
Unraveling the electronic control of hydride-ion diffusivity in oxyhydrides from model studies on BaTiO3−2xHx□x

Abstract
Mixed hydride–electronic conductors are technologically important materials, but the mechanism of hydride-ion diffusivity is generally not fully understood. The diffusivities of hydride-ions and oxygen vacancies are closely related because hydride-ions are accommodated in oxygen vacancies, and a neighbouring oxygen vacancy is required for the inter-site migration of a hydride-ion. Here, we investigate the impact of electron localization in the hydride-ion-accepting oxygen vacancy on the inter-site hydride-ion migration dynamics in the perovskite-type oxyhydride BaTiO3−2xHx□x (where □ denotes oxygen vacancies) using density functional theory (DFT). Supercell calculations were designed to model two (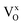 ), one
), one 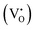 , and zero
, and zero 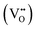 electrons localized in the hydride-ion-accepting site and correspondingly, zero, one, and two electrons delocalized in the conduction band formed from the Ti4+ 3d orbitals. It is found that the trapping of electrons causes the activation energy for inter-site migration to increase from 0.29 eV for
electrons localized in the hydride-ion-accepting site and correspondingly, zero, one, and two electrons delocalized in the conduction band formed from the Ti4+ 3d orbitals. It is found that the trapping of electrons causes the activation energy for inter-site migration to increase from 0.29 eV for 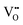 , to 0.39 eV for
, to 0.39 eV for 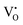 , to 0.60 eV when
, to 0.60 eV when 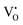 turns into
turns into 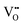 during the migration, and to 0.83 eV for
during the migration, and to 0.83 eV for 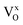 . In an analogous way, the mobility of oxygen vacancies becomes increasingly hindered with increased electron occupation in the vacancies. This suggests that the tailoring of the degree of electron localization by, e.g., bandgap engineering or the introduction of electron trapping impurity states may be effective in tuning the hydride-ion conductivity in oxyhydrides, not limited to BaTiO3−2xHx□x.
. In an analogous way, the mobility of oxygen vacancies becomes increasingly hindered with increased electron occupation in the vacancies. This suggests that the tailoring of the degree of electron localization by, e.g., bandgap engineering or the introduction of electron trapping impurity states may be effective in tuning the hydride-ion conductivity in oxyhydrides, not limited to BaTiO3−2xHx□x.

- This article is part of the themed collection: Materials Advances Covers
 Please wait while we load your content...
Please wait while we load your content...

