
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
ThDP-dependent enzyme catalyzed oxidation of aldehydes
Xiaoyang
Chen
 *a,
Meiting
Zhou
ac,
Xinyu
Duan
b,
Yuting
Zhang
a,
Xiaohe
Chu
*a and
Jian
Xu
*b
*a,
Meiting
Zhou
ac,
Xinyu
Duan
b,
Yuting
Zhang
a,
Xiaohe
Chu
*a and
Jian
Xu
*b
aCollaborative Innovation Center of Yangtze River Delta Region Green Pharmaceuticals, Zhejiang University of Technology, Hangzhou, 310014, China. E-mail: chenxychem@zjut.edu.cn; chuxhe@zjut.edu.cn
bCollege of Biotechnology and Bioengineering, Zhejiang University of Technology, Hangzhou 310014, China. E-mail: jianxu@zjut.edu.cn
cCollege of Biological, Chemical Science and Engineering, Jiaxing University, Jiaxing, 314001, China
First published on 6th August 2025
Abstract
Natural thiamine diphosphate (ThDP)-dependent enzymes are frequently utilized to catalyze the decarboxylation of β-keto acids and the benzoin condensation of aldehydes. Herein, we present a ThDP-dependent enzymatic oxidation of aldehydes mediated by sequential single electron transfer (SET) processes, utilizing hexachloroethane (C2Cl6) as the oxidant. The reaction exhibits high efficiency (yield up to 99% and turnover number up to 2000) and achieves effective stereoselective control for dynamic kinetic resolutions (e.e. up to 99%). This study uncovers a previously undiscovered capability of ThDP-dependent enzymes, thus broadening the functional repertoire of this enzyme class.
Introduction
Oxidation is a fundamental chemical reaction, with the conversion of aldehydes to carboxylic acids being one of the most established and widely employed methodologies.1 Although synthesizing carboxylic acids by oxidizing their corresponding aldehydes is straightforward, achieving efficient and environmentally friendly processes remains a significant challenge.2 Currently, many synthetic methods rely on stoichiometric amounts of hazardous oxidants, including dichromate,3 permanganate,4 periodate reagents,5 oxone6 and sodium chlorite (Pinnick oxidation)7 (Fig. 1a). | ||
| Fig. 1 Oxidation of aldehydes to carboxylic acids. (a) Oxidation with hazardous oxidants. (b) ALDHs and NHCs catalyzed oxidation. (c) This work: ThDP-dependent enzymes catalyzed oxidation. | ||
Nature employs a diverse array of strategies for carboxylic acid formation,8 with the enzymatic oxidation of aldehydes—exemplified by the remarkable activity of aldehyde dehydrogenases (ALDHs)—standing out as particularly efficient.9 These enzymes exhibit catalytic versatility toward a wide spectrum of both aliphatic and aromatic aldehydes, thereby establishing their potential as eco-compatible catalysts in organic synthetic transformations.10 Nevertheless, the inherent reliance on NAD(P)+ cofactor regeneration systems and the challenges in stereochemical control significantly limit their broader implementation in synthetic applications11 (Fig. 1b). On the other hand, the field of biocatalysis is experiencing a transformative revolution, driven by groundbreaking discoveries of enzymatic mechanisms. Recent advances have illuminated several remarkable biocatalytic pathways of oxidations and dehydrogenations, such as the monooxygenation capability of haem peroxygenase,12 the desymmetric dehydrogenation function of ene reductase,13 and the superoxide mechanism of haem catalase.14 Hence, the design and implementation of a more practical biocatalytic approach for aldehyde oxidation,15 particularly with precise stereoselective control, is a highly anticipated breakthrough in the advancement of biocatalysis.
Natural enzymes that employ thiamine diphosphate (ThDP) as a cofactor are capable of catalyzing decarboxylation reactions of β-keto acids as well as benzoin condensation reactions.16 ThDP is structurally categorized as an N-heterocyclic carbene (NHC) derivative.17 NHCs exhibit remarkable catalytic versatility, driving a wide range of chemical transformations,18 including the oxidation of aldehydes.19 The mechanism involves utilizing NHCs to convert the carbon of aldehydes from an electrophile to a nucleophile through the formation of a Breslow intermediate.20 Subsequently, various oxidizing agents can extract electrons from this electron-rich Breslow intermediate, resulting in the formation of oxidized products.21 However, NHC-catalyzed oxidation reactions face persistent challenges in enhancing catalytic efficiency (typically, catalyst loadings of 5–20 mol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) % are required. Fig. 1b).19d In recent years, the integration of significant intermediates from organic synthesis into enzymes to develop enzyme-catalyzed new-to-nature reactions has emerged as a prominent area of research.22 Inspired by the nucleophilicity of Breslow intermediates, our group has developed ThDP-dependent enzyme-catalyzed carbon–carbon bond formation23 and hydrogen–deuterium exchange.24 Huang pioneered the development of an innovative ThDP-dependent enzymatic system that facilitates radical coupling reactions via a key radical cation intermediate.25a,b Shortly afterward, Yang,25c Hayashi25d and Fasan25e further expanded this system. Very recently, Huang's group reported an electroenzymatic oxidation of aldehydes to carboxylic acids.26 In this study, we propose utilizing appropriate oxidants to convert Breslow intermediates via sequential single electron transfers (SETs) within the active site of ThDP-dependent enzymes. This transformation will enable nucleophilic attack by water, thereby facilitating aldehyde oxidation reactions catalyzed by ThDP-dependent enzymes (Fig. 1c). Furthermore, we can utilize the specific substrate pockets of enzymes to gain stereoselective control over this reaction.
% are required. Fig. 1b).19d In recent years, the integration of significant intermediates from organic synthesis into enzymes to develop enzyme-catalyzed new-to-nature reactions has emerged as a prominent area of research.22 Inspired by the nucleophilicity of Breslow intermediates, our group has developed ThDP-dependent enzyme-catalyzed carbon–carbon bond formation23 and hydrogen–deuterium exchange.24 Huang pioneered the development of an innovative ThDP-dependent enzymatic system that facilitates radical coupling reactions via a key radical cation intermediate.25a,b Shortly afterward, Yang,25c Hayashi25d and Fasan25e further expanded this system. Very recently, Huang's group reported an electroenzymatic oxidation of aldehydes to carboxylic acids.26 In this study, we propose utilizing appropriate oxidants to convert Breslow intermediates via sequential single electron transfers (SETs) within the active site of ThDP-dependent enzymes. This transformation will enable nucleophilic attack by water, thereby facilitating aldehyde oxidation reactions catalyzed by ThDP-dependent enzymes (Fig. 1c). Furthermore, we can utilize the specific substrate pockets of enzymes to gain stereoselective control over this reaction.
Results and discussion
To validate our proposal, we selected 4-methoxybenzaldehyde as the model substrate and conducted a series of screenings to identify ThDP-dependent enzymes and oxidants. The reactions were carried out with cell lysates. As shown in Table 1, enzymes with small binding pockets, such as pyruvate decarboxylase from Acetobacter pasteurianus (apPDC) and benzoylformate decarboxylase (BFD), demonstrated low to good reactivity with C2Cl6 as an oxidizing agent. Benzaldehyde lyase from Pseudomonas fluorescens biovar I (pfBAL), characterized by its large binding pocket, exhibited remarkable compatibility with a range of oxidizing agents. Different co-solvents, buffers and temperatures influenced the reaction slightly, and the highest yield was obtained with C2Cl6 as the oxidizing agent using 5% toluene as co-solvent in pH 8.0 MOPS buffer (Tables S1–S3). The enzyme had an excellent turnover number (TON) of 2000, which was higher than that of NHC organocatalysts (typically <100). Control experiments showed that the absence of the enzyme or oxidants prevented the reaction from occurring.|
|
|||
|---|---|---|---|
| Entry | Enzymes | Oxidants | Yield/% |
| a Reaction conditions: 5 mM 1a, 10 mM oxidants, 2.5 mM MgSO4, 0.15 mM ThDP in 50 μL DMSO and 950 μL crude cell extract (pH 8.0 MOPS buffer), 12 h, and 30 °C. DQ = 3,3′,5,5′-tetra-tert-butyldiphenylquinone. b 50 μL toluene instead of DMSO. | |||
| 1 | ApPDC | C2Cl6 | 43 |
| 2 | BFD | C2Cl6 | 83 |
| 3 | pfBAL | C2Cl6 | 88/99b |
| 4 | pfBAL | DQ | 93b |
| 5 | pfBAL | NaClO | 81b |
| 6 | pfBAL | H2O2 | 80b |
| 7 | pfBAL | CCl3Br | 90b |
| 8 | — | C2Cl6 | Trace |
| 9 | pfBAL | — | Trace |

With the optimized reaction conditions in hand, we assessed the scope of the ThDP-dependent enzyme-catalyzed aldehyde oxidation reaction. As shown in Table 2, most benzaldehyde derivatives containing substituents at different positions on the aryl group demonstrated favorable reaction outcomes (2a–2j). Furthermore, the electronic effects had a negligible impact, enabling good compatibility with both electron-withdrawing and electron-donating substituted substrates. The low conversion efficiency of 4-hydroxybenzaldehyde (1g) may be attributed to its structural incompatibility with the hydrophobic binding pocket of the enzyme.27 Notably, substrates containing alkynes (2h) could be effectively converted into their corresponding products while maintaining the integrity of the alkyne structure. Substrates substituted with ferrocene also exhibited good reactivity (2k). Encouraged by these results, we turned our attention to cinnamaldehyde derivatives (2l–2s). This reaction exhibited significant compatibility with this type of substrate, leading to a diverse array of products that possess substituents with differing positions and electronic properties on the benzene ring. In addition, we conducted a study on aliphatic aldehydes without conjugative effects, as their oxidation was generally considered more challenging. However, the reaction efficiency of hexanal was relatively low, resulting in only 9% yield (2t). The branched aliphatic aldehyde 1u reacted efficiently to achieve the corresponding product with 99% yield. Similarly, 3-phenylpropanal (2v) was also accepted by pfBAL, yielding 97% of the desired product. Furthermore, this reaction is easily scalable, enabling the efficient conversion of 100 mg of 1a into the corresponding acid in 91% isolated yield (102 mg 2a).
| a Reaction conditions: 5 mM 1, 10 mM C2Cl6, 2.5 mM MgSO4, 0.15 mM ThDP in 50 μL toluene and 950 μL crude cell extract (pH 8.0 MOPS buffer, containing about 0.5 mol% pfBAL), 4–12 h, and 30 °C. The yields were determined by GC or HPLC. |
|---|

|
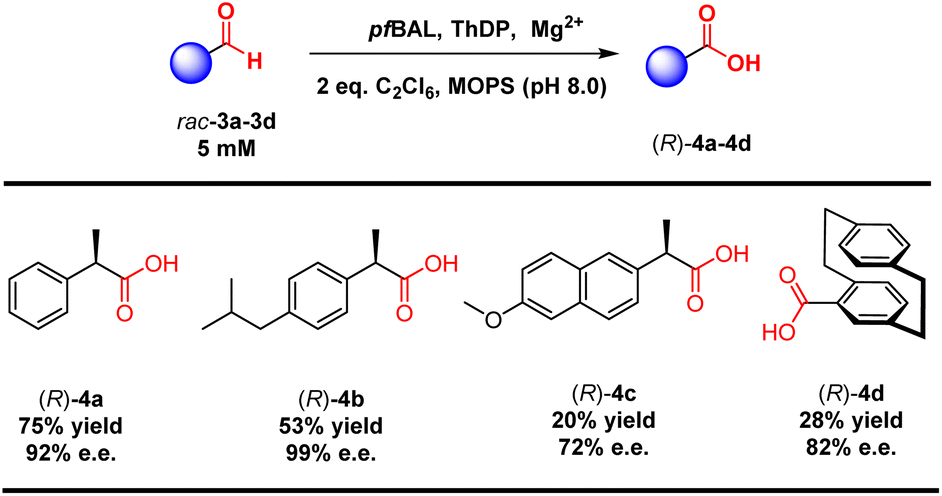
Considering the structural specificity of the enzymatic binding pocket, we endeavored to achieve stereoselective transformations in these reactions. 2-Phenylpropanal was chosen as the model substrate due to the rapid racemization of its chiral center under reaction conditions. Our objective was to utilize pfBAL-catalyzed aldehyde oxidation to achieve the dynamic kinetic resolution of this compound. To our delight, the use of 2 equivalents of C2Cl6 as the oxidant resulted in 40% yield and 90% e.e. of the corresponding product (R)-4a under the catalysis of a purified enzyme (Fig. 2a). Further increasing the oxidant concentration did not enhance the yield; instead, it led to a significant decrease in stereoselectivity. This decline was likely attributed to an increase in background oxidation reactions. We then investigated the influence of elevated reaction temperatures on the reaction. As shown in Fig. 2b, increasing the temperature significantly improved the reaction yield. The optimal temperature was determined to be 45 °C, where the yield reached 75% and the e.e. was 92%. The yield and selectivity decreased when the temperature exceeded 50 °C, likely due to the destabilization of the enzyme's structure at elevated temperatures (Fig. 2b).
 | ||
| Fig. 2 pfBAL catalyzed oxidation of 3a. (a) Optimization of the amount of C2Cl6. (b) Optimization of the reaction temperature. The yields and e.e. values were determined by GC. | ||
To validate our concept, we evaluated a range of substrate types, including those with isobutyl substitutions, which showed promising yields and stereoselectivity ((R)-4b, 53% yield and 99% e.e.). Notably, the planar chiral product [2.2]paracyclophane-4-carboxylic acid (4d) exhibited 28% yield and 82% e.e. via a kinetic resolution process. These results emphasized the ability of ThDP-dependent enzymes to control stereoselectivity in this reaction (Table 3).
| a Reaction conditions: 5 mM 3, 10 mM C2Cl6, 2.5 mM MgSO4, 0.15 mM ThDP, 0.3 mol% purified pfBAL in 100 μL toluene and 900 μL pH 8.0 MOPS buffer, 12 h, and 45 °C. The yields and e.e. values were determined by GC or HPLC. |
|---|
|
To elucidate the mechanism of this enzymatic oxidation reaction, we incorporated 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) and 1,1-diphenylethylene into the model reactions for the radical capture experiments and observed the adducts formed between radical intermediates and radical traps (Fig. 3a, S1 and S2). This result demonstrated that the reaction proceeded via a SET mechanism. Additionally, the reduction product C2Cl4 could be determined by GCMS in the model reaction (Fig. S3). We proposed the mechanism illustrated in Fig. 3b. The oxidation process initiated with the formation of a Breslow intermediate, which underwent two sequential SET processes in the presence of the oxidant C2Cl6. These processes resulted in the generation of an acyl azolium intermediate, which was subsequently hydrolyzed under aqueous conditions to yield the corresponding carboxylic acids, ultimately leading to the release of ThDP.
 | ||
| Fig. 3 (a) Mechanism experiments, (b) proposed mechanism, and (c) MD simulation of pfBAL with 3a; PDB: 3D7K.27 | ||
Finally, we conducted molecular docking calculations and molecular dynamics (MD) simulations to gain insight into the origin of stereoselectivity in the pfBAL-catalyzed aldehyde oxidation reaction (Fig. S4–S7). As illustrated in Fig. 3c, the (R)-enantiomer of 3a exhibited stable binding within the enzyme's active site, maintaining a catalytic distance of 3–4 Å (from ThDP to the carbonyl group of the substrate). In contrast, the (S)-enantiomer failed to achieve stable binding and was observed to dissociate from the enzyme's binding pocket during the MD simulations. From the substrate–protein interaction perspective (Fig. S5 and S7), pfBAL-Rgs forms stronger contacts than pfBAL-Sgs, ensuring tighter binding. In pfBAL-Rgs, a hydrogen bond between the ligand's carbonyl and the cofactor anchors the substrate in an optimal pre-reaction conformation. In contrast, the lack of this bond in pfBAL-Sgs allows the S-type substrate to quickly dissociate. In addition, binding free energy calculations revealed that (R)-3a exhibited enhanced stabilization when interacting with the enzyme (pfBAL-(R)-3a, −35.39 kcal mol−1vs. pfBAL-(S)-3a, −30.84 kcal mol−1). These findings aligned with the experimental results.
Conclusions
In conclusion, we have developed a ThDP-dependent enzyme-catalyzed aldehyde oxidation reaction. This study utilizes the ThDP cofactor in pfBAL to bind with the substrate and produce an electron-rich Breslow intermediate, with C2Cl6 acting as the oxidizing agent to facilitate the oxidation of aldehyde. The reaction exhibits high efficiency and offers stereoselective control for chiral substrates. Mechanistic experiments are consistent with a single electron oxidation mechanism, and MD simulations have been employed to elucidate the origin of stereoselectivity. This study reveals a previously unreported function of ThDP-dependent enzymes, thus broadening the functional repertoire of this enzyme class.Author contributions
J. X. and X. C. directed the project. X. C. and M. Z. developed the reactions and performed the majority of synthetic experiments. X. D. and Y. Z. assisted with synthetic experiments. J. X. and X. C. wrote the paper with input from all authors.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the SI. Experimental methods, optimization data, protein expression, computational details, chemical characterization and NMR spectra. See DOI: https://doi.org/10.1039/d5sc03250d.Acknowledgements
The financial support from the National Natural Science Foundation of China (No. 22201106, 22477112, 22322705, and 22171243), the National Key Research and Development Program of China (No. 2021YFC2102000) and the Zhejiang Provincial Natural Science Foundation (LQ22B020006) is gratefully acknowledged.Notes and references
- (a) J. March, Advanced Organic Chemistry: Reactions, Mechanisms and Structure, 4th edn, Wiley, New York, 1992 Search PubMed; (b) T. J. Collins, Designing Ligands for Oxidizing Complexes, Acc. Chem. Res., 1994, 27, 279–285 CrossRef; (c) M. B. Smith and J. March, Advanced Organic Chemistry: Reactions, Mechanisms and Structure, 6th edn, Wiley, Hoboken, 2007 Search PubMed; (d) G. Tojo and M. Fernández, Oxidation of Primary Alcohols to Carboxylic Acids, Springer, New York, 2010 Search PubMed; (e) P. Y. Bruice, Organic Chemistry, Prentice Hall, Englewood Cliffs, NJ, 2012 Search PubMed.
- (a) M. Liu and C.-J. Li, Angew. Chem., Int. Ed., 2016, 55, 10806–10810 CrossRef PubMed; (b) H. Yu, S. Ru, G. Dai, Y. Zhai, H. Lin, S. Han and Y. Wei, Angew. Chem., Int. Ed., 2017, 56, 3867–3871 CrossRef PubMed.
- J. K. Thottathil, J. L. Moniot, R. H. Mueller, M. K. Y. Wong and T. P. Kissick, J. Org. Chem., 1986, 51, 3140–3143 CrossRef.
- A. Mahmood, A. Robinson and L. Powell, Org. Process Res. Dev., 1999, 3, 363–364 CrossRef.
- M. Hunsen, Synthesis, 2005, 15, 2487–2490 CrossRef.
- B. R. Travis, M. Sivakumar, G. O. Hollist and B. Borhan, Org. Lett., 2003, 5, 1031–1034 CrossRef PubMed.
- B. S. Bal, W. E. Childers and H. W. Pinnick, Tetrahedron, 1981, 37, 2091–2096 CrossRef.
- (a) S. M. Glueck, S. Gümüs, W. M. F. Fabianb and K. Faber, Chem. Soc. Rev., 2010, 39, 313–328 RSC; (b) D. Troiano, V. Orsat and M. Dumont, ACS Catal., 2020, 10, 9145–9169 CrossRef CAS.
- (a) N. A. Sophos and V. Vasiliou, Chem.-Biol. Interact., 2003, 144, 5–22 CrossRef; (b) Z.-J. Liu, S. Y.-J. Sun, J. Rose, Y.-J. Chung, C.-D. Hsiao, W.-R. Chang, I. Kuo, J. Perozich, R. Lindahl, J. Hempel and B.-C. Wang, Nat. Struct. Biol., 1997, 4, 317–326 CrossRef CAS.
- (a) X.-Y. Zhang, X. Wang, N.-W. Li, Z.-W. Guo, M.-H. Zong and N. Li, ChemCatChem, 2020, 12, 3257–3264 CrossRef CAS; (b) T. Adachi, T. Miyata, F. Makino, H. Tanaka, K. Namba, K. Kano, K. Sowa, Y. Kitazumi and O. Shirai, ACS Catal., 2023, 13, 7955–7965 CrossRef CAS; (c) X. Jiao, Y. Zhang, Q. Chen, J. Pan and J. Xu, Catal. Sci. Technol., 2016, 6, 7094–7100 RSC; (d) Y. Han, N. Geng, J. Sha, H. Li, C. You, W. Liu, J. Zhang, J. Shi, X. Wu and W. Zhang, Adv. Synth. Catal., 2025, e202500027 CrossRef CAS.
- (a) J. P. S. Choo, F. L. Sirota, W. W. L. See, B. Eisenhaber and Z. Li, ACS Catal., 2023, 13, 11268–11276 Search PubMed; (b) S. Wu, Y. Zhou, D. Seet and Z. Li, Adv. Synth. Catal., 2017, 359, 2132–2141 CrossRef CAS; (c) W. W. L. See, J. P. S. Choo, L. Du and Z. Li, Adv. Synth. Catal., 2024, 366, 2967–2975 CrossRef CAS.
- D. Deng, Z. Jiang, L. Kang, L. Liao, X. Zhang, Y. Qiao, Y. Zhou, L. Yang, B. Wang and A. Li, Nat. Catal., 2025, 8, 20–32 CrossRef CAS.
- (a) H. Wang, B. Gao, H. Cheng, S. Cao, X. Ma, Y. Chen and Y. Ye, Nat. Chem., 2025, 17, 74–82 CrossRef CAS PubMed; (b) Y. Hu, J. Chen, S. Qi, H. Wang, Z. Zhu, Y. Peng, W. Wang, G. Huang, Z. Fang, Y. Ye, Z. Wang and K. Guo, Angew. Chem., Int. Ed., 2025, e202501425 CAS.
- C.-C. Chen, Z.-P. Yu, Z. Liu, Y. Yao, P.-L. Hagedoorn, R. A. Schmitz, L. Yang, L. Yu, A. Liu, X. Sheng, H. Su, Y. Ma, T. Wang, J.-W. Huang, L. Zhang, J. Yan, J. Bao, C. Cui, X. Li, P. Shen, W. Zhang, J. Min, C.-Y. Wang, R.-T. Guo and S.-S. Gao, Nature, 2025, 640, 840–846 CrossRef CAS PubMed.
- P. Könst, H. Merkens, S. Kara, S. Kochius, A. Vogel, R. Zuhse, D. Holtmann, I. W. C. E. Arends and F. Hollmann, Angew. Chem., Int. Ed., 2012, 51, 9914–9917 CrossRef PubMed.
- (a) L. Lanza, F. Rabe von Pappenheim, D. Bjarnesen, C. Leogrande, A. Paul, L. Krug, K. Tittmann and M. Müller, Angew. Chem., Int. Ed., 2024, 63, e202404045 CrossRef CAS; (b) K. Hernández, T. Parella, G. Petrillo, I. Usón, C. M. Wandtke, J. Joglar, J. Bujons and P. Clapés, Angew. Chem., Int. Ed., 2017, 56, 5304–5307 CrossRef; (c) R. Westphal, C. Vogel, C. Schmitz, J. Pleiss, M. Müller, M. Pohl and D. Rother, Angew. Chem., Int. Ed., 2014, 53, 9376–9379 CrossRef CAS; (d) Y. Zhang, Y. Li, Y. Chen, W. Liu, Q. Zhao, J. Feng, P. Yao, Q. Wu and D. Zhu, ACS Catal., 2024, 14, 9687–9700 CrossRef CAS; (e) H. Li, Z. Zhu, D. Wu, Y. Ju, D. Li, K. Zou, C. Zhu, J. Zhang, G. Zhu, L. Zhang, Y. R. Chi and Y. Xie, J. Am. Chem. Soc., 2025, 147, 3102–3109 Search PubMed.
- (a) J. Pletcher and M. Sax, Science, 1966, 154, 1331–1333 CrossRef CAS PubMed; (b) M. Bielecki and R. Kluger, Angew. Chem., Int. Ed., 2017, 56, 6321–6323 CrossRef CAS.
- D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307–9387 Search PubMed.
- (a) S. D. Sarkar, S. Grimme and A. Studer, J. Am. Chem. Soc., 2010, 132, 1190–1191 CrossRef PubMed; (b) E. E. Finney, K. A. Ogawa and A. J. Boydston, J. Am. Chem. Soc., 2012, 134, 12374–12377 CrossRef CAS; (c) J. Guin, S. D. Sarkar, S. Grimme and A. Studer, Angew. Chem., Int. Ed., 2008, 47, 8727–8730 Search PubMed; (d) W. Harnying, P. Sudkaow, A. Biswas and A. Berkessel, Angew. Chem., Int. Ed., 2021, 60, 19631–19636 Search PubMed.
- M. Pareek, Y. Reddi and R. B. Sunoj, Chem. Sci., 2021, 12, 7973–7992 RSC.
- S. Chakraborty, S. Barik and A. T. Biju, Chem. Soc. Rev., 2025, 54, 1102–1124 Search PubMed.
- (a) D. C. Miller, S. V. Athavale and F. H. Arnold, Nat. Synth., 2022, 1, 18–23 CrossRef CAS PubMed; (b) K. Chen and F. H. Arnold, Nat. Catal., 2020, 3, 203–213 CrossRef CAS.
- X. Chen, Z. Wang, Y. Lou, Y. Peng, Q. Zhu, J. Xu and Q. Wu, Angew. Chem., Int. Ed., 2021, 60, 9326–9329 CrossRef CAS PubMed.
- J. Xu, Y. Lou, L. Wang, Z. Wang, W. Xu, W. Ma, Z. Chen, X. Chen and Q. Wu, ACS Catal., 2021, 11, 13348–13354 CrossRef CAS.
- (a) Y. Xu, H. Chen, L. Yu, X. Peng, J. Zhang, Z. Xing, Y. Bao, A. Liu, Y. Zhao, C. Tian, Y. Liang and X. Huang, Nature, 2024, 625, 74–78 CrossRef CAS; (b) Z. Xing, F. Liu, J. Feng, L. Yu, Z. Wu, B. Zhao, B. Chen, H. Ping, Y. Xu, A. Liu, Y. Zhao, C. Wang, B. Wang and X. Huang, Nature, 2025, 637, 1118–1123 CrossRef CAS; (c) X. Liu, S. Xu, H. Chen and Y. Yang, ACS Catal., 2024, 14, 9144–9150 CrossRef CAS; (d) S. Kato, S. Fujisawa, Y. Adachi, M. Bandai, Y. Mori, S. Mori, T. Shirai and T. Hayashi, J. Am. Chem. Soc., 2025, 147, 14837–14844 CrossRef CAS PubMed; (e) Y. Lu, R. D. Adukure, S. Roy, D. L. Chien, M. J. McGill, S. Polara, G. A. Cisneros, K. A. Scheidt and R. Fasan, J. Am. Chem. Soc., 2025, 147, 17804–17816 CrossRef PubMed.
- B. Zhao, Y. Xu, Q. Zhu, A. Liu, X. Peng, T. Zhang, L. Yu, Y. Zhang and X. Huang, Nature, 2025, 643, 699–704 CrossRef CAS.
- G. S. Brandt, N. Nemeria, S. Chakraborty, M. J. McLeish, A. Yep, G. L. Kenyon, G. A. Petsko, F. Jordan and D. Ringe, Biochem., 2008, 47, 7734–7743 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |