
Fused hexacyclic thienoquinoids terminated by indandione for low bandgap organic semiconductors†
Yiyang
Xu
,
Tian
Du
,
Yunfeng
Deng
 * and
Yanhou
Geng
* and
Yanhou
Geng
School of Materials Science and Engineering and Tianjin Key Laboratory of Molecular Optoelectronic Science, Tianjin University, Key Laboratory of Organic Integrated Circuits, Ministry of Education, and Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300072, P. R. China. E-mail: yunfeng.deng@tju.edu.cn
First published on 7th May 2025
Abstract
Two indandione-terminated quinoidal compounds, Q4T and Q4T-4F, featuring a fused-ring thiophene-thieno[3,2-b]thiophene-thiophene (4T) as the central core, were designed and synthesized. The extended π-conjugation of the 4T unit, combined with the quinoidal framework, significantly broadens the absorption spectrum, achieving a low-energy onset of up to 1107 nm. Despite their extended π-conjugation, both Q4T and Q4T-4F exhibit good stability in air, even under UV irradiation. In organic thin-film transistors, both compounds demonstrate electron-transporting behavior, with electron mobilities of (6.3 ± 0.8) × 10−4 and (1.2 ± 0.3) × 10−2 cm2 V−1 s−1 for Q4T and Q4T-4F, respectively. Notably, Q4T-4F exhibits higher electron mobility than Q4T, which is mainly attributed to its closer three-dimensional molecular stacking, as revealed by single-crystal analysis. This unique packing facilitates more efficient electron hopping between molecules. These findings provide an effective strategy for designing and synthesizing low-bandgap n-type molecules with good stability.
Introduction
Structurally, conjugated molecules can exist in either an aromatic or quinoidal form.1–7 In the aromatic form, the units are connected by single carbon bonds, whereas in the quinoidal form, they are linked by double carbon bonds. Typically, the aromatic form is more stable. However, the quinoidal form can be maintained by introducing specific terminal groups at the ends of the conjugated framework to enforce the double-bond configuration.Compared to their aromatic counterparts, quinoidal compounds exhibit significant bond-length alternation, a smaller optical bandgap, and higher electron affinity. Additionally, the conjugated backbone of quinoidal compounds is more rigid and planar due to the inherent double-bond connectivity. These structural and electronic properties make quinoidal compounds a fascinating class of molecules for applications in organic electronics.8–15 Specifically, the strong intermolecular interactions of quinoidal structures is essential for efficient charge transport in organic thin-film transistors (OTFTs) (Chart 1).16–25 The intrinsic electron-accepting nature of quinoidal compounds, combined with their ability to stabilize radical anions enhance their suitability for stable n-type conduction in organic thermoelectrics (OTEs).26–30 The strong absorption in both the visible and near infrared (NIR) regions is beneficial for light-harvesting in organic photovoltaics (OPVs).31–33 Beyond these devices, quinoidal compounds are also being explored for use in emerging applications such as photodetector, spintronics, photothermal conversion, gas sensors and biological sensing.34–39 As a result, the design and synthesis of quinoidal compounds have opened new opportunities in materials science and continue to attract increasing interest in the field of chemistry.
 | ||
| Chart 1 The chemical structures of representative n-type small molecules. For comparison, the small molecules with aromatic structure are also illustrated.40–43 | ||
In principle, quinoidal molecules require electron-withdrawing end-groups to stabilize their termini in the double-bond state.14,18 To achieve this, dicyanomethylene group and its derivatives are commonly introduced at both termini of quinoidal molecules.9 Additionally, other quinoidal systems, such as isatin-terminated16,17,29,44 and carbonyl-terminated quinoids,21,30,45 pyrrolidone-fused quinoids,46–48 and p-azaquinodimethane,15,19 have also been developed. Recently, we introduced a new quinoidal system featuring indandione as the terminal group.18,20,26,49–51 By incorporating electron-donating or electron-withdrawing substituents on the terminal benzene rings and modifying the structure of the quinoidal framework, it is possible to fine-tune the electronic properties of these compounds. This tunability allows quinoidal molecules to meet the specific requirements of various applications. Herein, we reported two indandione-terminated quinoidal compounds, Q4T and Q4T-4F, with a fused hexacyclic thiophene unit as the quinoidal core (Scheme 1), in which thieno[3,2-b]thiophene is fused with another two thiophene units (4T) through a sp3 carbon. A fused ring was chosen as the quinoidal core because such compounds have been widely employed in designing high-performance OTFT and OPV materials.52–54 Besides, the four 4-hexylbenzene groups attached to the sp3 carbon can improve the solubility of the quinoidal compounds. Both compounds displayed strong absorption in NIR region and exhibited an optical bandgap as small as 1.02 eV. When used as the semiconductor layer in OTFTs, these compounds demonstrated n-type transport behaviour. Compared to Q4T, the fluorine-substituted terminal quinoidal compound Q4T-4F exhibited higher electron mobility, attributed to its favourable molecular packing.
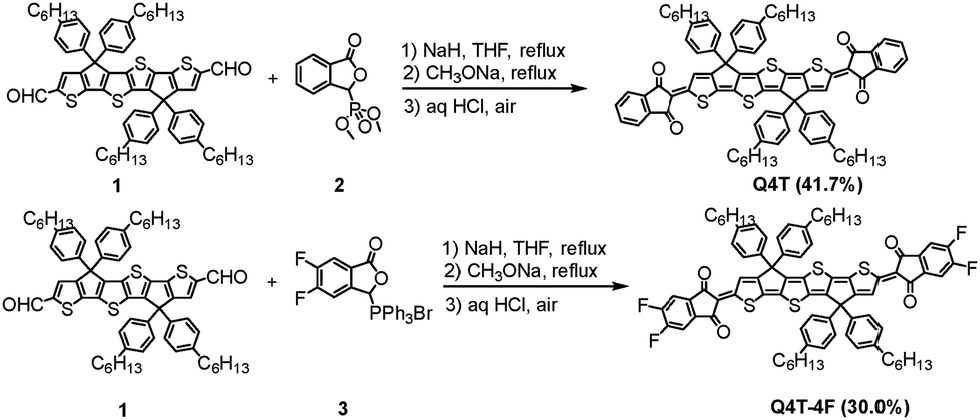 | ||
| Scheme 1 The synthetic route for Q4T and Q4T-4F. | ||
Results and discussions
The synthetic routes for Q4T and Q4T-4F are illustrated in Scheme 1. Compounds 1, 2 and 3 were synthesized according to previously reported methods.18,49,55 The synthesis of Q4T and Q4T-4F followed our previously established protocol,18 involving a Wittig reaction between compounds 1 and 2 or 3, followed by alkoxide-induced rearrangement and air oxidation. The separated yields for Q4T and Q4T-4F are 41.7% and 30.0%, respectively. To verified the low synthetic yield of the quinoidal compounds, we first carried out the Wittig–Horner reaction and found that the intermediate compound was obtained in a high yield of 85%. Moreover, considering that the rearrangement reaction proceeded efficiently under reported conditions,56 we speculate that the low overall yield of Q4T-4F is mainly limited by the oxidation step. The structures of Q4T and Q4T-4F were fully characterized by 1H nuclear magnetic resonance (NMR), 13C NMR, 19F NMR and high resolution matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry (Fig. S1–S6, in the ESI†), and unambiguously confirmed by X-ray crystallographic analysis (vide infra). Both compounds have good solubility in organic solvents such as chloroform and chlorobenzene, which is important for device fabrication by solution processing. Additionally, thermogravimetric analysis (TGA) revealed that Q4T and Q4T-4F exhibited good thermal stability, with decomposition temperatures above 330 °C under a nitrogen atmosphere (Fig. S7, in the ESI†). The melting points of Q4T and Q4T-4F are 237 and 213 °C, respectively, as revealed by differential scanning calorimetry (DSC) analysis (Fig. S8, in the ESI†).Single crystals of Q4T and Q4T-4F, suitable for X-ray crystallographic analysis, were obtained by the slow diffusion of methanol into their toluene solutions. The geometric structures and molecular packing arrangements are shown in Fig. 1 and 2. Both compounds exhibited two independent molecular conformations in the crystal: form I and form II. Conformational molecules in form I displayed a curvature with a wave shape, whereas those in form II exhibited a linear and planar geometry. Similar to the analogous molecules with aromatic structures,55,57 no significant intermolecular interactions were observed in the quinoidal core, due to the four hexylphenyl groups oriented almost perpendicularly to the quinoidal backbone, disrupting regular π–π stacking. Instead, stronger intermolecular π–π stacking interactions were found between the terminal indandione groups. For Q4T, the two molecular conformations formed individual slipped 1D π–π stacking assemblies with the distances of 3.19 Å (form I–form I) and 3.41 Å (form II–form II) (Fig. 1c). These assemblies were crosswise arranged without significant intermolecular interactions. In contrast, Q4T-4F displayed a more interconnected packing motif. Specifically, form I and form II of Q4T-4F each adopted a slipped 1D π–π stacking arrangement with the distances of 3.25 Å and 3.49 Å, respectively (Fig. 2d and f). Additionally, the π-columns formed by form I and form II molecules were bridged by the π–π interactions with a stacking distance of 3.35 Å (Fig. 2e). As a result, Q4T-4T can be packed into compact 3D “network” motif through the multiple π–π interactions (Fig. 2c). The closer 3D molecular stacking creates multiple electron transport channels, facilitating efficient electron transport between molecules.57,58 The electron transfer integrals (t) of the two quinoidal compounds were calculated by DFT using the single crystal packing structures (red values in Fig. 1 and 2). It clearly shows that Q4T-4F exhibits significantly larger t values than Q4T. The bond lengths between the terminal groups and the quinoidal core, as well as between the two β-carbon atoms in the thiophene units, are very close to the length of a typical C(sp2)–C(sp2) double bond (Fig. S9, in the ESI†), indicating that these two quinoidal had large contribution of closed-shell structures in the ground state The large contribution of closed-shell structures is also consistent with the distinct signals observed in the 1H NMR spectra (Fig. S1 and S2, ESI†) because the presence of open shell structures generally broaden 1H NMR signals.
 | ||
| Fig. 1 X-ray crystallographic structure of Q4T: (a) side view, (b) packing diagram π–π distance. Hydrogen atoms and hexylphenyl groups are omitted for the sake of clarity; the red number in parentheses is the calculated transfer integrals. | ||
 | ||
| Fig. 2 X-ray crystallographic structure of Q4T-4F: (a) side view, (b) packing diagram and (c) π–π distance. Hydrogen atoms and hexylphenyl groups are omitted for the sake of clarity; the red number in parentheses is the calculated transfer integrals. | ||
To further elucidate the ground-state electronic structure of Q4T and Q4T-4F, the diradical character index (y0) was calculated. The y0 index quantifies the fraction of the open-shell resonance form in the ground state, with y0 = 1 indicating a fully open-shell structure.59 Density functional theory (DFT) calculations revealed that Q4T and Q4T-4F exhibited small diradical character index (y0) values of approximately 0.04, confirming their predominantly closed-shell electronic structures. Interestingly, the y0 values of Q4T and Q4T-4F are substantially lower than that of an indandione-terminated quinoidal compound featuring a five-ring fused indacenodithiophene (where a benzene ring is fused with two thiophene units through a sp3 carbon) as the central core (y0 = 0.25),51 despite the latter has a shorter π-conjugation length. The reduced diradical character in Q4T and Q4T-4F can be attributed to the lower resonance stabilization energy of thieno[3,2-b]thiophene compared to benzene. This can be qualitative evaluated by the calculation of NICS(1)zz values of 4T and indacenodithiophene. It is shown that the terminal thiophene rings in both 4T and indacenodithiophene exhibit identical NICS(1)zz values (Fig. S10, in the ESI†). However, the central thieno[3,2-b]thiophene unit in 4T has a less negative NICS(1)zz value compared to the benzene ring in indacenodithiophene, suggesting that 4T possesses lower aromaticity and, consequently, lower resonance stabilization energy.
The ultraviolet-visible-NIR (UV-vis-NIR) absorption spectra of Q4T and Q4T-4F in dichlorobenzene solutions and thin films are presented in Fig. 3, and their absorption maxima (λmax) and optical bandgaps (Eg) are summarized in Table 1. In solution, both compounds showed a strong absorption band in 600–850 nm range with extinction coefficient of (ε) reaching up to 2.5 × 105 L mol−1 cm−1. The high ε value can be attributed to the rigid and planar structure of the quinoidal skeleton. In contrast, a weak absorption was observed in the 300 to 600 nm range. These absorption characteristics are consistent with the close-shell ground state of Q4T and Q4T-4F, as the presence of radical character typically resulted in a reduced ε value and the emergence of pronounced absorption in the ultraviolet region.60,61 The introduction of F atoms in the terminal benzene rings caused a slight bathochromic shift in the absorption, increasing from 827 nm for Q4T to 833 nm for Q4T-4F. This phenomenon can be attributed to the inductive effect of the F atoms. The trends observed in the variations of ε and λmax are consistent with the results of time-dependent DFT (Fig. S11, in the ESI†), which was performed using Gaussian 09 program at the B3LYP/6-31G(d,p) level. Notably, the λmax of Q4T are red-shifted by 65 nm compared to the reported aromatic molecules based on 4T end-capped with electron-deficient unit 1,1-dicyanomethylene-3-indanone.55,62,63 Besides, compared to the widely studied dicyanomethylene end-group, the terminal benzene rings in indandione can extend the π-conjugation of the quinoidal molecules. These results highlight the advantages of the indandione terminated quinoidal structure in the synthesis of low-bandgap small molecules. The λmax values for Q4T and Q4T-4F in thin films are 879 nm and 885 nm, respectively. Compared with the absorption in solution, significant redshifts of 52 nm are observed in the film states. Additionally, the absorption bands in the thin films became broader relative to their solution-phase spectra, covering from 600 to 1000 nm. The absorption onsets are 991 nm for Q4T and 1107 nm for Q4T-4F, corresponding to optical bandgaps of 1.26 eV and 1.12 eV, respectively.
 | ||
| Fig. 3 Absorption spectra of Q4T and Q4T-4F (a) in o-DCB solution and (b) in thin film; (c) solution cyclic voltammogram and (d) the HOMO/LUMO energy levels of Q4T and Q4T-4F. | ||
| Compounds | λ solmax (nm) (ε/105 L mol−1 cm−1) | λ filmmax (nm) |
E
optg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (V) a (V) |
E HOMO (V) | E LUMO (V) |
|---|---|---|---|---|---|
| a The optical bandgaps were calculated from the onset of thin film absorption spectra according to Eoptg = 1240/λonset. b The HOMO and LUMO energy levels were calculated according to the equations: EHOMO = −(4.8 + Eoxionset) eV, ELUMO = −(4.80 + Eredonset) eV, in which Eoxionset and Eredonset represent the oxidation and reduction onset-potentials against the half potential of Fc/Fc+ (E° = 0.42 eV). | |||||
| Q4T | 755, 827 (2.4) | 783, 879 | 1.26 | −5.24 | −4.21 |
| Q4T-4F | 757, 833 (2.5) | 788, 885 | 1.12 | −5.27 | −4.29 |
To assess the stability of Q4T and Q4T-4F, we conducted extended monitoring of their 1H NMR spectra and UV-vis-NIR absorption spectra in air. Both their 1H NMR spectra and UV-vis-NIR absorption spectra showed no significant changes after the solutions were stored in air for over 10 days (Fig. S12–S14, in the ESI†). Furthermore, even when the solutions were irradiated with UV light for 30 min, the UV-vis-NIR absorption spectra remained unchanged (Fig. S15, in the ESI†). These results confirm the good stability of both Q4T and Q4T-4F, which is consistent with their closed-shell ground state structures.
Cyclic voltammetry in anhydrous dichloromethane was used to investigate the electrochemical properties of Q4T and Q4T-4F. Both compounds showed distinctive redox peaks. The highest occupied molecular orbital (HOMO)/lowest unoccupied molecular orbital (LUMO) energy levels were calculated from the onset of the first oxidation and reduction peak, with values of −5.24/−4.21 eV for Q4T and −5.27/−4.29 eV for Q4T-4F. The slightly lower-lying HOMO and LUMO energy levels of Q4T-4F, compared to Q4T, can be attributed to the electron-withdrawing effect of F atoms. Moreover, the more pronounced lowering of the LUMO energy relative to the HOMO energy is consistent with the smaller optical bandgap observed for Q4T-4F. This also suggests that the terminal F modifications have a more significant impact on the LUMO energy level than on the HOMO energy level.
To further understand the frontier molecular orbital energy levels of Q4T and Q4T-4F, DFT calculations were performed at the B3LYP/6-31G(d,p) level (Fig. 4). The calculated trends in HOMO and LUMO energy levels between Q4T and Q4T-4F align with the experimental results from electrochemical studies. In both compounds, the HOMO and LUMO were found to be well-delocalized across the quinoidal skeleton, with partial electron density distributed on the terminal benzene rings. The delocalization of both the HOMO and LUMO at the termini contributes to narrowing the optical bandgap. Interestingly, this behaviour contrasts with analogous compounds featuring aromatic structures. In aromatic systems, the HOMO electron density is predominantly localized at the central donor unit, while the LUMO electron density is mainly distributed on the terminal acceptor units.55,63 The distinct delocalization observed in Q4T and Q4T-4F underscores the unique electronic properties imparted by their quinoidal structures.
 | ||
| Fig. 4 Calculated HOMO and LUMO distribution diagrams (a) Q4T and (b) Q4T-4F. The branched alkyl chains were replaced with methyl groups in the calculations. | ||
The charge transport properties of the two quinoidal molecules were evaluated using top-gate/bottom-contact (TG/BC) OTFTs fabricated on Si/SiO2 substrates, with Au as the source/drain electrodes. A spin-coated semiconductor layer, and subsequently a dielectric Cytop layer, were deposited on the top of Au electrodes. Both Q4T and Q4T-4F demonstrated n-channel transport characteristics, with electron mobility (μe) of (3.0 ± 0.7) × 10−3 and (8.7 ± 0.4) × 10−3 cm2 V−1 s−1, respectively, after annealing at 120 °C. In the output characteristics, a linear current increase was observed at low VGS and high VDS, and the transfer curves exhibited a current increase when the VGS is below turn-on voltage (Fig. 5). These observations suggest a weak hole injection and/or transport. However, no field effect could be detected under negative VGS for the OTFT devices, despite the relatively high-lying HOMO levels of Q4T and Q4T-4F. Similar phenomenon has also been reported in the previous studies on quinoidal compounds.18,64 Besides, the high VT presented in transfer curves could cause low reliability of the extract mobility.65 To address the above issues, a thin Ba(OH)2 layer was introduced between the Au source/drain electrodes and the semiconductor layer. This modification can reduce the electron injection barrier and increase the hole injection barrier by lowering the work function of the Au electrodes.66 After Ba(OH)2 modification, the weak hole injection and/or transport was entirely suppressed, as evidenced by the clear off-regime observed in the transfer characteristics. Meanwhile, the output characteristics displayed a linear increase at low VDS and saturation at higher VDS, indicating the realization of pure n-channel transport behaviour. The Ba(OH)2 modification also reduced the threshold voltage and eliminated the hysteresis in both the transfer and output curves. Following the modification, the μe of Q4T and Q4T-4F thin films annealed at 120 °C were (6.3 ± 0.8) × 10−4 and (1.2 ± 0.3) × 10−2 cm2 V−1 s−1, respectively. The two orders of magnitude higher μe observed for Q4T-4F compared to Q4T can be attributed to the formation of 3D transport channels in Q4T-4F, as elucidated in the single-crystal analysis. In addition, grazing-incidence X-ray diffraction (GIXRD) measurements revealed that Q4T-4F exhibits a higher degree of molecular packing order compared to Q4T, as evidenced by the presence of a distinct diffraction peak in the out-of-plane direction for Q4T-4F, while no such peak was observed for Q4T (Fig. S16, in the ESI†). Atomic force microscopy (AFM) measurements (Fig. S17, in the ESI†) revealed similar thin-film morphologies for both compounds, reinforcing that the enhanced charge transport in Q4T-4F arises from its intrinsic molecular packing properties rather than from differences in thin-film morphology. The air stability of the OTFTs was evaluated by monitoring the mobility in air without encapsulation (relative humidity = ∼40%). All devices showed good air stability with minor degradation in electron mobility after 10 days exposure to air (Fig. S18, in the ESI†). This can be attributed to the low-lying LUMO levels of the quinoidal compounds and the small current injection observed in Q4T and Q4T-4F based devices.22,67
 | ||
| Fig. 5 Typical (a)–(d) transfer and (e)–(h) output characteristics of OTFT devices based on (a), (e), (c) and (g) Q4T and (b), (f), (d) and (h) Q4T-4F without (a), (b), (e) and (f) and with (c), (d), (g) and (h) Ba(OH)2 modification. Prior to the deposition of dielectric layer, the thin films were annealed at 120 °C in an argon-filled glovebox for 10 min. | ||
Conclusions
We have synthesized two quinoidal compounds, Q4T and Q4T-4F, with indandione as the termini and thieno[3,2-b]thiophene fused core (4T) as the quinoidal core. The extended π-conjugation of the 4T core, combined with the quinoidal framework, resulted in strong absorption in the infrared region for both compounds. The thin-film absorption of Q4T showed a peak at 879 nm, which red-shifted to 885 nm upon the introduction of terminal F atoms in Q4T-4F. The closed-shell electronic structure, confirmed by single-crystal analysis and theoretical calculations, conferred good air stability to these compounds in solution. The stability was confirmed by monitoring their 1H NMR spectra and UV-vis-NIR absorption spectra over time. Both Q4T and Q4T-4F exhibited low-lying LUMO energy levels below −4.20 eV. As a result, they showed electron-transport characteristics in OTFTs, and the electron mobility of Q4T-4F was two orders of magnitude higher than that of Q4T attributed to the 3D interpenetrating transport network observed in the molecular packing of Q4T-4F and a higher degree of molecular packing order of Q4T-4F.Experimental
All air-sensitive reactions were performed using standard Schlenk techniques under nitrogen. All solvents and reagents were purchased from Innochem, Energy Chemical, etc., which were used directly unless stated otherwise.Synthesis of compound Q4T
To the solution of 1 (300.0 mg, 0.293 mmol) and 2 (155.9 mg, 0.644 mmol) in anhydrous THF (3.5 mL), sodium hydride (60% dispersion in mineral oil, 29.3 mg, 0.731 mmol) was added in portions at 0 °C. After stirring for 17 h at room temperature, sodium methoxide (5.4 mol L−1 in methanol, 1.22 mmol) was added. The resulting mixture was stirred under reflux for 2 h, then the solvent was removed under vacuum. Subsequently, chloroform (15 mL) and HCl aqueous solution (2 mol L−1, 15 mL) were added. The resulting mixture was stirred in air overnight and then extracted with chloroform. The combined organic layer was washed with brine and dried with anhydrous MgSO4 before filtration and concentration under vacuum. The crude product was purified by chromatography on silica gel using PE/dichloromethane (DCM) = 1/1 (v/v) as the eluent, affording Q4T as a dark green solid in the yield of 41.7% (153 mg). 1H NMR (400 MHz, C2D2Cl4, ppm): δ 8.02 (s, 2H), 7.76–7.72 (m, 4H), 7.65–7.62 (m, 4H), 7.14–7.07 (m, 16H), 2.55–2.51 (t, J = 4.0 Hz, 8H), 1.57–1.50 (m, 8H), 1.29–1.21 (s, 24H), 0.83–0.80 (t, J = 4.0 Hz, 12H); 13C NMR (100 MHz, CDCl3, ppm): δ 190.3, 187.8, 174.4, 163.8, 153.9, 153.2, 143.1, 142.9, 141.5, 137.8, 137.6, 134.2, 134.1, 130.5, 129.2, 127.7, 122.4, 122.1, 120.8, 117.7, 60.2, 35.6, 31.7, 31.2, 29.7, 29.1, 23.8, 22.6, 14.1; MS (MALDI-TOF): calcd for C82H78O4S4 [M]+: 1254.478; found: 1254.504.Synthesis of compound Q4T-4F
To the solution of 1 (300.0 mg, 0.293 mmol) and 3 (329.1 mg, 0.644 mmol) in anhydrous THF (3.5 mL), sodium hydride (60% dispersion in mineral oil, 29.3 mg, 0.731 mmol) was added in portions at 0 °C. After stirring for 17 h at room temperature, sodium methoxide (5.4 mol L−1 in methanol, 1.22 mmol) was added. The resulting mixture was stirred under reflux for 2 h, then the solvent was removed under vacuum. Subsequently, chloroform (15 mL) and HCl aqueous solution (2 mol L−1, 15 mL) were added. The resulting mixture was stirred in air overnight and then extracted with chloroform. The combined organic layer was washed with brine and dried with anhydrous MgSO4 before filtration and concentration under vacuum. The crude product was purified by chromatography on silica gel using PE/dichloromethane (DCM) = 1/1 (v/v) as the eluent, affording Q4T-4F as a dark green solid in the yield of 30.0% (103 mg). 1H NMR (400 MHz, C2D2Cl4, ppm) δ 7.98 (s, 2H), 7.56–7.50 (m, 4H), 7.14–7.06 (m, 16H), 2.55–2.51 (t, J = 8.0 Hz, 8H), 1.57–1.50 (m, 8H), 1.30–1.20 (m, 24H), 0.83–0.80 (t, J = 8.0 Hz, 12H); 13C NMR (100 MHz, CDCl3, ppm) δ 187.8, 185.3, 174.9, 164.3, 155.9, 155.7, 154.6, 153.6, 153.3, 153.2, 153.1, 143.2 143.0, 138.5, 138.4, 137.9, 137.4, 130.9, 129.2, 127.7, 120.7, 116.9, 111.8, 111.7, 111.6, 111.3, 68.2, 60.3, 38.8, 35.6, 31.7, 31.2, 30.3, 29.7, 29.1, 23.8, 22.6, 14.1, 10.9; 19F NMR (376 MHz, CDCl3, ppm): δ 125.66–125.70, 125.93–125.98; MS (MALDI-TOF): calcd for C82H74F4O4S4 [M]+: 1326.441; found: 1326.479.Author contributions
Y. D. and Y. G. convinced and supervised this work. Y. D. designed the experiments. Y. X. and T. D. performed the synthesis experiments, device fabrication and characterization. All authors were involved in the analysis of the results and prepared the manuscript.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC 2424255 (Q4T) and CCDC 2424254 (Q4T-4F).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Key R&D Program of China (no. 2021YFA0717900), National Natural Science Foundation of China (no. 22222506, 22475150 and 52121002) and the Fundamental Research Funds for the Central Universities.Notes and references
- P. M. Burrezo, J. L. Zafra, J. T. López Navarrete and J. Casado, Angew. Chem., Int. Ed., 2017, 56(9), 2250–2259 CrossRef CAS PubMed.
- P. Mayorga Burrezo, W. Zeng, M. Moos, M. Holzapfel, S. Canola, F. Negri, C. Rovira, J. Veciana, H. Phan, J. Wu, C. Lambert and J. Casado, Angew. Chem., Int. Ed., 2019, 58(41), 14467–14471 CrossRef CAS PubMed.
- Y. Zou, L. Jiao, Y. Han, L. Ren, Q. Zhou and J. Wu, J. Am. Chem. Soc., 2024, 146(40), 27293–27298 CrossRef CAS PubMed.
- T. Luo, Y. Wang, J. Hao, P.-A. Chen, Y. Hu, B. Chen, J. Zhang, K. Yang and Z. Zeng, Angew. Chem., Int. Ed., 2023, 62(5), e202214653 CrossRef CAS PubMed.
- L. Yuan, J. Yang, S. Qi, Y. Liu, X. Tian, T. Jia, Y. Wang and C. Dou, Angew. Chem., Int. Ed., 2023, 62(51), e202314982 CrossRef CAS PubMed.
- X.-X. Chen, J.-T. Li, Y.-H. Fang, X.-Y. Deng, X.-Q. Wang, G. Liu, Y. Wang, X. Gu, S.-D. Jiang and T. Lei, Nat. Commun., 2022, 13(1), 2258 CrossRef CAS PubMed.
- R. Gao, B. Wu, Z. Liang, X. Zhao, Y. Deng, H. Tian and Y. Geng, Mater. Chem. Front., 2020, 4(3), 891–898 RSC.
- J. Guo, Z. Li, T. Zhang, X. Tian, Y. Wang and C. Dou, Chin. Chem. Lett., 2024, 35(5), 109337 Search PubMed.
- C. Zhang and X. Zhu, Adv. Funct. Mater., 2020, 30(31), 2000765 CrossRef CAS.
- Y. Sun, Y. Guo and Y. Liu, Mater. Sci. Eng. Res. Chem. Intermed., 2019, 136, 13–26 CrossRef.
- C. Wang, Y. Yang, L. Lin, B. Xu, J. Hou, Y. Deng and Y. Geng, Angew. Chem., Int. Ed., 2023, 62(35), e202307856 CrossRef CAS PubMed.
- C. Wang, Y. Xin, H. Gu, L. Ye, Y. Liu, Y. Zhou, Y. Deng and Y. Geng, Angew. Chem., Int. Ed., 2025, 64(3), e202415440 CrossRef CAS PubMed.
- L. Wang, X. Shi, S. Feng, W. Liang, H. Fu and J. Yao, CCS Chem., 2022, 4(8), 2748–2756 CrossRef CAS.
- Y.-f Deng, T.-z Wang and X.-k Geng, Acta Polym. Sin., 2023, 54(6), 803–817 Search PubMed.
- X. Liu, C. L. Anderson and Y. Liu, Acc. Chem. Res., 2023, 56(12), 1669–1682 Search PubMed.
- Y. Kim, H. Hwang, N.-K. Kim, K. Hwang, J.-J. Park, G.-I. Shin and D.-Y. Kim, Adv. Mater., 2018, 30(22), 1706557 CrossRef PubMed.
- Y. Deng, B. Sun, Y. He, J. Quinn, C. Guo and Y. Li, Angew. Chem., Int. Ed., 2016, 55(10), 3459–3462 CrossRef CAS PubMed.
- T. Du, R. Gao, Y. Deng, C. Wang, Q. Zhou and Y. Geng, Angew. Chem., Int. Ed., 2020, 59(1), 221–225 CrossRef CAS PubMed.
- X. Liu, B. He, C. L. Anderson, J. Kang, T. Chen, J. Chen, S. Feng, L. Zhang, M. A. Kolaczkowski, S. J. Teat, M. A. Brady, C. Zhu, L.-W. Wang, J. Chen and Y. Liu, J. Am. Chem. Soc., 2017, 139(24), 8355–8363 Search PubMed.
- C. Wang, T. Du, Y. Deng, J. Yao, R. Li, X. Zhao, Y. Jiang, H. Wei, Y. Dang, R. Li and Y. Geng, Chem. Sci., 2021, 12(27), 9366–9371 RSC.
- K. Kawabata, M. Saito, I. Osaka and K. Takimiya, J. Am. Chem. Soc., 2016, 138(24), 7725–7732 CrossRef CAS.
- A. Velusamy, C.-H. Yu, S. N. Afraj, C.-C. Lin, W.-Y. Lo, C.-J. Yeh, Y.-W. Wu, H.-C. Hsieh, J. Chen, G.-H. Lee, S.-H. Tung, C.-L. Liu, M.-C. Chen and A. Facchetti, Adv. Sci., 2021, 8(1), 2002930 CrossRef CAS.
- A. Velusamy, Y.-Y. Chen, M.-H. Lin, S. N. Afraj, J.-H. Liu, M.-C. Chen and C.-L. Liu, Adv. Sci., 2024, 11(9), 2305361 CrossRef CAS.
- C. Zhang, Y. Zang, F. Zhang, Y. Diao, C. R. McNeill, C.-A. Di, X. Zhu and D. Zhu, Adv. Mater., 2016, 28(38), 8456–8462 CrossRef CAS PubMed.
- Y. Xiong, J. Tao, R. Wang, X. Qiao, X. Yang, D. Wang, H. Wu and H. Li, Adv. Mater., 2016, 28(28), 5949–5953 CrossRef CAS.
- T. Du, Y. Liu, C. Wang, Y. Deng and Y. Geng, Macromolecules, 2022, 55(14), 5975–5984 CrossRef CAS.
- K. Yang, X. Zhang, A. Harbuzaru, L. Wang, Y. Wang, C. Koh, H. Guo, Y. Shi, J. Chen, H. Sun, K. Feng, M. C. Ruiz Delgado, H. Y. Woo, R. P. Ortiz and X. Guo, J. Am. Chem. Soc., 2020, 142(9), 4329–4340 CrossRef CAS PubMed.
- D. Yuan, D. Huang, S. M. Rivero, A. Carreras, C. Zhang, Y. Zou, X. Jiao, C. R. McNeill, X. Zhu, C.-A. Di, D. Zhu, D. Casanova and J. Casado, Chemistry, 2019, 5(4), 964–976 CrossRef CAS.
- T. Wang, C. Xu, Y. Deng and Y. Geng, Adv. Funct. Mater., 2025, 35(2), 2412647 CrossRef CAS.
- H. Cai, H. Tang, T. Wang, C. Xu, J. Xie, M. Fu, X. Luo, Z. Hu, Y. Zhang, Y. Deng, G. Li, C. Liu, F. Huang and Y. Cao, Angew. Chem., Int. Ed., 2024, 63(25), e202402375 CrossRef CAS.
- L. Ren, H. Fan, D. Huang, D. Yuan, C.-A. Di and X. Zhu, Chem. – Eur. J., 2016, 22(48), 17136–17140 CrossRef CAS PubMed.
- H. Feng, B. Yin, L. Pan, X. Liu, S. Kim, Y. Zhao, X. Huang, C. Yang and C. Duan, Chem. Commun., 2023, 59(62), 9529–9532 RSC.
- L. Pan, T. Zhan, J. Oh, Y. Zhang, H. Tang, M. Yang, M. Li, C. Yang, X. Liu, P. Cai, C. Duan, F. Huang and Y. Cao, Chem. – Eur. J., 2021, 27(54), 13527–13533 CrossRef CAS PubMed.
- T. Mikie and I. Osaka, J. Mater. Chem. C, 2020, 8(41), 14262–14288 RSC.
- X. Zhao, H. Cai, Y. Deng, Y. Jiang, Z. Wang, Y. Shi, Y. Han and Y. Geng, Macromolecules, 2021, 54(7), 3498–3506 CrossRef CAS.
- Z. Zeng and J. Wu, Chem. Rec., 2015, 15(1), 322–328 CrossRef CAS.
- X. Shi and C. Chi, Top. Curr. Chem., 2017, 375(4), 68 CrossRef.
- X. Huo, X. Shan, Q. Pan, Z. Cao, Z. Cong, J. Song, J. Jiang, Q. Ma, L. Jia and J. Gao, Sens. Actuators, B, 2024, 403, 135184 CrossRef CAS.
- Q. He, A. Basu, H. Cha, M. Daboczi, J. Panidi, L. Tan, X. Hu, C. C. Huang, B. Ding, A. J. P. White, J.-S. Kim, J. R. Durrant, T. D. Anthopoulos and M. Heeney, Adv. Mater., 2023, 35(11), 2209800 CrossRef CAS.
- J. Youn, P.-Y. Huang, Y.-W. Huang, M.-C. Chen, Y.-J. Lin, H. Huang, R. P. Ortiz, C. Stern, M.-C. Chung, C.-Y. Feng, L.-H. Chen, A. Facchetti and T. J. Marks, Adv. Funct. Mater., 2012, 22(1), 48–60 CrossRef CAS.
- A. Velusamy, S. N. Afraj, S. Yau, C.-L. Liu, Y. Ezhumalai, P. Kumaresan and M.-C. Chen, J. Chin. Chem. Soc., 2022, 69(8), 1253–1275 CrossRef CAS.
- J.-H. Dou, Y.-Q. Zheng, Z.-F. Yao, Z.-A. Yu, T. Lei, X. Shen, X.-Y. Luo, J. Sun, S.-D. Zhang, Y.-F. Ding, G. Han, Y. Yi, J.-Y. Wang and J. Pei, J. Am. Chem. Soc., 2015, 137(50), 15947–15956 CrossRef CAS.
- Q. Zhou, Y. Jiang, T. Du, Z. Wang, Z. Liang, Y. Han, Y. Deng, H. Tian and Y. Geng, J. Mater. Chem. C, 2019, 7(44), 13939–13946 RSC.
- L. Lin, C. Wang, Y. Deng and Y. Geng, Chem. – Eur. J., 2023, 29(12), e202203336 CrossRef CAS PubMed.
- K. Kawabata, I. Osaka, M. Nakano, N. Takemura, T. Koganezawa and K. Takimiya, Adv. Electron. Mater., 2015, 1(6), 1500039 CrossRef.
- W. Cui and F. Wudl, Macromolecules, 2013, 46(18), 7232–7238 CrossRef CAS.
- J. W. Rumer, M. Levick, S.-Y. Dai, S. Rossbauer, Z. Huang, L. Biniek, T. D. Anthopoulos, J. R. Durrant, D. J. Procter and I. McCulloch, Chem. Commun., 2013, 49(40), 4465–4467 RSC.
- M. Yang, T. Du, X. Zhao, X. Huang, L. Pan, S. Pang, H. Tang, Z. Peng, L. Ye, Y. Deng, M. Sun, C. Duan, F. Huang and Y. Cao, Sci. China: Chem., 2021, 64(7), 1219–1227 CrossRef CAS.
- L. Sun, T. Du, C. Wang, D. Geng, L. Li, Y. Han and Y. Deng, Chem. – Eur. J., 2021, 27(69), 17437–17443 CrossRef CAS.
- T. Du, Y. Liu, Y. Deng and Y. Geng, Chin. J. Chem., 2023, 41(7), 776–782 CrossRef CAS.
- Y. Liu, C. Wang, T. Wang, F. Jiao, S. Dong, Y. Deng and Y. Geng, Chin. J. Chem., 2024, 42(9), 997–1003 CrossRef CAS.
- J. Hou, O. Inganäs, R. H. Friend and F. Gao, Nat. Mater., 2018, 17(2), 119–128 CrossRef CAS.
- S. N. Afraj, A. Velusamy, C.-Y. Chen, J.-S. Ni, Y. Ezhumalai, C.-H. Pan, K.-Y. Chen, S.-L. Yau, C.-L. Liu, C.-H. Chiang, C.-G. Wu and M.-C. Chen, J. Mater. Chem. A, 2022, 10(20), 11254–11267 RSC.
- A. Velusamy, S. N. Afraj, Y.-S. Guo, J.-S. Ni, H.-L. Huang, T.-Y. Su, Y. Ezhumalai, C.-L. Liu, C.-H. Chiang, M.-C. Chen and C.-G. Wu, ACS Appl. Mater. Interfaces, 2024, 16(5), 6162–6175 CrossRef CAS.
- X. Shi, L. Zuo, S. B. Jo, K. Gao, F. Lin, F. Liu and A. K. Y. Jen, Chem. Mater., 2017, 29(19), 8369–8376 CrossRef CAS.
- S. L. Shapiro, K. Geiger and L. Freedman, J. Org. Chem., 1960, 25(11), 1860–1865 CrossRef CAS.
- S. Dai, J. Zhou, S. Chandrabose, Y. Shi, G. Han, K. Chen, J. Xin, K. Liu, Z. Chen, Z. Xie, W. Ma, Y. Yi, L. Jiang, J. M. Hodgkiss and X. Zhan, Adv. Mater., 2020, 32(21), 2000645 CrossRef CAS PubMed.
- H. Lai, Q. Zhao, Z. Chen, H. Chen, P. Chao, Y. Zhu, Y. Lang, N. Zhen, D. Mo, Y. Zhang and F. He, Joule, 2020, 4(3), 688–700 CrossRef CAS.
- J. Huang, S. Lu, P.-A. Chen, K. Wang, Y. Hu, Y. Liang, M. Wang and E. Reichmanis, Macromolecules, 2019, 52(12), 4749–4756 CrossRef CAS.
- C. Wang, T. Du, Z. Liang, J. Zhu, J. Ren and Y. Deng, J. Mater. Chem. C, 2021, 9(6), 2054–2062 RSC.
- T. Takahashi, K.-i Matsuoka, K. Takimiya, T. Otsubo and Y. Aso, J. Am. Chem. Soc., 2005, 127(25), 8928–8929 CrossRef CAS PubMed.
- X. Shi, X. Liao, K. Gao, L. Zuo, J. Chen, J. Zhao, F. Liu, Y. Chen and A. K.-Y. Jen, Adv. Funct. Mater., 2018, 28(33), 1802324 CrossRef.
- W. Wang, C. Yan, T.-K. Lau, J. Wang, K. Liu, Y. Fan, X. Lu and X. Zhan, Adv. Mater., 2017, 29(31), 1701308 CrossRef PubMed.
- Y. Suzuki, M. Shimawaki, E. Miyazaki, I. Osaka and K. Takimiya, Chem. Mater., 2011, 23(3), 795–804 CrossRef CAS.
- H. H. Choi, K. Cho, C. D. Frisbie, H. Sirringhaus and V. Podzorov, Nat. Mater., 2018, 17(1), 2–7 CrossRef CAS.
- Z. Fei, Y. Han, J. Martin, F. H. Scholes, M. Al-Hashimi, S. Y. AlQaradawi, N. Stingelin, T. D. Anthopoulos and M. Heeney, Macromolecules, 2016, 49(17), 6384–6393 CrossRef CAS.
- Y. Sui, Y. Deng, T. Du, Y. Shi and Y. Geng, Mater. Chem. Front., 2019, 3(10), 1932–1951 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2424254 (Q4T-4F) and 2424255 (Q4T). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5tc00699f |
| This journal is © The Royal Society of Chemistry 2025 |