
Synthesis and applications of fluorous phosphines†‡
Chung-kay Edwin
Law
and
István T.
Horváth
*
Department of Biology and Chemistry, City University of Hong Kong, 83 Tat Chee Avenue, Kowloon, Hong Kong S.A.R, China. E-mail: istvan.t.horvath@cityu.edu.hk
First published on 14th June 2016
Abstract
Fluorous phosphines having one or more fluorous ponytails containing longer and shorter perfluoroalkyl substituents are reviewed, including their synthesis, some of their basic properties and their applications in biphasic, organometallic and organocatalysis.
 Chung-Kay Edwin Law | Chung-Kay Edwin Law obtained his BSc degree in Biochemistry from the University of Bristol, UK (2007) and his MSc degree in Chemistry from the University of Bristol, UK working with Prof. Russell J. Cox (2011). He then came to City University of Hong Kong in early 2012 as a Research Assistant and started his PhD studies on fluorous synthesis and catalysis with Professor István T. Horváth in 2013. |
 István T. Horváth | István T. Horváth is a Chair Professor of Chemistry at the City University of Hong Kong. He received his Diploma in chemical engineering (1977) and PhD. in chemistry (1979), both from the University of Pannonia, Veszprém, Hungary. He was a Postdoctoral Research Associate at Yale University (1982–1984), a Scientific Co-worker at ETH Zürich (1984–1987), and a Senior Staff Chemist at Exxon Corporate Research, Annandale, NJ (1987–1998). After spending 10 years at Eötvös University, Budapest, as a Professor, he moved to Hong Kong in May, 2009. He has published over 160 scientific papers, book chapters and patents. He was the Editor-in-Chief of the “Encyclopedia of Catalysis” (Wiley, 2002), Editor of “Fluorous Chemistry” (Springer, 2012), and a Co-Editor of Aqueous Organometallic Chemistry and Catalysis (Kluwer, 1995), Handbook of Fluorous Chemistry (Wiley-VCH, 2004), and Multiphase Homogeneous Catalysis (Wiley-VCH, 2005). The books on fluorous chemistry were based on the fluorous biphasic concept he invented in the early nineties. He received the 1st Fluorous Technology Award in 2005, the Senior Humboldt Research Award in 2006, and the Green Chemistry Lecture Award in 2008. He has been a Fellow of the Royal Society of Chemistry (2013) and the American Chemical Society (2014). |
Introduction
The successful combination of high catalyst activity and selectivity with facile catalyst recycling and product(s) separation/purification has been one of the most important goals of contemporary homogeneous catalysis research, which could significantly improve the atom economy of a reaction.1,2 Phosphines have been frequently used as ligands in transition metal homogeneous catalysis, especially to fine tune a wide range of catalytic reactions operating under different conditions in various liquid phases. Since the transition metal catalyst, the substrate(s), and the product(s) are typically used and formed in the same liquid phase, the product(s) separation could be cumbersome. An innovative approach to overcome the separation issue is the use of temperature-dependent biphasic liquid systems, in which the miscibility of the two liquids as well as the solubility of the catalyst and the product(s) are highly dependent on the temperature.1b Ideally, the two liquids are not miscible at a lower temperature and the catalyst and the substrate(s) are residues in the same liquid phase. At a higher temperature, the two liquids form a single liquid phase and the catalyst converts the substrate(s) to the product(s). After complete conversion of the substrate(s), the reaction mixture is cooled to form two liquid phases, among which one should preferentially contain the catalyst and the other the product(s). Simple decantation of the product phase could be used for facile catalyst recycling. A temperature-dependent solid–liquid immobilization method can be used, if the solid form of a homogeneous catalyst can be dissolved in the reaction medium at higher temperatures and separated from the product after the reaction at lower temperatures.1bThe fluorous biphasic concept, invented almost 25 years ago, was based on the temperature-dependent miscibility of perfluoroalkanes, perfluoro-dialkylethers, and perfluoro-trialkylamines (fluorous solvents) with typical organic solvents and on the preferential solubility of fluorous reagents or catalysts in fluorous solvents under ambient conditions (Scheme 1).3 The required high partition coefficients of the fluorous reagents or catalysts were achieved by the attachment of fluorous pony-tails of appropriate sizes and numbers.
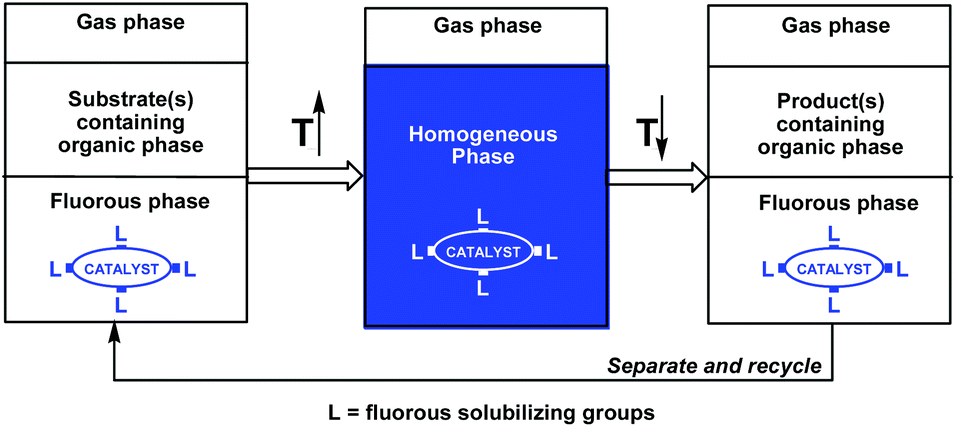 | ||
| Scheme 1 Fluorous biphase concept. | ||
Since phosphine ligands have been employed in numerous catalytic reactions in academia and industry, it should therefore not be surprising that significant efforts have been made to develop fluorous phosphine ligands. Similarly to other fluorous compounds, the properties of the fluorous phosphines and their transition metal complexes are governed by a number of factors. In order to ensure high partition of the fluorous phosphines into the fluorous phase, a number of requirements have to be met. The most efficient fluorous pony-tails are perfluoroalkyl groups, while heteroatom-containing tails are also possible, such as perfluoro-polyoxaalkyl groups.4a,b Because of the polar nature of the carbon–fluorine bonds in perfluoro-aryl groups, they have limited applicability in fluorous chemistry. The fluorine content should be more than 60 wt% and the fluorine atoms should be arranged in close proximity along the ponytails. A suitable spacer, an alkylene one in most cases, should be placed between the phosphorus atom and the perfluoroalkyl chains. This is important because of the strong electron-withdrawing effects of the perfluoroalkyl chains attached closely to the phosphorus atom that can significantly lower the basicity of the phosphine ligand.
A useful extension of the fluorous biphase concept was based on the temperature-dependent organic-liquid and fluorous-solid phase miscibilities (Scheme 2).5a–c Since many fluorous reagents and catalysts are solids at ambient temperature, they show thermomorphic behaviors, and can be dissolved in the organic phase at elevated temperatures. After cooling under ambient conditions, the solid fluorous catalysts precipitate from the reaction mixture, and they can be recycled by filtration.
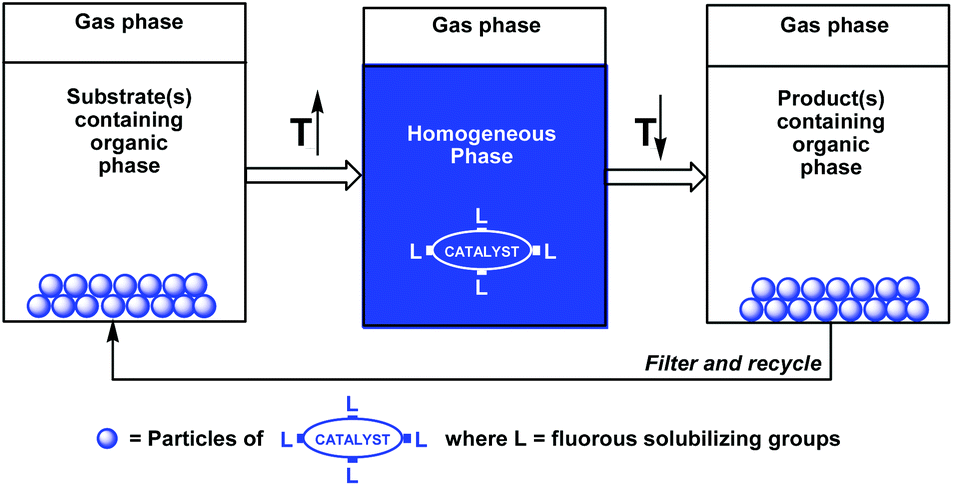 | ||
| Scheme 2 Temperature dependent solubilization or precipitation of a solid fluorous catalyst. | ||
Gladysz et al. have demonstrated an efficient fluorous release and catch concept by using a thermomorphic fluorous phosphine rhodium hydrosilylation catalyst absorbed to a Teflon© tape (Scheme 3).6 By placing the Teflon© tape in a reaction mixture at high temperature, the fluorous catalyst dissolves in the reaction environment to perform its catalytic function. By cooling the reaction mixture to a lower temperature, the fluorous catalyst is re-absorbed to the Teflon©.
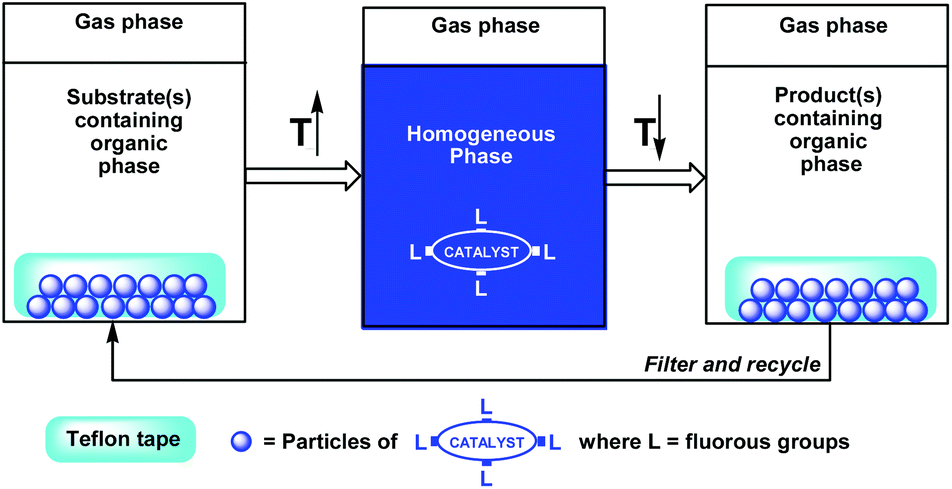 | ||
| Scheme 3 An immobilized fluorous catalyst on Teflon©. | ||
The fluorous pony-tail should be of a suitable length. Traditionally, the alkylene spacer should have 2–5 carbon atoms and the perfluoroalkyl pony-tail should have 6–10 carbon atoms. Finally, it should be noted that the fluorous partition coefficients of transition metal complexes of fluorous phosphines are expected to be higher when more than one fluorous phosphine is attached to the metal center. Most of the early synthesis of fluorous phosphines involved the attachment of long perfluoroalkyl chains (>C8) to the phosphorus atom with ethylene or trimethylene spacers. These have ensured a high enough fluorine content to achieve satisfactory fluorous partition and effective biphasic catalysis. Some success has been achieved to synthesize perfluoroalkyl groups containing triaryl phosphines, which could be characterized as ‘light fluorous’ phosphines, e.g. the fluorine content was lower than 50 wt%. While a fluorous extraction may not work for these light fluorous phosphines, fluorous solid phase extraction could be used for their recovery.
Many fluorous trialkyl phosphines have been used as organo-catalysts or ligands in the presence of transition metals. Since these fluorous phosphines generally consisted of one or two methylene spacers between the phosphorus atom and the C6–10-perfluoroalkyl chains, they were highly fluorous-soluble and active. However, the release of fluorous phosphines to the environment could lead to serious environmental and health problems.7 Low level leaching to the product(s) or accidental releases could result in oxidative and/or hydrolytic fragmentation and the formation of a long-chain perfluoroalkyl carboxylate (LCPFAC), among which perfluoro-octanoic acid (CF3(CF2)6-COOH) is the best known.8 These compounds have high bioaccumulations and are shown to lead to endocrine and liver disorders. The bioaccumulation and the toxicity of homologous perfluoroalkyl carboxylates with shorter perfluoroalkyl chains are lower than those with longer perfluoroalkyl chains. The combination of shorter C1–4-perfluoroalkyl-groups was suggested to maintain high fluorous partition and minimize bioaccumulation.9 It should be noted that the high oxygen solubility in fluorous solvents could lead to the oxidation of phosphines especially those trialkyl phosphines which have more than 3 methylene groups between the phosphorus atom and the perfluoroalkyl chain. While the application of fluorous solvents could lead to an ecological issue, temperature dependent solvent-free fluorous systems could overcome the environmental issues.
Fluorous trialkyl phosphines
Synthetic strategies
Nonafluorobutylethyl phosphine 1a was synthesized by reacting with perfluorobutyl iodide, elemental phosphorus, and a nickel salt as the catalyst (Scheme 4).10a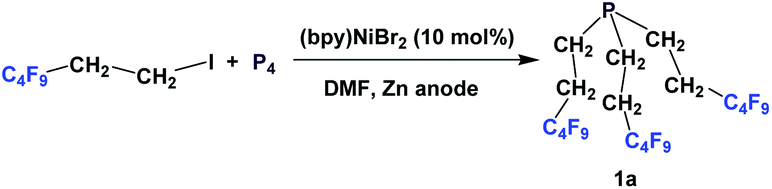 | ||
| Scheme 4 The synthesis of 1a. | ||
A number of methods involved either fluorous Grignard or organozinc reagents (Scheme 5). The first reported synthesis of tridecafluorohexylethyl phosphine 1b, (C6F13CH2CH2)3P, appeared in 1985, which utilized the fluorous organozinc reagent C6F13CH2CH2ZnI and afforded the phosphine through its reaction with phosphorus trichloride.10b Thereafter, Knochel et al. have reported the use of a fluorous dialkylzinc reagent, (C6F13CH2CH2)2Zn, to synthesize fluorous phosphine 1b (75% yield, protected as a borane complex).11 On the other hand, the Grignard reagent C6F13CH2CH2MgI has also been synthesized, upon activation of magnesium metal by 1,2-dibromoethane, by Hope et al. and it was used in the synthesis of the same fluorous phosphine, with a yield of 50%.12a The same fluorous Grignard reagent has also been used to synthesize fluorous phenyl phosphines containing C6F13CH2CH2-ponytails (vide infra).
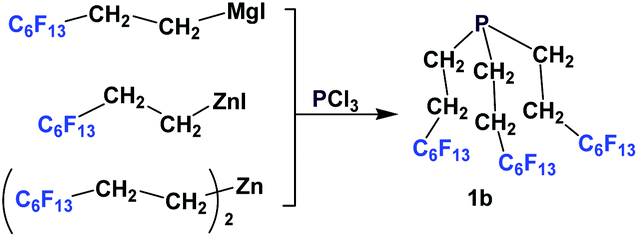 | ||
| Scheme 5 Grignard/organozinc approach to synthesize fluorous triethyl phosphine 1b. | ||
Another approach to synthesize perfluoroalkylethyl phosphines is to react PH3 with fluorous olefins (Scheme 6).13 The method is a free-radical mediated process, and a series of perfluoroalkylethyl phosphines, where RF = perfluorohexyl-ethyl (1b), perfluorooctylethyl (1c), and perfluorodecylethyl (1d), were synthesized.
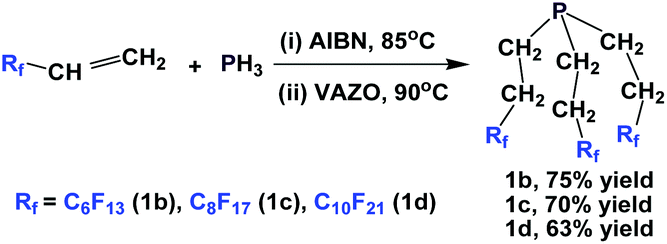 | ||
| Scheme 6 Free-radical mediated, PH3 addition to fluorous olefins to generate perfluoroalkylethyl phosphines 1b–1d. | ||
If a fluorous olefin is not commercially available, it can be synthesized by coupling the corresponding fluorous iodide with allyltributyltin through a photochemical process (Scheme 7). The resulting fluorous olefin then can be subject to the same type of PH3 addition reaction as shown in Scheme 4. Fluorous phosphines with longer hydrocarbon spacers, such as perfluorooctylpropyl phosphine 2c (73% yield), perfluorooctylbutyl phosphine 3 (66% yield) and perfluorooctylpentyl phosphine 4 can be synthesized.13
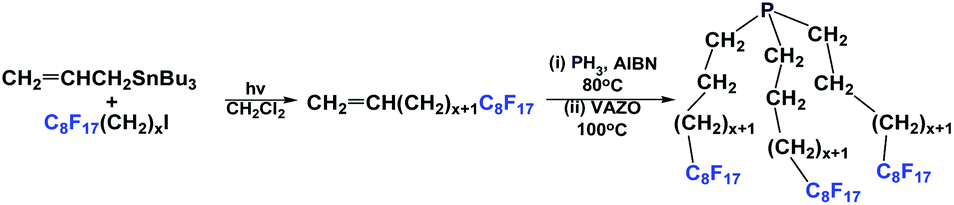 | ||
| Scheme 7 The synthesis of perfluorooctylalkyl phosphines. | ||
Since PH3 is toxic and extremely flammable when a trace of P2H4 is present, alternative approaches have also been developed to avoid its use. A modular synthesis of fluorous phosphine 12 was demonstrated by Gladysz et al. (Scheme 8).14a–b An Arbuzov reaction between tridecafluorohexylethyl iodide and triethyl phosphite at an elevated temperature led to a fluorous phosphonate. Upon reduction with lithium aluminium hydride, tridecafluorohexylethyl phosphine was formed, which was subjected to two consecutive free-radical reactions to form 12.
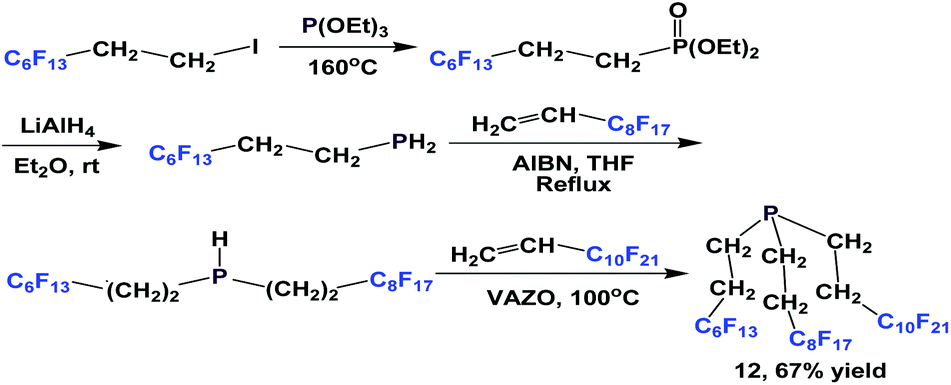 | ||
| Scheme 8 Modular synthesis of fluorous phosphine 12. | ||
A multi-stage method has been developed to synthesize a number of fluorous phosphines with different chains (Scheme 9).15 Fluorous olefins were reacted with phenyl phosphine through a free-radical mediated process. The bis-perfluoroalkyl phenyl phosphines were alkylated to give the corresponding phosphonium salts. Basic hydrolysis led to the corresponding fluorous phosphine oxides, which were then reduced with trichlorosilane to afford fluorous phosphines 2b, 2c, 5 and 6.
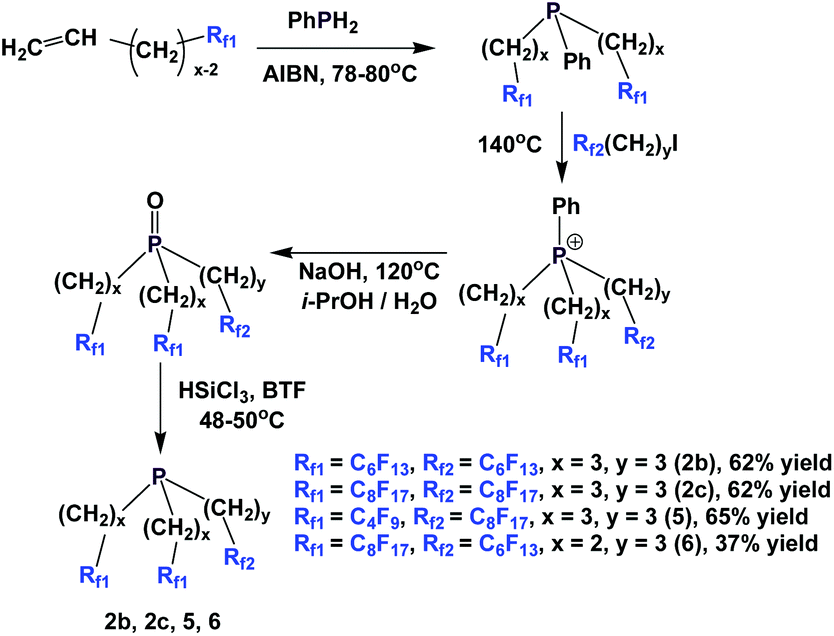 | ||
| Scheme 9 Fluorous phosphine synthesis with PhPH2. | ||
Perfluorooctylethyl iodide reacted with LiPH2·DME at −45 °C to give perfluorooctylethyl phosphine. This primary phosphine was reacted with various fluorous olefins to give fluorous phosphines 7–10, containing the perfluorooctyl ponytails and oligomethylene spacers of various lengths (Scheme 10).16 This approach was a safer and more economical method than the direct application of PH3.
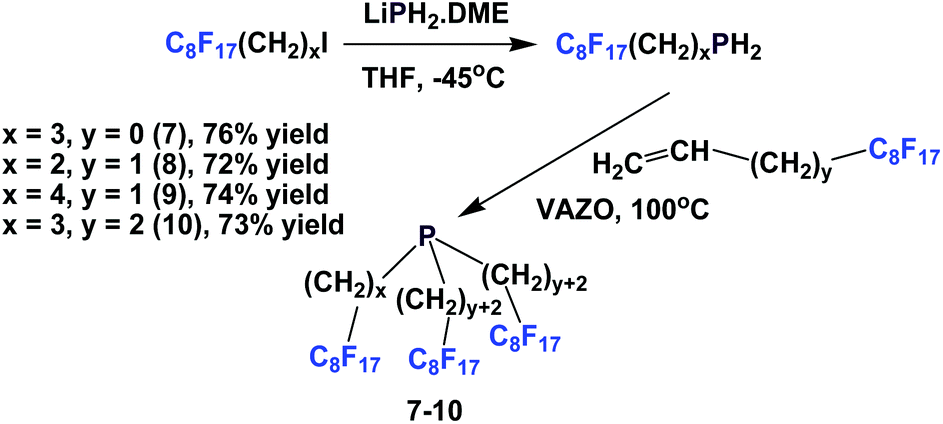 | ||
| Scheme 10 Synthesis of fluorous phosphines using LiPH2·DME. | ||
Fluorous phosphines 13b and 13c, which contain branched alkyl groups, were synthesized by using the corresponding fluorous Grignard reagents (Scheme 11).17
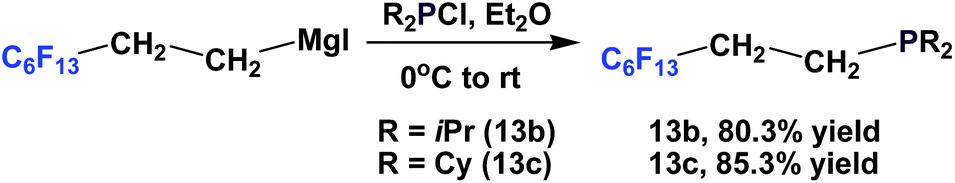 | ||
| Scheme 11 Synthesis of fluorous phosphines 13b and 13c. | ||
Fluorous phosphine 13a was synthesized by a radical reaction with Et2PH at elevated temperature (Scheme 12).18
 | ||
| Scheme 12 Synthesis of fluorous phosphine 13a. | ||
Fluorous diphosphine 14 was prepared by the reaction of a fluorous olefin and 1,2-diphosphinoethane in the presence of AIBN (Scheme 13).18
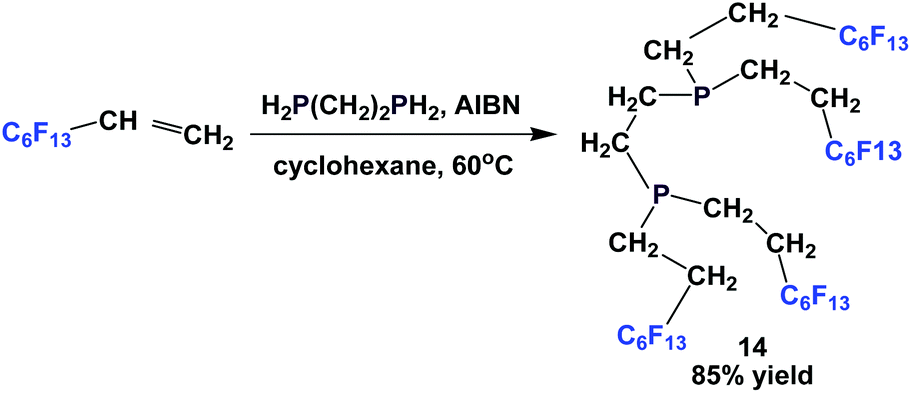 | ||
| Scheme 13 Synthesis of fluorous diphosphine 14. | ||
Fluorous diphosphines 15a–15c were prepared by the addition of 1,5-diphosphinopentane to 3 different fluorous olefins in the presence of AIBN (Scheme 14).19
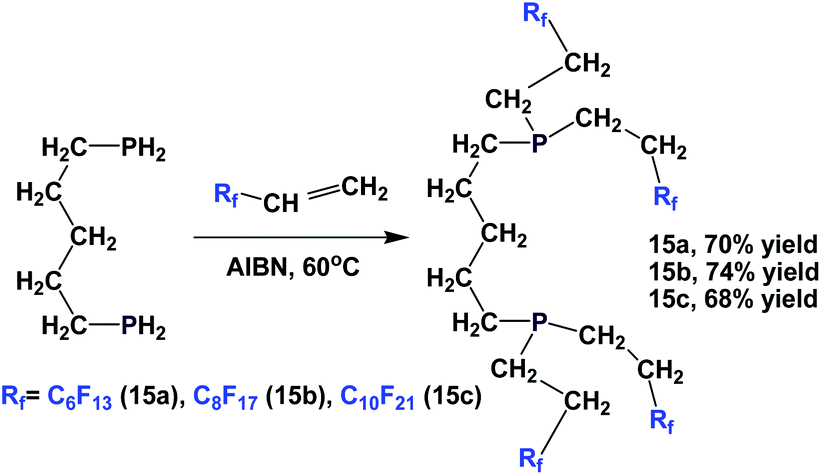 | ||
| Scheme 14 Synthesis of fluorous diphosphines 15a–15c. | ||
The synthesis of analogous fluorous diphosphines, 16a and 16b, is also reported (Scheme 15).20
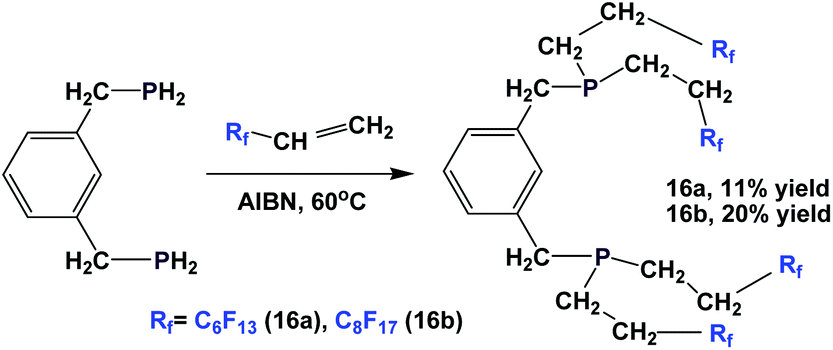 | ||
| Scheme 15 Synthesis of fluorous diphosphines 16a and 16b. | ||
The enantiopure phosphines 17a and 17b have been prepared by previously discussed methods, using fluorous Grignard reagents and fluorous olefins (Scheme 16).21 With the Grignard approach, the yields of both 17a and 17b are 30%, and with the olefin approach yields are in the range of 44–50%. 2a–2c, 3, 9, and 11 were synthesized by an iterative approach (Scheme 17).22
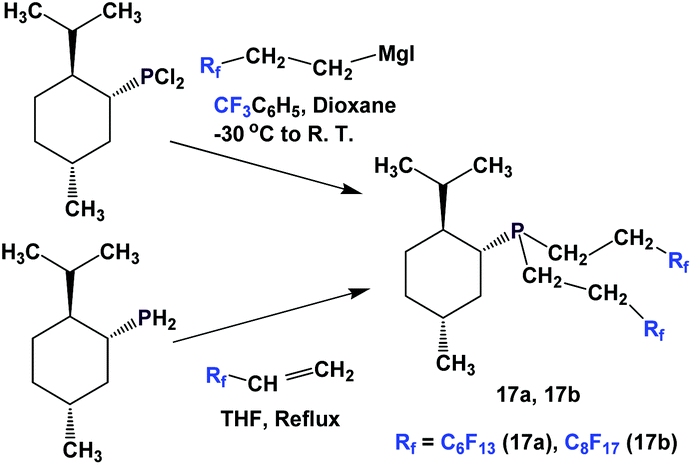 | ||
| Scheme 16 Synthesis of (menthyl)P(CH2CH2Rf)2. | ||
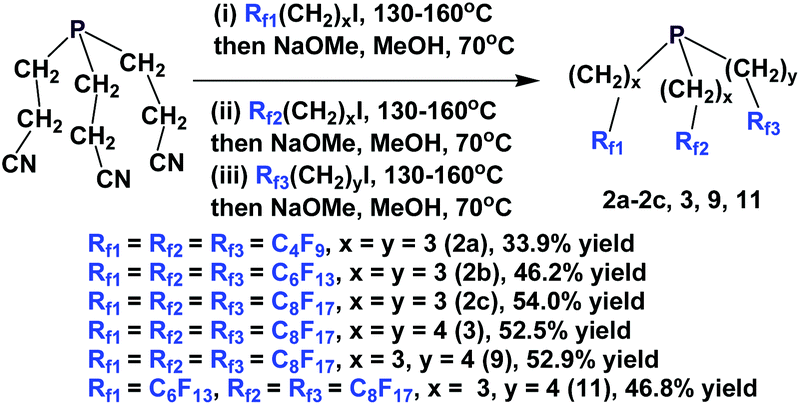 | ||
| Scheme 17 Iterative synthesis of fluorous phosphines. | ||
An interesting way to incorporate a fluorous ponytail into the diphosphine structure is to place the fluorous tail between the 2 phosphorus atoms, like the fluorous phosphine 18 (Scheme 18).23 A dibromide with a fluorous backbone is reacted with the lithium anion of di-tert-butyl phosphide.
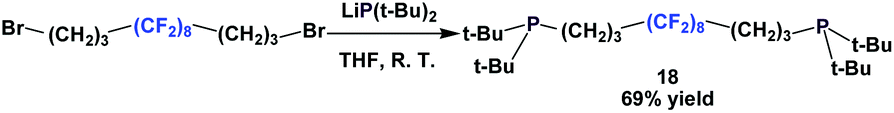 | ||
| Scheme 18 Fluorous diphosphine with (CH2)3(CF2)8(CH2)3-backbone. | ||
Perfluorotrialkyl phosphines have also been prepared by a 2-stage process (Scheme 19).24 The corresponding non-fluorinated trialkyl phosphine was subjected to exhaustive fluorination, yielding a difluoro-phosphorus (V) intermediate. Then, this intermediate was reduced by PSi(CH3)3, generating the desired perfluorotrialkyl phosphines 19a–19f. Using a similar method, perfluorinated di-phosphine 21, (C2F5)2PCF2CF2P(C2F5)2, was also synthesized. Perfluorotrialkyl-phosphine 19e could also be prepared by the nickel-catalyzed electrochemical reduction as illustrated in Scheme 4, using C6F13I and elemental phosphorus as the reactants.25
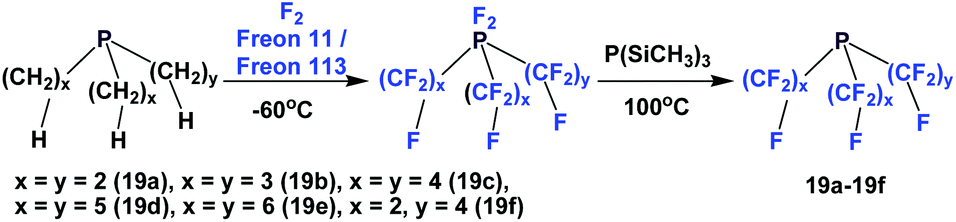 | ||
| Scheme 19 Perfluoroalkyl phosphine synthesis. | ||
A photochemical approach has been developed to make perfluorodecyl (bis)-tert-butyl phosphine such as 20a (Scheme 20).26 After forming its corresponding phosphine sulfide, the yield was 53%.
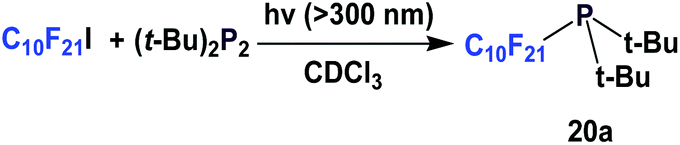 | ||
| Scheme 20 Photochemical way to make phosphine 20a. | ||
A recent modification of the conditions led to two other perfluorodecyl dialkyl phosphines, 20b and 20c (Scheme 21).27
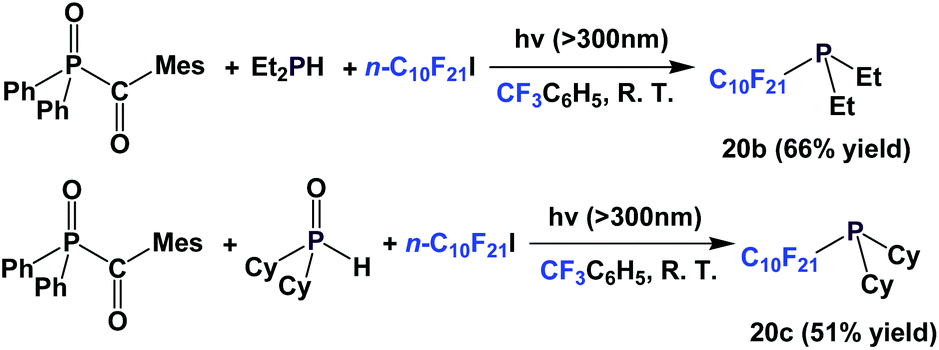 | ||
| Scheme 21 A modified approach to make phosphines 20b and 20c. | ||
Applications of fluorous trialkyl phosphines
Fluorous trialkyl phosphines have been used in many catalytic reactions, including both transition metal catalysis and organocatalysis. In many cases, transition metal complexes of fluorous phosphines have been prepared to gain further insights into the phosphines’ physical and chemical properties.The fluorous phosphine rhodium complex HRh(CO)P(CH2CH2C6F13)3 was prepared using ligand 1b, as a catalyst precursor for the hydroformylation of olefins in the presence of excess 1b (Scheme 22).3a This was the first thermo-regulated homogeneous bi-phasic catalyst system. The reaction proceeded in one phase at a higher temperature. The product aldehyde could be partitioned into the organic phase, and the rhodium catalyst remained in the fluorous phase at room temperature. Thus, the fluorous rhodium catalyst could be recycled and reused. Upon further development, the catalyst was used for a continuous hydroformylation of ethylene (R = H), and that allowed the continuous removal of product propanal. It was also shown that the stability of the fluorous phosphine rhodium complex was better than the triphenylphosphine-modified rhodium complex.28
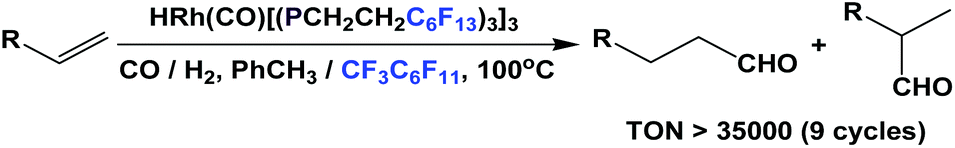 | ||
| Scheme 22 Hydroformylation catalyzed by a rhodium complex of phosphine 1b. | ||
The fluorous trialkyl phosphine-modified rhodium complex, derived from ligand 1c, has been used in the hydroboration of olefins, giving alcohols after an aqueous oxidative workup (Scheme 23).29 The reaction started as a biphasic system. At a higher reaction temperature the formation of a single phase was observed, containing all reactants, solvents and the catalyst. Upon cooling, the formation of two liquid phases occurred, providing a simple separation of the boronate product in the organic phase, from the precious fluorous phosphine rhodium catalyst in the fluorous phase.
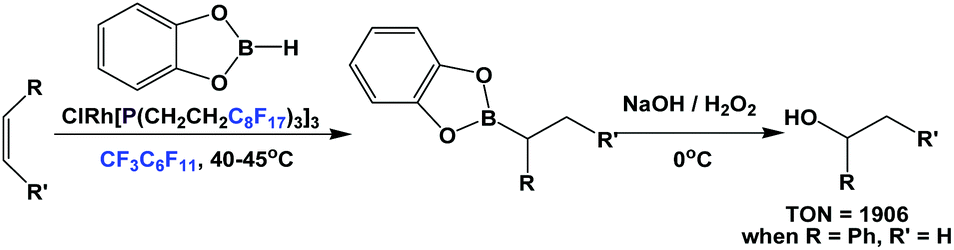 | ||
| Scheme 23 Hydroboration of olefins catalyzed by a rhodium complex of phosphine 1c. | ||
The fluorous Vaska complex of phosphine 1c was also prepared (Scheme 24).30 It has been found that its oxidative addition to alkyl halides is likely to involve a radical mechanism.
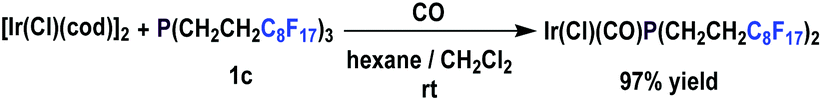 | ||
| Scheme 24 Formation of an iridium complex from phosphine 1c. | ||
A similar rhodium carbonyl chloride complex, RhCl(CO)[P(CH2CH2C8F17)3]2, was also prepared using 1c.31 On the other hand, various ruthenium, rhodium, and nickel complexes containing (C6F13CH2CH2)3P have been prepared.32a–c
Fluorous phosphine ligands 1b–1d have been attached to Grubbs type metathesis catalysts (Scheme 25). Gladysz et al. have shown that these fluorous Grubbs catalysts were active in ring-opening metathesis polymerization and ring-closing metathesis.33,34 These complexes led to rate enhancement in ring-closing metathesis, probably due to a phase-transfer activation – as the dissociated fluorous phosphine partitioned into the fluorous phase, so that the free fluorous phosphines would not compete with the substrates for the catalysts. The fluorous Grubbs complex with the fluorous trialkyl phosphine, two chlorides and the imidazolium group was structurally characterized.35,36
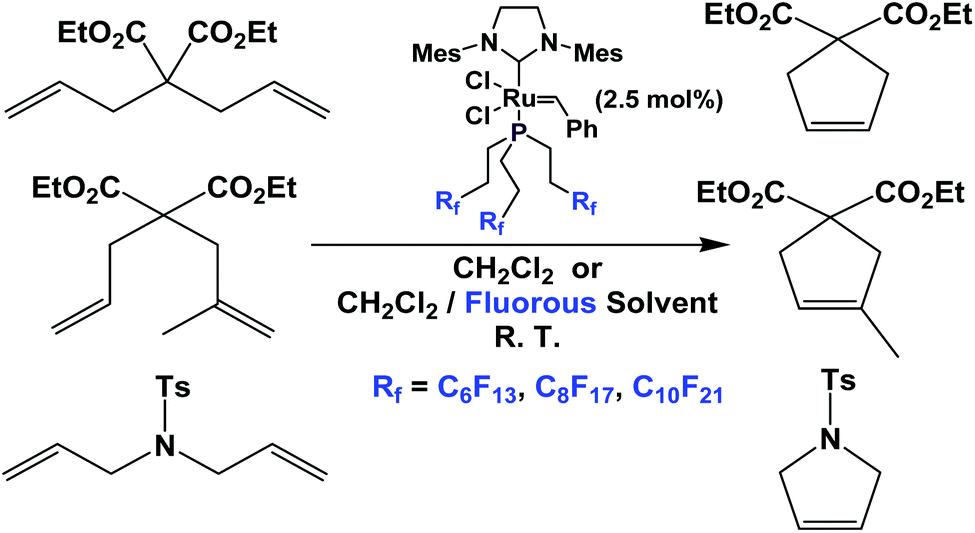 | ||
| Scheme 25 Ring closing metathesis catalyzed by a Grubbs complex derived from fluorous phosphines 1b–1d. | ||
The rhodium complex of fluorous phosphine 13c has been shown to catalyze the homogeneous hydrogenation of 1-hexene, under 1 atm. of hydrogen. A TON of 75 could be achieved (Scheme 26).17
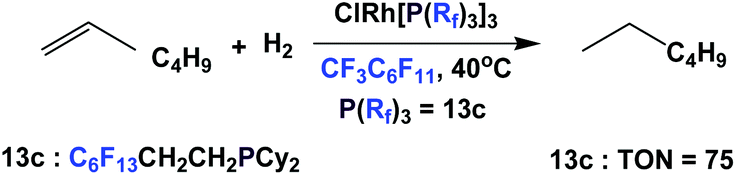 | ||
| Scheme 26 Homogeneous hydrogenation of 1-hexene. | ||
Another catalytic reaction for which fluorous trialkyl phosphines have found widespread use is the hydrosilylation of various substrates. A rhodium complex of fluorous phosphine 1c was prepared and it was used in the hydrosilylation of enones and ketones (Scheme 27).37 The hydrosilylations of enones were carried out at elevated temperatures in fluorous solvents, while the hydrosilylations of ketones were carried out under ambient conditions, using a mixed fluorous/organic solvent system.
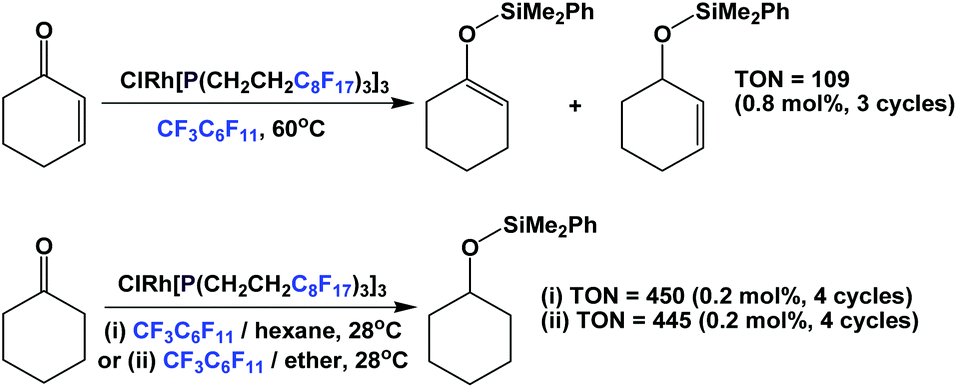 | ||
| Scheme 27 Hydrosilylation of enones and ketones using a rhodium complex derived from fluorous phosphine 1c. | ||
Lantos et al. have demonstrated that the fluorous phosphine 2c or 3 – a modified gold catalyst could be efficiently recycled for three further runs (Scheme 28).38 The source of gold could be a gold salt or gold nanoparticles.
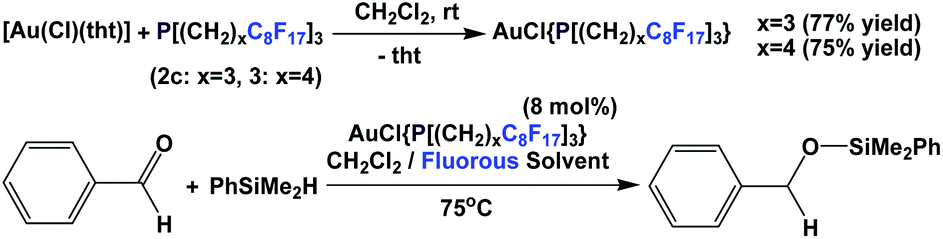 | ||
| Scheme 28 Gold-catalyzed hydrosilylation of aldehydes using phosphines 2c and 3. | ||
A number of other interesting metal complexes with trialkyl fluorous phosphines have also been synthesized. Sharpley et al. have found that the fluorous phosphine modified H2Os3(CO)10 complex was a ROMP catalyst for the synthesis of polynorbornene.39 Malosh et al. have synthesized tungsten carbonyl complexes with fluorous phosphine 1b.40 The same group has made a bi-nuclear ruthenium complex with fluorous phosphine 1b.41 This ruthenium complex has been shown to be a good hydrogenation catalyst, converting acetophenone to 1-phenylethanol in benzotrifluoride at an elevated temperature.
Recently, Gladysz et al. have prepared 3 novel platinum chloride complexes, using fluorous phosphines 1c, 2c and 2d (Scheme 29).42 The fluorophilic ponytails could lead to great fluorous phase partitions and these platinum chloride complexes were used to synthesize ‘insulated’ molecular wires, consisting of polyynediyl units.
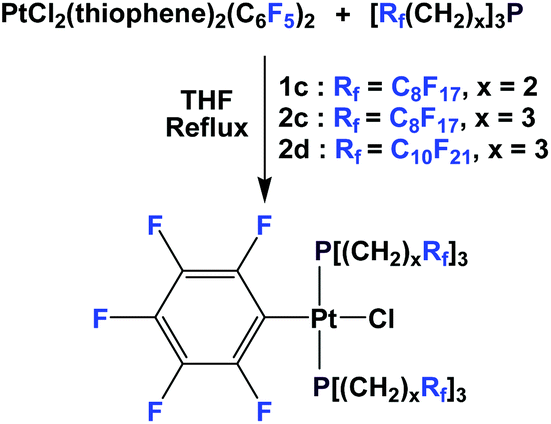 | ||
| Scheme 29 Fluorous platinum complexes. | ||
Fluorous trialkyl phosphines have also been used in organocatalysis. Fluorous phosphine 2c catalyzes the intramolecular type Morita–Baylis–Hillman reaction (Scheme 30).43 The catalyst loading is 10 mol%, and the reaction was carried out in a single phase at an elevated temperature. The fluorous phosphine ‘crashed out’ as a precipitate upon cooling, and thus could be recycled 4 more times.
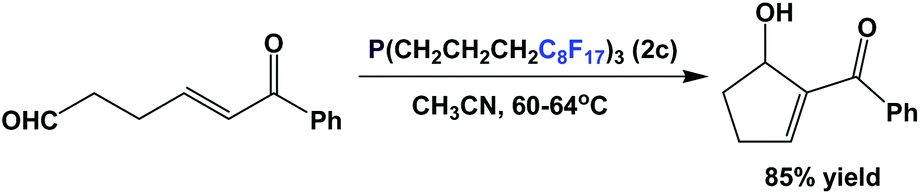 | ||
| Scheme 30 Morita–Baylis–Hillian reaction catalyzed by fluorous phosphine 2c. | ||
Fluorous phosphines 1c and 3 were used as catalysts in the conjugate additions of various alcohols to methyl propiolate at an elevated temperature (Scheme 31).5a
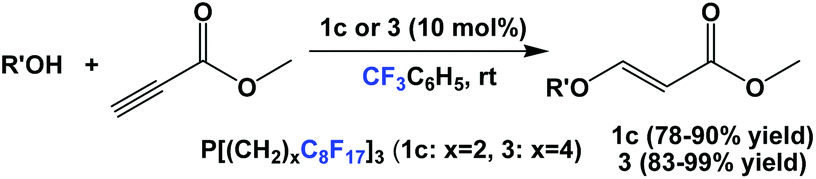 | ||
| Scheme 31 Addition of alcohol substrates to methyl propriolate by fluorous phosphines 1c and 3. | ||
The thermomorphic phosphines 2c and 2d have been employed in Michael addition reactions (Scheme 32).44 The catalyst loading was 10 mol%, and a number of electron-deficient olefins were used as substrates. The fluorous phosphines could be recycled four times.
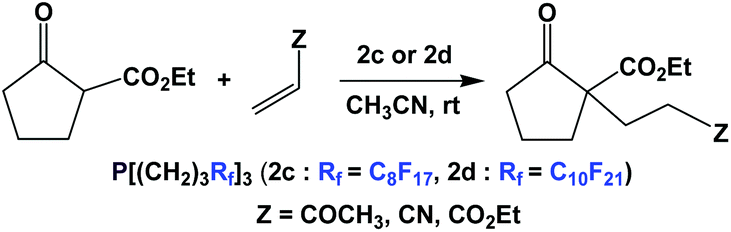 | ||
| Scheme 32 Michael additions catalyzed by fluorous phosphines 2c and 2d. | ||
Fluorous alkyl aryl phosphines
Synthetic strategies
Alkyl aryl phosphines containing one or two C6F13CH2CH2-ponytails, tridecafluoroethyl diphenyl phosphine 22 and tridecafluoroethyl phenyl phosphine 23, have been reported by Hope et al. and they were synthesized through a Grignard approach (Scheme 33).12a This general method was also employed in the synthesis of tridecafluoroethyl phosphine 1b, using PCl3 as the phosphorus source.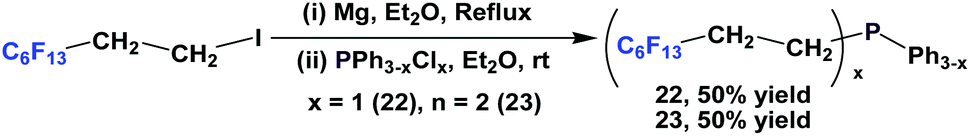 | ||
| Scheme 33 Synthesis of alkyl aryl phosphines 22 and 23. | ||
Perfluoroalkyl diphenylphosphines 24c and 24d were synthesized by nickel-catalyzed electrochemical reductions (Scheme 34).10a,25
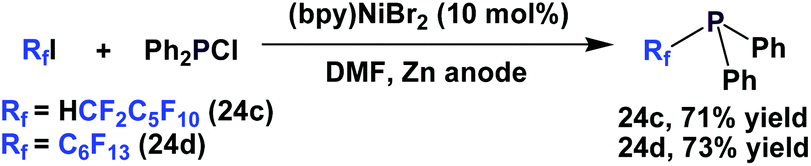 | ||
| Scheme 34 Electrochemical reduction to make perfluoroalkyl diphenylphosphines 24c and 24d. | ||
A photochemical reaction between perfluoroalkyl iodide and Ph2P–PPh2 led to perfluoroalkyl diphenylphosphines, 24a, 24b, 24d to 24g (Scheme 35).2624a and 24b were protected as their corresponding phosphine sulfides, with yields of 60% and 85%.
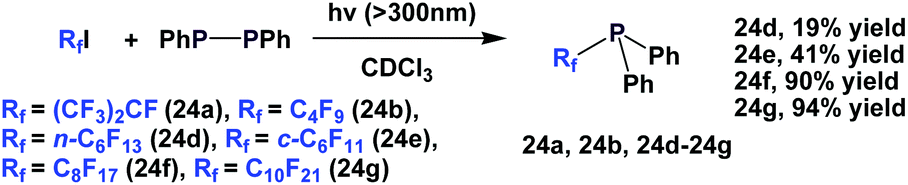 | ||
| Scheme 35 Synthesis of diphenylphosphines 24a, 24b, 24d to 24g. | ||
A similar and recently developed approach led to the synthesis of perfluoroalkyl diphenylphosphines 24d, 24f–24h (Scheme 36).2624d was protected as its phosphine sulfide in 31% yield.
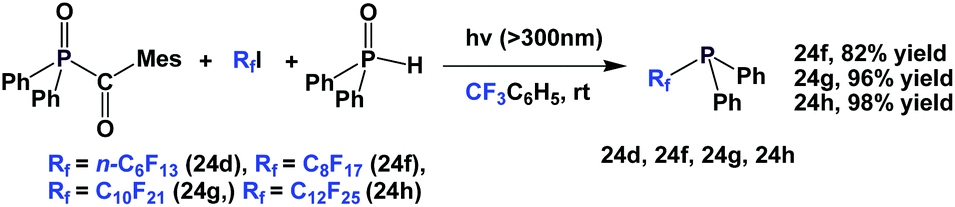 | ||
| Scheme 36 Alternative approaches to synthesize perfluoroalkyl diphenylphosphines 24d, 24f–24h. | ||
By keeping the fluorous ponytail as perfluorodecyl, a number of different aryl groups can be incorporated into the diarylphosphines 25a–25d (Scheme 37).27
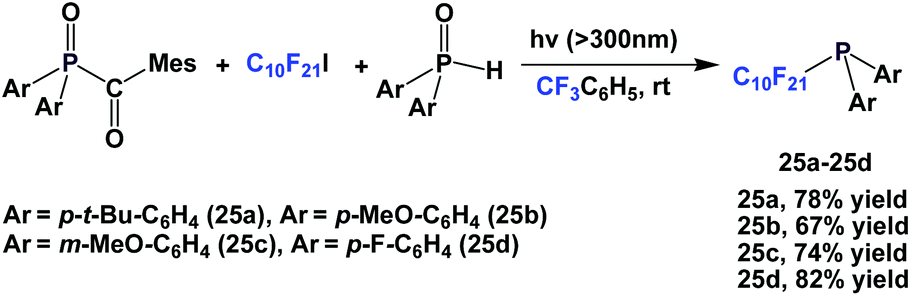 | ||
| Scheme 37 Synthesis of perfluorodecyl diarylphosphines 25a–25d. | ||
Fluorous diphosphine 26 could be synthesized by a two-step procedure (Scheme 38).12a
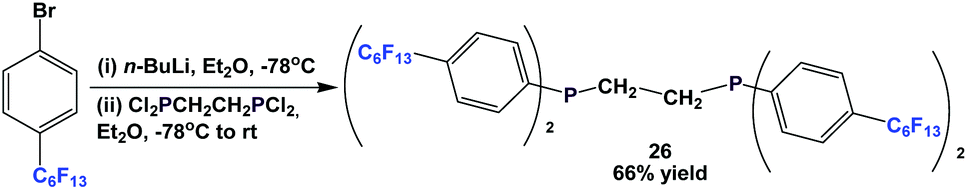 | ||
| Scheme 38 Synthesis of fluorous diphosphine 26. | ||
Through a nucleophilic substitution/reduction sequence, fluorous diphosphine 27 was prepared (Scheme 39).45
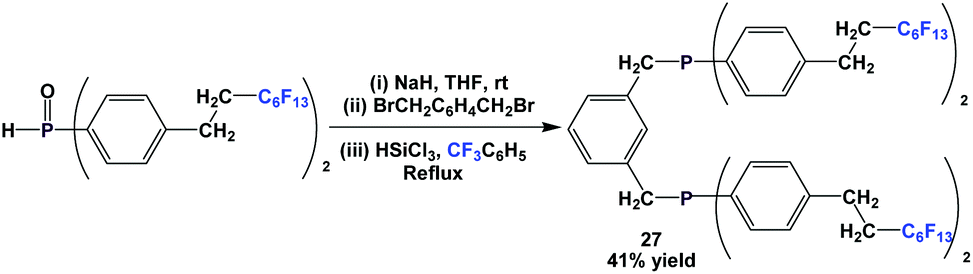 | ||
| Scheme 39 Synthesis of fluorous diphosphine 27. | ||
The different fluorous ponytails could also be attached to the aryl rings through a silicon spacer, as illustrated by the diphosphines 28–30 (Scheme 40).46a–c
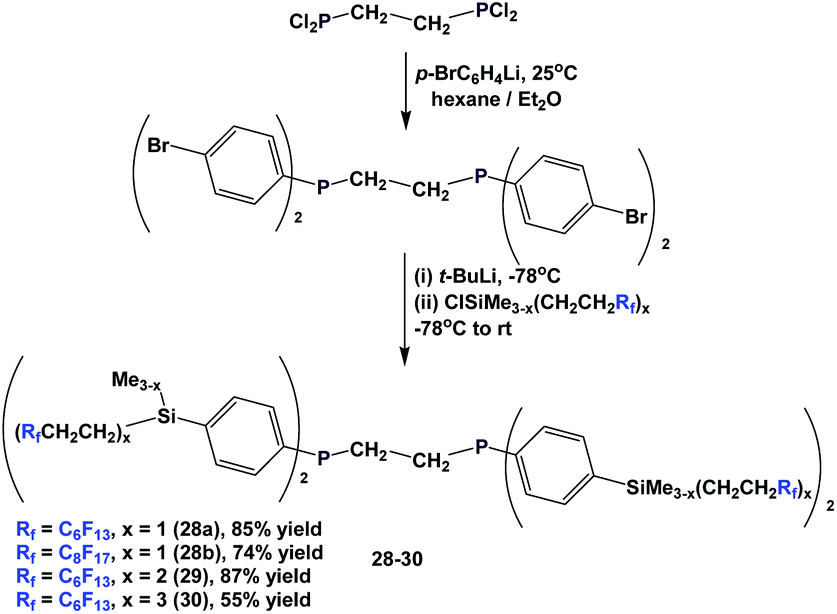 | ||
| Scheme 40 Synthesis of the fluorous diphosphines 28 to 30. | ||
Fluorous diphosphines 31a and 31b were synthesized in an analogous manner to 18 (Scheme 41).23
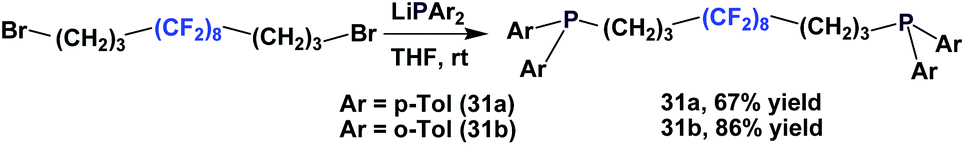 | ||
| Scheme 41 Synthesis of alkyl diarylphosphines 31a and 31b. | ||
Fluorous diphosphines 32 and 33 were synthesized by two different approaches, similar to Schemes 30 and 31 (Scheme 42).26,27 Both 32 and 33 were protected as their phosphine sulfides, with the Ph2P–PPh2 approach yields were 76% and 78% respectively. With the Ph2P(O)C(O)Mes approach, the yield of the protected 33 was 96%.
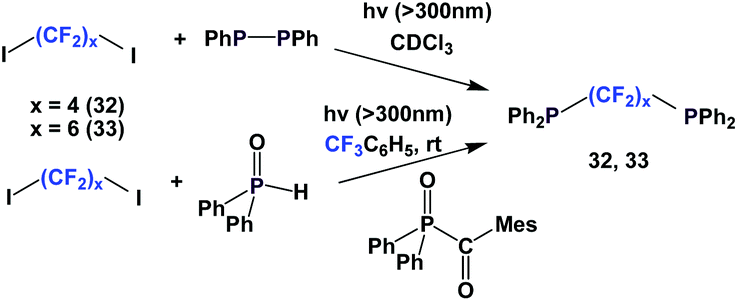 | ||
| Scheme 42 Synthesis of diphosphines 32 and 33. | ||
Applications of fluorous alkyl aryl phosphines
Perfluorodecyl diphenylphosphine 24g was used as a ligand for a Pd-catalyst for various coupling reactions (Scheme 43).26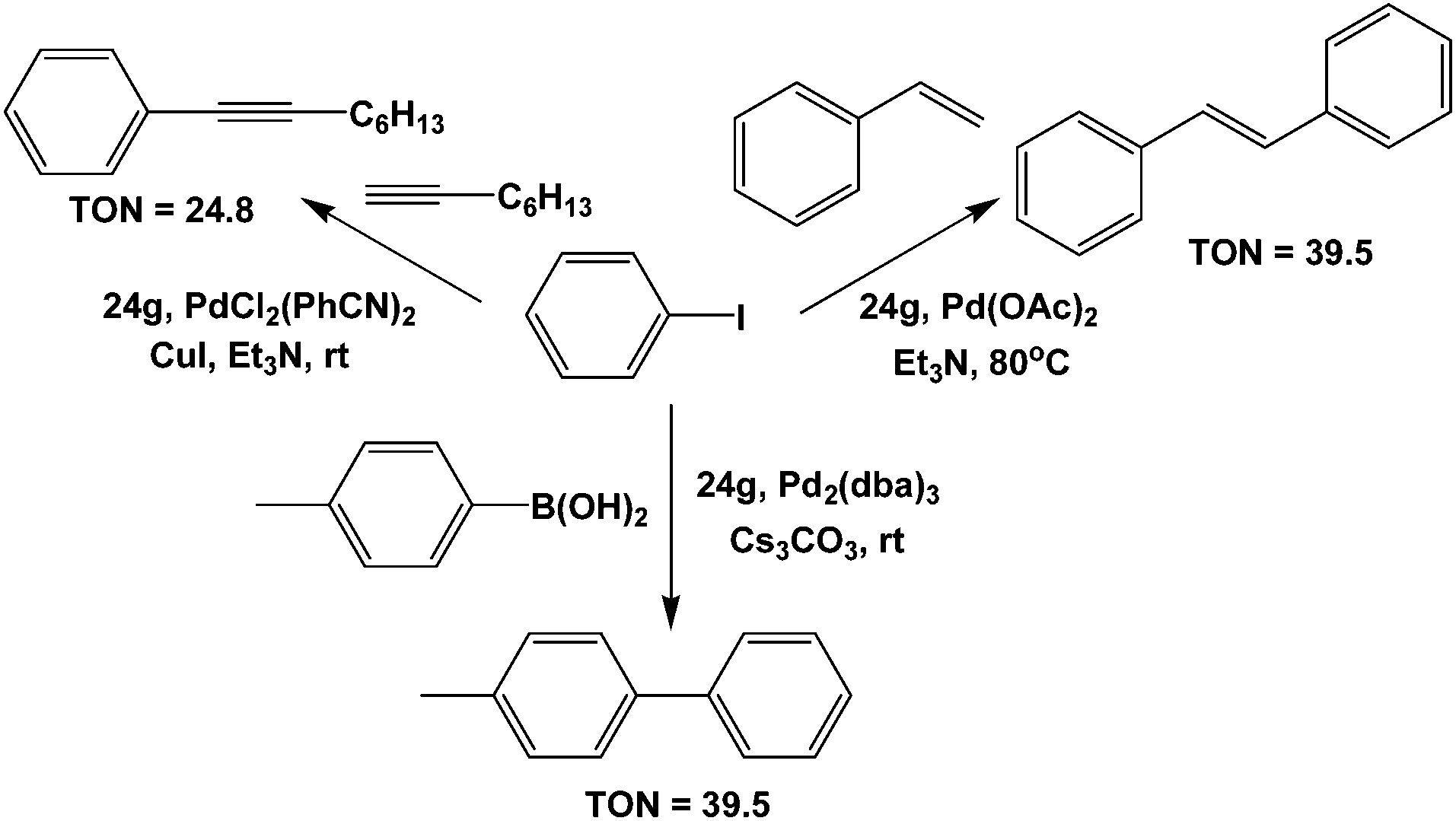 | ||
| Scheme 43 Coupling reactions catalyzed by Pd complexes of 24g. | ||
Duncan et al. have made the Ni, Pd and Pt complexes from fluorous diphosphine 27. The Pd complex catalyzed the Heck coupling reaction between methyl acrylate and an aryl halide (Scheme 44).45 The catalyst was recovered by a fluorous solid-phase extraction, and reused at least 4 times without any loss in catalytic activity.
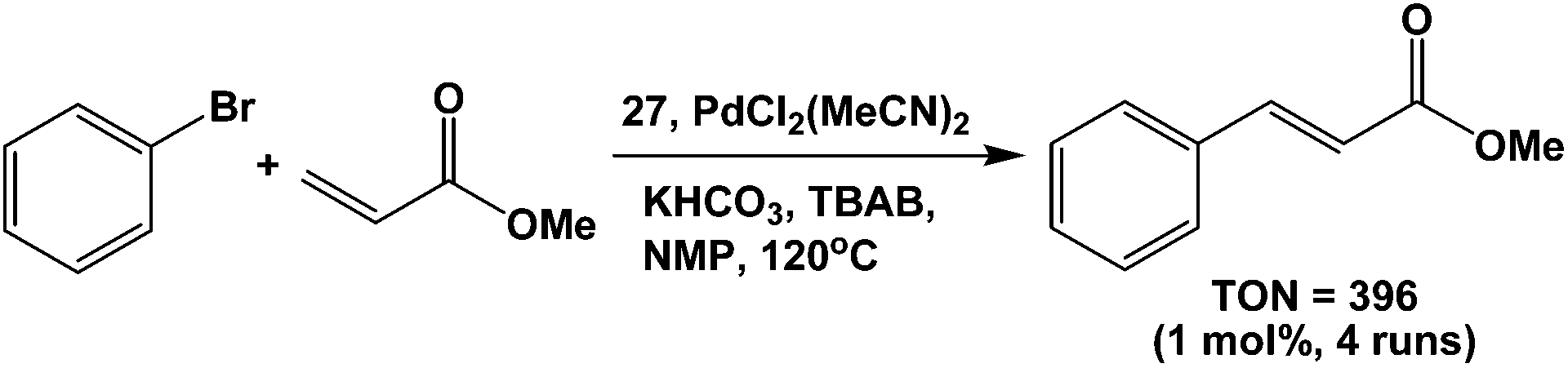 | ||
| Scheme 44 Heck coupling catalyzed by a palladium complex of 27. | ||
Fluorous triaryl phosphines
Fluorous triaryl phosphines have aryl groups attached to the phosphorus atom and they contain numerous fluorous ponytails attached to the aryl rings.The first fluorous triaryl phosphine was 34, which had a perfluorooctyl group in the para-position of the perfluorophenyl ring (Scheme 45).47
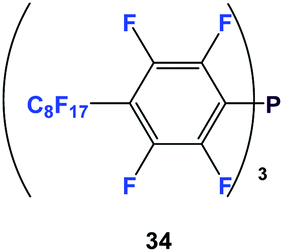 | ||
| Scheme 45 Fluorous triaryl phosphine 34. | ||
Synthetic strategies
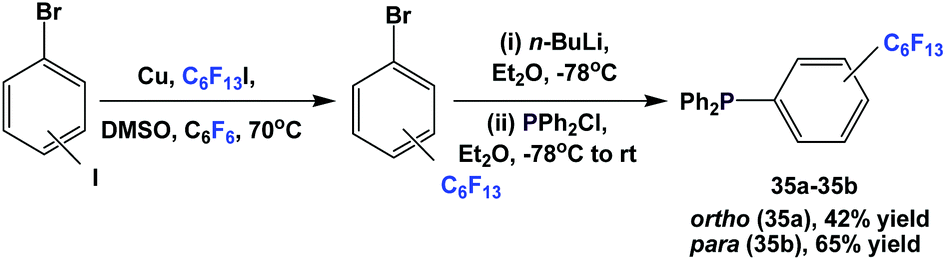 | ||
| Scheme 46 Synthesis of fluorous triaryl phosphines 35a and 35b. | ||
Fluorous triaryl phosphines 36a and 36b were synthesized by a two-step procedure (Scheme 47).48a Two ponytails were attached to one of the aryl rings. The authors reported the yields to be in the range of 40–70%.
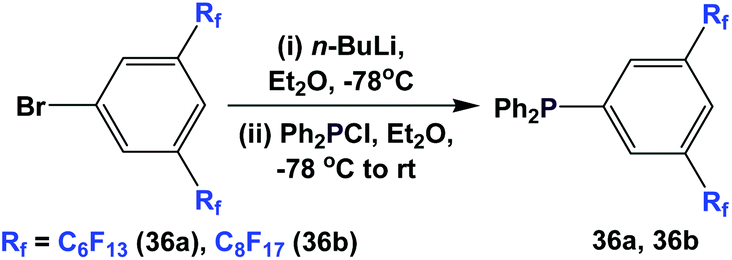 | ||
| Scheme 47 Synthesis of fluorous triaryl phosphines 36a and 36b. | ||
Fluorous triaryl phosphine 37, which contains a C6F13(CH2)2-ponytail, was made using a four-step procedure (Scheme 48).49 A fluorous organozinc reagent was generated and it was coupled with a di-halobenzene using a Pd(0) catalyst at 45 °C to give 1-bromo-4-(perfluoroethyl)benzene. Subsequent lithiation and phosphination led to 37.
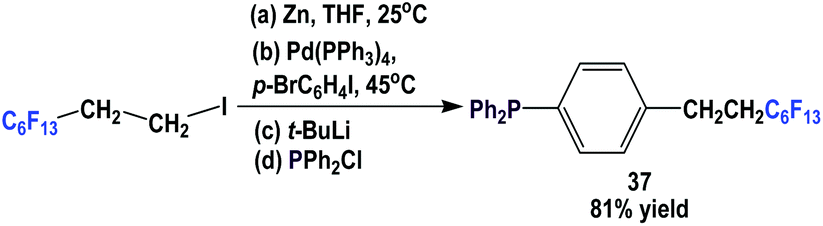 | ||
| Scheme 48 Synthesis of fluorous triaryl phosphine 37. | ||
Fluorous triaryl phosphine 38 possessed a highly branched ponytail, and it was synthesized by a four-step process (Scheme 49).49 1,1,1,3,4,4,5,5,5-Nonafluoro-2-(trifluoro-methyl)pent-2-ene was reacted with caesium fluoride at an elevated temperature, and then with a benzyl bromide. The resulting fluorous aryl bromide was subjected to lithiation followed by phosphination to give 38.
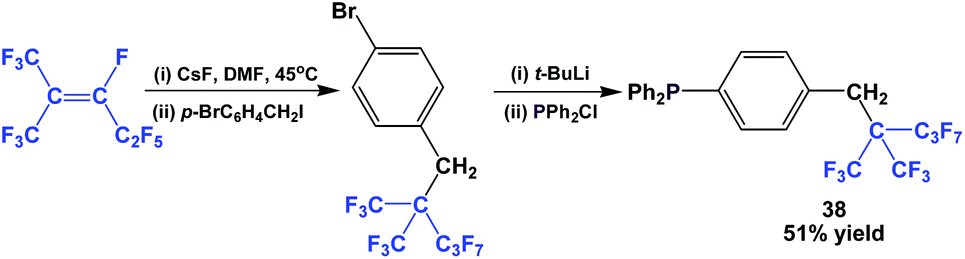 | ||
| Scheme 49 Fluorous triaryl phosphine 38 with a branched ponytail. | ||
Fluorous triaryl phosphines PhP-[C6H4(CH2)x(Rf)n]2
Fluorous triaryl phosphine 39 was synthesized in an analogous manner as shown in Scheme 46 (Scheme 50).12a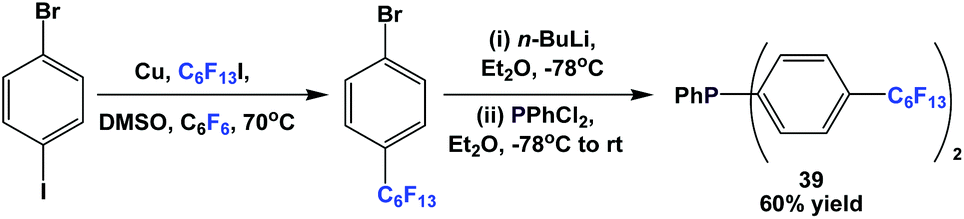 | ||
| Scheme 50 Synthesis of fluorous triaryl phosphine 39. | ||
Fluorous triaryl phosphines 40a and 40b were synthesized analogously to the approach in Scheme 47 (Scheme 51).48a Two types of ponytails could be attached onto the aryl rings. The reported yields were also in the range of 40–70%.
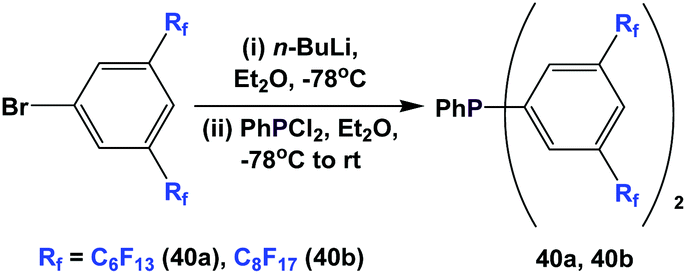 | ||
| Scheme 51 Synthesis of fluorous triaryl phosphines 40a and 40b. | ||
Fluorous triaryl phosphine 41 was synthesized in a four-step procedure similar to Scheme 48 (Scheme 52).49 The yield was better than 37, reaching 90%.
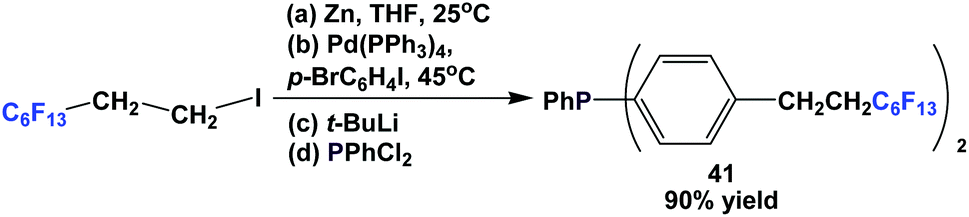 | ||
| Scheme 52 Synthesis of fluorous triaryl phosphine 41. | ||
The fluorous triaryl phosphine 42, with a branched ponytail, was synthesized similarly to Scheme 49 (Scheme 53).49 Again, the yield was much higher than for 38.
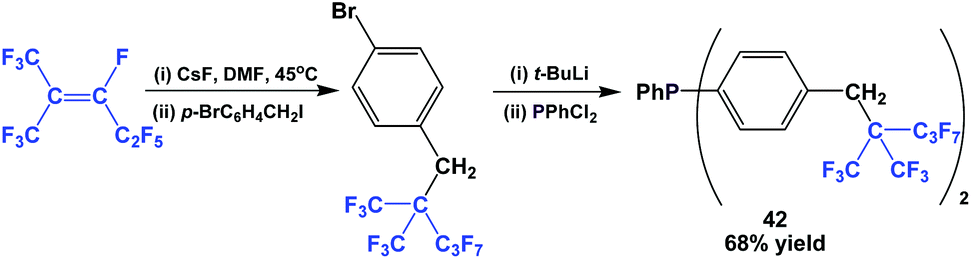 | ||
| Scheme 53 Synthesis of fluorous phenyl phosphine 42. | ||
Fluorous triaryl phosphines P-[C6H4-(CH2)x(Rf)n]3
Fluorous triaryl phosphine 43 was synthesized by the reaction of 1-bromo-4-(perfluorohexyl)benzene with n-BuLi at a low temperature, and then with phosphorus trichloride (Scheme 54).12a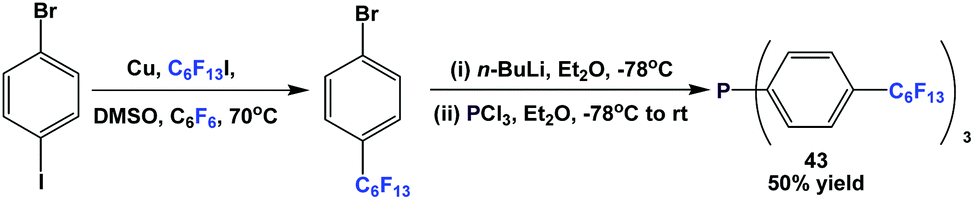 | ||
| Scheme 54 Synthesis of fluorous triaryl phosphine 43. | ||
Mixed fluorous triaryl phosphines with 2 different types of ponytails, 44a and 44b, were synthesized using a two-step procedure (Scheme 55).48a Yields were in the range of 40–70%.
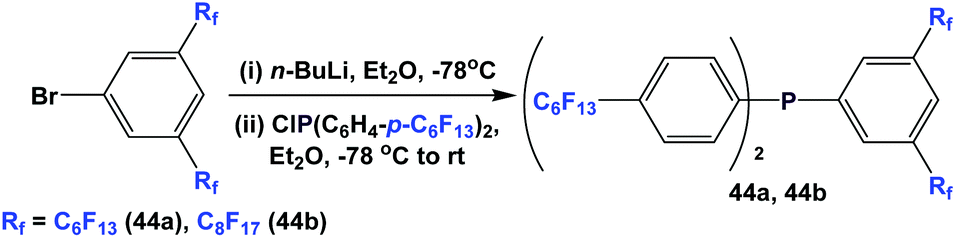 | ||
| Scheme 55 Synthesis of fluorous triaryl phosphines 44a and 44b. | ||
The fluorous triaryl phosphine 45a, with three C6F13CH2CH2-ponytails, was synthesized similarly to the approaches presented in Schemes 48 and 52 (Scheme 56).49
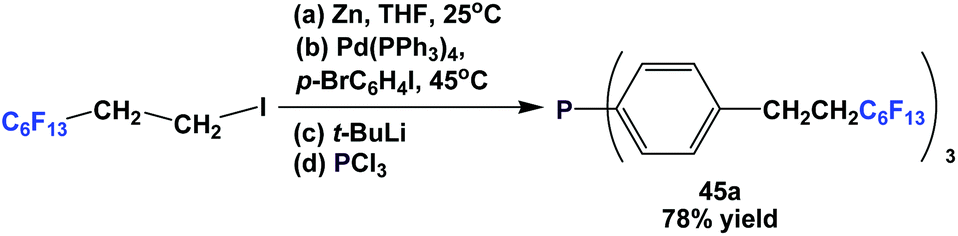 | ||
| Scheme 56 Synthesis of fluorous triaryl phosphine 45a. | ||
The fluorous triaryl phosphine 46, containing a branched ponytail, was synthesized (Scheme 57).49
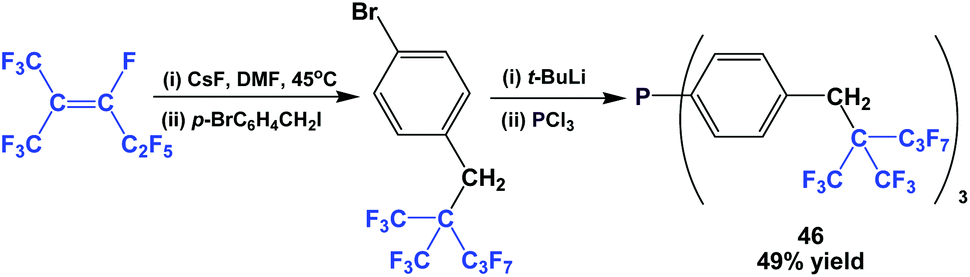 | ||
| Scheme 57 Fluorous triaryl phosphine 46 with a branched ponytail. | ||
Fluorous triaryl phosphines 47a–47c, which contained propylene spacers, were synthesized through a multi-step process. Wittig olefination, homogeneous hydrogenation, lithiation, and phosphination afforded the phosphines with three different types of ponytails (Scheme 58).50
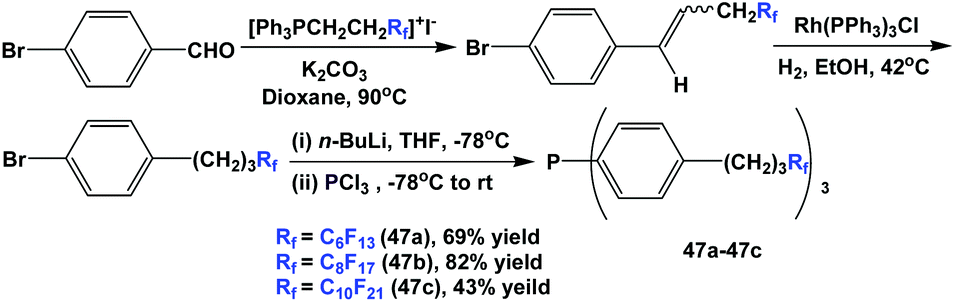 | ||
| Scheme 58 Synthesis of fluorous triaryl phosphines 47a–47c with (CH2)3 spacers. | ||
An even longer spacer can be used: this is illustrated by the synthesis of fluorous triaryl phosphine 48 (Scheme 59).51 A Heck coupling was the key C–C bond-forming reaction. Fluorous triaryl phosphine 48 was protected as its borane complex, with a yield of 64%.
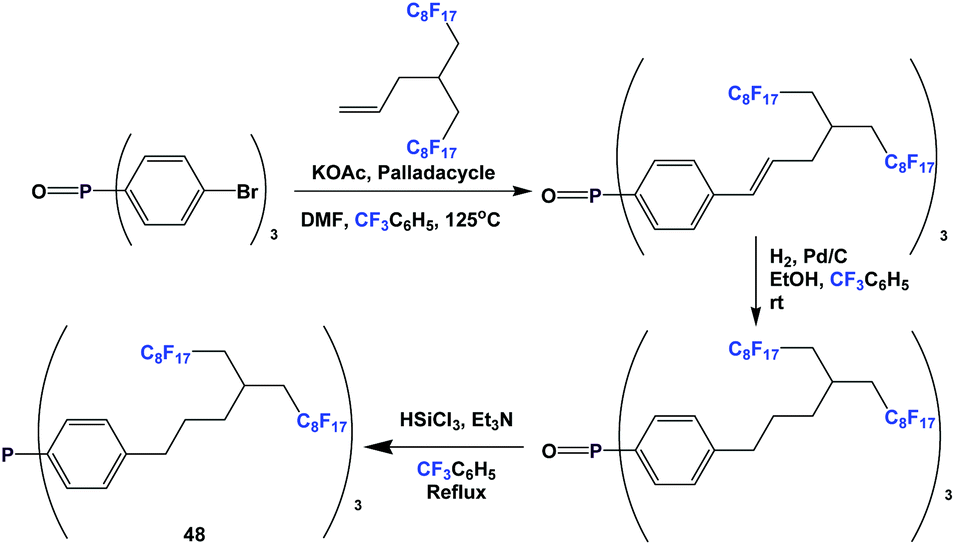 | ||
| Scheme 59 Synthesis of fluorous triaryl phosphine 48. | ||
Fluorous triaryl phosphines with silicon-containing spacers, 49–51, were also synthesized using 2 similar approaches (Scheme 60).52a–c,53 The attachment of the fluorous, silicon-containing spacers could be carried out before or after the lithiation–phosphination sequence, demonstrating the flexibility of the approach. Extensive investigations, including the application of combinatorial chemistry, have been conducted to optimize reaction yields.
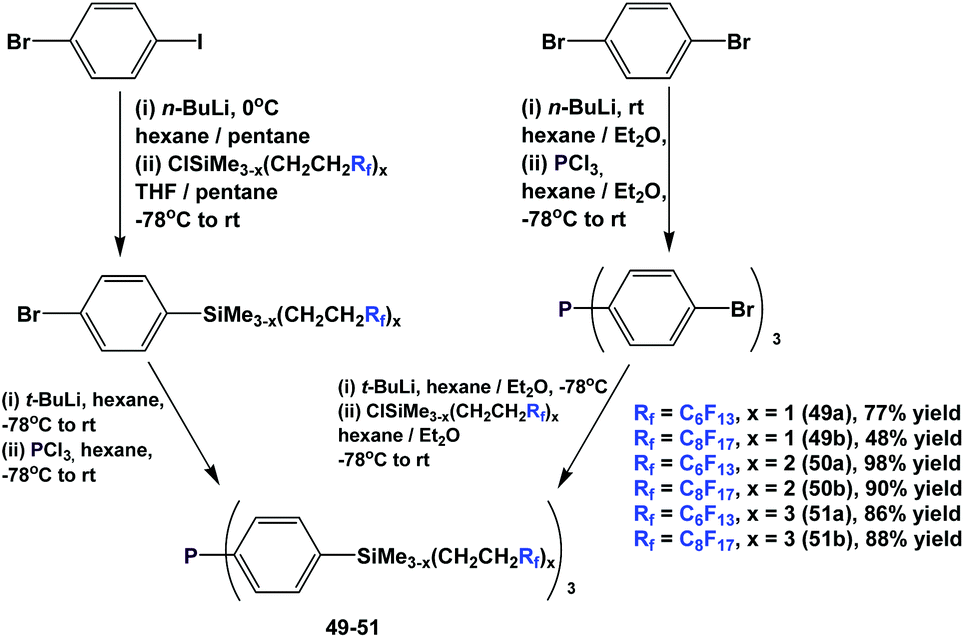 | ||
| Scheme 60 Synthesis of fluorous triaryl phosphines 49–51. | ||
Fluorous BINAP-type chiral phosphine 52a was synthesized through a nickel-catalyzed coupling reaction (Scheme 61).54a
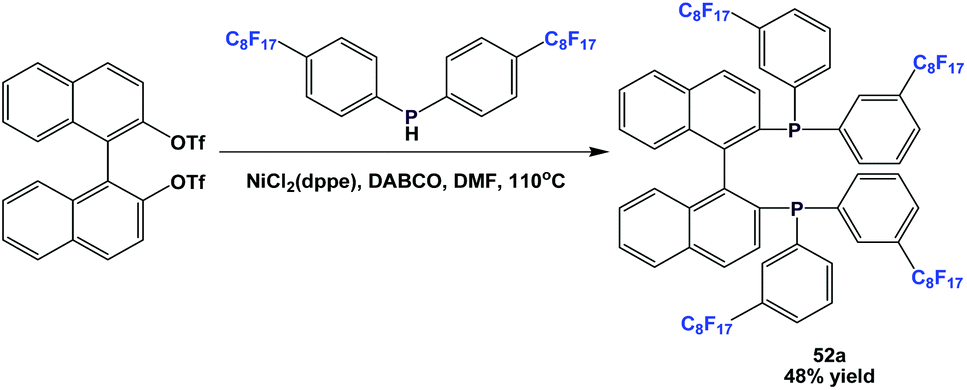 | ||
| Scheme 61 Synthesis of fluorous BINAP-type phosphine 52a. | ||
More variants, 52b and 52c, were synthesized by a similar approach (Scheme 62).54a
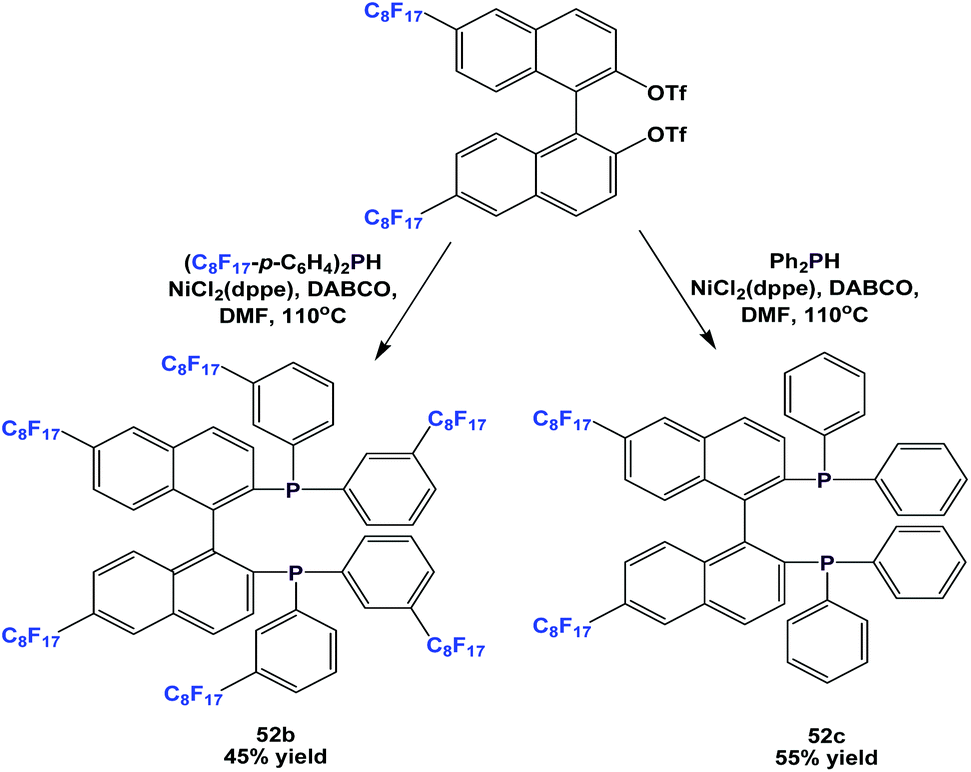 | ||
| Scheme 62 Synthesis of fluorous phosphines 52b and 52c. | ||
There are other variants of fluorous BINAP and MOP ligands, with perfluoroalkyl, perfluoroether, or perfluoroalkyl-silicon spacers attached (52d, 52e, 53–57) (Scheme 63).55,56a–d
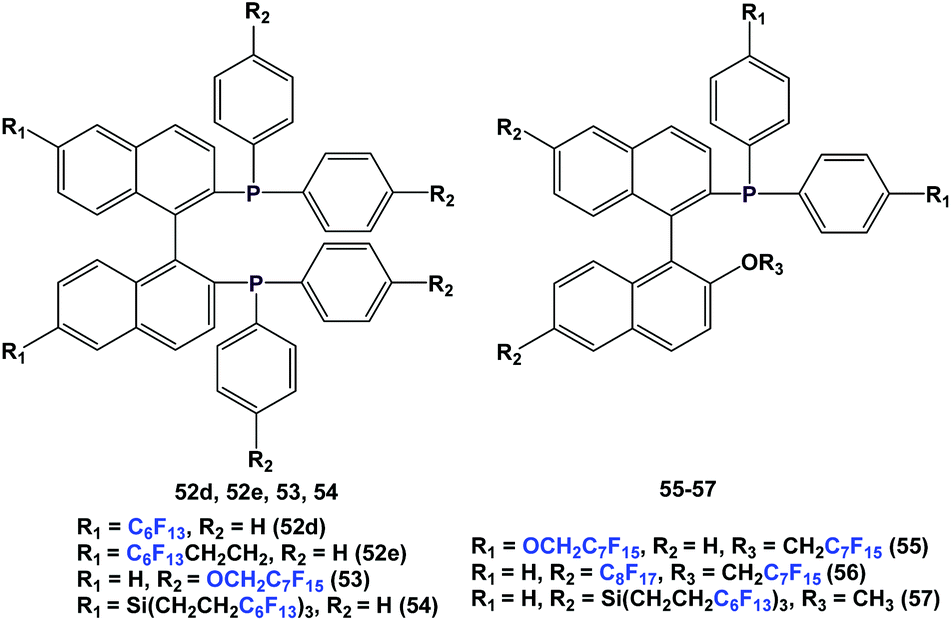 | ||
| Scheme 63 Fluorous BINAP and MOP ligands. | ||
Applications of fluorous triaryl phosphines
Fluorous triaryl phosphines, like their trialkyl counterparts, were also studied in a number of important catalytic reactions, and many transition metal complexes of these phosphine ligands were prepared and used as catalyst precursors.The rhodium complexes of fluorous triaryl phosphines 36a, 39 and 44a were tested in the hydroformylation of 1-octene in perfluorocarbon solvents at 20 bar CO/H2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (Scheme 64).48a,57
:1) (Scheme 64).48a,57
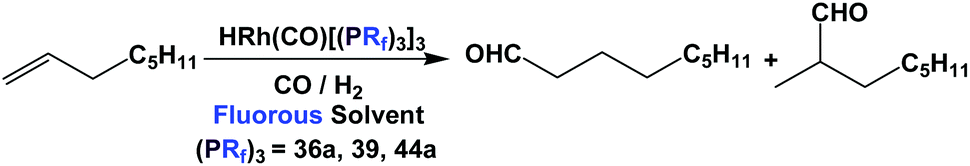 | ||
| Scheme 64 Hydroformylation of 1-octene in a fluorous solvent. | ||
It is interesting to compare the performance of the three fluorous triaryl phosphines, which contained two phenyl rings (36a), one phenyl ring (39) and a mixed triaryl combination (44a), respectively. While all three fluorous phosphines gave comparable results in terms of conversion and rate, the bis-meta-perfluoroalkyl substitution in 39 was less selective to the desired linear aldehyde product than the other two. It was proposed that the bulkiness of 39 led to a different catalytic mechanism, resulting in lower linear selectivity.48a
The fluorous triaryl phosphine 43 was used as a ligand in the hydroformylation of 1-octene at 20 bar CO/H2 (1:1).58a With a rhodium loading of 2 mmol dm−3 (P:Rh ratio = 1:10), the catalyst achieved a TOF of 29800 at 90 °C.
Aghmiz et al. have used fluorous phosphine 58 as a ligand in the hydroformylation of 1-octene (Scheme 65).58c Using 0.05 mol% of catalyst and 40 bar CO/H2 (1:1), they achieved a TOF of 1040 h−1 with the normal-to-iso product ratio as 74:26.
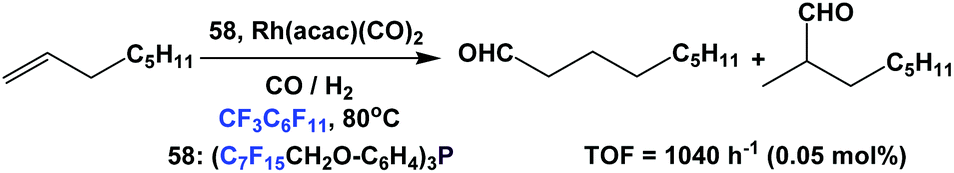 | ||
| Scheme 65 Hydroformylation of 1-octene. | ||
Koch and Leitner have used fluorous triaryl phosphine 45a as a ligand in the hydroformylation of 1-octene at 20 bar CO/H2 (1:1) in supercritical carbon dioxide (Scheme 66).58d The TON reached 2440 when 0.038 mol% of a rhodium catalyst was used.
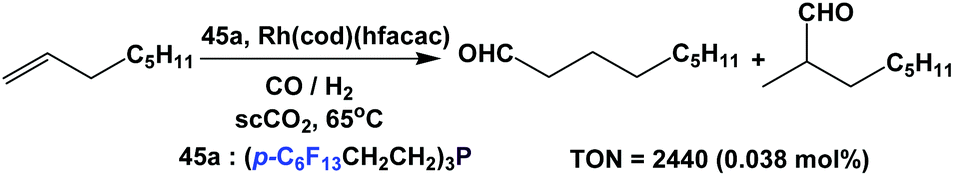 | ||
| Scheme 66 Hydroformylation of 1-octene in scCO2. | ||
The rhodium complex of fluorous phosphine 47b could catalyze the homogeneous hydrogenation of cyclohexenone to cyclohexanone under either mono-phasic or bi-phasic conditions (Scheme 67).50 On the other hand, Friesen et al. have synthesized a rhodium complex which incorporates a phenyl phosphine with a perfluoro-polyalkylether (PFPAE) tail.59 The resulting phenyl phosphine rhodium complex was also a great catalyst for homogeneous hydrogenation of similar substrates. Using 0.5 mol% of catalyst, a TOF of 23.45 h−1 was achieved at 45 °C.59
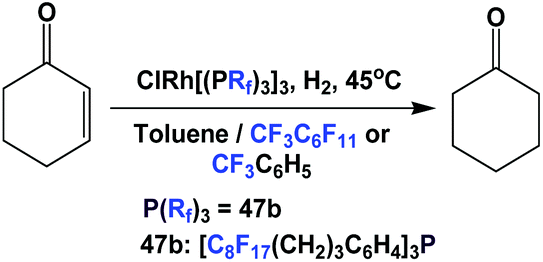 | ||
| Scheme 67 Homogeneous hydrogenation of cyclohexenone. | ||
The palladium complex of fluorous phosphine 50a was used to catalyze the methoxycarbonylation of styrene (Scheme 68).60 It is interesting to see that the fluorous phosphine modified palladium catalyst showed higher selectivity towards the branched product. A TOF of 250 h−1 was achieved.
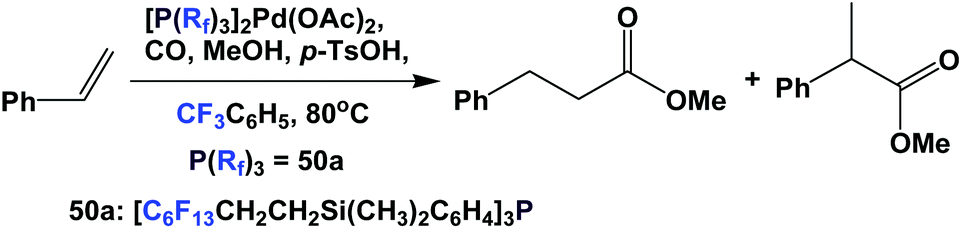 | ||
| Scheme 68 Methoxycarbonylation of styrene. | ||
Rhodium complexes of fluorous triaryl phosphines 49a and 50b catalyzed the hydrosilylation of n-hexene (Scheme 69).61 For both fluorous phosphine rhodium catalysts, they were recycled twice without noticeable loss of activity, though the authors discovered that losses of rhodium occurred in both cases – 12% for 49a and 1.7% for 50b.
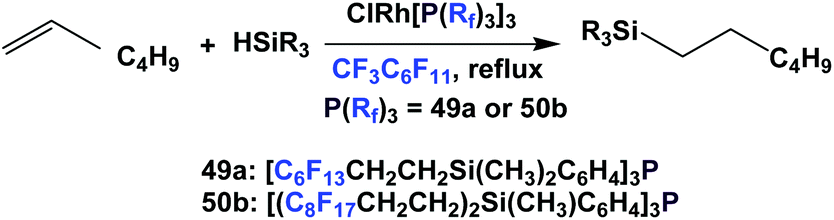 | ||
| Scheme 69 Hydrosilylation of n-hexene. | ||
The ‘fluorous Wilkinson's catalysts’ prepared from fluorous phosphines 46a and 46b were used to catalyze the hydrogenation of 1-octene to octane with 1 bar H2 (Scheme 70).62 The catalysis using 46b could achieve a cumulative TON of 3117 after 9 cycles, and the rhodium leaching in both cases was less than 0.5%.
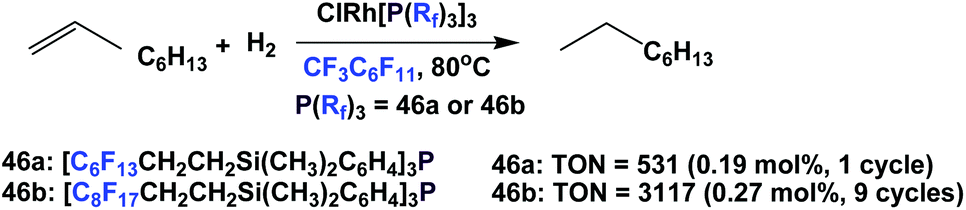 | ||
| Scheme 70 Hydrogenation of 1-octene. | ||
The cationic rhodium complexes of BINAP ligands 52a and 52b were synthesized. These fluorous BINAP rhodium complexes catalyzed the hydrogenation of styrene in supercritical CO2 (Scheme 71).54a They found that by adding methanol as a co-solvent into the system, the conversions improved, which was likely due to an increase of the polarity of the reaction media.
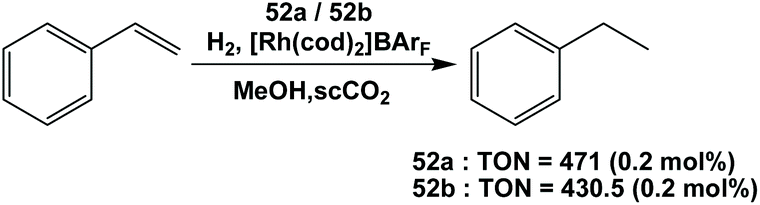 | ||
| Scheme 71 Hydrogenation of styrene. | ||
Fluorous BINAP chiral phosphines 52d and 52e were used in the asymmetric hydrogenation of dimethyl itaconate (Scheme 72).58c Enantiomeric excesses higher than 95% could be achieved for both fluorous BINAP chiral phosphines.
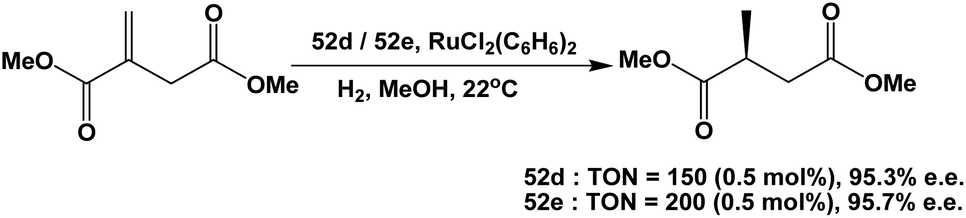 | ||
| Scheme 72 Homogeneous hydrogenation of dimethyl itaconate. | ||
The fluorous phosphine 54 has been used in the asymmetric Heck reaction between an aromatic chloro-triflate and a dihydrofuran (Scheme 73).55 The enantiomeric excess could reach as high as 93%. The fluorous BINAP palladium catalyst solution, residing in the fluorous phase, could be recycled for further catalytic use.
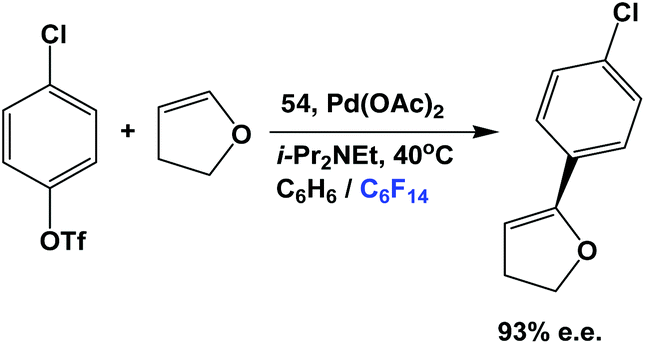 | ||
| Scheme 73 Asymmetric Heck reaction of a chloro-triflate and a dihydrofuran. | ||
Recently, Gladysz et al. have prepared two novel nickel salicylaldiminato phosphine complexes from fluorous phosphines 45b and 47b, and they catalyzed ethylene polymerization under both monophasic (toluene) and biphasic (fluorous/toluene) conditions (Scheme 74).63 The mechanism dictates the dissociation of the phosphine ligand from the nickel complex, which allows an ethylene molecule to bind to the active site and effects polymerization. The fluorous nature of the phosphine ligand led to its partition into the fluorous phase. The two fluorous phosphine nickel complexes proved to be faster catalysts than their Ni(Ph)(PPh3) analogue, and in both cases, the turnover frequency was better when the catalysis was carried out in a biphasic rather than a monophasic system.
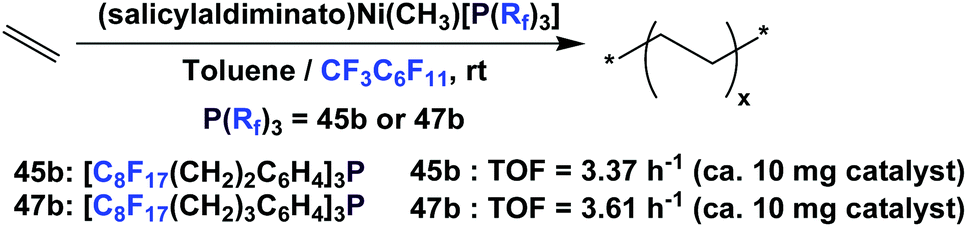 | ||
| Scheme 74 Fluorous phosphine nickel complex-catalyzed ethylene polymerization. | ||
Conclusions
The typical synthesis and basic properties of around 90 phosphines with one or more perfluoroalkyl substituents were reviewed. Their selected applications in biphasic, organometallic, or organo-catalysis were demonstrated. The advantages of fluorous phosphines are good to excellent recyclability and the tunability of catalytic activity.Acknowledgements
This work was funded by the City University Hong Kong, project number 9667113 (2015).Notes and references
- (a) B. Cornils and W. A. Herrmann, Applied Homogeneous Catalysis with Organometallic Compounds: A Comprehensive Handbook in Three Volumes (2nd Edition), Wiley-VCH, 2002 CrossRef; (b) A. E. C. Collis and I. T. Horváth, Catal. Sci. Technol., 2011, 1, 912 RSC.
- B. M. Trost, Science, 1991, 254, 1471 CrossRef CAS PubMed.
- (a) I. T. Horváth and J. Rábai, Science, 1994, 266, 72 CrossRef PubMed; (b) I. T. Horváth, Acc. Chem. Res., 1998, 31, 641 CrossRef; (c) J. A. Gladysz, D. P. Curran and I. T. Horváth, Handbook of Fluorous Chemistry, Wiley-VCH, 2004 CrossRef; (d) Fluorous Chemistry in Topics in Current Chemistry, ed. I. T. Horváth, Springer, Heidelberg, 2012 Search PubMed.
- (a) M. Skalický, et al. , Organometallics, 2012, 31, 1524 CrossRef; (b) M. Babuněk, et al. , J. Fluorine Chem., 2014, 161, 66 CrossRef.
- (a) M. Wende and J. A. Gladysz, J. Am. Chem. Soc., 2003, 125, 5861 CrossRef CAS PubMed; (b) M. Wende, R. Meier and J. A. Gladysz, J. Am. Chem. Soc., 2001, 123, 11490 CrossRef CAS PubMed; (c) K. Ishihara, S. Kondo and H. Yamamoto, Synlett, 2001, 1371 CrossRef CAS.
- L. V. Dinh and J. A. Gladysz, Angew. Chem., Int. Ed., 2005, 44, 4095 CrossRef CAS PubMed.
- (a) N. Kudo, N. Bandi, E. Suzuki, M. Katakura and Y. Kawashima, Chem.-Biol. Interact., 2000, 124, 119 CrossRef CAS PubMed; (b) K. P. Das, B. E. Grey, R. D. Zehr, C. R. Wood, J. L. Butenhoff, S.-C. Chang, D. J. Ehresman, Y.-M. Tan and C. Lau, Toxicol. Sci., 2008, 105, 173 CrossRef CAS PubMed.
- US EPA Long-Chain Perfluorinated Chemicals (PFCs) Action Plan (2009): http://www.epa.gov/oppt/existingchemicals/ pubs/actionplans/pfcs.html.
- I. T. Horváth, Changing Designer Issues in Fluorous Chemistry, 1st International Symposium of Fluorous Technologies, 2005 Search PubMed.
- (a) D. Y. Mikhaylov, T. V. Gryaznova, Y. B. Dudkina, F. M. Polyancev, S. K. Latypov, O. G. Sinyashin and Y. H. Budnikova, J. Fluorine Chem., 2013, 153, 178 CrossRef CAS; (b) S. Benefice-Malouet, H. Blancou and A. Commeyras, J. Fluorine Chem., 1985, 30, 171 CrossRef CAS.
- F. Langer, K. Puntener, R. Sturmer and P. Knochel, Tetrahedron: Asymmetry, 1997, 8, 715 CrossRef CAS.
- (a) P. Battacharyya, D. Gudmunsen, E. G. Hope, R. D. W. Kemmitt, D. R. Paige and A. M. Stuart, J. Chem. Soc., Perkin Trans. 1, 1997, 1, 3609 RSC; (b) E. G. Hope, J. Sherrington and A. M. Stuart, Adv. Synth. Catal., 2006, 348, 1365 CrossRef; (c) N. Audic, P. W. Dyer, E. G. Hope and A. M. Stuart, et al. , Adv. Synth. Catal., 2010, 352, 2241 CrossRef CAS.
- L. J. Alvey, D. Rutherford, J. J. J. Juliette and J. A. Gladysz, J. Org. Chem., 1998, 63, 6302 CrossRef CAS PubMed.
- (a) C. Emnet, R. Tuba and J. A. Gladysz, Adv. Synth. Catal., 2005, 347, 1819 CrossRef CAS; (b) C. Emnet and J. A. Gladysz, Synthesis, 2005, 1012 CAS.
- G. Vlád, F. U. Richter and I. T. Horváth, Tetrahedron Lett., 2005, 46, 8605 CrossRef.
- L. J. Alvey, R. Meier, T. Soós, P. Bernatis and J. A. Gladysz, Eur. J. Inorg. Chem., 2000, 1975 CrossRef CAS.
- D. C. Smith, Jr., E. D. Stevens and S. P. Nolan, Inorg. Chem., 1999, 38, 5277 CrossRef.
- M. F. Sellin, I. Bach, J. M. Webster, F. Montilla, V. Rosa, T. Avilés, M. Poliakoff and D. J. Cole-Hamilton, J. Chem. Soc., Dalton Trans., 2002, 4569 RSC.
- C. S. Consorti, F. Hampel and J. A. Gladysz, Inorg. Chim. Acta, 2006, 359, 4874 CrossRef CAS.
- R. Tuba, V. Tesevic, L. V. Dinh, F. Hampel and J. A. Gladysz, Dalton Trans., 2005, 2275 RSC.
- A. Klose and J. A. Gladysz, Tetrahedron: Asymmetry, 1999, 10, 2665 CrossRef CAS.
- G. Vlád, F. U. Richter and I. T. Horváth, Org. Lett., 2004, 6, 4559 CrossRef PubMed.
- L. de Quadras, J. Stahl, F. Zhuravlev and J. A. Gladysz, J. Organomet. Chem., 2007, 692, 1859 CrossRef CAS.
- J. J. Kampa, J. W. Nail and R. J. Lagow, Angew. Chem., Int. Ed., 1995, 34, 1241 CrossRef CAS.
- D. W. Allen, Organophosphorus Chem., 2014, 43, 1 CAS.
- S. Kawaguchi, Y. Minamida, T. Ohe, A. Nomoto, M. Sonoda and A. Ogawa, Angew. Chem., Int. Ed., 2013, 52, 1748 CrossRef CAS PubMed.
- Y. Sato, S.-I. Kawaguchi and A. Ogawa, Chem. Commun., 2015, 51, 10385 RSC.
- I. T. Horváth, G. Kiss, R. A. Cook, J. E. Bond, P. A. Stevens, J. Rábai and E. J. Mozeleski, J. Am. Chem. Soc., 1998, 120, 3133 CrossRef.
- J. J. J. Juliette, D. Rutherford, I. T. Horváth and J. A. Gladysz, J. Am. Chem. Soc., 1999, 121, 2696 CrossRef CAS.
- M.-A. Guillevic, C. Rocaboy, A. M. Arif, I. T. Horváth and J. A. Gladysz, Organometallics, 1998, 17, 707 CrossRef CAS.
- J. Fawcett, E. G. Hope, R. D. W. Kemmitt, D. R. Paige, D. R. Russell, A. M. Stuart, D. J. Cole-Hamilton and M. J. Payneb, Chem. Commun., 1997, 1127 RSC.
- (a) C. Li, S. P. Nolan and I. T. Horváth, Organometallics, 1998, 17, 452 CrossRef CAS; (b) V. Herrera, P. J. F. de Rege, I. T. Horváth, T. L. Husebo and R. P. Hughes, Inorg. Chem. Commun., 1998, 1, 197 CrossRef CAS; (c) F. G. Fontaine, M. A. Dubois and D. Zargarian, Organometallics, 2001, 20, 5156 CrossRef CAS.
- R. Tuba, R. Correea da Costa, H. S. Bazzi and J. A. Gladysz, ACS Catal., 2012, 2, 155 CrossRef CAS.
- R. Correa da Costa and J. A. Gladysz, Chem. Commun., 2006, 2619 RSC.
- R. Correa da Costa, F. Hampel and J. A. Gladysz, Polyhedron, 2007, 26, 581 CrossRef CAS.
- R. Tuba, E. N. Brothers, J. H. Reibenspies, H. S. Bazzi and J. A. Gladysz, Inorg. Chem., 2012, 51, 9943 CrossRef CAS PubMed.
- L. V. Dinh and J. A. Gladysz, New J. Chem., 2005, 29, 173 RSC.
- D. Lantos, M. Contel, A. Larreac, D. Szabó and I. T. Horváth, QSAR Comb. Sci., 2006, 25, 719 Search PubMed.
- T. J. Malosh and J. R. Shapley, J. Organomet. Chem., 2010, 695, 1776 CrossRef CAS.
- T. J. Malosh, S. R. Wilson and J. R. Shapley, Inorg. Chim. Acta, 2009, 362, 2849 CrossRef CAS.
- T. J. Malosh, S. R. Wilson and J. R. Shapley, J. Organomet. Chem., 2009, 694, 3331–3337 CrossRef CAS.
- M. C. Clough, T. Fiedler, N. Bhuvanesh and J. A. Gladysz, J. Organomet. Chem., 2015 Search PubMed , asap.
- F. O. Seidel and J. A. Gladysz, Adv. Synth. Catal., 2008, 350, 2443 CrossRef CAS.
- C. Gimbert, A. Vallribera, J. A. Gladysz and M. Jurisch, Tetrahedron Lett., 2010, 51, 4662 CrossRef CAS.
- D. Duncan, E. G. Hope, K. Singh and A. M. Stuart, Dalton Trans., 2011, 40, 1998 RSC.
- (a) E. de Wolf, B. Richter, B.-J. Deelman and G. van Koten, J. Org. Chem., 2000, 65, 5424 CrossRef CAS PubMed; (b) J. Van den Broeke, E. de Wolf, B. J. Deelman and G. Van Koten, Adv. Synth. Catal., 2003, 345, 625 CrossRef CAS; (c) E. de Wolf, A. L. Spek, B. W. M. Kuipers and A. P. Philipse, et al. , Tetrahedron, 2002, 58, 3911 CrossRef CAS.
- H. Gopal, C. E. Snyder Jr. and C. Tamborski, J. Fluorine Chem., 1979, 14, 511 CrossRef CAS.
- (a) D. J. Adams, J. A. Bennett, D. J. Cole-Hamilton, E. G. Hope, J. Hopewell, J. Kight, P. Pogorzelec and A. M. Stuart, Dalton Trans., 2005, 3862 RSC; (b) D. J. Adams, D. J. Cole-Hamilton, D. A. J. Harding and E. G. Hope, et al. , Tetrahedron, 2004, 60, 4079 CrossRef CAS.
- Q. Zhang, Z. Luo and D. P. Curran, J. Org. Chem., 2000, 65, 8866 CrossRef CAS PubMed.
- T. Soós, B. L. Bennett, D. Rutherford, L. P. Barthel-Rosa and J. A. Gladysz, Organometallics, 2001, 20, 3079 CrossRef.
- M. Wende, F. Seidel and J. A. Gladysz, J. Fluorine Chem., 2003, 124, 45 CrossRef CAS.
- (a) B. Richter, E. de Wolf, G. van Koten and B.-J. Deelman, J. Org. Chem., 2000, 65, 3885 CrossRef CAS PubMed; (b) E. L. V. Goetheer, A. W. Verkerk, L. J. P. Van den Broeke and E. De Wolf, et al. , J. Catal., 2003, 219, 126 CrossRef CAS; (c) B. Richter, B. J. Deelman and G. Van Koten, J. Mol. Catal. A: Chem., 1999, 145, 317 CrossRef CAS.
- E. de Wolf, E. Riccomagno, J. J. M. de Pater, B.-J. Deelman and G. van Koten, J. Comb. Chem., 2004, 6, 363 CrossRef CAS PubMed.
- (a) H. Altinel, G. Avsarb, M. K. Yilmaza and B. Guzel, J. Supercrit. Fluids, 2009, 51, 202 CrossRef CAS; (b) D. J. Birdsall, E. G. Hope, A. M. Stuart and W. Chen, et al. , Tetrahedron Lett., 2001, 42, 8551 CrossRef CAS; (c) Y. Hu, D. J. Birdsall, A. M. Stuart and E. G. Hope, et al. , J. Mol. Catal. A: Chem., 2004, 219, 57 CrossRef CAS.
- Y. Nakamura, S. Takeuchi, S. Zhang, K. Okumura and Y. Ohgo, Tetrahedron Lett., 2002, 43, 3053 CrossRef CAS.
- (a) M. Cavazzini, S. Quici, G. Pozzi, D. Maillard and D. Sinou, Chem. Commun., 2001, 1220 RSC; (b) D. Sinou, D. Maillard, A. Aghmiz and A. M. M. Bulto, Adv. Synth. Catal., 2003, 345, 603 CrossRef CAS; (c) J. Bayardon, M. Cavazzini, D. Maillard, G. Pozzi, S. Quici and D. Sinou, Tetrahedron: Asymmetry, 2003, 14, 2215 CrossRef CAS; (d) D. Maillard, J. Bayardon, J. D. Kurichiparambil, C. Nguefack-Fournier and D. Sinou, Tetrahedron: Asymmetry, 2002, 13, 1449 CrossRef CAS.
- B. Croxtall, J. Fawcett, E. G. Hope and A. M. Stuart, J. Chem. Soc., Dalton Trans., 2002, 491 RSC.
- (a) D. F. Foster, D. J. Adams, D. Gudmunsen, A. M. Stuart, E. G. Hope and D. J. Cole-Hamilton, Chem. Commun., 2002, 722 RSC; (b) E. G. Hope and A. M. Stuart, J. Fluorine Chem., 1999, 100, 75 CrossRef CAS; (c) A. Aghmiz, C. Claver, A. M. Masdeu-Bulto and D. Maillard, et al. , J. Mol. Catal. A: Chem., 2004, 208, 97 CrossRef CAS; (d) D. Koch and W. Leitner, J. Am. Chem. Soc., 1998, 120, 13398 CrossRef CAS.
- C. M. Friesen, C. D. Montgomery and S. A. J. U. Temple, J. Fluorine Chem., 2012, 144, 24 CrossRef CAS.
- J. J. M. de Pater, B.-J. Deelman, C. J. Elsevier and G. van Koten, J. Mol. Catal. A: Chem., 2006, 258, 334 CrossRef CAS.
- E. de Wolf, E. A. Speets, B.-J. Deelman and G. van Koten, Organometallics, 2001, 20, 3686 CrossRef CAS.
- B. Richter, A. L. Spek, G. van Koten and B.-J. Deelman, J. Am. Chem. Soc., 2000, 122, 3945 CrossRef CAS.
- Z. Xi, H. S. Bazzi and J. A. Gladysz, J. Am. Chem. Soc., 2015, 137, 10930 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Prof. Trost on the occasion of his 75th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qo00115g |
| This journal is © the Partner Organisations 2016 |