DOI:
10.1039/C6QO00037A
(Research Article)
Org. Chem. Front., 2016,
3, 412-422
Efficient synthesis of 2-acylquinolines based on an aza-vinylogous Povarov reaction†
Received
21st January 2016
, Accepted 15th February 2016
First published on 15th February 2016
Abstract
The aza-vinylogous Povarov reaction between α-ketoimines and α,β-unsaturated dimethylhydrazones in the presence of indium trichloride affords 2-acyl-4-alkyl-4-dimethylhydrazonomethyl-1,2,3,4-tetrahydroquinolines with good cis/trans diastereoselectivities. These compounds were readily transformed into highly substituted 2-acylquinolines using a two-reaction sequence involving the oxidative generation of a C-4 nitrile group, followed by its elimination under thermal conditions. Alternatively, a one-pot protocol based on the reaction of hydrazones with magnesium monoperoxyphthalate hexahydrate in refluxing methanol afforded the target 2-acylquinolines in good to excellent yields. This methodology was also extended to the preparation of 2-arylquinolines.
Introduction
Quinoline is one of the most interesting heterocycles, present in a wide range of natural products1 and pharmaceutically relevant compounds.2,3 Furthermore, quinoline derivatives also have interesting applications as agrochemicals, metal-coordinating moieties in new materials such as OLED phosphorescent compounds and structural fragments of ligands useful in organocatalysis.4 Due to its importance, the synthesis of the quinoline nucleus has been studied for well over a century, the classical methods5 including the well-known Skraup,6 Doebner–Miller,7 Friedländer8 and Combes9 reactions, among others. However, these methods often suffer from disadvantages such as the need for a large excess of certain reagents, strong acids as promoters, harsh reaction conditions and complex experimental protocols, besides poor regioselectivities in some cases. In recent years, many new approaches have been developed,10 most of which are based on transition-metal-catalyzed reactions11 but the use of expensive and often toxic metal catalysts can be viewed as a disadvantage. Furthermore, neither the classical nor the modern methods have sufficient generality to provide access to certain types of quinoline derivatives. In this context, the development of mild and efficient routes to previously inaccessible quinoline frameworks, preferably avoiding the use of transition metals, is still a worthwhile and challenging task. One relevant example is the case of quinolines bearing a carbonyl substituent at C-2, which comprises compounds with a variety of pharmacological activities,12 but whose preparation with the currently available methodologies either requires multi-step sequences12d,13 or is low-yielding and/or very restricted in scope, being limited to the case R2 = CO2Et14 or to very few examples (in some cases, a single one) of 2-acyl substituents.15 The Povarov reaction, i.e., the formal [4 + 2] cycloaddition between N-arylimines and electron-rich olefins,16 is one of the main methods for the synthesis of tetrahydroquinolines.17 It can be adapted for the preparation of quinolines by coupling cycloaddition to an oxidation step,18 or by employing acetylenes as dienophiles. Neither these methods nor some more general, transition metal-catalysed Povarov-like reactions,11h,i have been applied for the preparation of quinolines bearing a C-2 carbonyl substituent other than an ester group and, furthermore, the former strategy is restricted in most cases to the synthesis of quinolines with a 4-aryl substituent.19
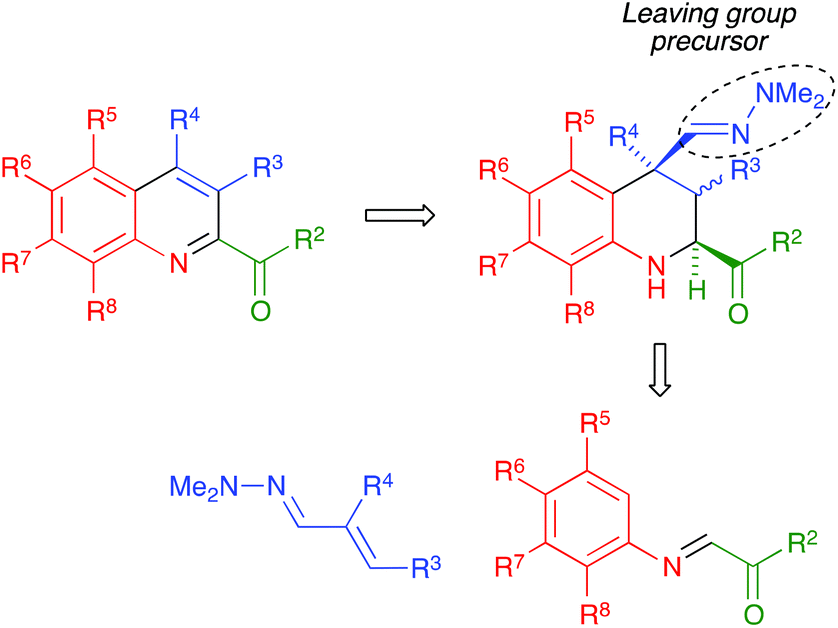
We present in this article a general solution to the problem of 2-acylquinoline synthesis, using the strategy summarized in Scheme 1. We have recently reported that the indium trichloride-catalyzed reaction between aromatic imines and α,β-unsaturated N,N-dimethylhydrazones affords 1,2,3,4-tetrahydroquinolines bearing a hydrazone function at the quaternary C-4 position via a one-pot diastereoselective process that can be viewed as an aza-vinylogous Povarov reaction and constitutes the first example of an α,β-unsaturated dimethylhydrazone behaving as a dienophile in a hetero Diels–Alder reaction.20 We describe here the generalization of this reaction to include the case of α-ketoimines, and a simple and efficient process allowing the transformation of the dimethylhydrazone moiety at C-4 of the resulting tetrahydroquinolines into a good leaving group and its subsequent elimination with concomitant dehydrogenation, to furnish 2-acylquinolines. In the cases when C-4 was not quaternary, the aromatization was compatible with the retention of a functional group at this position.
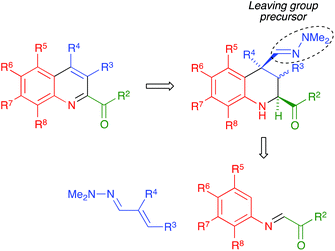 |
| | Scheme 1 Our synthetic plan for the preparation of quinolines bearing a C-2 carbonyl substituent. | |
Results and discussion
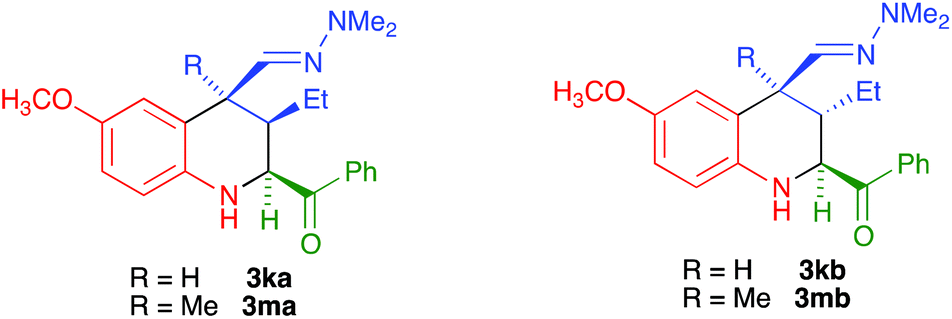
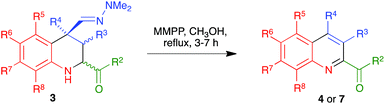
While the use of ethyl glyoxylate in Povarov chemistry is relatively well established,21 to our knowledge glyoxal derivatives have only been previously employed by Zhu,22 although with a limited structural variation in the glyoxal component. On the other hand, the use of these substrates in our reaction was almost unexplored.20 Therefore, our first task was the generalization of our aza-vinylogous Povarov reaction in the presence of a variety of α-ketoimines. As summarized in Scheme 2 and Table 1, α-oxoimines 1, which were prepared by mixing the corresponding aniline and α-dicarbonyl starting materials, were treated with several α,β-unsaturated N,N-dimethylhydrazones 2 at room temperature, in acetonitrile containing indium trichloride as a Lewis acid catalyst, to afford a variety of structurally varied 2-acyl-1,2,3,4-tetrahydroquinolines 3, which could be substituted at all the quinoline positions. The R2 substituent is normally a phenyl moiety, either unsubstituted (entries 1, 4, 5 and 11–13) or bearing electron-withdrawing (entries 2 and 6) or electron-releasing (entry 3) groups. Heteroaryl substituents could also be accommodated, as shown by the existence of a 2-furyl unit in entry 10. The presence of an ester group at C-2 was also possible (entries 7–9). The reactions leading to 3-unsubstituted tetrahydroquinolines (entries 1–10 and 12) show an 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 to 1:0 diastereoselection in favour of the isomer having a cis relationship between the 4-dimethylhydrazonomethyl and 2-acyl groups, with both substituents being equatorial. The only exception was compound 3f (entry 6), for which diastereoselection was lost; this was attributed to steric interference between the C-5 and C-4 substituents when the latter is equatorial. In the cases of compounds 3k and 3m, bearing a C-3 ethyl substituent, the C-2 and C-4 substituents were cis, with complete selectivity, and the group at C-3 can be cis or trans, with no selectivity. Therefore, these compounds were obtained as ca. 1:1 mixtures of compounds 3ka/3kb and 3ma/3mb. Since our final goal was the preparation of aromatic compounds, we only carried out the full characterization of one of these diastereomers, namely compound 3ka (see the ESI† for a summary of the NOE studies leading to these stereochemical conclusions).
:2 to 1:0 diastereoselection in favour of the isomer having a cis relationship between the 4-dimethylhydrazonomethyl and 2-acyl groups, with both substituents being equatorial. The only exception was compound 3f (entry 6), for which diastereoselection was lost; this was attributed to steric interference between the C-5 and C-4 substituents when the latter is equatorial. In the cases of compounds 3k and 3m, bearing a C-3 ethyl substituent, the C-2 and C-4 substituents were cis, with complete selectivity, and the group at C-3 can be cis or trans, with no selectivity. Therefore, these compounds were obtained as ca. 1:1 mixtures of compounds 3ka/3kb and 3ma/3mb. Since our final goal was the preparation of aromatic compounds, we only carried out the full characterization of one of these diastereomers, namely compound 3ka (see the ESI† for a summary of the NOE studies leading to these stereochemical conclusions).
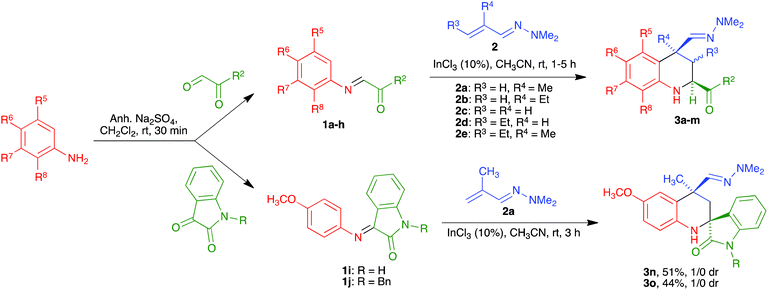 |
| | Scheme 2 Synthesis of 1,2,3,4-tetrahydroquinolines 3a–o. | |
Table 1 Scope of the synthesis of 1,2,3,4-tetrahydroquinolines bearing a C-2 carbonyl substituent
| Entry |
Starting imine |
Comp. |
R2 |
R3 |
R4 |
R5 |
R6 |
R7 |
R8 |
Time, h |
Yield (%) |
dr |
|
Cis:trans diastereoselection in 2,4-disubstituted compounds.
Known in the literature.20
The C-2 and C-4 substituents are cis and the group at C-3 can be cis or trans, with no selectivity.
|
| 1 |
1a
|
3a
|
C6H5 |
H |
CH3 |
H |
OCH3 |
H |
H |
3 |
72 |
82:18a |
| 2 |
1b
|
3b
|
4-FC6H4 |
H |
CH3 |
H |
OCH3 |
H |
H |
3 |
75 |
84:16a |
| 3 |
1c
|
3c
|
4-OCH3C6H4 |
H |
CH3 |
H |
OCH3 |
H |
H |
3 |
70 |
83:17a |
| 4 |
1a
|
3d
|
C6H5 |
H |
CH2CH3 |
H |
OCH3 |
H |
H |
3 |
63 |
91:09a |
| 5 |
1d
|
3e
|
C6H5 |
H |
CH3 |
H |
CH3 |
H |
CH3 |
4 |
95 |
98:02a |
| 6 |
1e
|
3f
|
4-FC6H4 |
H |
CH3 |
CH3 |
H |
CH3 |
H |
5 |
50 |
50:50a |
| 7 |
1f
|
3g
|
OEt |
H |
CH3 |
H |
OCH3 |
H |
H |
2 |
88b |
100:0a |
| 8 |
1g
|
3h
|
OEt |
H |
CH3 |
H |
CH3 |
H |
CH3 |
2 |
86 |
84:16a |
| 9 |
1f
|
3i
|
OEt |
H |
CH2CH3 |
H |
OCH3 |
H |
H |
2 |
74 |
87:13a |
| 10 |
1h
|
3j
|
2-Furyl |
H |
CH3 |
H |
OCH3 |
H |
H |
3 |
88b |
100:0a |
| 11 |
1a
|
3k
|
C6H5 |
CH2CH3 |
H |
H |
OCH3 |
H |
H |
1.5 |
66 |
50:50c |
| 12 |
1a
|
3l
|
C6H5 |
H |
H |
H |
OCH3 |
H |
H |
1 |
72 |
100:0a |
| 13 |
1a
|
3m
|
C6H5 |
CH2CH3 |
CH3 |
H |
OCH3 |
H |
H |
1.5 |
73 |
57:43c |
Finally, although it was not directly applied to our planned quinoline synthesis, in order to complete the study of the scope of the aza-vinylogous Povarov reaction with α-oxoimines we also verified the viability of the reaction involving isatin-derived imines 1i and 1j (Scheme 2). This reaction afforded spiro compounds 3n and 3o as single diastereomers.23
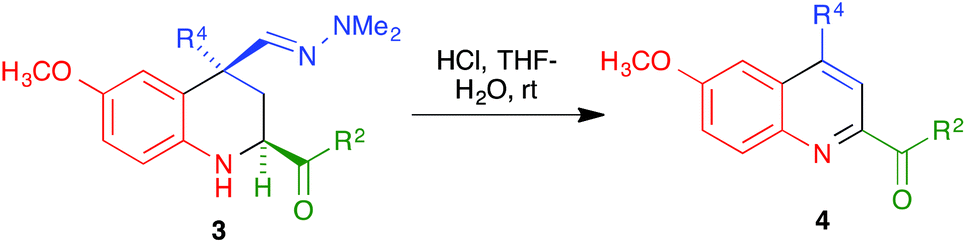
With compounds 3 in hand, we examined several routes potentially allowing their transformation into quinoline derivatives. In the first place, we studied the acid hydrolysis of the hydrazine group in a few representative tetrahydroquinolines 3 in order to obtain the corresponding aldehyde, which we planned to aromatize subsequently. Interestingly, as shown in Scheme 3 and Table 2, treatment of compounds 3a–d and 3g with hydrochloric acid in a THF–water reaction medium in an air-open flask afforded directly the corresponding quinolines 4, although the yields were only moderate in most cases and poor for the compound bearing a bulkier substituent at C-4.
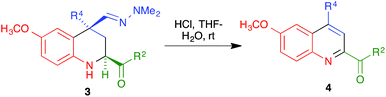 |
| | Scheme 3 Acid-promoted transformation of compounds 3 into quinolines 4. | |
Table 2 Yields for the preparation of quinolines 4 under acidic conditions
| Entry |
Comp. |
R2 |
R4 |
Time, h |
Yield, % |
| 1 |
4a
|
C6H5 |
CH3 |
24 |
76 |
| 2 |
4b
|
4-FC6H4 |
CH3 |
7 |
55 |
| 3 |
4c
|
4-OCH3C6H4 |
CH3 |
24 |
48 |
| 4 |
4d
|
C6H5 |
CH2CH3 |
24 |
25 |
| 5 |
4g
|
OEt |
CH3 |
20 |
95 |
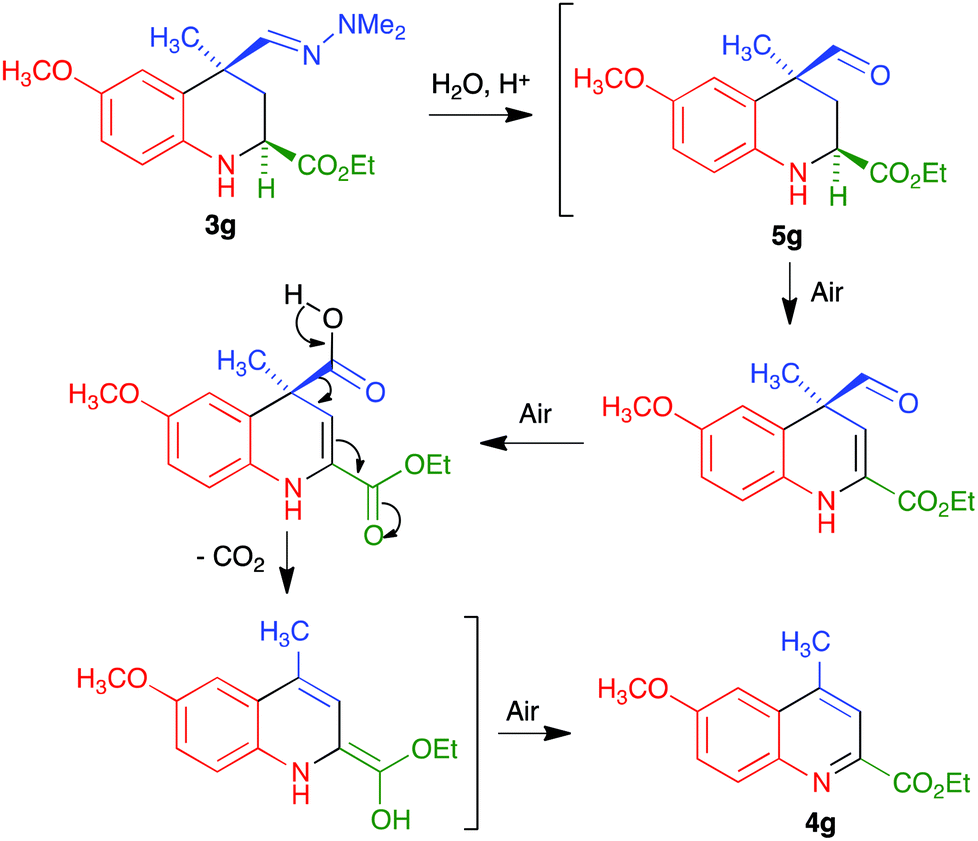
We propose the mechanism summarized in Scheme 4 to explain the one-pot transformation of compounds 3 into 4 in the presence of an acid, particularized for the case of 3g. Acidic hydrolysis to aldehyde 5g would be followed by dehydrogenation in the presence of atmospheric oxygen, probably assisted by captodative stabilization24 of an intermediate C-2 radical due to the presence of the adjacent carbonyl group, and oxidation of the aldehyde to a carboxylic acid. This intermediate would then evolve to the final product by decarboxylation followed by keto–enol tautomerism and aromatization by air-promoted dehydrogenation.
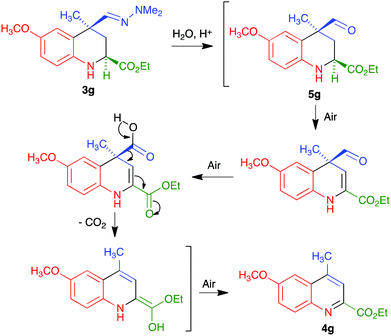 |
| | Scheme 4 Mechanism proposed to explain the transformation of 3 into 4. | |
Although we did not carry out a detailed mechanistic study, the above proposal has some experimental support. Thus, we isolated compound 5g by hydrolyzing the hydrazone function in 3g under mild conditions (CuCl2 hydrate at room temperature in THF–water25), and verified that this compound was readily transformed into 4g under our reaction conditions. The assistance from the C-2 carbonyl substituent for both the dehydrogenation and aromatization steps was confirmed by the fact that the aldehyde similar to 5g but bearing a C-2 phenyl substituent instead of an ester, which is known in the literature,25b was stable under the same conditions.
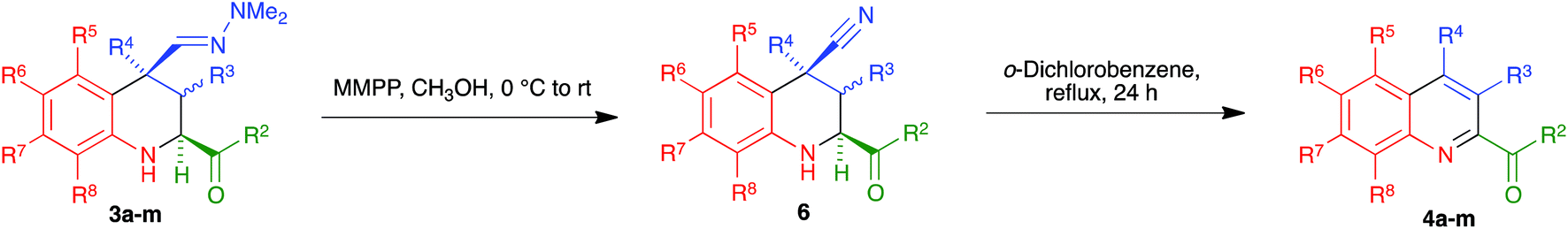
In an effort to improve the yields of our quinoline synthesis, we resorted to an alternative aromatization strategy, based on the oxidation of the hydrazone function to a nitrile followed by elimination and dehydrogenation. To this end, after some experimentation, we discovered that treatment of compounds 3 with magnesium monoperoxyphthalate hexahydrate (MMPP)26 in methanol at room temperature allowed the clean preparation of the corresponding nitriles 6. These compounds were then transformed into the target quinolines 4 by refluxing them in o-dichlorobenzene, via air-induced dehydrogenation with loss of hydrogen cyanide (Scheme 5 and Table 3). Importantly from the point of view of efficiency and user-friendliness, both the hydrazones 3 and the nitriles 6 could be employed as cis–trans mixtures without any loss in yield of the final quinolines 4.
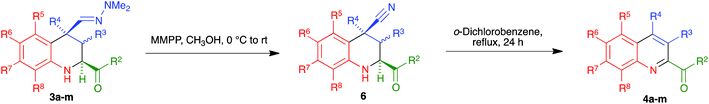 |
| | Scheme 5 Synthesis of quinolines 4a–m from tetrahydroquinolines 3. | |
Table 3 Scope of the synthesis of 1,2,3,4-tetrahydroquinolines bearing a C-2 carbonyl substituent
| Entry |
Comp. |
R2 |
R3 |
R4 |
R5 |
R6 |
R7 |
R8 |
Compounds 6 |
Compounds 4 |
| Time, h |
Yield, % |
Yield, % |
|
Together with 14% of compound 4b.
Compound 6d was not isolated. Instead, the MMPP oxidation afforded directly the corresponding quinoline 4d.
Together with 32% of compound 4i.
These reactions afforded the 2-acyl-4-cyanoquinolines 7a and 7b (see Scheme 6 below).
|
| 1 |
a
|
C6H5 |
H |
CH3 |
H |
OCH3 |
H |
H |
2 |
72 |
90 |
| 2 |
b
|
4-FC6H4 |
H |
CH3 |
H |
OCH3 |
H |
H |
2 |
70a |
97 |
| 3 |
c
|
4-OCH3C6H4 |
H |
CH3 |
H |
OCH3 |
H |
H |
2 |
89 |
88 |
| 4 |
d
|
C6H5 |
H |
CH2CH3 |
H |
OCH3 |
H |
H |
1.5 |
—b |
98 |
| 5 |
e
|
C6H5 |
H |
CH3 |
H |
CH3 |
H |
CH3 |
0.5 |
98 |
95 |
| 6 |
f
|
4-FC6H4 |
H |
CH3 |
CH3 |
H |
CH3 |
H |
1 |
93 |
91 |
| 7 |
g
|
OEt |
H |
CH3 |
H |
OCH3 |
H |
H |
1 |
60 |
97 |
| 8 |
h
|
OEt |
H |
CH3 |
H |
CH3 |
H |
CH3 |
2 |
83 |
97 |
| 9 |
i
|
OEt |
H |
CH2CH3 |
H |
OCH3 |
H |
H |
1 |
63c |
98 |
| 10 |
j
|
2-Furyl |
H |
CH3 |
H |
OCH3 |
H |
H |
0.4 |
78 |
98 |
| 11 |
k
|
C6H5 |
CH2CH3 |
H |
H |
OCH3 |
H |
H |
4 |
Not isolated |
—d |
| 12 |
l
|
C6H5 |
H |
H |
H |
OCH3 |
H |
H |
2 |
Not isolated |
—d |
| 13 |
m
|
C6H5 |
CH2CH3 |
CH3 |
H |
OCH3 |
H |
H |
4 |
69 |
87 |
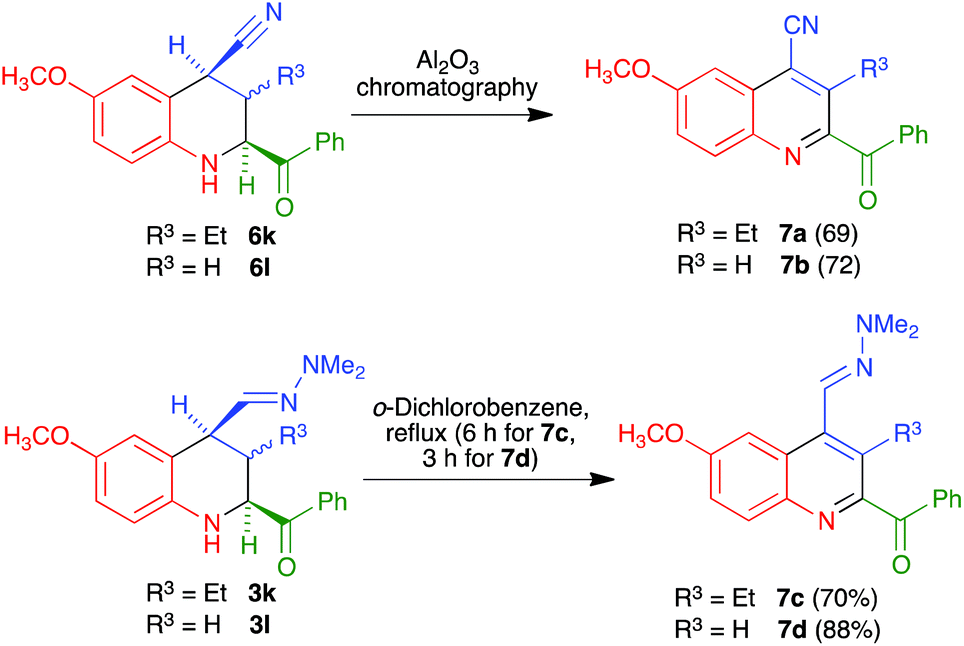
Interestingly, for the case R4 = H an alternative aromatization process took place, which allowed the preparation of quinolines that maintained the C-4 nitrile substituent. Thus, when crude nitriles 6k and 6l were subjected to our usual protocol of heating in refluxing o-dichlorobenzene, they afforded the quinoline-4-carbonitrile derivatives 7a and 7b in excellent yields. We subsequently found that the heating step could be avoided, since simple chromatography of the crude nitriles in basic alumina afforded the quinoline-4-carbonitrile derivatives in similar yields. The peculiar behaviour of these compounds can be attributed to the acidity of their H-4 proton, which is simultaneously benzylic and adjacent to the nitrile group. The formation of a C-4 anion in the presence of basic alumina in the chromatographic column may explain their easy dehydrogenation by atmospheric oxygen. On the other hand, in order to achieve the presence of a further functional group at C-4, we also examined the dehydrogenation of hydrazones 3k and 3l to furnish quinolines 7c and 7d, and found that it required harsher conditions involving reflux in o-dichlorobenzene (Scheme 6). This is consistent with the proposed mechanism, in view of the lower acidity of the protons α to a dimethylhydrazone group in comparison with those α to a nitrile.
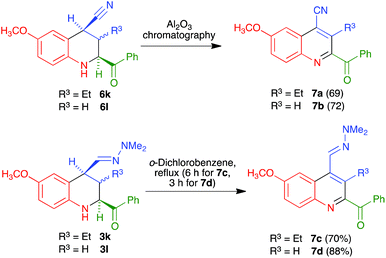 |
| | Scheme 6 Synthesis of C4-functionalized 2-acylquinolines. | |
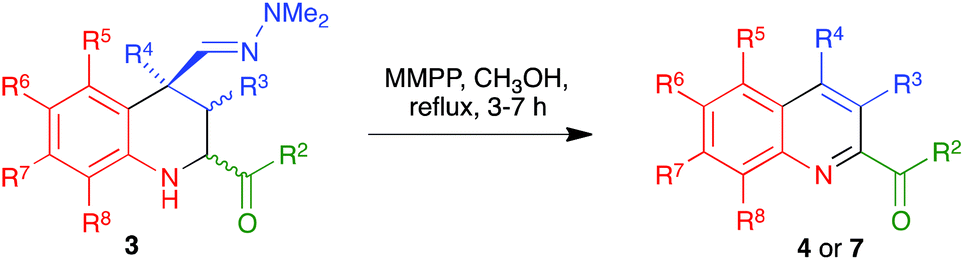
The reactions in refluxing o-dichlorobenzene were very efficient, but had the disadvantage of requiring high temperatures and a tedious procedure for the elimination of this high-boiling solvent. In an effort to overcome these shortcomings, we examined the use of alternative oxidants for the last step of our sequence. Furthermore, we reasoned that performing both reactions with the same oxidant, besides simplifying the experimental protocol, might set the stage for the development of a method for the transformation of the Povarov products into the target 2-acylquinolines in a single operation. With this goal in mind, we examined the use of MMPP for the aromatization step, eventually discovering that the use of 2 eq. of this reagent in refluxing methanol allowed carrying out the whole procedure in a single operation (Scheme 7). As summarized in Table 4, this new protocol generally afforded improved overall yields and was experimentally more convenient than the two-step procedure described above.
 |
| | Scheme 7 Direct, one-pot synthesis of 2-acylquinolines 4 and 7 from Povarov products. | |
Table 4 Results of the one-pot synthesis of 2-acylquinolines 4 and 7
| Entry |
Compound |
Time, h |
Yield, % |
| 1 |
4a
|
7 |
74 |
| 2 |
4b
|
3 |
78 |
| 3 |
4c
|
4 |
76 |
| 4 |
4d
|
5 |
79 |
| 5 |
4e
|
3 |
74 |
| 6 |
4f
|
5 |
80 |
| 7 |
4g
|
6 |
75 |
| 8 |
4h
|
5 |
81 |
| 9 |
4i
|
4 |
77 |
| 10 |
4j
|
6 |
85 |
| 11 |
4m
|
7 |
80 |
| 12 |
7a
|
7 |
82 |
| 13 |
7b
|
7 |
78 |
Finally, we have studied the preparation of one of the acylquinolines on a ca. 10-fold larger scale. Thus, the preparation of compound 4a was carried out in 40% overall yield for the whole process starting from 4.5 mmol of imine 1a, while the yield on our usual working scale was 53%. At this scale, an increase in the reaction time from 7 h to 18 h was necessary for the one-pot oxidation process in the presence of MMPP.
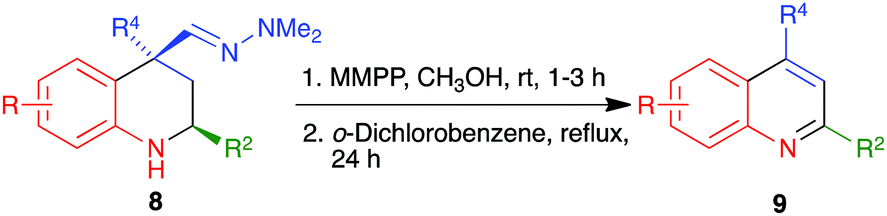
Compounds bearing 2-arylquinoline structural fragments have a broad range of biological activities that include antiproliferative27 and antifungal28 properties, modulation of HIV transcription29 and inhibition of hepatitis C NS3/4A protease,30 among many others. For this reason, we examined the application of our method to the synthesis of these compounds from the known20 2-aryl Povarov adducts (compounds 8). As shown in Scheme 8 and Table 5, the two-step protocol consisting of the oxidation of the dimethylhydrazones 8 with MMPP at room temperature followed by aromatization of the crude nitriles by prolonged heating in refluxing o-dichlorobenzene afforded excellent yields of the target 2-arylquinolines 9. In this case, the one-pot protocol involving heating the starting materials 8 with MMPP in refluxing methanol gave complex mixtures. For the characterization of the intermediate nitriles, see the ESI.†
 |
| | Scheme 8 Synthesis of 2-arylquinolines 9 from Povarov products 8. | |
Table 5 Results of the synthesis of 2-arylquinolines 9
| Entry |
Comp. |
R |
R2 |
R4 |
Yield, % |
| 1 |
9a
|
6-OCH3 |
C6H5 |
CH3 |
92 |
| 2 |
9b
|
6-OCH3 |
4-ClC6H4 |
CH3 |
86 |
| 3 |
9c
|
6-OCH3 |
4-OCH3C6H4 |
CH3 |
87 |
| 4 |
9d
|
6-OCH3 |
4-CH3C6H4 |
CH3 |
86 |
| 5 |
9e
|
6,8-(CH3)2 |
C6H5 |
CH3 |
95 |
| 6 |
9f
|
6-CH3 |
4-OCH3C6H4 |
CH3 |
92 |
| 7 |
9g
|
6,8-(CH3)2 |
C6H5 |
CH2CH3 |
89 |
| 8 |
9h
|
5,7-(OCH3)2 |
4-CH3C6H4 |
CH3 |
95 |
Experimental
General experimental details
All reagents and solvents were of commercial quality and were used as received. Reactions were monitored by thin layer chromatography on aluminium plates coated with silica gel and a fluorescent indicator. Separations by flash chromatography were performed using a Combiflash Teledyne automated flash chromatograph or on conventional silica gel columns. Melting points were measured with a Kofler-type heating plate microscope, model 723 from Reichert, and are uncorrected. Infrared spectra were recorded with a Perkin-Elmer FTIR Paragon-1000 spectrophotometer as thin films on NaCl disks; wavenumbers are given in cm−1. NMR spectroscopy data were recorded using spectrometers at the CAI de Resonancia Magnética, UCM, operating at 250 MHz for 1H NMR and 63 or 125 MHz for 13C NMR; chemical shifts are given in (δ) parts per million and coupling constants (J) in hertz. Elemental analyses were performed at the CAI de Microanálisis, Universidad Complutense, using a Leco CHNS-932 combustion microanalyzer.
General procedure for the synthesis of imines 1.
A solution of aniline (1 eq.) and aldehyde (1 eq.) in CH2Cl2 was stirred vigorously in the presence of anhydrous Na2SO4 (5 g) for 30 min. The filtered solution was evaporated to dryness affording the desired imines, which were used for the next step without further purification. Some commercially available glyoxals (4-methoxyphenyl and 4-fluorophenyl) are available as hydrates containing an unknown amount of water and therefore the amount of aldehyde used in these cases was optimized by gradual addition until no aniline was detected by 1H-NMR. Characterization data for imines 1 can be found in the ESI.†
General procedure for the aza-vinylogous Povarov reactions.
To a stirred solution of imine 1 (1 eq., 1.0 mmol) and InCl3 (10 mol%) in acetonitrile (20 ml) was added hydrazone 2 (1.2 eq., 1.2 mmol). Stirring was continued for the time period specified in Table 1 and after the completion of the reaction, as indicated by TLC, the reaction mixture was diluted with water (10 ml), extracted with CH2Cl2 (4 × 10 ml), dried and evaporated. Purification of the resulting crude was achieved through flash chromatography using a Combiflash Teledyne automated chromatograph, eluting with a gradient from neat petroleum ether to 9:1 petroleum ether–ethyl acetate. This purification afforded cis–trans mixtures, with the dr values specified in Table 1, which were used as such for the subsequent reactions. The major cis isomers were isolated and characterized in most cases, and to this end the mixtures were purified by silica gel column chromatography, using the mixture of solvents specified in each case. Characterization data for representative compounds are given below. For the complete data, see the ESI.† Compounds 3g and 3j are known in the literature.20
(±)-(2S*,4S*)-2-Benzoyl-4-[(2,2-dimethylhydrazono)methyl]-6-methoxy-4-methyl-1,2,3,4-tetrahydroquinoline (3a).
Reaction time: 3 h. Purification of the cis isomer: flash chromatography with petroleum ether:ethyl acetate (10:1, v/v). Yellow solid, mp: 130–131 °C. 1H-NMR (CDCl3, 250 MHz) δ: 1.65 (s, 3H, CH3); 1.73 (t, J = 12.6 Hz, 1H, H-3ax); 2.07 (dd, J = 13.0, 2.8 Hz, 1H, H-3eq); 2.72 (s, 6H, N(CH3)2); 3.73 (s, 3H, OCH3); 4.60 (br s, 1H, NH); 5.09 (dd, J = 12.2, 2.7 Hz, 1H, H-2); 6.53 (s, 1H, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N); 6.64–6.66 (m, 1H, H-5); 6.70–6.73 (m, 2H, H-7 and H-8); 7.49 (t, J = 7.4 Hz, 2H, H-3′ and H-5′); 7.61 (t, J = 7.4 Hz, 1H, H-4′); 7.92–7.98 (m, 2H, H-2′ and H-6′). 13C-NMR (CDCl3, 63 MHz) δ: 26.6 (CH3); 39.2 (C-3); 41.4 (C-4); 43.6 (N(CH3)2); 54.6 (C-2); 56.1 (OCH3); 113.8 (C-5); 113.9 (C-7); 117.4 (C-8); 128.5 (C-4a); 128.6 (C-2′ and C-6′); 129.2 (C-3′ and C-5′); 133.9 (C-4′); 135.0 (C-1′); 136.8 (C-8a); 143.6 (CHN); 152.5 (C-6); 199.9 (CO). IR (NaCl) ν: 3370.9 (N–H), 2954.1 (C–H), 1685.3 (CO), 1597.0 (CN) cm−1. Elemental analysis (%): Calc. for C21H25N3O2 (M = 351.44): C, 71.77; H, 7.17; N, 11.96. Found: C, 71.48; H, 7.03; N, 11.80.
N); 6.64–6.66 (m, 1H, H-5); 6.70–6.73 (m, 2H, H-7 and H-8); 7.49 (t, J = 7.4 Hz, 2H, H-3′ and H-5′); 7.61 (t, J = 7.4 Hz, 1H, H-4′); 7.92–7.98 (m, 2H, H-2′ and H-6′). 13C-NMR (CDCl3, 63 MHz) δ: 26.6 (CH3); 39.2 (C-3); 41.4 (C-4); 43.6 (N(CH3)2); 54.6 (C-2); 56.1 (OCH3); 113.8 (C-5); 113.9 (C-7); 117.4 (C-8); 128.5 (C-4a); 128.6 (C-2′ and C-6′); 129.2 (C-3′ and C-5′); 133.9 (C-4′); 135.0 (C-1′); 136.8 (C-8a); 143.6 (CHN); 152.5 (C-6); 199.9 (CO). IR (NaCl) ν: 3370.9 (N–H), 2954.1 (C–H), 1685.3 (CO), 1597.0 (CN) cm−1. Elemental analysis (%): Calc. for C21H25N3O2 (M = 351.44): C, 71.77; H, 7.17; N, 11.96. Found: C, 71.48; H, 7.03; N, 11.80.
(±)-(2S*,4S*)-4-[(2,2-Dimethylhydrazono)methyl]-6-methoxy-2-(4-methoxybenzoyl)-4-methyl-1,2,3,4-tetrahydroquinoline (3c).
Reaction time: 3 h. Purification of the cis isomer: flash chromatography with petroleum ether:ethyl acetate (20:1, v/v). Yellow solid, mp: 127–128 °C. 1H-NMR (CDCl3, 250 MHz) δ: 1.64 (s, 3H, CH3); 1.72 (t, J = 12.7 Hz, 1H, H-3ax); 2.06 (dd, J = 13.0, 2.8 Hz, 1H, H-3eq); 2.73 (s, 6H, N(CH3)2); 3.73 (s, 3H, OCH3); 3.88 (s, 3H, OCH3); 4.58 (bs, 1H, NH); 5.04 (dd, J = 12.3, 2.7 Hz, 1H, H-2); 6.53 (s, 1H, CHN); 6.63–6.66 (m, 1H, H-5); 6.69–6.73 (m, 2H, H-7 and H-8); 6.96 (d, J = 9.0 Hz, 2H, H-3′ and H-5′); 7.94 (d, J = 9.0 Hz, 2H, H-2′ and H-6′). 13C-NMR (CDCl3, 63 MHz) δ: 26.5 (CH3); 39.5 (C-3); 41.4 (C-4); 43.6 (N(CH3)2); 54.1 (C-2); 55.9 (OCH3); 56.1 (OCH3); 113.9 (C-5); 113.9 (C-7); 114.4 (C-3′ and C-5′); 117.5 (C-8); 127.8 (C-1′); 128.6 (C-4a); 131.0 (C-2′ and C-6′); 137.0 (C-8a); 143.4 (CHN); 152.5 (C-6); 164.1 (C-4′); 198.2 (CO). IR (NaCl) ν: 3375.9 (N–H), 2960.7 (C–H), 1676.3 (CO), 1600.5 (CN) cm−1. Elemental analysis (%): Calc. for C22H27N3O3 (M = 381.47): C, 69.27; H, 7.13; N, 11.02. Found: C, 68.90; H, 6.90; N, 10.77.
(±)-(2S*,4S*)-Ethyl 4-[(2,2-dimethylhydrazono)methyl]-4,6,8-trimethyl-1,2,3,4-tetrahydroquinoline-2-carboxylate (3h).
Reaction time: 2 h. Purification: flash chromatography with petroleum ether:ethyl acetate (10:1, v/v). Yellow oil, consisting of a ca. 9:1 mixture of the cis and trans diastereomers. Spectral data correspond to the major cis compound. 1H-NMR (CDCl3, 250 MHz) δ: 1.34 (t, J = 7.1 Hz, 3H, OCH2CH3); 1.51 (s, 3H, CH3); 1.93 (t, J = 12.4 Hz, 1H, H-3ax); 2.10 (dd, J = 13.2, 4.0 Hz, 1H, H-3eq); 2.18 (s, 3H, CH3); 2.21 (s, 3H, CH3); 2.79 (s, 6H, N(CH3)2); 4.19 (m, 1H, H-2); 4.24–4.32 (m, 3H, OCH2CH3 and NH); 6.64 (s, 1H, CHN); 6.75 (s, 1H, H-7); 6.79 (s, 1H, H-5). 13C-NMR (CDCl3, 63 MHz) δ: 14.3 (CH2CH3); 17.4 (ArCH3); 20.5 (ArCH3); 26.8 (CH3); 37.3 (C-3); 40.2 (C-4); 43.5 (N(CH3)2); 51.1 (C-2); 61.5 (CH2CH3); 122.2 (C-4a); 126.1 (C-8); 126.3 (C-6); 126.4 (C-5); 129.6 (C-7); 138.1 (C-8a); 144.3 (CHN); 173.6 (CO). IR (NaCl) ν: 3405 (N–H), 2930 (C–H), 1730 (CO) cm−1. Elemental analysis (%): Calc. for C18H27N3O2 (M = 317.43): C, 68.11; H, 8.57; N, 13.24. Found: C, 67.98; H, 8.32; N, 12.99.
(±)-(2′S*,4′S*)-1-Benzyl-4′-[(2,2-dimethylhydrazono)methyl]-6′-methoxy-4′-methyl-3′,4′-dihydro-1′H-spiro[indoline-3,2′-quinolin]-2-one (3o).
Purification: flash chromatography with petroleum ether:ethyl acetate (9:1, v/v). Orange solid, mp: 173–174 °C. 1H-NMR (CDCl3, 250 MHz) δ: 1.60 (s, 3H, CH3); 2.30 (d, J = 13.6 Hz, 1H, CH2Ph); 2.59 (s, 6H, N(CH3)2); 2.64 (d, J = 13.6 Hz, 1H, CH2Ph); 3.79 (s, 3H, OCH3); 3.87 (br s, 1H, NH); 4.70 (d, J = 15.6 Hz, 1H, H-3′); 5.09 (d, J = 15.6 Hz, 1H, H-3′); 6.58 (d, J = 8.6 Hz, 1H, H-8′); 6.67–6.74 (m, 3H, H-4, H-5 and H-6); 6.85–6.89 (m, 2H, H-5′ and H-7′); 7.07–7.12 (m, 2H, H-7 and H-4′′); 7.30–7.33 (m, 5H, H-2′′, H-3′′, H-5′′, H-6′′ and CHN). 13C-NMR (CDCl3, 63 MHz) δ: 29.3 (CH3); 39.6 (C-4′); 43.2 (N(CH3)2); 43.9 (C-3′); 55.9 (OCH3); 60.7 (CH2Ph); 77.4 (C-2′); 109.3 (C-5′*); 113.1 (C-7′*); 114.0 (C-8′*); 116.8 (C-7*); 122.4 (C-4*); 125.2 (C-6*); 127.4 (C-2′′ and C-6′′); 127.8 (C-5); 128.9 (C-4′′); 129.0 (C-3′′ and C-5′′); 132.9 (C-4a′); 136.0 (C-8a′*); 136.5 (C-7a*); 141.9 (C-3a); 143.7 (CHN); 145.2 (C-1′′*); 153.2 (C-6′); 178.8 (CO). IR (NaCl) ν: 3332.1 (N–H), 2927.0 (C–H), 1709.6 (CO) cm−1. Elemental analysis (%): Calc. for C28H30N4O2 (M = 454.56): C, 73.98; H, 6.65; N, 12.33. Found: C, 73.58; H, 6.38; N, 12.09.
General procedure for the synthesis of nitriles 6
To a solution of hydrazone 3 (0.5 mmol, 1 eq., as a cis–trans mixture) in methanol (10 mL) was added dropwise a suspension of MMPP·6H2O (0.62 mmol, 1.25 eq.) in methanol (5 mL), and the mixture was stirred until consumption of the starting material. The mixture was then diluted with water (20 mL) and extracted with CH2Cl2 (3 × 20 mL). The organic layer was washed with water (20 mL) and brine (20 mL), dried (Na2SO4), concentrated and the crude residue was regarded to be sufficiently pure to be used for the next step without further purification. The major cis compounds were purified by flash chromatography on silica gel, eluting with a mixture of petroleum ether:ethyl acetate (9:1, v/v), and their characterization data follow. Compound 6d could not be isolated, as it spontaneously aromatized to quinoline 4d. Flash chromatography on alumina of derivatives 6k and 6l, eluting with a mixture of petroleum ether:ethyl acetate (9.5:0.5, v/v), afforded directly the aromatic derivatives 7a and 7b. Compounds 6ka and 6kb could be isolated by fast chromatography on silica gel. Characterization data for representative compounds are given below. For the complete data, see the ESI.†
(±)-(2S*,4S*)-2-Benzoyl-6-methoxy-4-methyl-1,2,3,4-tetrahydroquinoline-4-carbonitrile (6a).
Reaction time: 2 h. Yellow oil. 1H-NMR (CDCl3, 250 MHz) δ: 1.87 (s, 3H, CH3); 2.07 (t, J = 12.7 Hz, 1H, H-3ax); 2.50 (dd, J = 13.0, 3.2 Hz, 1H, H-3eq); 3.78 (s, 3H, OCH3); 4.97 (dd, J = 12.5, 3.2 Hz, 1H, H-2); 6.72 (d, J = 8.7 Hz, 1H, H-8); 6.80 (dd, J = 8.8, 2.7 Hz, 1H, H-7); 6.97 (d, J = 2.6 Hz, 1H, H-5); 7.48–7.59 (m, 2H, H-3′ and H-5′); 7.62–7.72 (m, 1H, H-4′); 7.87–7.96 (m, 2H, H-2′ and H-6′). 13C-NMR (CDCl3, 63 MHz) δ: 28.6 (CH3); 35.6 (C-4); 36.7 (C-3); 52.9 (OCH3); 56.0 (C-2); 112.2 (C-7); 116.5 (C-5); 117.8 (C-8); 120.6 (CN); 123.7 (C-4a); 128.3 (C-2′ and C-6′); 129.3 (C-3′ and C-5′); 134.1 (C-4′); 134.4 (C-1′); 135.2 (C-8a); 152.7 (C-6); 198.0 (CO). IR (NaCl) ν: 3373.4 (C–N), 2926.2 (C–H), 2235.0 (CN), 1691.6 (CO) cm−1. Elemental analysis (%): Calcd for C19H18N2O2 (M = 306.36): C, 74.49; H, 5.92; N, 9.14. Found: C, 74.23; H, 5.75; N, 8.98.
(±)-(2S*,4S*)-2-(4-Methoxybenzoyl)-6-methoxy-4-methyl-1,2,3,4-tetrahydroquinoline-4-carbonitrile (6c).
Reaction time: 2 h. Yellow oil. 1H-NMR (CDCl3, 250 MHz) δ: 1.87 (s, 3H, CH3); 2.07 (t, J = 12.8 Hz, 1H, H-3ax); 2.49 (dd, J = 13.0, 3.0 Hz, 1H, H-3eq); 3.78 (s, 3H, OCH3); 3.91 (s, 3H, OCH3); 4.61 (bs, 1H, NH); 4.91 (d, J = 10.7 Hz, 1H, H-2); 6.71 (d, J = 8.8 Hz, 1H, H-8); 6.79 (dd, J = 8.8, 2.6 Hz, 1H, H-7); 6.96–7.02 (m, 3H, H-5, H-3′ and H-5′); 7.92 (d, J = 8.9 Hz, 2H, H-2′ and H-6′). 13C-NMR (CDCl3, 63 MHz) δ: 28.7 (CH3); 35.6 (C-4); 37.0 (C-3); 52.4 (C-2); 55.8 (OCH3); 56.0 (OCH3); 112.2 (C-7*); 114.5 (C-3′ and C-5′); 116.5 (C-5*); 118.0 (C-8*); 120.6 (CN*); 123.8 (C-4a*); 127.1 (C-1′); 130.8 (C-2′ and C-6′); 135.4 (C-8a); 152.7 (C-6); 164.4 (C-4′); 196.2 (CO). IR (NaCl) ν: 3362.8 (N–H), 2924.9 (C–H), 2234.4 (CN), 1680.4 (CO) cm−1. Elemental analysis (%): Calcd for C20H20N2O3: (M = 336.38): C, 71.41; H, 5.99; N, 8.33. Found: C, 71.23; H, 5.78; N, 8.17.
(±)-(2S*,4S*)-Ethyl 4-cyano-4,6,8-trimethyl-1,2,3,4-tetrahydroquinoline-2-carboxylate (6h).
Reaction time: 2 h. Yellow oil. 1H-NMR (CDCl3, 250 MHz) δ: 1.34 (t, J = 7.1 Hz, 3H, OCH2CH3); 1.72 (s, 3H, CH3); 2.15 (s, 3H, ArCH3); 2.24 (s, 3H, ArCH3); 2.31 (t, J = 10.0 Hz, 1H, H-3ax); 2.50 (dd, J = 13.3, 3.6 Hz, 1H, H-3eq); 4.08 (ddd, J = 11.5, 3.7, 1.6 Hz, 1H, H-2); 4.33 (q, J = 7.1 Hz, 2H, OCH2CH3 and NH); 6.86 (s, 1H, H-7); 7.08 (s, 1H, H-5). 13C-NMR (CDCl3, 63 MHz) δ: 14.3 (OCH2CH3); 17.3 (CH3); 20.5 (CH3); 29.0 (CH3); 34.7 (C-4); 35.4 (C-3); 50.0 (C-2); 62.2 (OCH2CH3); 119.0 (CN); 122.7 (C-4a); 124.2 (C-8); 125.7 (C-5); 127.2 (C-6); 131.3 (C-7); 136.9 (C-8a); 172.0 (CO). IR (NaCl) ν: 3348.9 (N–H), 2921.4 (C–H), 2241.9 (CN), 1733.2 (CO) cm−1. Elemental analysis (%): Calcd for C16H20N2O2 (M = 272.34): C, 70.56; H, 7.40; N, 10.29. Found: C, 70.43; H, 7.18; N, 10.12.
(±)-(2S*,4S*)-2-(2-Furylcarbonyl)-6-methoxy-4-methyl-1,2,3,4-tetrahydroquinoline-4-carbonitrile (6j).
Reaction time: 0.4 h. Yellow solid, mp: 101–102 °C. 1H-NMR (CDCl3, 250 MHz) δ: 1.87 (s, 3H, CH3); 2.15 (t, J = 12.5, 1H, H-3ax); 2.71 (dd, J = 12.7, 3.2 Hz, 1H, H-3eq); 3.81 (s, 3H, OCH3); 4.57 (bs, 1H, NH); 4.76 (dd, J = 12.3, 3.0 Hz, 1H, H-2); 6.66 (dd, J = 3.6, 1.7 Hz, 1H, H-4′); 6.72 (d, J = 8.7 Hz, 1H, H-8); 6.81 (dd, J = 8.8, 2.7 Hz, 1H, H-7); 6.99 (d, J = 2.7 Hz, 1H, H-5); 7.41 (dd, J = 3.6, 0.6 Hz, 1H, H-3′); 7.69 (dd, J = 1.7, 0.6 Hz, 1H, H-5′). 13C-NMR (CDCl3, 63 MHz) δ: 28.4 (CH3); 35.3 (C-4); 35.7 (C-3); 53.3 (C-2); 55.8 (OCH3); 111.9 (C-4′); 112.8 (C-7); 116.2 (C-5); 117.5 (C-8); 118.9 (C-3′); 120.4 (CN); 123.7 (C-4a); 134.9 (C-8a); 146.8 (C-5′); 150.8 (C-2′); 152.5 (C-6); 186.5 (CO). IR (NaCl) ν: 3370.3 (N–H), 2932.8 (C–H), 2232.4 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), 1676.1 (CO) cm−1. Elemental analysis (%): Calc. for C17H16N2O3 (M = 296.32): C, 68.91; H, 5.44; N, 9.45. Found: C, 68.65; H, 5.45; N, 9.39.
N), 1676.1 (CO) cm−1. Elemental analysis (%): Calc. for C17H16N2O3 (M = 296.32): C, 68.91; H, 5.44; N, 9.45. Found: C, 68.65; H, 5.45; N, 9.39.
General procedures for the synthesis of 2-acylquinolines 4
Synthesis of 2-acylquinolines 4 from nitriles 6.
A solution of compound 6 (0.4 mmol) in o-dichlorobenzene (5 mL) was refluxed for 24 h. The reaction mixture was allowed to reach room temperature and then it was concentrated under reduced pressure. The oily residue was purified by flash column chromatography, eluting with mixtures of petroleum ether and ethyl acetate, to give compounds 4.
Direct synthesis of 2-acylquinolines 4 from Povarov products 3 under acidic conditions.
To a solution of the starting hydrazone 3 (1 mmol) in THF (5 mL) was added dropwise 1 M HCl (5 mL) with stirring, and the resulting solution was vigorously stirred at room temperature for the times specified in Table 2. Then, the reaction mixture was quenched with 3 M aqueous NH4OH, and the basic solution was extracted with ethyl acetate (10 mL). The aqueous layer was then extracted with CH2Cl2 (2 × 10 mL), and the combined organic layers were washed with water (1 × 20 mL) and brine (1 × 20 mL), and dried over anhydrous Na2SO4. Removal of the solvent and column chromatography on silica gel eluting with a mixture of petroleum ether:ethyl acetate (9:1, v/v) gave quinolines 4a–d and 4g in the yields shown in Table 2. Characterization data for these compounds can be found below.
Direct synthesis of 2-acylquinolines 4 from Povarov products 3 under oxidative conditions.
To a solution of compound 3 (0.5 mmol) in methanol (5 mL) was added magnesium monoperoxyphthalate hexahydrate, MMPP (247 mg, 0.5 mmol). The solution was refluxed for 1 h, an additional amount of MMPP (247 mg, 0.5 mmol) was added and the reflux was maintained until the completion of the reaction, as judged by TLC (see the overall reaction times in Table 4). Water (10 mL) was added to the cooled reaction mixture, and the resulting suspension was extracted with CH2Cl2 (3 × 15 mL). The combined extracts were dried over anhydrous Na2SO4 and evaporated. The residue was purified as described above.
The characterization data for representative compounds 4 are given below. For the complete data, see the ESI.†
2-Benzoyl-6-methoxy-4-methylquinoline (4a).
Purification: flash chromatography with petroleum ether:ethyl acetate (9:1, v/v). Yellow solid, mp: 117–119 °C. 1H-NMR (CDCl3, 250 MHz) δ: 2.75 (s, 3H, CH3); 4.00 (s, 3H, OCH3); 7.24 (d, J = 2.7 Hz, 1H, H-5); 7.42 (dd, J = 9.2, 2.4 Hz, 1H, H-7); 7.50 (t, J = 7.4 Hz, 2H, H-3′ and H-5′); 7.61 (t, J = 7.3 Hz, 1H, H-4′); 7.96 (s, 1H, H-3); 8.11 (d, J = 9.2 Hz, 1H, H-8); 8.22 (d, J = 7.3 Hz, 2H, H-2′ and H-6′). 13C-NMR (CDCl3, 63 MHz) δ: 19.3 (CH3); 55.8 (OCH3); 101.8 (C-5); 121.9 (C-3); 122.5 (C-7); 128.2 (C-3′ and C-5′); 130.4 (C-4a); 131.6 (C-2′ and C-6′); 132.8 (C-8); 133.0 (C-4′); 136.6 (C-1′); 142.5 (C-4); 144.0 (C-8a); 152.0 (C-2); 159.4 (C-6); 194.1 (CO). IR (NaCl) ν: 2924.3 (C–H), 1657.9 (CO), 1619.8 cm−1. Elemental analysis (%): Calc. for C18H15NO2 (M = 277.32): C, 77.96; H, 5.45; N, 5.05. Found: C, 77.69; H, 5.41; N, 4.95.
6-Methoxy-4-methoxybenzoyl-4-methylquinoline (4c).
Purification: flash chromatography with petroleum ether:ethyl acetate (9:1, v/v). White solid, mp: 142–144 °C. 1H-NMR (CDCl3, 250 MHz) δ: 2.75 (s, 3H, CH3); 3.90 (s, 3H, OCH3); 4.00 (s, 3H, OCH3); 6.99 (d, J = 8.7 Hz, 2H, H-3′ and H-5′); 7.24 (d, J = 2.2 Hz, 1H, H-5); 7.42 (dd, J = 9.2, 2.2 Hz, 1H, H-7); 7.91 (s, 1H, H-3); 8.11 (d, J = 9.2 Hz, 1H, H-8); 8.28 (d, J = 8.7 Hz, 2H, H-2′ and H-6′). 13C-NMR (CDCl3, 63 MHz) δ: 19.3 (CH3); 55.6 (OCH3); 55.8 (OCH3); 101.8 (C-5); 113.6 (C-3′ and C-5′); 122.0 (C-3*); 122.4 (C-7*); 129.4 (C-4a*); 130.3 (C-1′*); 132.7 (C-8); 134.0 (C-2′ and C-6′); 142.5 (C-4*); 143.9 (C-8a*); 152.8 (C-2); 159.2 (C-6); 163.7 (C-4′); 192.6 (CO). IR (NaCl) ν: 2922.1 (C–H), 1646.5 (CO), 1620.4 (CN) cm−1. Elemental analysis (%): Calc. for C19H17NO3 (M = 307.34): C, 74.25; H, 5.58; N, 4.56. Found: C, 74.08; H, 5.84; N, 4.31.
Ethyl 4,6,8-trimethylquinoline-2-carboxylate (4h).
Purification: flash chromatography with petroleum ether:ethyl acetate (9:1, v/v). Yellow solid, mp: 125–127 °C. 1H-NMR (CDCl3, 250 MHz) δ: 1.48 (t, J = 7.1 Hz, 3H, OCH2CH3); 2.53 (s, 3H, CH3); 2.71 (s, 3H, CH3); 2.85 (s, 3H, CH3); 4.50 (q, J = 7.1 Hz, 2H, OCH2CH3); 7.45 (s, 1H, H-5); 7.61 (s, 1H, H-7); 7.96 (s, 1H, H-3). 13C-NMR (CDCl3, 63 MHz) δ: 14.5 (OCH2CH3); 18.2 (CH3); 19.3 (CH3); 22.2 (CH3); 61.9 (OCH2CH3); 120.6 (C-3*); 121.6 (C-5*); 129.5 (C-4a); 132.4 (C-7); 138.2 (C-6*); 139.0 (C-8*); 144.8 (C-4); 145.3 (C-8a); 145.8 (C-2); 166.2 (CO). IR (NaCl) ν: 2929.7 (C–H), 1704.5 (CO), 1118.0 (C–O) cm−1. Elemental analysis (%): Calc. for C15H17NO2 (M = 243.30): C, 74.05; H, 7.04; N, 5.76. Found: C, 73.72; H 6.83; N, 6.06.
2-Furylcarbonyl-6-methoxy-4-methylquinoline (4j).
Purification: flash chromatography with petroleum ether:ethyl acetate (9:1, v/v). Yellow solid, mp: 150–152 °C. 1H-NMR (CDCl3, 250 MHz) δ: 2.75 (s, 3H, CH3); 4.01 (s, 3H, OCH3); 6.67 (dd, J = 3.5, 1.7 Hz, 1H, H-4′); 7.23 (d, J = 2.7 Hz, 1H, H-5); 7.45 (dd, J = 9.2, 2.7 Hz, 1H, H-7); 7.79 (dd, J = 1.6, 0.7 Hz, 1H, H-5′); 8.09 (s, 1H, H-3); 8.13 (d, J = 9.2 Hz, 1H, H-8); 8.29 (dd, J = 3.5, 0.7 Hz, 1H, H-3′). 13C-NMR (CDCl3, 63 MHz) δ: 19.0 (CH3); 55.6 (OCH3); 101.6 (C-5); 112.4 (C-4′); 120.8 (C-3); 122.3 (C-7); 124.4 (C-3′); 130.6 (C-4a); 132.5 (C-8); 142.7 (C-4); 143.6 (C-8a); 147.5 (C-5′); 150.8 (C-2); 151.2 (C-2′); 159.3 (C-6); 179.6 (CO). IR (NaCl) ν: 2939.4 (C–H), 1620.4 (CN) cm−1. Elemental analysis (%): Calc. For C16H13NO3 (M = 267.28): C, 71.90; H, 4.90; N, 5.24. Found: C, 71.76; H, 4.87; N, 5.14.
General procedure for the synthesis of 2-acylquinoline-4-carbonitriles 7a,b
The crude nitriles 6k,l obtained by the general method described above, were dissolved in CH2Cl2 (5 mL), to which alumina (100 mg) was added. The solvent was evaporated and the resulting solid was loaded onto a cartridge that was attached to the flash chromatography system. Chromatography on alumina furnished quinolines 7a and 7b, respectively.
2-Benzoyl-3-ethyl-6-methoxyquinoline-4-carbonitrile (7a).
Reaction time: 4 h. Purification: flash chromatography on alumina with petroleum ether:ethyl acetate (9:1, v/v). White solid, mp: 138–141 °C. 1H-NMR (CDCl3, 250 MHz) δ: 1.34 (t, J = 7.5 Hz, 3H, CH2CH3); 3.10 (q, J = 7.5 Hz, 2H, CH2CH3); 4.03 (s, 3H, OCH3); 7.37–7.53 (m, 4H, H-3′, H-5′, H-5 and H-7); 7.63 (m, 1H, H-4′); 7.91 (d, J = 7.1 Hz, 2H, H-2′ and H-6′); 8.02 (d, J = 9.1 Hz, 1H, H-8). 13C-NMR (CDCl3, 63 MHz) δ: 15.9 (CH3); 25.2 (CH2); 56.2 (OCH3); 102.1 (C-5); 115.1 (C-4); 117.6 (CN); 124.2 (C-7); 128.5 (C-4a); 128.7 (C-3′ and C-5′); 130.9 (C-2′ and C-6′); 132.1 (C-8*); 134.2 (C-4′); 135.9 (C-3*); 140.8 (C-1′*); 141.3 (C-8a*); 153.2 (C-2); 161.1 (C-6); 193.8 (CO). IR (NaCl) ν: 2927.5 (C–H), 2225.1 (CN), 1664.8 (CO) cm−1. Elemental analysis (%): Calc. for C20H16N2O2 (M = 316.35): C, 75.93; H, 5.10; N, 8.86. Found: C, 75.65; H, 5.31; N, 8.52.
2-Benzoyl-6-methoxyquinoline-4-carbonitrile (7b).
Reaction time: 2 h. Purification: flash chromatography on alumina with petroleum ether:ethyl acetate (9:1, v/v). Yellow solid, mp: 140–142 °C. 1H-NMR (CDCl3, 250 MHz) δ: 4.06 (s, 3H, OCH3); 7.45 (d, J = 2.7 Hz, 1H, H-5); 7.50–7.57 (m, 3H, H-3′, H-5′ and H-7); 7.64 (m, 1H, H-4′); 8.17 (d, J = 9.3 Hz, 1H, H-8); 8.20–8.27 (m, 2H, H-2′ and H-6′); 8.48 (s, 1H, H-3). 13C-NMR (CDCl3, 63 MHz) δ: 56.3 (OCH3); 102.3 (C-5); 115.8 (C-4); 118.0 (CN); 125.3 (C-7*); 125.6 (C-3*); 128.4 (C-3′ and C-5′); 128.6 (C-4a); 131.5 (C-2′ and C-6′); 133.2 (C-8*); 133.5 (C-4′*); 135.7 (C-1′); 142.9 (C-8a); 151.3 (C-2); 161.8 (C-6); 191.8 (CO). IR (NaCl) ν: 2920.4 (C–H), 2229.9 (CN), 1664.4 (CO) cm−1. Elemental analysis (%): Calc. for C18H12N2O2 (M = 288.30): C, 74.99; H, 4.20; N, 9.72. Found: C, 74.77; H, 4.32; N, 9.64.
General procedure for the synthesis of 2-acyl-4-(dimethylhydrazonomethyl)quinolines 7c,d
A solution of hydrazone 3k or 3l in o-dichlorobenzene (5 mL) was refluxed until no starting material was detected by TLC. The reaction mixture was allowed to reach room temperature and the solvent was evaporated under reduced pressure. The oily residue was purified by flash column chromatography, eluting with a 9:1 (v/v) mixture of petroleum ether and ethyl acetate, to give compounds 7c,d.
2-Benzoyl-4-[(2,2-dimethylhydrazono)methyl]-3-ethyl-6-methoxyquinoline (7c).
Reaction time: 6 h. Yellow solid, mp: 109–111 °C. 1H-NMR (CDCl3, 250 MHz) δ: 1.23 (t, J = 7.5 Hz, 3H, CH2CH3); 2.87 (q, J = 7.5 Hz, 2H, CH2CH3); 3.17 (s, 6H, N(CH3)2); 3.95 (s, 3H, OCH3); 7.32 (dd, J = 9.2, 2.8 Hz, 1H, H-7); 7.43 (d, J = 7.3 Hz, 2H, H-3′ and H-5′); 7.56 (m, 1H, H-4′); 7.64 (s, 1H, CHN); 7.88–7.95 (m, 3H, H-2′, H-6′ and H-8); 8.28 (d, J = 2.8 Hz, 1H, H-5). 13C-NMR (CDCl3, 63 MHz) δ: 15.5 (CH3); 22.7 (CH2); 42.7 (N(CH3)2); 55.5 (OCH3); 104.5 (C-5); 121.5 (C-7); 127.1 (C-4a); 127.4 (CHN); 128.6 (C-3′ and C-5′); 130.8 (C-2′ and C-5′); 131.4 (C-8); 131.9 (C-3*); 133.8 (C-4′); 136.5 (C-4*); 137.4 (C-1′*); 142.4 (C-8a); 154.9 (C-2); 158.9 (C-6); 196.2 (CO). IR (NaCl) ν: 2920.9 (C–H), 1669.6 (CO); 1616.3 (CN) cm−1. Elemental analysis (%): Calc. for C22H23N3O2 (M = 361.44): C, 73.11; H, 6.41; N, 11.63. Found: C, 73.27; H, 6.61; N, 11.34.
2-Benzoyl-4-[(2,2-dimethylhydrazono)methyl]-6-methoxyquinoline (7d).
Reaction time: 3 h. Yellow oil. 1H-NMR (CDCl3, 250 MHz) δ: 3.19 (s, 6H, N(CH3)2); 3.99 (s, 3H, OCH3); 7.40 (dd, J = 9.3, 2.8 Hz, 1H, H-7); 7.50 (t, J = 7.3 Hz, 2H, H-3′ and H-5′); 7.58–7.61 (m, 2H, H-4′ and CHN); 8.08 (d, J = 9.2 Hz, 1H, H-8); 8.20–8.24 (m, 3H, H-2′, H-6′ and H-3); 8.27 (d, J = 2.8 Hz, 1H, H-5). 13C-NMR (CDCl3, 63 MHz) δ: 42.7 (N(CH3)2); 55.6 (OCH3); 103.5 (C-5); 118.8 (C-3); 122.1 (C-7); 127.0 (C-4a); 127.5 (CHN); 128.2 (C-3′ and C-5′); 131.6 (C-2′ and C-6′); 132.6 (C-8); 132.9 (C-4′); 136.7 (C-4*); 139.9 (C-1′*); 144.0 (C-8a); 152.3 (C-2); 159.6 (C-6); 194.4 (CO). IR (NaCl) ν: 2922.8 (C–H), 1653.9 (CO); 1616.3 (CN) cm−1. Elemental analysis (%): Calc. for C20H19N3O2 (M = 333.38): C, 72.05; H, 5.74; N, 12.60. Found: C, 72.10; H, 5.86; N, 12.83.
General procedure for the synthesis of 2-arylquinolines 9
To a cooled (0 °C) solution of tetrahydroquinoline 820b (0.5 mmol) in methanol (5 mL) was added magnesium monoperoxyphthalate hexahydrate, MMPP (247 mg, 0.5 mmol). The solution was stirred at room temperature for 1–3 h. Water (10 mL) was added to the cooled reaction mixture, and the resulting suspension was extracted with CH2Cl2 (3 × 15 mL). The combined extracts were dried over anhydrous Na2SO4 and evaporated. A solution of the crude nitrile thus obtained in o-dichlorobenzene (5 mL) was refluxed for 24 h (48 h in the case of 9h). The reaction mixture was allowed to reach room temperature and the solvent was evaporated under reduced pressure. The oily residue was purified by flash column chromatography, eluting with a 9:1 (v/v) mixture of petroleum ether and ethyl acetate, to give compounds 9, some of which are known in the literature. The characterization data for representative compounds 9 are given below. For the complete data, see the ESI.†
2-(4-Chlorophenyl)-6-methoxy-4-methylquinoline (9b).
White solid, mp: 119–122 °C. 1H-NMR (CDCl3, 250 MHz) δ: 2.72 (s, 3H, CH3); 3.97 (s, 3H, OCH3); 7.20 (d, J = 2.8 Hz, 1H, H-5); 7.38 (dd, J = 9.2, 2.8 Hz, 1H, H-7); 7.45–7.49 (m, 2H, H-3′ and H-5′); 7.65 (s, 1H, H-3); 8.04–8.09 (m, 3H, H-8, H-2′ and H-6′). 13C-NMR (CDCl3, 63 MHz) δ: 19.4 (CH3); 55.7 (OCH3); 102.0 (C-5); 119.7 (C-3); 121.8 (C-7); 128.3 (C-4a); 128.6 (C-2′ and C-6′); 129.0 (C-3′ and C-5′); 131.9 (C-8); 135.1 (C-4′); 138.4 (C-1′); 143.6 (C-8a); 144.2 (C-4); 153.5 (C-2); 157.8 (C-6). IR (NaCl) ν: 2918.0 (C–H), 1624.6 (CN), 717.2 (C–Cl) cm−1. Elemental analysis (%): Calc. for C17H14ClNO (M = 283.75): C, 71.96; H, 4.97; N, 4.94. Found: C, 72.08; H, 5.27; N, 5.00.
4-Ethyl-6,8-dimethyl-2-phenylquinoline (9g).
White solid, mp: 116–118 °C. 1H-NMR (CDCl3, 250 MHz) δ: 1.44 (t, J = 7.5 Hz, 3H, CH2CH3); 2.53 (s, 3H, CH3); 2.87 (s, 3H, CH3); 3.14 (q, J = 7.3 Hz, 2H, CH2CH3); 7.41 (s, 1H, H-5); 7.44–7.56 (m, 3H, H-3′, H-4′ and H-5′); 7.65 (s, 1H, H-7); 7.73 (s, 1H, H-3); 8.23–8.27 (m, 2H, H-2′ and H-6′). 13C-NMR (CDCl3, 63 MHz) δ: 14.5 (CH3); 18.5 (CH3); 22.1 (CH3); 25.8 (CH2); 117.2 (C-3); 120.2 (C-5); 126.4 (C-4a); 127.5 (C-2′ and C-6′); 128.8 (C-3′ and C-5′); 129.0 (C-4′); 131.7 (C-7); 135.4 (C-6*); 138.0 (C-8*); 140.4 (C-1′); 146.0 (C-4); 149.8 (C-8a); 154.5 (C-2). IR (NaCl) ν: 2917.4 (C–H), 1596.8 (C–C), 1029.1 (C–N) cm−1. Elemental analysis (%): Calc. for C19H19N (M = 261.36): C, 87.31; H, 7.33; N, 5.36. Found: C, 87.06; H, 7.35; N, 5.51.
5,7-Dimethoxy-4-methyl-2-(4-methylphenyl)quinoline (9h).
Reaction time, 48 h. Yellow solid, mp: 130–133 °C. 1H-NMR (CDCl3, 250 MHz) δ: 2.42 (s, 3H, CH3); 2.87 (s, 3H, CH3); 3.92 (s, 3H, OCH3); 3.95 (s, 3H, OCH3); 6.47 (d, J = 2.4 Hz, 1H, H-6); 7.11 (d, J = 2.4 Hz, 1H, H-8); 7.30 (d, J = 7.9 Hz, 2H, H-3′ and H-5′); 7.41 (s, 1H, H-3); 8.01 (d, J = 8.2 Hz, 2H, H-2′ and H-6′). 13C-NMR (CDCl3, 63 MHz) δ: 21.5 (CH3); 24.7 (CH3); 55.6 (OCH3); 55.7 (OCH3); 98.3 (C-6*); 101.1 (C-8*); 115.6 (C-4a); 119.1 (C-3); 127.4 (C-3′ and C-5′); 129.6 (C-2′ and C-6′); 136.9 (C-4′); 139.2 (C-1′); 146.3 (C-4); 151.9 (C-8a); 157.3 (C-5*); 158.7 (C-7*); 160.6 (C-2). IR (NaCl) ν: 2928.3 (C–H), 1620.1 (C–C), 1135.4 (C–O) cm−1. Elemental analysis (%): Calc. for C19H19NO2 (M = 293.36): C, 77.79; H, 6.53; N, 4.77. Found: C, 77.78; H, 6.71; N, 4.87.
Conclusions
2-Acyl-4-alkyl-1,2,3,4-tetrahydroquinolines functionalized at C-4 with a dialkylhydrazone group were available in good to excellent yields and cis/trans diastereoselectivities via the InCl3-catalyzed aza-vinylogous Povarov reaction between α-ketoimines and α,β-unsaturated dimethylhydrazones. These compounds were readily transformed into synthetically and biologically relevant 2-acylquinolines with a high degree of substitution by means of a sequence that involves the oxidative generation of a nitrile group at C-4, followed by its thermal elimination. Alternatively, a one-pot protocol based on the reaction of the hydrazones with magnesium monoperoxyphthalate hexahydrate in refluxing methanol afforded the target 2-acylquinolines in good to excellent yields. This methodology was also extended to the preparation of 2-arylquinolines.
Acknowledgements
Financial support from MINECO (grant CTQ2012-33272-BQU) is gratefully acknowledged.
Notes and references
- J. P. Michael, Nat. Prod. Rep., 2008, 25, 166 RSC.
- For representative reviews of the importance of quinolines in drug discovery, see:
(a) V. R. Solomon and H. Lee, Curr. Med. Chem., 2011, 18, 1488 CrossRef CAS PubMed;
(b) A. Marella, O. P. Tanwar, R. Saha, M. R. Ali, S. Srivastava, M. Akhter, M. Shaquiquzzaman and M. M. Alam, Saudi Pharm. J., 2013, 21, 1 CrossRef PubMed;
(c) B. Pati and S. Banerjee, J. PharmaSciTech, 2014, 3, 59 CAS.
-
(a) B. D. Bax, P. F. Chan, D. S. Eggleston, A. Fosberry, D. R. Gentry, F. Gorrec, I. Giordano, M. M. Hann, A. Hennessy, M. Hibbs, J. Huang, E. Jones, J. Jones, K. K. Brown, C. J. Lewis, E. W. May, M. R. Saunders, O. Singh, C. E. Spitzfaden, C. Shen, A. Shillings, A. J. Theobald, A. Wohlkonig, N. D. Pearson and M. N. Gwynn, Nature, 2010, 466, 935 CrossRef PubMed;
(b) M. Rouffet, C. A. F. de Oliveira, Y. Udi, A. Agrawal, I. Sagi, J. A. McCammon and S. M. Cohen, J. Am. Chem. Soc., 2010, 132, 8232 CrossRef CAS PubMed;
(c) S. Andrews, S. J. Burgess and D. Skaalrud, J. Med. Chem., 2010, 53, 916 CrossRef CAS PubMed;
(d) Y. L. Chen, Y. L. Zhao, C. M. Lu, C. C. Tzeng and J. P. Wang, Bioorg. Med. Chem., 2006, 14, 4373 CrossRef CAS PubMed;
(e) O. Billker, V. Lindo, M. Panico, A. E. Etienne, T. Paxton, A. Dell, M. Rogers, R. E. Sinden and H. R. Morris, Nature, 1998, 392, 289 CrossRef CAS PubMed.
-
(a) C. Palacio and S. J. Connon, Org. Lett., 2011, 13, 1298 CrossRef CAS PubMed;
(b) S. J. Connon, Chem. Commun., 2008, 2499 RSC;
(c)
H. W. Haesslin and C. Krueger, WO2002034048A1, 2002 Search PubMed;
(d) J. I. Kim, I. Shin, H. Kim and J. Lee, J. Am. Chem. Soc., 2003, 46, 1242 Search PubMed.
- For a review of the traditional quinoline syntheses, see:
G. Jones, Comprehensive Heterocyclic Chemistry II, ed. A. Katritzky, C. W. Rees and E. F. V. Scriven, Pergamon Press, 1996, ch. 5.05, vol. 5, p. 167 Search PubMed.
- Z. H. Skraup, Ber. Dtsch. Chem. Ges., 1880, 13, 2086 Search PubMed.
- O. Doebner and W. Miller, Ber. Dtsch. Chem. Ges., 1881, 14, 2812 CrossRef.
-
(a) P. Friedländer, Ber. Dtsch. Chem. Ges., 1882, 15, 2572 CrossRef;
(b) For a review, see: J. L. Marco-Contelles, E. Pérez-Mayoral, A. Samadi, M. C. Carreiras and E. Soriano, Chem. Rev., 2009, 109, 2652 CrossRef CAS PubMed.
- A. Combes, Bull. Soc. Chim. Fr., 1883, 49, 89 Search PubMed.
- For reviews of more recent methods, see:
(a) V. Kouznetsov, L. Y. Vargas Méndez and C. M. Meléndez Gómez, Curr. Org. Chem., 2005, 9, 141 CrossRef CAS;
(b) S. Madapa, Z. Tusi and S. Batra, Curr. Org. Chem., 2008, 12, 1116 CrossRef CAS;
(c) S. M. Prajapati, K. D. Patel, R. H. Vekariya, S. N. Panchal and H. D. Patel, RSC Adv., 2014, 4, 24463 RSC.
-
(a) H. Li, X. Xu, J. Yang, X. Xie, H. Huang and Y. Li, Tetrahedron Lett., 2011, 52, 530 CrossRef CAS;
(b) H. Li, C. Wang, H. Huang, X. Xu and Y. Li, Tetrahedron Lett., 2011, 52, 1108 CrossRef CAS;
(c) S. Cai, J. Zeng, Y. Bai and X. Liu, J. Org. Chem., 2012, 77, 801 CrossRef CAS PubMed;
(d) O. M. Kuzmina, A. K. Steib, D. Flubacher and P. Knochel, Org. Lett., 2012, 14, 4818 CrossRef CAS PubMed;
(e) M. L. Neo and R. J. Dhanorkar, Tetrahedron, 2015, 71, 338 CrossRef;
(f) L. Kong, Y. Zhou, H. Huang, Y. Yang, Y. Liu and Y. Li, J. Org. Chem., 2015, 80, 1275 CrossRef CAS PubMed;
(g) J. Zhou, G. Wu, M. Zhang, X. Jie and W. Su, Chem. – Eur. J., 2012, 18, 8032 CrossRef CAS PubMed;
(h) X. Zhang, B. Liu, X. Shu, Y. Gao, H. Lv and J. Zhu, J. Org. Chem., 2012, 77, 501 CrossRef CAS PubMed;
(i) X. Ji, H. Huang, Y. Li, H. Chen and H. Jiang, Angew. Chem., Int. Ed., 2012, 51, 7292 CrossRef CAS PubMed.
-
(a) Y. F. Zhu, X. C. Wang, P. Connors, K. Wilcoxen, Y. H. Gao, R. Gross, N. Strack, T. Gross, J. R. McCarthy, Q. Xie, N. Ling and C. Chen, Bioorg. Med. Chem., 2003, 13, 1931 CrossRef CAS;
(b) S. Manfredini, S. Vertuani, B. Pavan, F. Vitali, M. Scaglianti, F. Bortolotti, C. Biondi, A. Scatturin, P. Prasad and A. Dalpiaz, Bioorg. Med. Chem., 2004, 12, 5453 CrossRef CAS PubMed;
(c) C. L. Horchler, J. P. McCauley, J. E. Hall, D. H. Snyder, W. C. Moore, T. J. Hudzik and M. J. Chapdelaine, Bioorg. Med. Chem., 2007, 15, 939 CrossRef CAS PubMed;
(d) B. Reux, T. Nevalainen, K. H. Raitio and A. M. P. Koskinen, Bioorg. Med. Chem., 2009, 17, 4441 CrossRef CAS PubMed;
(e) J. McNulty, R. Vemula, C. Bordón, R. Yolke and L. Jones-Brando, Org. Biomol. Chem., 2014, 12, 255 RSC.
- Z. Mao, H. Qu, Y. Zhao and X. Lin, Chem. Commun., 2012, 48, 9927 RSC.
-
(a) E. Vicente-García, F. Catti, R. Ramo and R. Lavilla, Org. Lett., 2010, 12, 860 CrossRef PubMed;
(b) H. Richter and O. G. Mancheño, Org. Lett., 2011, 13, 6066 CrossRef CAS PubMed. For two additional reports, describing a single example of the synthesis of 2-alkoxycarbonylquinolines via Friedländer reactions, see:
(c) B. R. McNaughton and B. L. Miller, Org. Lett., 2003, 5, 4257 CrossRef CAS PubMed;
(d) Y. Chen, J. Huang, T. L. Hwang, T. J. Li, S. Cui, J. Chan and M. Bio, Tetrahedron Lett., 2012, 53, 3237 CrossRef CAS.
-
(a) Z. Yin, Z. Zhang, J. F. Kadow, N. A. Meanwell and T. Wang, J. Org. Chem., 2004, 69, 1364 CrossRef CAS PubMed;
(b) K. Matcha and A. P. Antonchick, Angew. Chem., Int. Ed., 2013, 52, 2082 CrossRef CAS PubMed.
- For reviews of the Povarov reaction, see:
(a) V. A. Glushkov and A. G. Tolstikov, Russ. Chem. Rev., 2008, 77, 137 CrossRef CAS;
(b) V. V. Kouznetsov, Tetrahedron, 2009, 65, 2721 CrossRef CAS;
(c) D. Bello, R. Ramón and R. Lavilla, Curr. Org. Chem., 2010, 14, 332 CrossRef CAS;
(d) M. Fochi, L. Caruana and L. Bernardi, Synthesis, 2014, 135 Search PubMed.
- For a review of the chemistry of tetrahydroquinolines, see: V. Sridharan, P. A. Suryavanshi and J. C. Menéndez, Chem. Rev., 2011, 111, 7157 CrossRef CAS PubMed.
- For representative examples, see:
(a) Dehydrogenation with DDQ: V. Sridharan, C. Avendaño and J. C. Menéndez, Tetrahedron, 2009, 65, 2087 CrossRef CAS;
(b)
In situ hydrogen transfer from excess imine: N. Shindoh, H. Tokuyama, Y. Takemoto and K. Takasu, J. Org. Chem., 2008, 73, 7451 CrossRef CAS PubMed.
-
(a) H. Huang, H. Jiang, K. Chen and H. Liu, J. Org. Chem., 2009, 74, 5476 CrossRef CAS PubMed;
(b) X. Li, Z. Mao, Y. Wang, W. Chen and X. Lin, Tetrahedron, 2011, 67, 3858 CrossRef CAS.
-
(a) V. Sridharan, P. T. Perumal, C. Avendaño and J. C. Menéndez, Org. Biomol. Chem., 2007, 5, 1351 RSC;
(b) V. Sridharan, P. Ribelles, V. Estévez, M. Villacampa, M. T. Ramos, P. T. Perumal and J. C. Menéndez, Chem. – Eur. J., 2012, 18, 5056 CrossRef CAS PubMed.
- For a representative recent example, see: G.-M. Nan and W. Liu, Chin. Chem. Lett., 2015, 26, 1289 CrossRef CAS.
- A. Bunescu, Q. Wang and J. Zhu, Org. Lett., 2014, 16, 1756 CrossRef CAS PubMed.
- For precedent of the use of isatin-derived imines in non-vinylogous Povarov reactions, see:
(a) V. V. Kouznetsov, J. S. Nello Forero and D. F. Amado Torres, Tetrahedron Lett., 2008, 49, 5855 CrossRef CAS;
(b) F. Shi, G.-J. Xing, R.-Y. Zhu, W. Tan and S. Tu, Org. Lett., 2013, 15, 128 CrossRef CAS PubMed.
- For a review of the captodative effect, see: J. C. Easton, Chem. Rev., 1997, 97, 53 CrossRef PubMed.
-
(a) T. Mino, S. Fukui and M. Yamashita, J. Org. Chem., 1997, 62, 734 CrossRef CAS PubMed;
(b) P. Ribelles, M. T. Ramos and J. C. Menéndez, Org. Lett., 2012, 14, 1402 CrossRef CAS PubMed.
- E. Díez, R. Fernández, C. Gasch, J. M. Lassaletta, J. M. Llera, E. Martín-Zamora and J. J. Vázquez, J. Org. Chem., 1997, 62, 5144 CrossRef.
- Y.-L. Chen, C.-J. Huang, Z.-Y. Huang, C.-H. Tseng, F.-S. Chang, S.-H. Yang, S.-R. Lin and C.-C. Tzeng, Bioorg. Med. Chem., 2006, 14, 3098 CrossRef CAS PubMed.
- C. M. Meléndez Gómez, V. V. Kouznetsov, M. A. Sortino, S. L. Álvarez and S. A. Zacchino, Bioorg. Med. Chem., 2008, 16, 7908 CrossRef PubMed.
- L. M. Bedoya, M. J. Abad, E. Calonge, L. Astudillo Saavedra, M. Gutiérrez, V. V. Kouznetsov, J. Alcamia and P. Bermejo, Antiviral Res., 2010, 87, 338 CrossRef CAS PubMed.
- N. J. Liverton, M. K. Holloway, J. A. McCauley, M. T. Rudd, J. W. Butcher, S. S. Carroll, J. DiMuzio, C. Fandozzi, K. F. Gilbert, S. S. Mao, C. J. McIntyre, K. T. Nguyen, J. J. Romano, M. Stahlhut, B. L. Wan, D. B. Olsen and J. P. Vacca, J. Am. Chem. Soc., 2008, 130, 4607 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental procedures and characterization data, copies of spectra. See DOI: 10.1039/c6qo00037a |
|
| This journal is © the Partner Organisations 2016 |