
Atroposelective synthesis of axially chiral P,S-ligands based on arynes†‡
Anaïs
Berthelot-Bréhier
ab,
Armen
Panossian
*a,
Françoise
Colobert
b and
Frédéric R.
Leroux
 *a
*a
aCNRS – Université de Strasbourg (ECPM), UMR 7509, COHA, 25 Rue Becquerel, 67087 Strasbourg, France. E-mail: armen.panossian@unistra.fr; frederic.leroux@unistra.fr; Web: http://www.coha-lab.org
bCNRS – Université de Strasbourg (ECPM), UMR 7509, SynCat, 25 Rue Becquerel, 67087 Strasbourg, France
First published on 9th April 2015
Abstract
The first atropo-selective aryl–aryl coupling based on arynes in the presence of a tert-butylsulfinyl group as the chiral auxiliary on the aryllithium nucleophile is described. The approach allows for the efficient access to a novel family of atropo-enantiopure biphenyl-based phosphine–thioether ligands. The new P,S heterodonor ligands were assessed in model palladium-catalyzed allylic substitution reactions.
Introduction
The importance of biaryls in natural products, drugs, catalysis, and organic materials has led to the development of numerous strategies for their synthesis, particularly in the case of atropo-enriched biaryls.1,2 In our laboratory, we developed the potential of the known reaction of an aryllithium and an ortho-dihaloarene bearing at least one exchangeable halogen, leading to functionalizable 2-halobiaryls.3–6 We studied its regioselectivity4–8 and applied it to the synthesis of various C1-symmetric phosphorus ligands.9–12 Recently, we described the use of this so-called ‘ARYNE coupling’ in the modular synthesis of highly atropo-enriched biphenyls (Scheme 1),13 following Clayden's ‘traceless resolving agent’ method based on sulfoxides.14,15 We started from achiral or racemic polyhalobiphenyls obtained by ‘ARYNE coupling’, which were then desymmetrized or deracemized. We showed that 2,2′,6-tribromo-1,1′-biphenyl could serve as a platform for accessing atropo-enriched biphenyls, variously derivatizable at positions 2, 2′ and 6. In the present paper, we report on an improved alternative method, where axial stereoenrichment is installed during the aryl–aryl coupling step by means of a covalently attached chiral sulfoxide auxiliary, i.e. an atropo-diastereoselective ‘ARYNE coupling’.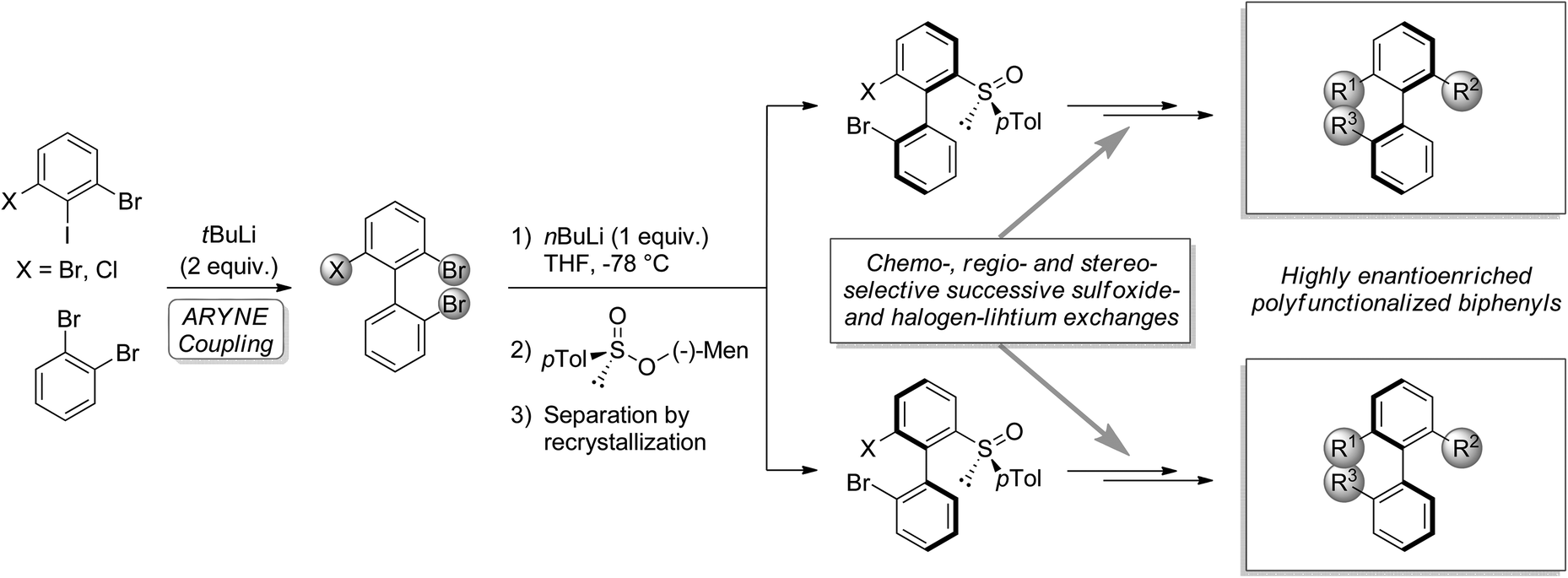 | ||
| Scheme 1 Post-‘ARYNE coupling’ desymmetrization (X = Br) or deracemization (X = Cl) and functionalization of biphenyls. | ||
Results and discussion
In the target transformation, the position of the chiral auxiliary would be the key for an efficient atropo-selective coupling. On the one hand, the auxiliary could be located on the aryllithium nucleophile, closest to the reacting center, namely ortho to lithium. Using a chiral coordinating group at the ortho position would produce a more rigid metallacycle, increasing the chances for a high stereoinduction. On the other hand, when the aryne bears the auxiliary ortho to the triple bond, complex regioselectivity issues could arise due to steric and electronic effects of the aryne substituents,7 including the chiral auxiliary. In particular, a lithium-coordinating auxiliary could favor the addition of the nucleophile alternately on both termini of the triple bond depending on the reaction conditions, as shown by Meyers,16–18 leading to competing kinetic and thermodynamic controls. Consequently, we chose to locate the chiral auxiliary on the nucleophile. The already proposed mechanism5 would be modified as depicted in Scheme 2. Diastereoselection could proceed either by a favored approach of the aryne to the chiral nucleophile B, or by formation of the more stable atropo-diastereomer of the resulting biaryllithium F, depending on the early or late nature of the transition state. Arynes being high energy intermediates, and considering the successful prediction of the regioselectivity of nucleophilic addition onto arynes based on the ground state distortion model,19–24 it is more probable that the transition state would be early. This implies in our case that atropo-diastereoselection arises from the dissymmetric approach of the arylmetal to the aryne.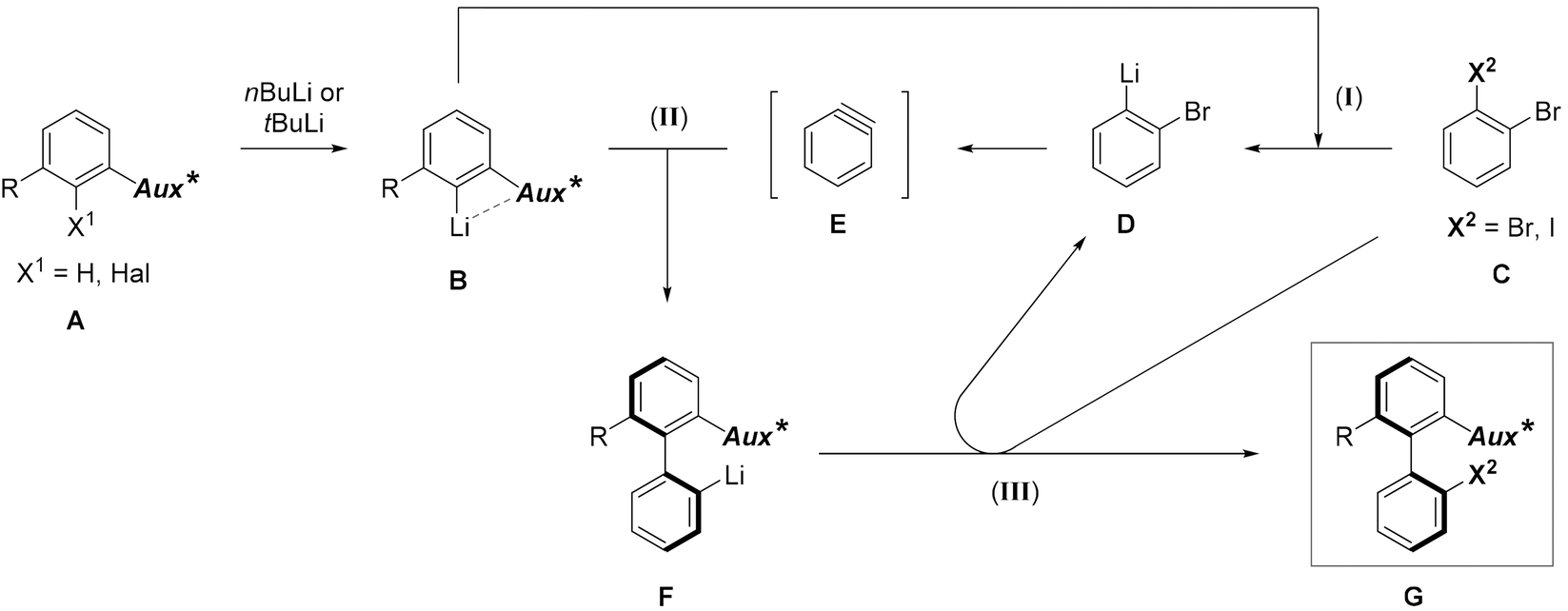 | ||
| Scheme 2 Proposed mechanism of the atropo-diastereoselective ‘ARYNE coupling’ (Aux* = coordinating chiral auxiliary). | ||
Due to the successful results obtained in the literature using sulfoxides as chiral auxiliaries in directed lithiation–diastereoselective trapping sequences on aromatic compounds,25–34 as well as in our precedent work,35–40 and due to the robustness of the tert-butyl sulfinyl group, we chose the latter as the stereoinducer in our targeted transformation.
Atropo-diastereoselective ‘ARYNE coupling’ with aryl tert-butyl sulfoxides
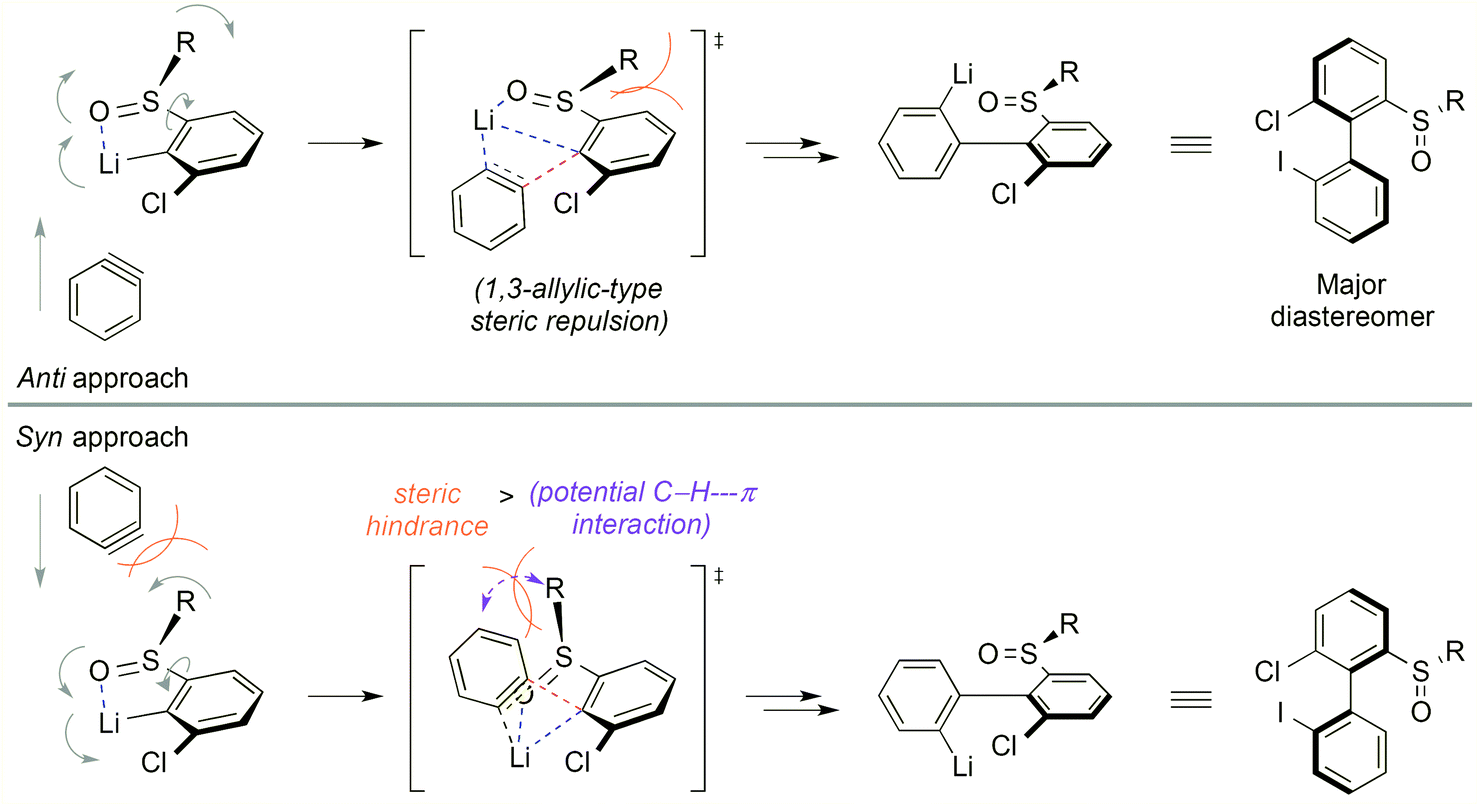
Aryl tert-butyl sulfoxides 1a–c were therefore prepared following Ellman's method,41,42 after enantioselective oxidation of di-tert-butyl disulfide and treatment with the desired aryllithium reagents (Scheme 3). Sulfoxide 1a was chosen as the test substrate in the ‘ARYNE coupling’ with 1,2-dibromobenzene due to its easy access, its completely regioselective lithiation with nBuLi,43,44 and the ease of detection of its derivatives by 1H and 13C NMR thanks to the OMe signals. Preliminary experiments showed that under the previously reported conditions (treatment of the pronucleophile with nBuLi at −78 °C, followed by addition of 1,2-dibromobenzene at −78 °C and stirring while warming up to room temperature) no coupling product was obtained (Table 1, entry 1). We suspected a lack of reactivity of the lithiated arylsulfoxide in the halogen/lithium exchange with 1,2-dibromobenzene (stage (I) in Scheme 2). Accordingly, 0.2 equivalents of nBuLi were added after introduction of the benzyne precursor so as to trigger the generation of ortho-bromo-lithiobenzene and the chain reaction (Table 1, entry 2). Here again, the formation of the coupling product was not observed, and only the starting sulfoxide, 1,2-dibromobenzene, and its homocoupling product, 2,2′-dibromobiphenyl, were recovered. Increasing the temperature before introduction of 1,2-dibromobenzene for a facile elimination of LiBr did not lead to the desired biarylsulfoxide either (entry 3). Finally, in order to facilitate further the formation of the aryne, in both stages (I) and (III) of the mechanism (Scheme 2), 1,2-dibromobenzene was replaced with 1-bromo-2-iodobenzene, since halogen/lithium exchanges are faster with iodine than with bromine. In this case, starting from sulfoxides 1a–c, the corresponding coupling products 2b–d were indeed obtained, in various yields and atropo-selectivities (Table 1, entries 4–6). Only one diastereomer of 2b could be isolated in 20% yield from the complex mixture, although formation of the other atropo-isomer could not be ruled out (entry 4). The benzodioxole analogue 2d was obtained in a ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric ratio, yet with a good 82% yield (entry 6). Most interestingly, biaryl 2c could be obtained with a promising diastereomeric ratio of 79:21, and the atropo-diastereomers could be separated by column chromatography with respective yields of 40% and 11% (entry 5). After recrystallization from ethyl acetate, the absolute configuration of the major isomer could be determined by X-ray diffraction crystallography and was shown to be (SS,aS) (Fig. 1).45 Moreover, we could also show that each atropo-diastereomer of 2c was obtained with high enantioenrichment (er = 97:3 for (SS,aS)-2c, 99:1 for (SS,aR)-2c) by chiral SFC analysis after synthesis of both racemic diastereomers starting from (±)-di-tert-butyl-thiosulfinate, confirming the configurational stability of the sulfinyl auxiliary during the ‘ARYNE coupling’.
:1 diastereomeric ratio, yet with a good 82% yield (entry 6). Most interestingly, biaryl 2c could be obtained with a promising diastereomeric ratio of 79:21, and the atropo-diastereomers could be separated by column chromatography with respective yields of 40% and 11% (entry 5). After recrystallization from ethyl acetate, the absolute configuration of the major isomer could be determined by X-ray diffraction crystallography and was shown to be (SS,aS) (Fig. 1).45 Moreover, we could also show that each atropo-diastereomer of 2c was obtained with high enantioenrichment (er = 97:3 for (SS,aS)-2c, 99:1 for (SS,aR)-2c) by chiral SFC analysis after synthesis of both racemic diastereomers starting from (±)-di-tert-butyl-thiosulfinate, confirming the configurational stability of the sulfinyl auxiliary during the ‘ARYNE coupling’.
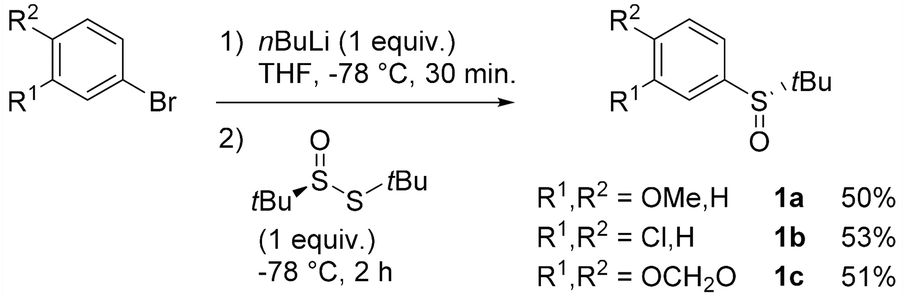 | ||
| Scheme 3 Synthesis of the starting aryl tert-butyl sulfoxides. | ||
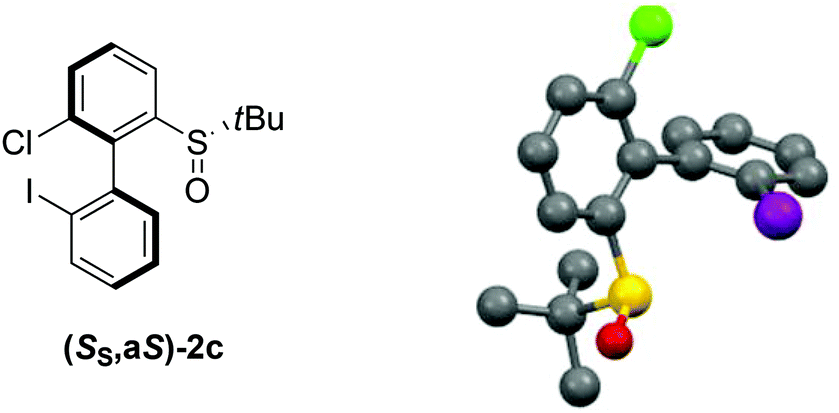 | ||
| Fig. 1 X-ray diffraction of the major atropo-diastereomer (SS,aS)-2c.45 | ||
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | S.M. | R1 | R2 | T (°C) | X | Product | n | Yielda (%) | drb |
| a Combined yield of both atropo-diastereomers. b Diastereomeric ratio measured by 1H NMR of the crude mixture. c Only one atropo-diastereomer could be isolated, whose configuration could not be ascertained; formation of the other atropo-isomer could not be ruled out. d The absolute configuration of the major atropo-diastereomer was shown to be (SS,aS) (see the text). | |||||||||
| 1 | 1a | OMe | H | −78 | Br | 2a | 0 | 0 | — |
| 2 | 1a | OMe | H | −78 | Br | 2a | 0.2 | 0 | — |
| 3 | 1a | OMe | H | −35 | Br | 2a | 0.2 | 0 | — |
| 4 | 1a | OMe | H | −78 | I | 2b | 0.2 | 20c | 100:0c |
| 5 | 1b | Cl | H | −78 | I | 2c | 0.2 | 51 | 79:21d |
| 6 | 1c | OCH2O | −78 | I | 2d | 0.2 | 82 | 54:46 |
|
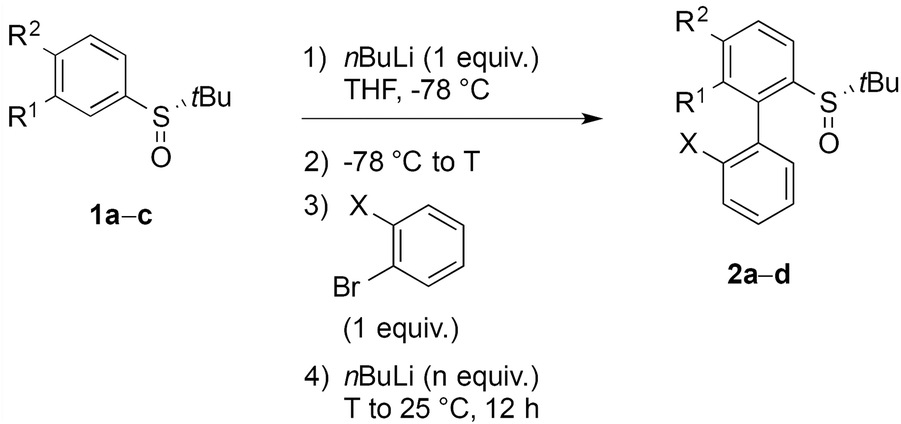
The stereochemical outcome of the ‘ARYNE coupling’ could be explained by the following considerations (Scheme 4). During the attack of the aryllithium nucleophile onto the aryne, both aromatic rings should be perpendicular to minimize steric interactions. Due to the strong lithium-coordinating properties of sulfinyl groups, the ortho-lithioaryl sulfoxide intermediate forms a metallacycle, where the tert-butyl substituent and the non-bonding electron pair point away from each side of the benzene ring, respectively, towards the top and the bottom in Scheme 4. If the aryne approaches the carbon–lithium bond anti to the tBu group, i.e. from the bottom face, the sulfinyl group is forced to rotate “clockwise” in the transition state, thus pushing the tert-butyl towards the ortho-proton. This 1,3-allylic-type repulsion would hence defavor the anti transition state. On the other hand, in the syn approach, the rotation of the sulfinyl moiety would be “counter-clockwise”, pushing the tBu group into a pseudo-axial position. Despite the flatness of the aryne positioned perpendicularly to the aryllithium, the tBu/aryne steric repulsion would dramatically increase the energy of the syn transition state. A weak stabilizing electrostatic interaction between the π cloud of the aryne and the C–H bonds of the tBu group, whose electron density is affected by the electron-withdrawing sulfinyl function,46 could be expected and would oppose the steric repulsion. Given the unambiguous absolute configuration of the major atropo-diastereomer (Fig. 1), it appears that both this potential stabilizing C–H⋯π interaction and the 1,3-allylic-type repulsion of the anti approach are overpowered by the tBu/aryne repulsion in the syn approach. Nevertheless, the imperfect stereoselectivity observed in this reaction may be due to a delicate balance between these various steric and stereoelectronic parameters. This could be also suggested by the poor stereoselectivity of the coupling leading to 2d, which could be ascribed to a competitive coordination of lithium to the oxygen atoms of the dioxole motif, thus upsetting the relative weight of stabilizing or destabilizing interactions discussed above.
 | ||
| Scheme 4 Stereochemical rationale for the atropo-diastereoselective ‘ARYNE coupling’ of aryl tert-butyl sulfoxides (R = tBu). | ||
Synthesis of P,S ligands and evaluation in catalysis
As 2c bears a functionalisable iodo substituent ortho to the biphenyl junction, and as both of its atropo-diastereomers are separable by column chromatography, we reasoned that it could be easily converted into a set of enantiomeric P,S ligands. Indeed, the strong affinity of phosphorus and sulfur for transition metals has motivated the synthesis of many phosphine–thioether bidentate ligands.47,48 Yet, among them, very few are based on an atropo-enriched 1,1′-biaryl-2,2′-diyl phosphine–thioether backbone, and the latter are exclusively binaphthyl-derived ligands.49–58 We were hence interested in the properties of atropo-enriched biphenyl-linked phosphine–thioethers derived from 2c.Each atropomer of sulfoxide 2c was therefore converted into the corresponding thioether, before undergoing iodine/lithium exchange with tBuLi and trapping with chlorodiphenyl- or chlorodicyclohexylphosphine (Scheme 5). Gratifyingly, the corresponding biarylphosphines were obtained without loss of atropo-enrichment. The enantiomeric purity of sulfides 3 and of P,S ligands 4a,b was determined by SFC in comparison with the racemic counterparts obtained by a similar synthesis starting from racemic di-tert-butyl thiosulfinate. The retention of axial configuration was confirmed by X-ray diffraction analysis of monocrystals of compound (aS)-4a (Fig. 2).45
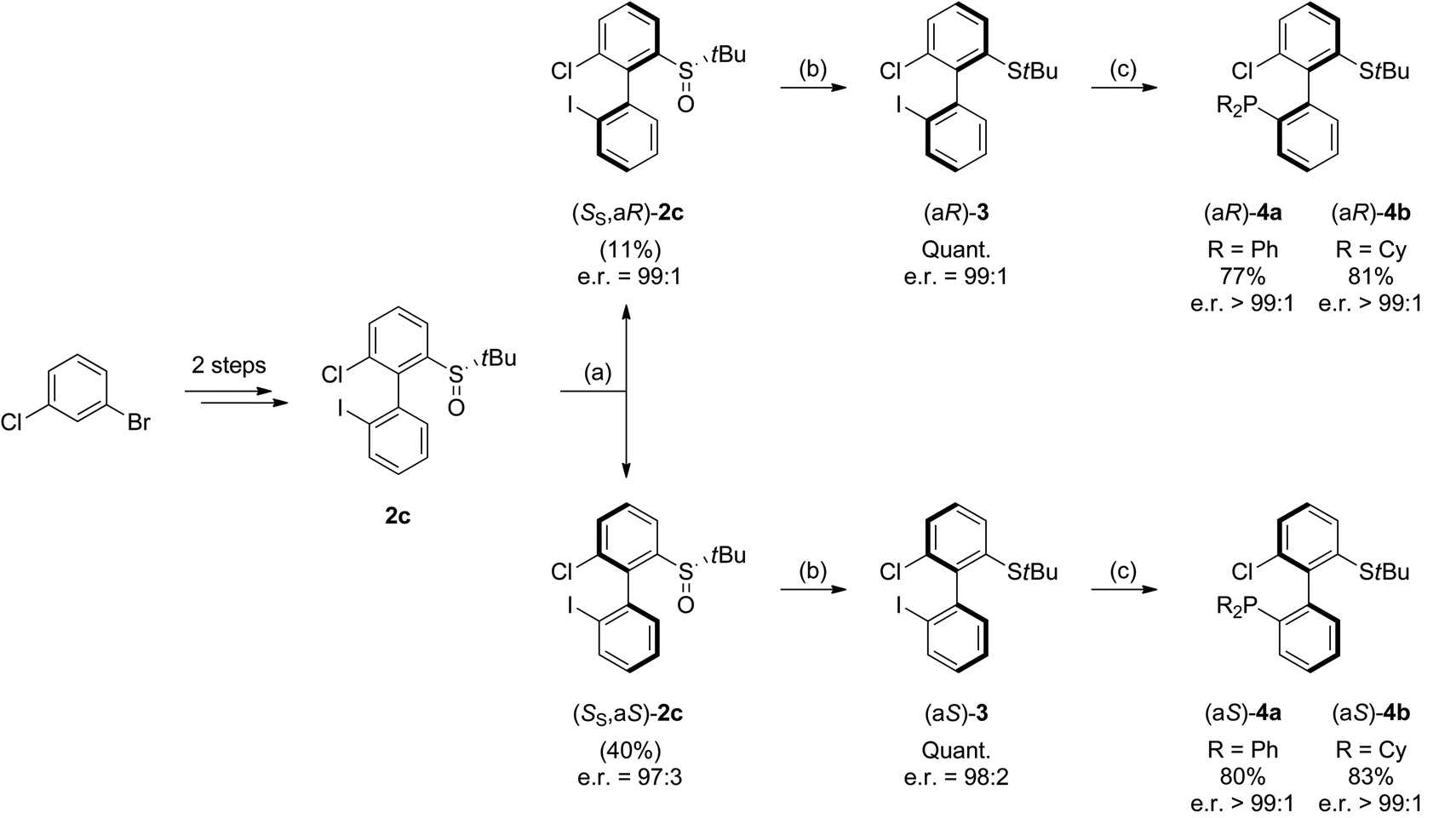 | ||
| Scheme 5 Synthesis of atropo-enantiopure biphenylic P,S ligands. (a) Separation by silica gel column chromatography. (b) TFAA (5 equiv.), NaI (3 equiv.), acetone, −40 °C, 30 min. (c) i. tBuLi (2 equiv.), THF, −100 °C, 5 min.; ii. ClPR2 in toluene, −100 to 25 °C. | ||
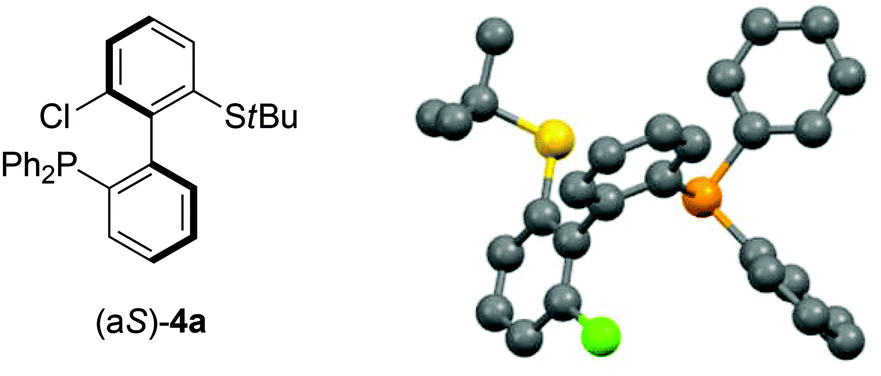 | ||
| Fig. 2 X-ray diffraction of (aS)-4a.45 | ||
The coordination of (aS)-4a with a transition metal was then studied by reacting it with bis(benzonitrile)dichloropalladium (Scheme 6). The expected complex ((aS)-4a)PdCl2 (5) was indeed obtained and characterized by 1H, 13C and 31P NMR as well as HRMS. A clear shift in 31P NMR from −14.1 ppm (free ligand 4a) to +29.4 ppm in 5 attested the coordination of the phosphine moiety to palladium. Similarly, the singlet corresponding to the protons of the tert-butyl group moved downfield, from 1.12 to 1.63 ppm, indicative of electron depletion at sulfur and hence of coordination of the thioether to the metal. In addition, the 13C NMR signal of the quaternary carbons in the tBu group moved upfield from 47.4 ppm in 4a to 30.4 ppm in 5, which is coherent with back-donation from the metal. The structure of complex 5 was further unambiguously determined by X-ray diffraction analysis of single crystals of 5 (Fig. 3).45 The metal is effectively chelated by the bidentate ligand, affording a distorted square-planar geometry. The Cl–Pd–Cl angle is almost equal to 90° (89.60°), but other angles deviate from the ideal case; the P–Pd–S and P–Pd–Cl3 angles are smaller than 90° (respectively 87.61° and 87.97°), while the S–Pd–Cl2 angle is 95.59°, i.e. wider than a right angle. These distortions are explained in part by the difference in trans influence between the phosphine and thioether moieties; indeed, the Pd–Cl2 bond measures 2.339 Å and the Pd–Cl3 bond measures 2.316 Å, demonstrating the stronger trans influence of the phosphine. Last but not least, the axial configuration of the free ligand is preserved in complex 5, and coordination of sulfur to palladium is effected so as to place the tBu group in an equatorial position, away from the phenyl substituents at phosphorus.
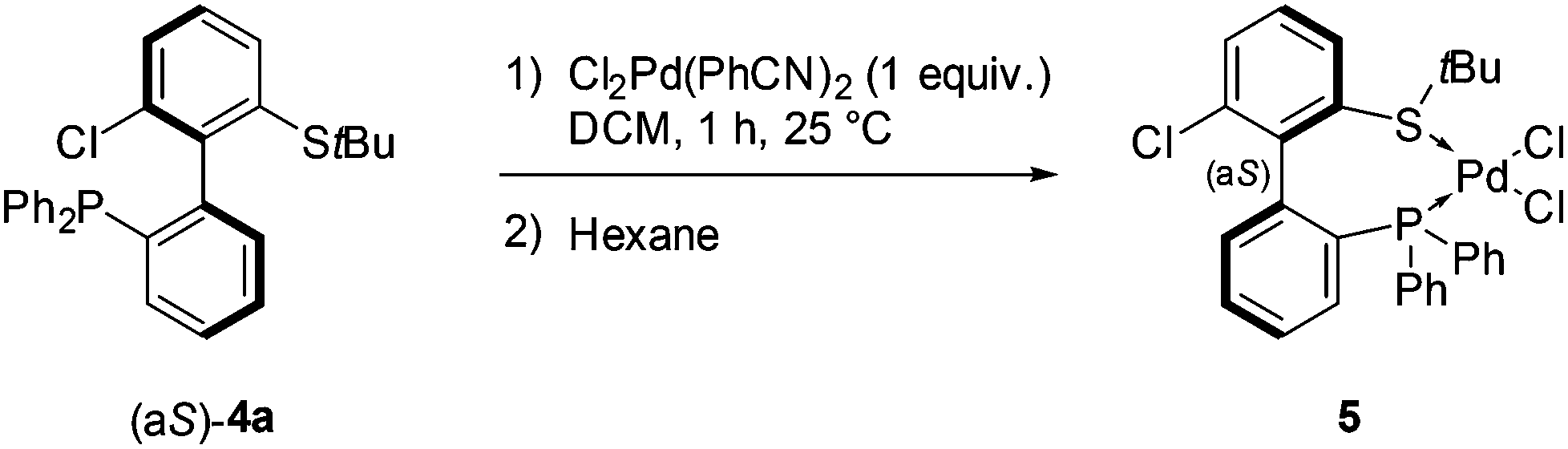 | ||
| Scheme 6 Synthesis of a palladium complex from atropo-enantiopure P,S ligand (aS)-4a. | ||
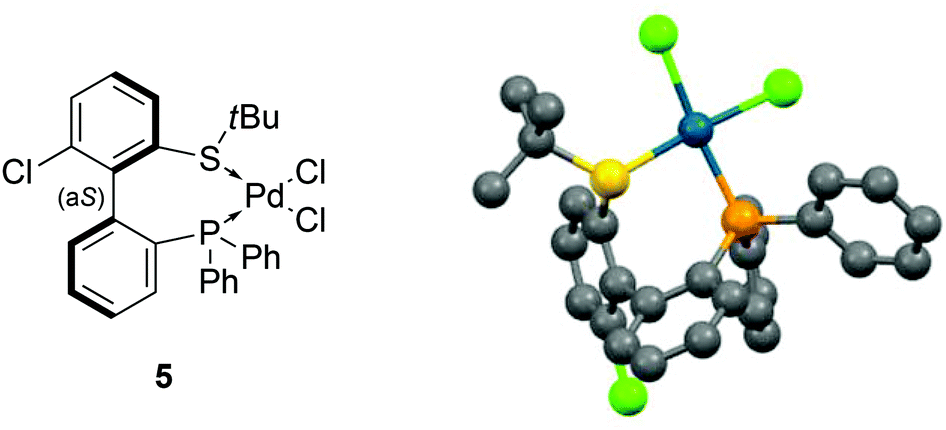 | ||
| Fig. 3 X-ray diffraction of 5.45 | ||
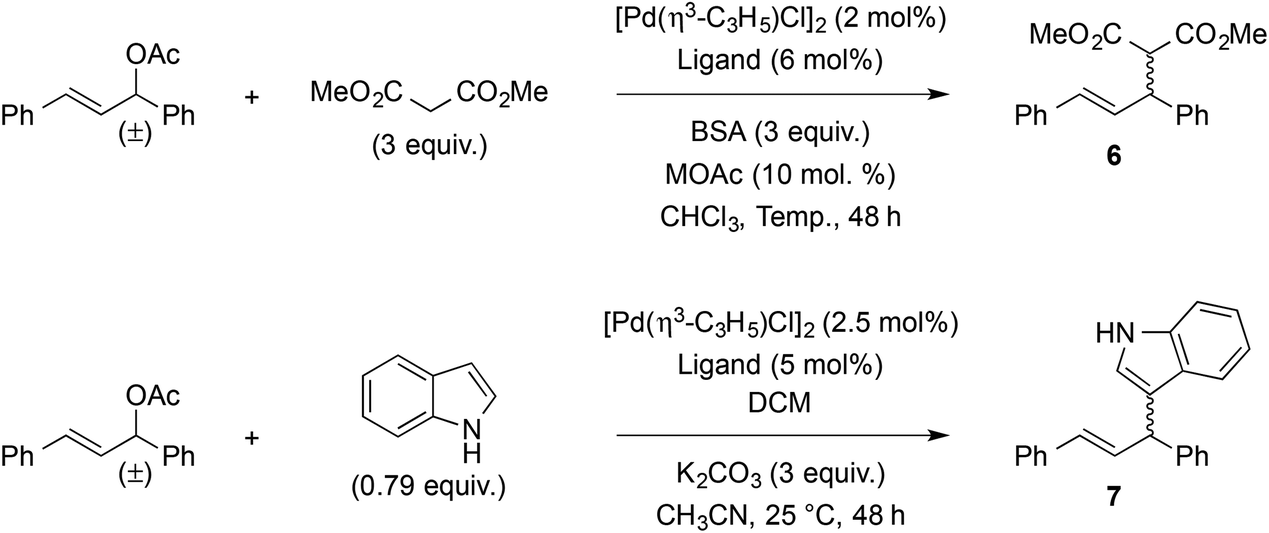
As palladium-catalyzed asymmetric allylic substitution constitutes a benchmark reaction for the assessment of chiral bidentate ligands, compounds 4a,b were evaluated in the reaction of 1,3-diphenylallyl acetate with dimethyl malonate and indole (Table 2). In the reaction with dimethyl malonate, ligands (aS)-4a and (aS)-4b gave similar results in terms of yield and enantioselectivity (entries 1 and 2), indicating negligible influence of the phosphorus substituents. Both enantiomers of ligand 4b also gave similar results as expected (entries 2 and 3). Running the reaction at a higher temperature (25 °C instead of 10 °C) did not bring any improvement (entry 4), but a slight increase in enantioselectivity was observed when decreasing the reaction temperature to −25 °C (entry 5). On the other hand, switching from potassium to lithium in the acetate salt proved markedly advantageous, as it increased the er from 90:10 (80% ee) to 97:3 (94% ee) at 25 °C, and from 93:7 (86% ee) to 99:1 (98% ee) at −25 °C (entries 4–7). For comparison purposes, binaphthyl-derived phosphine–thioether ligands afforded 68–96% yield and 7–96% ee in the same reaction.56 Ligand (aS)-4a was also evaluated in the reaction with indole, using Hoshi and Hagiwara et al.'s conditions developed for related sulfur-MOP ligands.58 In our case (entry 8), 83% yield and 92:8 er (i.e. 84% ee) were achieved, which compares honorably with the cited sulfur-MOP ligands (58–89% yield and 48–92% ee).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Ligand | Product | MOAc | T (°C) | Yielda (%) | erb,c (abs. config.) |
| a Isolated yield. b Enantiomeric ratio measured by chiral SFC analysis. c When possible, absolute configuration was assigned by comparison with literature data. | ||||||
| 1 | (aS)-4a | 6 | KOAc | 10 | 98 | 90:10 (R) |
| 2 | (aS)-4b | 6 | KOAc | 10 | 99 | 91:9 (R) |
| 3 | (aR)-4b | 6 | KOAc | 10 | 93 | 90:10 (S) |
| 4 | (aS)-4a | 6 | KOAc | 25 | 99 | 90:10 (R) |
| 5 | (aS)-4a | 6 | KOAc | −25 | 99 | 93:7 (R) |
| 6 | (aS)-4a | 6 | LiOAc | 25 | 99 | 97:3 (R) |
| 7 | (aS)-4a | 6 | LiOAc | −25 | 99 | 99:1 (R) |
| 8 | (aS)-4a | 7 | — | 25 | 83 | 92:8 |
Conclusions
This work describes the first attempt at developing an unprecedented asymmetric version of the so-called ‘ARYNE coupling’, i.e. the coupling of an aryllithium and an aryne generated in situ from an ortho-dihaloarene by halogen/lithium exchange in a chain reaction. A tert-butylsulfinyl group was used as the chiral auxiliary on the aryllithium nucleophile. In one case, both atropo-diastereomers of the resulting 2-(tert-butylsulfinyl)-6-chloro-2-iodo-1,1′-biphenyl could be separated by column chromatography, and derivatized into atropo-enantiopure biphenyl-based phosphine–thioether ligands. The latter are the first of their kind in the family of phosphine–thioethers, where congeners had been built up to now from a binaphthyl scaffold. The new P,S heterodonor ligands were assessed in model palladium-catalyzed allylic substitution reactions, where they showed comparable efficiency with regard to their binaphthyl homologues of the literature. Other developments of the atropo-diastereoselective ‘ARYNE coupling’ will soon be reported.Experimental section
General information
Starting materials, if commercially available, were purchased and used as received after the check of purity. When known compounds had to be prepared according to literature procedures, pertinent references are given. Air and moisture-sensitive materials were stored in Schlenk tubes under argon. Et2O, 1,4-dioxane and THF were dried by distillation over sodium/benzophenone after the characteristic blue color of sodium diphenyl ketyl (benzophenone sodium “radical–anion”) had been found to persist. CH2Cl2 was dried over CaH2 under argon. Diisopropylamine and triethylamine were dried over KOH under argon. Melting ranges (m.p.) given were found to be reproducible after recrystallization, unless stated otherwise (“decomp.”), and are uncorrected. Thin-layer chromatography (TLC) was carried out on 0.25 mm Merck silica-gel (60-F254). Column chromatography was carried out on silica gel by using MERCK silica (40–63 μm). n-Butyllithium (1.6 M in hexanes, Aldrich) was used as a solution and its concentration was determined following the Wittig–Harborth Double Titration method ((total base) − (residual base after reaction with 1,2-dibromoethane)). NMR-spectra were recorded on Bruker Avance 300 (1H NMR = 300 MHz, 13C NMR = 75.5 MHz) and Bruker Avance 400 (1H NMR = 400 MHz, 13C NMR = 100.6 MHz). Chemical shifts were referred on partial deuterated chloroform (δ[1H] = 7.26 and accordingly δ[13C] = 77.16 ppm) and given in ppm on the δ-scale. Multiplicities were abbreviated as s (singlet), d (doublet), t (triplet), q (quartet) and m (multiplet). Coupling constants were given in Hz. The spectra were processed with the programs Bruker TOPSPIN 2.1 or MestReNova 5.2.5. Mass spectra and elementary analysis were carried out by the Analytical Service of the University of Strasbourg. The angles of rotation were measured on a Perkin Elmer Polarimeter 341 and denoted as specific rotations: [α]20D. Crystal X-ray diffraction analysis was carried out by the Radiocrystallography Service of the University of Strasbourg.General procedure for the synthesis of tert-butylsulfinyl benzene derivatives
At −78 °C, n-butyllithium (1.6 M in hexanes, 1.1 equiv., 12.5 mL, 20.0 mmol) was added dropwise to a solution of the substrate (1 equiv., 20.0 mmol) in THF (100 mL, C = 0.2 mol L−1). The resulting mixture was stirred for 30 min at −78 °C and a solution of (S)-tert-butyl-tert-butanethiosulfinate (1 equiv., 3.89 g, 20.0 mmol) in THF (18 mL, C = 1.1 mol L−1) was added. The resulting mixture was stirred for 2 h at −78 °C and then MeOH was added (100 mL) at −78 °C, followed by water (100 mL) at 25 °C. The aqueous and organic layers were separated and the aqueous layer was extracted with ethyl acetate (3 × 100 mL). The combined organic layers were dried over Na2SO4, filtered and evaporated to dryness.Atropo-diastereoisomer (SS,aR)-2c (colorless solid) (50.7 mg, 0.121 mmol, 11%). 1H NMR (CDCl3, 300 MHz): δ = 7.94 (app. td, J = 7.5, 1.8 Hz, 2 H), 7.65–7.53 (m, 2 H), 7.48–7.38 (m, 2 H), 7.10 (app. td, J = 8.1, 2.1 Hz, 1 H), 1.12 (s, 9 H, S(O)CMe3) ppm; 13C NMR (CDCl3, 75 MHz): δ = 142.8 (CIV), 141.9 (CIV), 139.8, 139.8 (CIV), 134.9 (CIV), 132.3, 132.3, 129.9, 129.6, 128.2, 126.2, 102.1 (CIV, CI), 57.4 (CIV, S(O)CMe3), 23.6 (3 C, S(O)CMe3) ppm. ee = 97% (SFC, CHIRALCEL OD).
Atropo-diastereoisomer (SS,aS)-2c (crystallization from ethyl acetate, colorless crystal) (184 mg, 0.439 mmol, 40%). Mp = 130–132 °C; 1H NMR (CDCl3, 300 MHz): δ = 7.98 (app. d, J = 8.1 Hz, 1 H), 7.94 (dd, J = 7.8, 1.2 Hz, 1 H), 7.68–7.63 (m, 1 H), 7.56 (app. t, J = 7.8 Hz, 1 H), 7.42 (app. t, J = 7.8 Hz, 1 H), 7.21–7.05 (m, 2 H), 1.08 (s, 9 H, S(O)CMe3) ppm; 13C NMR (CDCl3, 75 MHz): δ = 143.0 (CIV), 142.1 (CIV), 140.2 (CIV), 139.4, 134.1 (CIV), 133.1, 132.5, 130.0, 129.6, 127.4, 125.9, 100.9 (CIV, CI), 57.9 (C4, S(O)CMe3), 23.5 (3 C, S(O)CMe3) ppm; [α]20D = −264.1 (c 1, CHCl3). ee = 94% (SFC, CHIRALCEL OD).
Atropo-diastereoisomer 1. 1H NMR (CDCl3, 300 MHz): δ = 7.96 (dd, J = 7.8, 1.0 Hz, 1 H), 7.54 (d, J = 8.1 Hz, 1 H), 7.50 (dd, J = 6.3, 1.5 Hz, 1 H), 7.43 (app. td, J = 7.5, 1.1 Hz, 1 H), 7.09–7.02 (m, 2 H), 6.08 (AB, ΔνAB = 15.0 Hz, J = 1.3 Hz, 2 H, OCH2O), 1.06 (s, 9 H, S(O)CMe3) ppm; 13C NMR (CDCl3, 75 MHz): δ = 149.9 (CIV), 145.2 (CIV), 139.9, 137.1(CIV), 133.2(CIV), 131.8, 129.9, 128.0, 125.0 (CIV), 122.2, 108.7, 102.0 (CII, OCH2O), 100.9 (CIV, CI), 57.2 (CIV, S(O)CMe3), 23.2 (3 C, S(O)CMe3) ppm.
Atropo-diastereoisomer 2. 1H NMR (CDCl3, 300 MHz): δ = 7.99 (dd, J = 8.1, 1.2 Hz, 1 H), 7.55 (d, J = 8.4 Hz, 1 H), 7.41 (app. td, J = 7.5, 1.2 Hz, 1 H), 7.24 (dd, J = 7.5, 1.5 Hz, 1 H), 7.10 (app. td, J = 7.8, 1.8 Hz, 1 H), 7.05 (d, J = 8.1 Hz, 1 H), 6.02 (AB, ΔνAB = 7.1 Hz, J = 1.2 Hz, 2 H, OCH2O), 1.03 (s, 9 H, S(O)CMe3) ppm; 13C NMR (CDCl3, 75 MHz): δ = 150.2 (CIV), 145.3 (CIV), 139.6, 137.3 (CIV), 132.6 (CIV), 132.4, 130.0, 127.6, 125.2 (CIV), 121.9, 108.5, 102.0 (CII, OCH2O), 101.4 (CIV, CI), 57.4 (CIV, S(O)CMe3), 23.3 (3 C, S(O)CMe3) ppm.
Synthesis of (aS)-3. The product was prepared according to the general procedure and starting from (aS)-2-((S)-tert-butylsulfinyl)-6-chloro-2′-iodo-1,1′-biphenyl (SS,aS)-2c. Purification by column chromatography afforded (aS)-3 as a colorless solid (191 mg, 0.474 mmol, 99%). [α]20D = −127.5 (c 1, CHCl3). ee = 96% (SFC, CHIRALCEL OJ-H).
Synthesis of (aR)-3. The product was prepared according to the general procedure and starting from (aR)-2-((S)-tert-butylsulfinyl)-6-chloro-2′-iodo-1,1′-biphenyl (SS,aR)-2c. Purification by column chromatography afforded (aR)-3 as a colorless solid (191 mg, 0.474 mmol, 99%). ee = 98% (SFC, CHIRALCEL OJ-H).
Synthesis of (aS)-4a. The product was prepared according to the general procedure and starting from (aS)-tert-butyl(6-chloro-2′-iodo-(1,1′-biphenyl)-2-yl)sulfane (aS)-3. Purification by column chromatography afforded (aS)-4a as a colorless solid (88.5 mg, 0.192 mmol, 77%). [α]20D = −117.8 (c 1, CHCl3). ee > 99% (SFC, CHIRALPAK AZ).
Synthesis of (aR)-4a. The product was prepared according to the general procedure and starting from (aR)-tert-butyl(6-chloro-2′-iodo-(1,1′-biphenyl)-2-yl)sulfane (aR)-3. Purification by column chromatography afforded (aR)-4a as a colorless solid (92.2 mg, 0.200 mmol, 80%). ee > 99% (SFC, CHIRALPAK AZ).
Synthesis of (aS)-4b. The product was prepared according to the general procedure and starting from (aS)-tert-butyl(6-chloro-2′-iodo-(1,1′-biphenyl)-2-yl)sulfane (aS)-3. Purification by column chromatography afforded (aS)-4b as a colorless solid (98.4 mg, 0.208 mmol, 83%). [α]20D = −125.2 (c 1, CHCl3). ee > 99% (SFC, CHIRALPAK AD).
Synthesis of (aR)-4b. The product was prepared according to the general procedure and starting from (aR)-tert-butyl(6-chloro-2′-iodo-(1,1′-biphenyl)-2-yl)sulfane (aR)-3. Purification by column chromatography afforded (aR)-4b as a colorless solid (96.0 mg, 0.203 mmol, 81%). ee > 99% (SFC, CHIRALPAK AD).
:4). Complex 5 and one molecule of toluene crystallize in the same packing: 1H NMR (CDCl3, 400 MHz) (complex 5 + toluene): δ = 8.07 (d, J = 7.6 Hz, 1 H), 7.83–7.71 (m, 4 H), 7.67 (app. t, J = 7.6 Hz, 1 H), 7.54–7.36 (m, 5 H), 7.35 (dd, J = 7.2, 3.6 Hz, 1 H), 7.30–7.22 (m, 4 H), 7.21–7.13 (m, 4 H), 7.00–6.93 (m, 2 H), 2.36 (s, 3 H, Me of toluene), 1.63 (s, 9 H, SCMe3) ppm; 13C NMR (CDCl3, 101 MHz) (complex 5+ toluene): δ = 141.25 (CIV, d, J = 7.1 Hz), 137.9 (CIV of toluene), 136.9 (CIV, d, J = 2.0 Hz), 136.2, 136.0, 134.6 (d, J = 11.2 Hz), 134.3 (CIV), 134.1 (d, J = 7.1 Hz), 132.6, 131.8–131.7 (m), 131.5–131.4 (m), 130.2, 129.6, 129.5, 129.0, 128.7, 128.5, 128.5, 128.4, 128.2, 127.6 (CIV, d, J = 9.2 Hz), 127.2, 125.3, 123.5 (CIV), 122.9 (CIV), 30.4 (SCMe3 + SCMe3), 21.5 (Me of toluene) ppm; 31P NMR (161 MHz, CDCl3): δ = +29.35; HRMS [M + Na]+ for C28H26Cl3PPdSNa calcd 658.949; found 658.949.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 167.8 (CIV, CO), 140.2 (CIV), 136.8 (CIV), 131.9, 129.1, 128.7, 128.5, 127.9, 127.6, 127.2, 126.4, 57.7, 52.6, 52.5, 49.2 ppm. Determination of enantioenrichment by chiral SFC (CHIRALPAK AD column).
O), 167.8 (CIV, CO), 140.2 (CIV), 136.8 (CIV), 131.9, 129.1, 128.7, 128.5, 127.9, 127.6, 127.2, 126.4, 57.7, 52.6, 52.5, 49.2 ppm. Determination of enantioenrichment by chiral SFC (CHIRALPAK AD column).
Acknowledgements
This work was supported by the CNRS (Centre National de la Recherche Scientifique) and the “Ministère de l'Education Nationale et de la Recherche” (France). A.B.B. thanks the “Ministère de l'Education Nationale et de la Recherche” (France) for an MENRT Ph.D. grant. We are very much grateful to Dr J. Graff and Prof. A. Alexakis, University of Geneva (Switzerland) for SFC and GC analysis.Notes and references
- G. Bringmann, A. J. Price Mortimer, P. A. Keller, M. J. Gresser, J. Garner and M. Breuning, Angew. Chem., Int. Ed., 2005, 44, 5384–5427 CrossRef CAS PubMed.
- G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563–639 CrossRef CAS PubMed.
- H. Gilman and B. Gaj, J. Org. Chem., 1957, 22, 447–449 CrossRef CAS.
- F. Leroux and M. Schlosser, Angew. Chem., Int. Ed., 2002, 41, 4272–4274 CrossRef CAS.
- F. R. Leroux, L. Bonnafoux, C. Heiss, F. Colobert and D. A. Lanfranchi, Adv. Synth. Catal., 2007, 349, 2705–2713 CrossRef CAS.
- L. Bonnafoux, F. Colobert and F. R. Leroux, Synlett, 2010, 2953–2955 CAS.
- V. Diemer, M. Begaud, F. R. Leroux and F. Colobert, Eur. J. Org. Chem., 2011, 341–354 CrossRef CAS.
- V. Diemer, F. R. Leroux and F. Colobert, Eur. J. Org. Chem., 2011, 327–340 CrossRef CAS.
- L. Bonnafoux, R. Gramage-Doria, F. Colobert and F. R. Leroux, Chem. – Eur. J., 2011, 17, 11008–11016 CrossRef CAS PubMed.
- L. Bonnafoux, F. R. Leroux and F. Colobert, Beilstein J. Org. Chem., 2011, 7, 1278–1287 CrossRef CAS PubMed.
- J. Bayardon, H. Laureano, V. Diemer, M. Dutartre, U. Das, Y. Rousselin, J.-C. Henry, F. Colobert, F. R. Leroux and S. Juge, J. Org. Chem., 2012, 77, 5759–5769 CrossRef CAS PubMed.
- V. Diemer, A. Berthelot, J. Bayardon, S. Juge, F. R. Leroux and F. Colobert, J. Org. Chem., 2012, 77, 6117–6127 CrossRef CAS PubMed.
- F. R. Leroux, A. Berthelot, L. Bonnafoux, A. Panossian and F. Colobert, Chem. – Eur. J., 2012, 18, 14232–14236 CrossRef CAS PubMed.
- J. Clayden, P. M. Kubinski, F. Sammiceli, M. Helliwell and L. Diorazio, Tetrahedron, 2004, 60, 4387–4397 CrossRef CAS.
- T. Thaler, F. Geittner and P. Knochel, Synlett, 2007, 2655–2658 CAS.
- A. I. Meyers and W. Rieker, Tetrahedron Lett., 1982, 23, 2091–2094 CrossRef CAS.
- A. I. Meyers and P. D. Pansegrau, Tetrahedron Lett., 1983, 24, 4935–4938 CrossRef CAS.
- P. D. Pansegrau, W. F. Rieker and A. I. Meyers, J. Am. Chem. Soc., 1988, 110, 7178–7184 CrossRef CAS.
- P. Maurin, M. Ibrahim-Ouali, J. L. Parrain and M. Santelli, J. Mol. Struct. (THEOCHEM), 2003, 637, 91–100 CrossRef CAS.
- P. H. Cheong, R. S. Paton, S. M. Bronner, G. Y. Im, N. K. Garg and K. N. Houk, J. Am. Chem. Soc., 2010, 132, 1267–1269 CrossRef CAS PubMed.
- G. Y. J. Im, S. M. Bronner, A. E. Goetz, R. S. Paton, P. H. Y. Cheong, K. N. Houk and N. K. Garg, J. Am. Chem. Soc., 2010, 132, 17933–17944 CrossRef CAS PubMed.
- S. M. Bronner, J. L. Mackey, K. N. Houk and N. K. Garg, J. Am. Chem. Soc., 2012, 134, 13966–13969 CrossRef CAS PubMed.
- A. E. Goetz, S. M. Bronner, J. D. Cisneros, J. M. Melamed, R. S. Paton, K. N. Houk and N. K. Garg, Angew. Chem., Int. Ed., 2012, 51, 2758–2762 CrossRef CAS PubMed.
- A. E. Goetz and N. K. Garg, Nat. Chem., 2013, 5, 54–60 CrossRef CAS PubMed.
- S. Ogawa and N. Furukawa, J. Org. Chem., 1991, 56, 5723–5726 CrossRef CAS.
- T. Shibutani, H. Fujihara and N. Furukawa, Tetrahedron Lett., 1991, 32, 2943–2946 CrossRef CAS.
- J. L. García Ruano, M. T. Aranda and M. Puente, Tetrahedron, 2005, 61, 10099–10104 CrossRef.
- P. B. Hitchcock, G. J. Rowlands and R. J. Seacome, Org. Biomol. Chem., 2005, 3, 3873–3876 CAS.
- N. Le Fur, L. Mojovic, N. Plé, A. Turck, V. Reboul and P. Metzner, J. Org. Chem., 2006, 71, 2609–2616 CrossRef CAS PubMed.
- J. L. G. Ruano and A. M. M. Castro, Heteroat. Chem., 2007, 18, 537–548 CrossRef CAS.
- C. Spitz, J. F. Lohier, V. Reboul and P. Metzner, Org. Lett., 2009, 11, 2776–2779 CrossRef CAS PubMed.
- V. M. Mastranzo, F. Yuste, B. Ortiz, R. Sanchez-Obregon, R. A. Toscano and J. L. G. Ruano, J. Org. Chem., 2011, 76, 5036–5041 CrossRef CAS PubMed.
- Y. Arroyo, M. A. Sanz-Tejedor, A. Parra and J. L. G. Ruano, Chem. – Eur. J., 2012, 18, 5314–5318 CrossRef CAS PubMed.
- J. L. Garcia Ruano, L. Marzo, V. Marcos, C. Alvarado and J. Aleman, Chem. – Eur. J., 2012, 18, 9775–9779 CrossRef CAS PubMed.
- A. Novodomska, M. Dudicova, F. R. Leroux and F. Colobert, Tetrahedron: Asymmetry, 2007, 18, 1628–1634 CrossRef CAS.
- F. Colobert, V. Valdivia, S. Choppin, F. R. Leroux, I. Fernandez, E. Alvarez and N. Khiar, Org. Lett., 2009, 11, 5130–5133 CrossRef CAS PubMed.
- T. Leermann, P.-E. Broutin, F. R. Leroux and F. Colobert, Org. Biomol. Chem., 2012, 10, 4095–4102 CAS.
- T. Wesch, A. Berthelot-Brehier, F. R. Leroux and F. Colobert, Org. Lett., 2013, 15, 2490–2493 CrossRef CAS PubMed.
- T. Wesch, F. R. Leroux and F. Colobert, Adv. Synth. Catal., 2013, 355, 2139–2144 CrossRef CAS.
- B. Yalcouye, S. Choppin, A. Panossian, F. R. Leroux and F. Colobert, Eur. J. Org. Chem., 2014, 6285–6294 CrossRef CAS.
- G. Liu, D. A. Cogan and J. A. Ellman, J. Am. Chem. Soc., 1997, 119, 9913–9914 CrossRef CAS.
- D. J. Weix and J. A. Ellman, Org. Lett., 2003, 5, 1317–1320 CrossRef CAS PubMed.
- D. M. Freudendahl, M. Iwaoka and T. Wirth, Eur. J. Org. Chem., 2010, 3934–3944 CrossRef CAS.
- J. B. Liu, G. H. Chen, J. W. Xing and J. Liao, Tetrahedron: Asymmetry, 2011, 22, 575–579 CrossRef CAS.
- The structures of products (SS,aS)-2c, (aS)-4a and 5 were determined by single crystal X-ray diffraction (see ESI‡). Summary of data: CCDC 1051048, 1051049 and 1051050, respectively.
- R. Robiette, G. Y. Fang, J. N. Harvey and V. K. Aggarwal, Chem. Commun., 2006, 741–743 RSC.
- J. R. Dilworth and N. Wheatley, Coord. Chem. Rev., 2000, 199, 89–158 CrossRef CAS.
- M. Gulea and S. Masson, Top. Curr. Chem., 2003, 229, 161–198 CrossRef CAS.
- S. Gladiali, A. Dore and D. Fabbri, Tetrahedron: Asymmetry, 1994, 5, 1143–1146 CrossRef CAS.
- J. Kang, S. H. Yu, J. I. Kim and H. G. Cho, Bull. Korean Chem. Soc., 1995, 16, 439–443 CAS.
- L. Kollar, S. Gladiali, M. J. Tenorio and W. Weissensteiner, J. Cluster Sci., 1998, 9, 321–328 CrossRef CAS.
- C. Baillie, W. P. Chen and J. L. Xiao, Tetrahedron Lett., 2001, 42, 9085–9088 CrossRef CAS.
- S. Gladiali, S. Medici, G. Pirri, S. Pulacchini and D. Fabbri, Can. J. Chem., 2001, 79, 670–678 CrossRef CAS.
- S. Gladiali, F. Grepioni, S. Medici, A. Zucca, Z. Berente and L. Kollár, Eur. J. Inorg. Chem., 2003, 2003, 556–561 CrossRef.
- C. Baillie and J. Xiao, Tetrahedron, 2004, 60, 4159–4168 CrossRef CAS.
- W. Zhang and M. Shi, Tetrahedron: Asymmetry, 2004, 15, 3467–3476 CrossRef CAS.
- T. Hoshi, T. Hayakawa, T. Suzuki and H. Hagiwara, J. Org. Chem., 2005, 70, 9085–9087 CrossRef CAS PubMed.
- T. Hoshi, K. Sasaki, S. Sato, Y. Ishii, T. Suzuki and H. Hagiwara, Org. Lett., 2011, 13, 932–935 CrossRef CAS PubMed.
- J. M. Chen, D. Li, H. F. Ma, L. F. Cun, J. Zhu, J. G. Deng and J. Liao, Tetrahedron Lett., 2008, 49, 6921–6923 CrossRef CAS.
- Q.-A. Chen, X. Dong, M.-W. Chen, D.-S. Wang, Y.-G. Zhou and Y.-X. Li, Org. Lett., 2010, 12, 1928–1931 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Professor Ei-ichi Negishi for his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 1051048–1051050. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00067j |
| This journal is © the Partner Organisations 2015 |