolved in boiling triethyl orthoformate (ca. 5 mL mmol−1) and ammonium tetrafluoroborate (1 equiv.) was added all at once. The solution was stirred at reflux for 16–48 hours then treated as specified below.:hexane, 1:7). Intermediate 15a was isolated as a shiny brown powder (0.670 g, 28%). M.p. 106–107 °C. 1H NMR (300 MHz, benzene-d6) δ 7.74 (m, 2H, Ar–H), 7.47 (s, 1H, N–H), 7.02–7.23 (m, 7H, Ar–H), 6.82 (m, 3H, Ar–H), 6.39 (d, 2H, J = 8.4 Hz, Ar–H), 4.63 (s, 1H, N–H), 2.03 (s, 3H, CH3). N′′-(4–Methylphenyl)-N′-phenylbenzimidohydrazide 15a (0.643 g, 2.13 mmol), triethyl orthoformate (10 mL), ammonium tetrafluoroborate (0.233 g, 2.22 mmol), 48 hours. A dark solution was afforded, allowed to cool to room temperature and the solvent was removed under reduced pressure. The black solid was purified using flash column chromatography (ethyl acetate:hexane, 1:1, to remove impurities followed by methanol:dichloromethane, 1:9). The resultant beige powder was washed with ether (2 × 10 mL) and water (2 × 10 mL) and triturated in boiling ethyl acetate, which was cooled to room temperature and filtered to afford the title compound 9a as a white powder (0.089 g, 10%). M.p. 147–148 °C. 1H NMR (600 MHz, dimethyl sulfoxide-d6) δ 11.29 (s, 1H, C5–H), 8.09 (d, 2H, J = 8.1 Hz, Ar–H), 7.78 (t, 2H, J = 7.7 Hz (Ar–H), 7.70 (t, 1H, J = 7.4 Hz, Ar–H), 7.66–7.61 (m, 1H, Ar–H), 7.58–7.52 (m, 6H, Ar–H), 7.48 (d, 2H, J = 8.1 Hz, Ar–H), 2.42 (s, 3H, CH3). 13C NMR (125 MHz, dimethyl sulfoxide-d6) δ 153.2 (C3), 143.2 (C5), 141.5, 134.8, 132.3, 130.8, 130.5, 130.4, 129.5, 129.2, 126.2, 122.4, 120.6, 20.8 (CH3). 19F NMR (285 MHz, dimethyl sulfoxide-d6) δ −148.25 (0.8F, BF4−), −148.31 (3.2F, BF4−). IR (solid): vmax 3068, 1737, 1558, 1514, 1485, 1462, 1344, 1230, 1027, 824, 758, 683 cm−1. Found HR-MS (ESI) m/z: 312.1497 (M+, 100%). C21H18N3+ requires m/z: 312.1495 (M+, 100%).
:hexane, 1:7). Intermediate 15c was not isolated, material was carried through as it was. Triethyl orthoformate (20 mL), ammonium tetrafluoroborate (1.40 g, 13.4 mmol), 16 hours. A dark solution was afforded and allowed to cool to room temperature. The beige precipitate was collected through filtration and washed with ether (2 × 20 mL) and water (2 × 20 mL) affording the title compound 9c as a beige powder (1.06 g, 20%). M.p. 207–208 °C. 1H NMR (600 MHz, dimethyl sulfoxide-d6) δ 11.36 (s, 1H, C5–H), 8.12–8.08 (m, 2H, Ar–H), 7.83–7.76 (m, 4H, Ar–H), 7.71 (t, 1H, J = 7.5 Hz, Ar–H), 7.68–7.62 (m, 1H, Ar–H), 7.60–7.53 (m, 6H, Ar–H). 13C NMR (125 MHz, dimethyl sulfoxide-d6) δ 163.2 (d, 1JCF = 250.0 Hz, C–F), 153.4 (C3), 143.3 (C5), 134.8, 132.4, 130.9, 130.5, 129.32, 129.25 (d, 3JCF = 9.5 Hz, C2′), 128.3 (d, 4JCF = 3.0 Hz, C1′), 122.2, 120.6, 117.3 (d, 2JCF = 23.6 Hz, C3′). 19F NMR (285 MHz, dimethyl sulfoxide-d6) δ −108.41 (1.29, Ar–F), −148.27 (0.8F, BF4−), −148.36 (3.2F, BF4−). IR (solid): vmax 3114, 1598, 1560, 1509, 1340, 1289, 1215, 1022, 849, 762, 737, 685 cm−1. Found HR-MS (ESI) m/z: 316.1243 (M+, 100%). C20H15FN3+ requires m/z: 316.1245 (M+, 100%).
:hexane, 1:6). Intermediate 15d was isolated as a pale yellow powder (1.30 g, 28%). M.p. 157–158 °C (melting with decomposition). 1H NMR (300 MHz, benzene-d6) δ 7.64 (m, 2H, Ar–H), 7.35 (br, 1H, N–H), 7.09–7.23 (m, 5H, Ar–H), 7.03 (m, 4H, Ar–H), 6.84 (t, 1H, J = 7.3 Hz, Ar–H), 6.06 (d, 2H, J = 8.8 Hz, Ar–H), 4.42 (br, 1H, N–H). N′′-(4-Bromophenyl)-N′-phenylbenzimidohydrazide 15d (1.18, 3.22 mmol). Triethyl orthoformate (15 mL), ammonium tetrafluoroborate (0.343 g, 3.28 mmol), 16 hours. The resultant mixture was filtered to give a beige residue which was washed with ether (3 × 15 mL), water (3 × 15 mL) and ethyl acetate (3 × 15 mL) to afford the title compound 9d as a white powder (1.18 g, 79%). M.p. 287–289 °C. 1H NMR (600 MHz, dimsthyl sulfoxide-d6) δ 11.35 (s, 1H, C5–H), 8.08 (d, 2H, J = 8.1 Hz, Ar–H), 7.93 (d, 2H, J = 8.7 Hz, Ar–H), 7.79 (t, 2H, J = 8.1 Hz, Ar–H), 7.61–7.74 (m, 4H, Ar–H), 7.57 (m, 4H, Ar–H). 13C NMR (125 MHz, dimethyl sulfoxide-d6) δ 153.2 (C3), 143.2 (C5), 134.7, 133.3, 132.4, 131.3, 131.0, 130.5, 129.3, 128.6, 124.9, 122.1, 120.6. 19F NMR (285 MHz, dimethyl sulfoxide-d6) δ −148.28 (0.8F, BF4−), −148.34 (3.2F, BF4−). IR (solid): vmax 3064, 1560, 1483, 1338, 1234, 1015, 839, 764, 682 cm−1. Found HR-MS (ESI) m/z: 376.0445, 378.0418 (M+, 100%). C20H1579/81BrN3+ requires m/z: 376.0444, 378.0423 (M+, 100%).
:hexane, 1:6). Intermediate 15e was isolated as a bright orange powder (0.280 g, 9%). M.p. 151–152 °C (melting with decomposition). 1H NMR (300 MHz, dimethyl sulfoxide-d6) δ 10.82 (br s, 1H, NH), 10.20 (s, 1H, NH), 8.29 (d, 2H, J = 9.2 Hz, Ar–H), 8.04–8.71 (m, 3H, Ar–H), 7.99 (d, 2H, J = 6.8 Hz, Ar–H), 7.54–7.74 (m, 6H, Ar–H), 7.18 (m, 1H, Ar–H). N′′-(4-Nitrophenyl)-N′-phenylbenzimidohydrazide 15e (0.280 g, 0.842 mmol), triethyl orthoformate (20 mL), ammonium tetrafluoroborate (0.088 g, 0.84 mmol), 20 hours. The mixture was filtered to give a beige residue which was washed with ether (3 × 10 mL), water (3 × 10 mL) and ethyl acetate (3 × 10 mL) to afford the title compound 9e as a beige powder (0.283 g, 78%). M.p. 285–287 °C. 1H NMR (600 MHz, dimethyl sulfoxide-d6) δ 11.46 (s, 1H, C5–H), 8.55 (d, 2H, J = 9.0 Hz, Ar–H), 8.09 (d, 2H, J = 7.5 Hz, Ar–H), 7.94 (d, 2H, J = 9.0 Hz, Ar–H), 7.80 (t, 2H, J = 7.5 Hz, Ar–H), 7.73 (t, 1H, J = 7.5 Hz, Ar–H), 7.67 (m, 1H, Ar–H), 7.56 (m, 4H, Ar–H). 13C NMR (125 MHz, dimethyl sulfoxide-d6) δ 153.2 (C3), 148.9 (C5), 143.3, 136.7, 134.7, 132.6, 131.1, 130.5, 129.46, 129.44, 128.2, 125.6, 121.9, 120.6. 19F NMR (285 MHz, dimethyl sulfoxide-d6) δ −148.25 (0.8F, BF4−), −148.31 (3.2F, BF4−). IR (solid): vmax 3091, 1561, 1522, 1485, 1344, 1236, 1010, 854, 765, 751, 722, 682 cm−1. Found HR-MS (ESI) m/z: 343.1191 (M+, 100%). C20H15N4O2+ requires m/z: 343.1190 (M+, 100%).
General methodology for pKa titrations
Each titration was carried out in a manner analogous to that which we have reported previously.46 For each titration, a solution of the appropriate indicator (ca. 1 × 10−4–6 × 10−4 M) and the triazolium salt in question (ca. 0.05–0.2 M) was prepared in dry DMSO (distilled over calcium hydride under reduced pressure and stored over activated 3 Å molecular sieves, <50 ppm water by Karl Fischer methodology). The spectrophotometric cell was sealed and dried under reduced pressure and was subsequently charged with dry nitrogen, followed by addition of the desired indicator solution (3 mL). The first UV spectrum of the solution was recorded to determine the baseline absorbance. Aliquots of sodium methylsulfinylmethylide (DIMSYL, ca. 2.5 μL each) in DMSO (prepared through the addition of sodium hydride to DMSO, 60 °C, 40 min) were injected into the cell and the UV spectrum was recorded for each solution composition until no further increase in absorbance was observed upon addition of DIMSYL at the wavelength monitored for each indicator (see ESI†). Aliquots of the desired triazolium salt in DMSO (ca. 2.5–10 μL each) were then injected into the UV cell and the UV spectrum was recorded after each injection, noting the absorbance at the wavelength of interest until either no further decrease in absorbance was noted with further salt addition, or until 300 μL had been added. Scans were taken in duplicate after each injection in order to ensure the system had reached equilibrium before further additions were made. With an appropriate indicator (one with a pKa value within 2 of that of the triazolium salt), an equilibrium was established resulting in the decrease of the absorbance value, indicating the reprotonation of the chosen indicator. The equilibrium constant for each titration was determined by fitting using Datafit 9 (Oakdale Engineering). For details of the fitting process, see our previous publication.46Conflicts of interest
There are no conflicts to declare.Acknowledgements
NK, MHD, MSG and CDB all acknowledge the support of the Australian government through the receipt of Australian Postgraduate Awards. The authors would also like to acknowledge the NMR facility within the Mark Wainwright Analytical Centre at the University of New South Wales for NMR support, along with Dr T. R. Unky, Jr., who is thanked for useful discussion.References
- T. Dröge and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 6940–6952 CrossRef.
- X. Bugaut and F. Glorius, Chem. Soc. Rev., 2012, 41, 3511–3522 RSC.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS PubMed.
- M. H. Wang and K. A. Scheidt, Angew. Chem., Int. Ed., 2016, 55, 14912–14922 CrossRef CAS.
- Q. Ren, M. Li, L. Yuan and J. Wang, Org. Biomol. Chem., 2017, 15, 4731–4749 RSC.
- S. Mondal, S. R. Yetra, S. Mukherjee and A. T. Biju, Acc. Chem. Res., 2019, 52, 425–436 CrossRef CAS PubMed.
- R. Breslow, J. Am. Chem. Soc., 1958, 80, 3719–3726 CrossRef CAS.
- Y. Hachisu, J. W. Bode and K. Suzuki, J. Am. Chem. Soc., 2003, 125, 8432–8433 CrossRef CAS PubMed.
- G. Q. Li, L. X. Dai and S. L. You, Chem. Commun., 2007, 852–854 RSC.
- H. Stetter, Angew. Chem., Int. Ed. Engl., 1976, 15, 639–647 CrossRef.
- D. Enders, K. Breuer, J. Runsink and J. H. Teles, Helv. Chim. Acta, 1996, 79, 1899–1902 CrossRef CAS.
- A. E. Mattson, A. R. Bharadwaj, A. M. Zuhl and K. A. Scheidt, J. Org. Chem., 2006, 71, 5715–5724 CrossRef CAS.
- M. S. Kerr, J. R. de Alaniz and T. Rovis, J. Org. Chem., 2005, 70, 5725–5728 CrossRef CAS.
- Y. Ma, S. Wei, J. Lan, J. Wang, R. Xie and J. You, J. Org. Chem., 2008, 73, 8256–8264 CrossRef CAS.
- J. P. Moerdyk and C. W. Bielawski, in Contemporary Carbene Chemistry, ed. R. A. Moss and M. P. Doyle, John Wiley & Sons, Inc., 2014, pp. 40–74 Search PubMed.
- H. V. Huynh, Chem. Rev., 2018, 118, 9457–9492 CrossRef CAS.
- A. Berkessel, S. Elfert, V. R. Yatham, J.-M. Neudörfl, N. E. Schlörer and J. H. Teles, Angew. Chem., Int. Ed., 2012, 51, 12370–12374 CrossRef CAS.
- A. Berkessel, V. R. Yatham, S. Elfert and J. M. Neudörfl, Angew. Chem., Int. Ed., 2013, 52, 11158–11162 CrossRef CAS.
- M. Paul, M. Breugst, J. M. Neudörfl, R. B. Sunoj and A. Berkessel, J. Am. Chem. Soc., 2016, 138, 5044–5051 CrossRef CAS.
- M. Paul, P. Sudkaow, A. Wessels, N. E. Schlörer, J. M. Neudörfl and A. Berkessel, Angew. Chem., Int. Ed., 2018, 57, 8310–8315 CrossRef CAS.
- M. Paul, J. M. Neudörfl and A. Berkessel, Angew. Chem., Int. Ed., 2019, 58, 10596–10600 CrossRef CAS.
- J. L. Moore, A. P. Silvestri, J. R. de Alaniz, D. A. DiRocco and T. Rovis, Org. Lett., 2011, 13, 1742–1745 CrossRef CAS.
- S. C. Cullen and T. Rovis, Org. Lett., 2008, 10, 3141–3144 CrossRef CAS.
- Y. Niu, N. Wang, A. Muñoz, J. Xu, H. Zeng, T. Rovis and J. K. Lee, J. Am. Chem. Soc., 2017, 139, 14917–14930 CrossRef CAS PubMed.
- C. J. Collett, R. S. Massey, J. E. Taylor, O. R. Maguire, A. C. O'Donoghue and A. D. Smith, Angew. Chem., Int. Ed., 2015, 54, 6887–6892 CrossRef CAS PubMed.
- C. J. Collett, R. S. Massey, O. R. Maguire, A. S. Batsanov, A. C. O'Donoghue and A. D. Smith, Chem. Sci., 2013, 4, 1514–1522 RSC.
- O. Hollóczki, Z. Kelemen and L. Nyulászi, J. Org. Chem., 2012, 77, 6014–6022 CrossRef.
- J. Mahatthananchai and J. W. Bode, in Contemporary Carbene Chemistry, ed. R. A. Moss and M. P. Doyle, John Wiley & Sons, Inc., 2014, pp. 237–273 Search PubMed.
- M. J. White and F. J. Leeper, J. Org. Chem., 2001, 66, 5124–5131 CrossRef CAS.
- N. Wang, J. Xu and J. K. Lee, Org. Biomol. Chem., 2018, 16, 6852–6866 CAS.
- J. N. Brønsted and K. J. Pedersen, Z. Phys. Chem., 1924, 108, 185–235 Search PubMed.
- A. M. Magill, K. J. Cavell and B. F. Yates, J. Am. Chem. Soc., 2004, 126, 8717–8724 CrossRef CAS.
- M. Paul, E. Detmar, M. Schlangen, M. Breugst, J. M. Neudörfl, H. Schwarz, A. Berkessel and M. Schäfer, Chem. – Eur. J., 2019, 25, 2511–2518 CrossRef CAS.
- Y.-J. Kim and A. Streitwieser, J. Am. Chem. Soc., 2002, 124, 5757–5761 CrossRef CAS.
- T. L. Amyes, S. T. Diver, J. P. Richard, F. M. Rivas and K. Toth, J. Am. Chem. Soc., 2004, 126, 4366–4374 CrossRef CAS.
- E. M. Higgins, J. A. Sherwood, A. G. Lindsay, J. Armstrong, R. S. Massey, R. W. Alder and A. C. O'Donoghue, Chem. Commun., 2011, 47, 1559–1561 RSC.
- D. J. Nelson and S. P. Nolan, Chem. Soc. Rev., 2013, 42, 6723–6753 RSC.
- R. S. Massey, C. J. Collett, A. G. Lindsay, A. D. Smith and A. C. O'Donoghue, J. Am. Chem. Soc., 2012, 134, 20421–20432 CrossRef CAS.
- D. E. Tucker, P. Quinn, R. S. Massey, C. J. Collett, D. J. Jasiewicz, C. R. Bramley, A. D. Smith and A. C. O'Donoghue, J. Phys. Org. Chem., 2015, 28, 108–115 CrossRef CAS.
- R. S. Massey, P. Quinn, S. Zhou, J. A. Murphy and A. C. O'Donoghue, J. Phys. Org. Chem., 2016, 29, 735–740 CrossRef CAS.
- R. W. Alder, P. R. Allen and S. J. Williams, J. Chem. Soc., Chem. Commun., 1995, 1267–1268 RSC.
- Y. Chu, H. Deng and J.-P. Cheng, J. Org. Chem., 2007, 72, 7790–7793 CrossRef CAS.
- Z. Li, X. Li and J.-P. Cheng, J. Org. Chem., 2017, 82, 9675–9681 CrossRef CAS PubMed.
- J. R. Wright, P. C. Young, N. T. Lucas, A. L. Lee and J. D. Crowley, Organometallics, 2013, 32, 7065–7076 CrossRef CAS PubMed.
- M. H. Dunn, N. Konstandaras, M. L. Cole and J. B. Harper, J. Org. Chem., 2017, 82, 7324–7331 CrossRef CAS PubMed.
- D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS.
- D. Enders and T. Balensiefer, Acc. Chem. Res., 2004, 37, 534–541 CrossRef CAS.
- D. Enders, J. Han and A. Henseler, Chem. Commun., 2008, 34, 3989–3991 RSC.
- F. G. Bordwell, Pure Appl. Chem., 1977, 49, 963–968 CAS.
- X. M. Zhang, F. G. Bordwell, M. Van Der Puy and H. E. Fried, J. Org. Chem., 1993, 58, 3060–3066 CrossRef CAS.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456–463 CrossRef CAS.
- F. G. Bordwell, Bordwell pKa Table (Acidity in DMSO), http://www.chem.wisc.edu/areas/reich/pkatable/.
- W. S. Matthews, J. E. Bares, J. E. Bartmess, F. G. Bordwell, F. J. Cornforth, G. E. Drucker, Z. Margolin, R. J. McCallum, G. J. McCollum and N. R. Vanier, J. Am. Chem. Soc., 1975, 97, 7006–7014 CrossRef CAS.
- Y.-J. Kim and A. Streitwieser, J. Am. Chem. Soc., 2002, 124, 5757–5761 CrossRef CAS PubMed.
- W. Olmstead and F. G. Bordwell, J. Org. Chem., 1980, 45, 3299–3305 CrossRef CAS.
- D. Enders, K. Breuer, U. Kallfass and T. Balensiefer, Synthesis, 2003, 1292–1295 CAS.
- B. Bostai, Z. Nováky, A. C. Bényei and A. Kotschy, Org. Lett., 2007, 9, 3437–3439 CrossRef CAS PubMed.
- F. G. Bordwell, A. V. Satish, F. Jordan, C. B. Rios and A. C. Chung, J. Am. Chem. Soc., 1990, 112, 792–797 CrossRef CAS.
- F. G. Bordwell and J. A. J. Harrelson, Can. J. Chem., 1990, 68, 1714–1718 CrossRef CAS.
- A. C. O'Donoghue and R. S. Massey, in Contemporary Carbene Chemistry, John Wiley & Sons, Inc., 2014, pp. 75–106 Search PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- N. S. Isaacs, Physical Organic Chemistry, Longman Limited, Essex, 1987 Search PubMed.
- L. Hammett, J. Am. Chem. Soc., 1937, 59, 96–103 CrossRef CAS.
- R. H. Crabtree, Coord. Chem. Rev., 2013, 257, 755–766 CrossRef CAS.
- T. Nakamura, T. Terashima, K. Ogata and S.-I. Fukuzawa, Org. Lett., 2011, 13, 620–623 CrossRef CAS PubMed.
- S. Inomata, H. Hiroki, T. Terashima, K. Ogata and S.-i. Fukuzawa, Tetrahedron, 2011, 67, 7263–7267 CrossRef CAS.
- H. Mayr and A. Ofial, Acc. Chem. Res., 2016, 49, 952–965 CrossRef CAS PubMed.
- L. M. N. Meseroll, J. R. McKee and M. Zanger, Synth. Commun., 2011, 41, 2557–2568 CrossRef CAS.
- S. Ni, Y. Yuan, J. Huang, X. Mao, M. Lv, J. Zhu, X. Shen, J. Pei, L. Lai, H. Jiang and J. Li, J. Med. Chem., 2009, 52, 5295–5298 CrossRef CAS.
- C. G. Neochoritis, T. Zarganes-Tzitzikas, S. Stotani, A. Domling, E. Herdtweck, K. Khoury and A. Domling, ACS Comb. Sci., 2015, 17, 493–499 CrossRef CAS.
- R. S. Bon, C. Hong, M. J. Bouma, R. F. Schmitz, F. J. J. de Kanter, M. Lutz, A. L. Spek and R. V. A. Orru, Org. Lett., 2003, 5, 3759–3762 CrossRef CAS PubMed.
- F. G. Bordwell and H. E. Fried, J. Org. Chem., 1981, 46, 4327–4331 CrossRef CAS.
- M. E. Bowen, B. R. Aavula and E. A. Mash, J. Org. Chem., 2002, 67, 9087–9088 CrossRef CAS PubMed.
- G. C. Vougioukalakis, M. M. Roubelakis and M. Orfanopoulos, J. Org. Chem., 2010, 75, 4124–4130 CrossRef CAS PubMed.
- F. Romanov-Michailidis, C. Besnard and A. Alexakis, Org. Lett., 2012, 14, 4906–4909 CrossRef CAS PubMed.
- K. Huang, G. Li, W.-P. Huang, D.-G. Yu and Z.-J. Shi, Chem. Commun., 2011, 47, 7224–7226 RSC.
- H. G. O. Becker, G. Hoffmann, K. M. Gwan and L. Knupfer, J. Prakt. Chem., 1988, 330, 325–337 CrossRef CAS.
- H. Kakuta, X. Zheng, H. Oda, S. Harada, Y. Sugimoto, K. Sasaki and A. Tai, J. Med. Chem., 2008, 51, 2400–2411 CrossRef CAS PubMed.
- R. Huisgen, J. Sauer and M. Seidel, Chem. Ber., 1961, 94, 2503–2509 CrossRef CAS.
- J. E. Thomson, C. D. Campbell, C. Concellón, N. Duguet, K. Rix, A. M. Z. Slawin and A. D. Smith, J. Org. Chem., 2008, 73, 2784–2791 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis of the fluorenes 1–4, absorbance data for the deprotonated forms of the fluorenes 1–5, synthesis of the triazolium salt 6 and pKa data, synthesis of the thione 11, pKa data for triazolium salts 7, 8 and 10, general procedure for the synthesis of the N-phenylbenzamides 14a–e, pKa data for triazolium salts 9a–e, synthesis of the triazolium salt 19 and pKa data, Hammett analysis of series 12, 13 and 16, NMR spectra of all prepared triazolium salts. See DOI: 10.1039/c9ob02258a |
| ‡ This salt is numbered as 9b because it forms part of a series of related salts 9a–e which will be examined subsequently. |
| § The electron donating properties of alkyl substituents have been determined to be similar, as quantified by their Hammett σ values,43,63 noting that this assumes that there are limited steric constraints in the cases presented here. |
| ¶ See footnote above regarding the similarity of the Hammett σ values of alkyl substituents. |
| || No uncertainties were reported in the original manuscript. However, using the data presented and the Hammett σ values used here, the slopes of the plots of pKa values vs. Hammett σ constants of the para-aryl substituents were determined to be (−2.12 ± 0.10) and (−1.87 ± 0.17) for series 12 and 13, respectively (see ESI† for plots). |
| ** An alternative origin for the decrease in electronic susceptibility from series 12 to series 16 is a significant change in charge delocalisation in the azolium core. However, this explanation seems unlikely given the electronic nature of the 3,4-substitution in each case is similar and the similar electronic susceptibilities of series 12 and 13 in DMSO. |
| †† The anion effects were examined in previous studies and shown to be negligible;43,46 hence the effects on pKa values of different electronic substituents can be compared between the case reported and the case introduced here. |
| ‡‡ This salt has a different anion to those considered elsewhere in this manuscript; at the concentrations used in this work (<ca. 0.01 M), pKa values in DMSO are not altered by ion pairing effects of the anion.43,46,56 Hence the pKa values of azolium salts with different anions can be compared based on the cation alone. |
| §§ As discussed previously,46 this increase in pKa is a result of both the electron donating properties of the methyl groups, and the steric hindrance about the nitrogen atom preventing the aryl rings from lying perpendicular to the imidazolium ring and therefore allowing stabilisation through resonance. |
| This journal is © The Royal Society of Chemistry 2020 |