
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Intramolecular arylsulfide-coordinated diboraanthracenes: effect of B–S coordination on ground-state and excited-state behavior†
Hiroki
Narita
 a,
Alexander
Virovets
b,
Hans-Wolfram
Lerner
b,
Matthias
Wagner
b and
Shigehiro
Yamaguchi
*ac
a,
Alexander
Virovets
b,
Hans-Wolfram
Lerner
b,
Matthias
Wagner
b and
Shigehiro
Yamaguchi
*ac
aDepartment of Chemistry, Graduate School of Science, Research Center for Materials Science (RCMS), Integrated Research Consortium on Chemical Sciences (IRCCS), Nagoya University, Furo-cho, Chikusa, Nagoya 464-8602, Japan. E-mail: yamaguchi.shigehiro.r9@f.mail.nagoya-u.ac.jp
bInstitut für Anorganische und Analytische Chemie, Goethe-Universität Frankfurt, Max-von-Laue-Straße 7, 60438 Frankfurt am Main, Germany
cInstitute of Transformative Bio-Molecules (WPI-ITbM), Nagoya University, Furo-cho, Chikusa, Nagoya 464-8601, Japan
First published on 14th April 2025
Abstract
Controlling boron–heteroatom interactions in triarylborane scaffolds can lead to stimuli-responsive photophysical properties. A key molecular design to this end is the utilization of a labile coordination bond between the boron atom and a Lewis basic heteroatom. Herein, we report the synthesis of a series of 9,10-dihydro-9,10-diboraanthracenes (DBAs) bearing ortho-arylthiomethyl-substituted phenyl groups on the boron atom as a new family of stimuli-responsive boron-containing π-conjugated molecules. The two ortho-arylthiomethyl groups coordinate to the boron atoms by forming five-membered rings in the DBA scaffolds to produce the cis isomers predominantly, where the strength of the boron–sulfur bonds can be tuned by structural and electronic modifications of the aryl groups. In the ground state, the B–S bond is cleaved upon heating in solution. In the excited state, the B–S bond undergoes dissociation, resulting in emission from tricoordinate species. The aryl groups on the sulfur atom also play a role in forming an intramolecular charge-transfer state, whereby the emissions are bathochromically shifted with large apparent Stokes shifts. Moreover, the B–S bonds are sensitive to solvent polarity and temperature, resulting in multiple emission properties depending on the surrounding environment.
Introduction
Organic π-conjugated compounds containing tricoordinate boron atoms have attracted much attention owing to their potential utilities in a wide range of applications,1 such as nonlinear optical materials,2,3 light-emitting materials,4 and fluorescent probes for bioimaging.5–8 The vacant p orbital of the boron atom in these molecules plays crucial roles in furnishing not only electron-accepting properties but also stimuli-responsive properties. In particular, boron-containing π-conjugated skeletons can form intermolecular Lewis acid–base complexes with various types of Lewis bases, for example, fluoride ions,9 cyanide ions,10 pyridine derivatives,11 and phosphine derivatives.12 The complexation impairs the electron-accepting ability of the boron moiety, resulting in substantial changes in their electronic structures and thereby photophysical properties, typically, hypsochromic shifts in the absorption and fluorescence spectra. By adjusting the balance between the Lewis acidity of the boron atom and the Lewis basicity of the base, the reversible switching between the tri- and tetracoordinate states is realized in some complexes in response to the surrounding environment or external stimuli, giving rise to various intriguing phenomena, such as thermochromism,13 solubility tuning,14 and photodissociation-induced dual emission.15A design strategy to form labile Lewis acid–base complexes is the introduction of a weak Lewis basic moiety into a triarylborane scaffold in an intramolecularly coordinating fashion. For this purpose, various coordinating groups have been utilized, such as –NR2,16 –OR,17 –C(R′)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O,18 –C(R′)NR,19 or –P(O)R2.20 As for intramolecularly sulfur-coordinated organoboranes, Rupar and coworkers reported a tetracoordinate borafluorene with a pincer-like aryl group (A), in which one of the sulfur atoms was coordinated to the boron center in the ground state, and the resulting B–S bond underwent dissociation in the excited state (Fig. 1a).17 Recently, we reported donor–π–acceptor (D–π–A)-type organoborane fluorophores bearing ortho-PX-substituted phenyl groups on the boron atom (B; X = O or S).20d The PX groups also underwent photodissociation in the excited state, where the PS derivative facilitated the dissociation compared with the PO congeners. Thus, the B–S coordination bond can be expected to enable the formation of labile Lewis acid–base complexes with stimuli- or environment-responsiveness; however, examples of such complexes are still limited. To expand the application scope of this compound class, more in-depth knowledge of the boron–sulfur interaction is required.
O,18 –C(R′)NR,19 or –P(O)R2.20 As for intramolecularly sulfur-coordinated organoboranes, Rupar and coworkers reported a tetracoordinate borafluorene with a pincer-like aryl group (A), in which one of the sulfur atoms was coordinated to the boron center in the ground state, and the resulting B–S bond underwent dissociation in the excited state (Fig. 1a).17 Recently, we reported donor–π–acceptor (D–π–A)-type organoborane fluorophores bearing ortho-PX-substituted phenyl groups on the boron atom (B; X = O or S).20d The PX groups also underwent photodissociation in the excited state, where the PS derivative facilitated the dissociation compared with the PO congeners. Thus, the B–S coordination bond can be expected to enable the formation of labile Lewis acid–base complexes with stimuli- or environment-responsiveness; however, examples of such complexes are still limited. To expand the application scope of this compound class, more in-depth knowledge of the boron–sulfur interaction is required.
 | ||
| Fig. 1 (a) Examples of boron-containing π-conjugated compounds with boron–sulfur coordination bonds. (b) Chemical structures of 1 and 2 and their reference compounds 3, 4, and cis-5 studied in this work. | ||
In addition, as for the boron-containing π-conjugated scaffold, most of the intramolecular borane–Lewis base complexes reported so far contain only one boron atom, with only a few examples bearing more than two boron atoms.19 In this context, 9,10-dihydro-9,10-diboraanthracene (DBA) derivatives are promising scaffolds because of their rigid framework, in which the vacant p orbitals on two boron atoms are effectively π-conjugated with the 1,2-phenylene moieties.21 The photophysical properties of some DBA derivatives have been studied. For instance, Cheng and coworkers reported DBA-based D–A–D-type compounds exhibiting highly efficient thermally activated delayed fluorescence properties.22 Recently, one of our groups demonstrated that two laterally π-expanded DBA derivatives exhibited ultralong room-temperature phosphorescence in a rigid poly(methyl methacrylate) matrix.23 Although a few examples of DBAs complexed with externally added Lewis bases, such as a fluoride ion,24 pyridine,25 1,2-diazine derivatives,26 and dimethylsulfide (C)26 have been reported, those are limited to intermolecular Lewis acid–base complexed systems.
To elucidate the impact of the B–S coordination bonds on the properties of boron-based π-electron systems, we synthesized in this study arylsulfide-substituted DBAs 1 and 2 as a new family of boron-based π-electron systems with intramolecular B–S coordination bonds (Fig. 1b). Arylthiomethyl groups were attached to the ortho position of the phenyl group on the boron atom to form a coordination bond in a five-membered ring fashion. Since the DBAs have two boron atoms, the intramolecular B–S coordination would form cis and trans isomers. To tune the intramolecular B–S coordination strength, an electron-withdrawing CF3 group was introduced at the para position of the arylthio moiety with respect to the sulfur atom. A comparison of their photophysical properties with those of mesityl-substituted tricoordinate congeners 3 and 4 and methylsulfide-coordinated DBA cis-5 confirmed that the arylsulfide–boron coordination perturbs their electronic structures. The fundamental behavior of the B–S-coordinated compounds upon heating or light irradiation is discussed in this article.
Results and discussion
Intramolecularly arylsulfide-coordinated DBAs 1 and 2 were synthesized by employing 9,10-dibromo-9,10-dihydro-9,10-diboraanthracene (6) and its π-extended analogue 7 as key precursors, respectively (Scheme 1).23,27 Thus, compound 6, which was prepared in situ according to the literature method,28 was treated with 2 equiv. of [2-(phenylthiomethyl)-6-methylphenyl]lithium. This reaction gave a mixture of cis and trans isomers of 1a, which could be separated by silica gel column chromatography. CF3-substituted derivative 1b and π-extended analogues 2a and 2b were prepared in a similar manner, and all cis and trans isomers were successfully separated by silica gel column chromatography or HPLC on silica gel. In both scaffolds, the cis isomers were predominantly obtained. These compounds thus obtained were sufficiently stable to be handled under ambient conditions without special precautions. In particular, the cis and trans isomers showcased sufficient configurational stability at room temperature, whereas cis–trans isomerization proceeded at higher temperatures (vide infra). | ||
| Scheme 1 Synthesis of intramolecular arylsulfide-coordinated diboraanthracenes. | ||
The structures of some of the compounds were unequivocally determined by single-crystal X-ray diffraction analysis. Crystal structures of cis-1a, cis-1b, trans-2a, and trans-2b are shown in Fig. 2, which clearly showcases that the coordination of the sulfur atoms to the boron atoms formed 5-membered rings irrespective of their cis- or trans-configurations. The central dibora-hexagon rings in the DBA skeletons of cis-1a and cis-1b adopted slightly distorted boat-like conformations, while trans-2a and trans-2b retained planar structures. The B–S distances in these compounds (cis-1a: 2.161(1) Å, cis-1b: 2.146(3)–2.381(3) Å, trans-2a: 2.228(4) Å, and trans-2b: 2.289(2) Å, Table 1) are much shorter than the sum (3.72 Å) of the van der Waals radii of the boron and sulfur atoms.29 The B–S distances are slightly longer compared to those of hitherto-known sulfur-coordinate compounds such as A (2.029(1) Å),17B (2.104(6) Å),20d and C (2.031(2) Å),26 suggesting that the B–S interaction in 1 and 2 are rather weak. The slightly longer B–S distances observed for the CF3-substituted derivatives cis-1b and trans-2b relative to cis-1a and trans-2a, respectively, demonstrated that the B–S interaction is weakened by decreasing the Lewis basicity of the sulfur atom. As a result of the coordination, the boron atoms adopted tetrahedral geometries with the sum of the bond angles around the boron atoms of 350.2° for cis-1a, 350.6–354.4° for cis-1b, 353.5° for trans-2a, and 355.1° for trans-2b. The tetrahedral characters (THCs)30 of their boron centers were calculated to be 31.1% for cis-1a, 17.8–29.8% for cis-1b, 20.7% for trans-2a, and 15.6% for trans-2b. It should be also noted that the B–S coordination resulted in the face-to-face orientation of the arylthio group against the DBA skeleton with the interfacial distances of 3.05–3.45 Å, although the overlaps between the π-planes were too small to form strong π–π interactions.
 | ||
| Fig. 2 Crystal structures of (a) cis-1a, (b) cis-1b, (c) trans-2a, and (d) trans-2b with thermal ellipsoids at 50% probability. Only one of the two crystallographically independent molecules of cis-1b is shown. Hydrogen atoms and solvent molecules are omitted for clarity. | ||
| Compound | B–S bond lengths/Å | Σ(C–B–C)/° | THC/% | ||
|---|---|---|---|---|---|
| a cis-1b contains two crystallographically independent molecules A and B in the unit cell. | |||||
| cis-1a | B1–S1 | 2.161(1) | 350.2 | 31.1 | |
| B2–S2 | 2.161(1) | 350.2 | 31.1 | ||
| cis-1ba | A | B1–S1 | 2.181(2) | 350.6 | 29.8 |
| B2–S2 | 2.231(3) | 353.4 | 21.0 | ||
| B | B1–S1 | 2.146(3) | 351.0 | 28.6 | |
| B2–S2 | 2.381(3) | 354.4 | 17.8 | ||
| trans-2a | B1–S1/B2–S2 | 2.228(4) | 353.5 | 20.7 | |
| trans-2b | B1–S1/B2–S2 | 2.289(2) | 355.1 | 15.6 | |
To gain insight into the intramolecular B–S coordination bond, natural bond orbital (NBO) analyses were conducted at the B3LYP-D3/6-311+G(d,p) level of theory on cis-1a, cis-1b, trans-1a, and trans-1b using their optimized structures obtained at the PBE0/6-31G(d) level.31 For comparison, NBO analyses were also conducted for methylsulfide-coordinated borafluorene A and DBA cis-5 (Fig. 1) as model compounds. The obtained Wiberg Bond Index (WBI) values are summarized in Table 2. In comparison with the WBI value of A, derived from antiaromatic and highly Lewis acidic borafluorene, those of the DBA compounds were rather small, indicating that their B–S coordination bonds are weak. A comparison between cis-1a, cis-1b, and cis-5 with different substituents on the sulfur atom demonstrated that the WBI values decreased in the order of cis-5 > cis-1a > cis-1b, indicating that the arylsulfide group renders the B–S coordination bond more labile. Moreover, the smaller WBI values of trans-1a and trans-1b compared with those of cis-1a and cis-1b, respectively, indicate that the B–S coordination bonds in the trans-isomers are weaker, suggesting that the configuration also affects the strength of the B–S coordination bonds. Although the origin of this difference remains unclear, it might be related to the fact that the trans isomers retain the planar conformation of the DBA moiety, whereas the cis isomers adopt a bent conformation deviated from the planar structures, which most likely decreases the steric congestion (Fig. S6†).
| Compound | B–S bond lengths/Å | Wiberg bond index | |
|---|---|---|---|
| a NBO calculations were conducted at the B3LYP-D3/6-311+G(d,p) level of theory. | |||
| cis-1a | B1–S1 | 2.233 | 0.537 |
| B2–S2 | 2.232 | 0.539 | |
| cis-1b | B1–S1 | 2.296 | 0.484 |
| B2–S2 | 2.296 | 0.484 | |
| trans-1a | B1–S1 | 2.284 | 0.504 |
| B2–S2 | 2.285 | 0.503 | |
| trans-1b | B1–S1 | 2.382 | 0.429 |
| B2–S2 | 2.347 | 0.452 | |
| cis-5 | B1–S1 | 2.145 | 0.625 |
| B2–S2 | 2.145 | 0.625 | |
| A | B1–S1 | 2.053 | 0.714 |
The intramolecular B–S coordination was also observed in solution via11B NMR spectroscopy (Fig. S1†). Thus, the 11B NMR spectra of 1 and 2 in CDCl3 showed relatively sharp signals at around 20–40 ppm at room temperature. These results contrast with the broad signals observed at around 70 ppm for the tricoordinate congeners 3 and 4, indicating that 1 and 2 adopt tetracoordinate structures even in solution. However, the chemical shifts of 1 and 2 appeared in a relatively low magnetic field region for tetracoordinate boron species (for instance, 5.3 ppm in CDCl3 for A), which suggests a relative weakness of the B–S coordination bonds in 1 and 2. For derivatives 1 and 2, the trans configuration and the introduction of an electron-withdrawing CF3 group shifted the signals to a lower magnetic field, implying that the strength of the B–S coordination was further perturbed to some extent due to these structural and electronic modifications.
As a consequence of the weak bonding character of the B–S coordination, the DBA derivatives underwent thermal cis–trans isomerization. For example, cis-1a isomerized to form some of the corresponding trans isomer upon increasing the temperature in solution (Fig. S2†). Using the intensity ratios of their 1H NMR spectra in toluene-d8, the equilibrium constants (Keq) for the cis–trans isomerization of cis-1a and cis-1b at 363 K were determined to be 0.28 and 0.48, respectively (Table S1†). Similar NMR measurements were conducted at various temperatures to determine the thermodynamic parameters for the cis–trans isomerization of cis-1a and cis-1b. Based on the corresponding van't Hoff plots (Fig. S3†), enthalpy changes (ΔH) of 11.8 and 8.4 kJ mol−1 and entropy changes (ΔS) of 21.9 and 16.9 J mol−1 K−1 were determined for cis-1a and cis-1b, respectively (Table S2†). Using these values, the Gibbs free energy changes for the cis → trans conversions of cis-1a and cis-1b at 298 K were calculated to be 5.30 and 3.32 kJ mol−1, respectively, demonstrating that the isomerization from the cis isomer to the trans isomer was slightly endergonic. Furthermore, the introduction of an electron-withdrawing CF3 group to the phenyl group on the sulfur atom reduced the energy difference between the cis and trans isomers.
The intramolecular B–S coordination affects considerably the photophysical properties of the DBAs. Thus, the UV-vis absorption spectra of cis-1a and cis-1b in cyclohexane showed absorption bands with a maximum wavelength (λabs) of around 290 nm (Fig. 3a and b), which were blue-shifted compared with that of mesityl-substituted DBA 3 (λabs = 406 nm).24a Similar blue shifts were also observed for π-expanded analogues cis-2a and cis-2b (cis-2a: λabs = 382 nm; cis-2b: λabs = 386 nm in cyclohexane; Fig. 3c and d) compared with that of 4 (λabs = 435 nm).23 These shifts can be attributed to the disruption of the p–π* conjugation through the vacant p orbital of the boron atom due to the coordination of the sulfur atom.
 | ||
| Fig. 3 UV-vis absorption (solid lines) and fluorescence (dashed lines) spectra of (a) cis-1a, (b) cis-1b, (c) cis-2a, and (d) cis-2b in cyclohexane (red) and CH2Cl2 (blue). | ||
In stark contrast, in the fluorescence spectra, the B–S coordination gave rise to red shifts in the emission maxima. Thus, in cyclohexane, cis-1a and cis-1b showed substantially red-shifted emissions with maximum wavelengths (λem) of 526 and 506 nm, respectively (Fig. 3a and b), while their fluorescence quantum yields (ΦF) were low (0.02–0.03). Notably, their λem values were more than or nearly 100 nm longer compared with that of tricoordinate compound 3 (λem = 413 nm).24a As a consequence, cis-1a and cis-1b exhibited considerably large apparent Stokes shifts (Δν = νabs − νem) of 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 and 13800 cm−1, respectively, even in nonpolar cyclohexane. The emission bands of cis-1a and cis-1b were further shifted to 572 and 533 nm, respectively, in CH2Cl2, resulting in even larger apparent Stokes shifts of 18600 and 15400 cm−1. Several boron-based fluorophores with a weakly coordinated Lewis base are known to undergo photodissociation of the boron–Lewis base coordination bond in the excited state, resulting in an emission from the tricoordinate species.15–17,20d However, the red-shifted emission of cis-1a and cis-1b relative to 3 cannot be explained by simply considering the photodissociation behavior. The broad shape of the emission bands and the dependence of the emission wavelength on solvent polarity suggest that intramolecular charge transfer (ICT) character in the excited state is likely responsible for these red-shifted emissions.
500 and 13800 cm−1, respectively, even in nonpolar cyclohexane. The emission bands of cis-1a and cis-1b were further shifted to 572 and 533 nm, respectively, in CH2Cl2, resulting in even larger apparent Stokes shifts of 18600 and 15400 cm−1. Several boron-based fluorophores with a weakly coordinated Lewis base are known to undergo photodissociation of the boron–Lewis base coordination bond in the excited state, resulting in an emission from the tricoordinate species.15–17,20d However, the red-shifted emission of cis-1a and cis-1b relative to 3 cannot be explained by simply considering the photodissociation behavior. The broad shape of the emission bands and the dependence of the emission wavelength on solvent polarity suggest that intramolecular charge transfer (ICT) character in the excited state is likely responsible for these red-shifted emissions.
Meanwhile, cis-2a and cis-2b showed weak broad emission bands with λem at 540 and 516 nm in cyclohexane (Fig. 3c and d), respectively, which were comparable to that of tricoordinate congener 4 (λem = 520 nm),23 while the fluorescence quantum yields of cis-2a (0.02) and cis-2b (0.04) were lower than that of 4 (0.26). In CH2Cl2, cis-2a showed broad emission bands likely consisting of two bands at around 500 and 600 nm, while the emission band of cis-2b (λem = 532 nm) was only slightly red-shifted compared to that in cyclohexane. These results indicate that while the π-expanded DBA skeleton also undergoes photodissociation, the ICT transition character is not always involved. Instead, it depends on the electronic effect of the aryl group on the sulfur atom.
The difference in the cis/trans configuration also affected the photophysical properties of the arylsulfide-coordinated DBAs. Thus, in the UV-vis absorption spectra in cyclohexane (Fig. S4†), trans-1a showed a slightly red-shifted absorption band relative to that of cis-1a (trans-1a: λabs = 300 nm; cis-1a: λabs = 282 nm). In the emission spectrum in cyclohexane, the emission band of trans-1a was slightly blue-shifted with a decreased apparent Stokes shift compared to cis-1a (trans-1a: λem = 512 nm, Δν = 13800 cm−1; cis-1a: λem = 526 nm, Δν = 16500 cm−1). Thus, the large apparent Stokes shift observed for cis-1a likely results partly from the cis-configuration structure. A similar trend was observed for other derivatives except for 1b (Table S3†).
To gain more insight into the photodissociation process, time-dependent density functional theory (TD DFT) calculations were conducted on 1 and 2 at the PBE0/6-31G(d) level of theory. The structural optimization in cis-1a in the lowest excited singlet state (S1) only gave a tricoordinate structure where both B–S bonds are dissociated, even when starting the optimization from an initial B–S-coordinated structure (Fig. 4). Similar results were obtained for cis-1b, cis-2a, and cis-2b. In the optimized structure of cis-1a in S1, the DBA skeleton became planar and the phenylthio groups were displaced from the DBA skeleton compared with the optimized structure in S0. While the optimized structure of cis-1b in S1 was similar to that of cis-1a, the phenylthio groups in cis-2a and cis-2b were oriented in closer proximity to the DBA skeletons in their optimized structures in S1 (B⋯S distances in S1: cis-1a, 3.54, 3.56 Å; cis-2a, 3.42, 3.46; cis-2b, 3.25, 3.25 Å). Notably, in the structures of cis-1a, cis-1b, and cis-2a in S1, the HOMOs were localized on the phenylthio groups and the LUMOs on the DBA skeletons. In contrast, both HOMO and LUMO of CF3-substituted cis-2b were localized on the π-expanded DBA skeleton, indicating that the emission of cis-2b has mainly a π–π* transition character, which is consistent with the fact that the emission band of cis-2b did not show solvent polarity dependence.
 | ||
| Fig. 4 Kohn–Sham molecular orbitals for cis-1a, cis-1b, cis-2a, and cis-2b in the S1 optimized structures. TD-DFT calculations were carried out at the PBE0/6-31G(d) level of theory. | ||
In the structure optimized in S1, cis-1a has a rather high-lying HOMO of −5.30 eV despite the fact that it is mainly localized in the phenylthio moiety. In this structure, the bond length between the sulfur atom and the ipso-carbon atom of the phenyl group was shorter compared to that in S0 (Fig. S9†). In addition, a larger bond-length alternation was observed in the phenyl moiety in the S1 optimized structure, indicating that the sulfur atom donates a lone pair electron to the phenyl group upon photodissociation.
Although both the HOMO and HOMO−1 of cis-1a were localized on different phenylthio moieties, the HOMO level was higher by 0.77 eV than the HOMO−1 level (Fig. S10†). For comparison, the TD-DFT calculation was also conducted for phenylmethylsulfide. The HOMO level in the S1 optimized structure was estimated to be −6.08 eV, which was comparable to the HOMO−1 of cis-1a. While the lone pair orbital of the sulfur atom and π-orbital of the phenyl group were almost orthogonal in the HOMO−1 of cis-1a, these orbitals were parallel in the HOMO of cis-1a, which most likely contributes to the high-lying HOMO localized in the phenylthio moiety of cis-1a. In contrast, such a high energy level of the arylthio moiety was not observed in cis-2b, suggesting that the energy-level balance between the DBA skeleton and the arylthio moiety is crucial for the unusual emission properties observed in the phenylthio-substituted derivatives.
As mentioned above, the cis–trans isomerization of arylsulfide-coordinated DBAs occurred in toluene-d6 upon heating, suggesting that the B–S bond dissociates at high temperatures. This behavior was confirmed via temperature-dependent UV-vis absorption and fluorescence measurements, which were performed for cis-2a and cis-2b because cis-1a and cis-1b lacked absorption bands in the visible region. Upon heating toluene solutions of cis-2a and cis-2b from 293 to 373 K, bathochromic shifts of the absorption bands were observed with isosbestic points (Fig. 5a and b). Since the absorption spectra of cis-2 and trans-2 are nearly identical (Fig. S4c and d†), these changes are unlikely to arise solely from cis–trans isomerization. We therefore attribute them to partial dissociation of the B–S coordination bond in the ground state. In the fluorescence spectra, the fluorescence intensity of cis-2a decreased with increasing temperature, which was accompanied by a hypsochromic shift of the emission maximum wavelength (Fig. 5c). In contrast to cis-2a, the fluorescence intensity of cis-2b was enhanced with increasing temperature (Fig. 5d). It should be noted that these spectral changes were reversible: the original spectra of both compounds were observed upon cooling from 373 to 293 K (Fig. S5†).
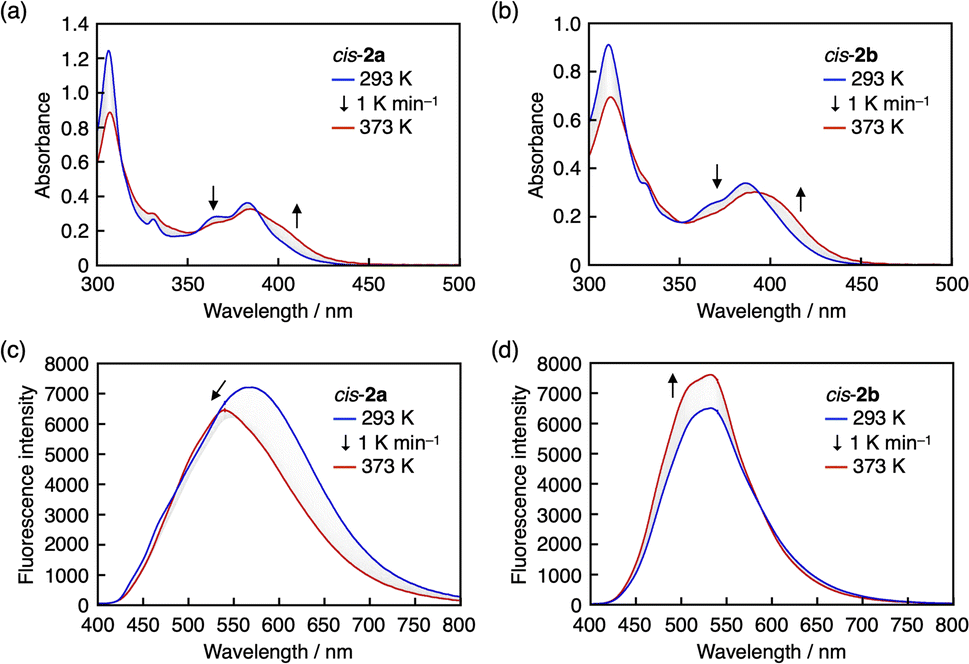 | ||
| Fig. 5 (a and b) UV-vis absorption and (c and d) fluorescence spectra of (a and c) cis-2a and (b and d) cis-2b upon heating from 293 to 373 K at 1 K min−1 in toluene. | ||
In both π-expanded cis-2a and cis-2b in 2-MeTHF, when the temperature was decreased from 290 to 80 K, vibronically structured emission bands with increased intensity appeared at around 420–550 nm (Fig. 6a and b). These emission bands likely stem from tetracoordinate species in which the sulfur atoms remain coordinated to the boron atoms. The fact that these new bands appeared above the glass transition temperature of the solvent suggests that the decrease in temperature instead of the increased viscosity of the medium is responsible for the retardation of the B–S dissociation in the excited state. At temperatures lower than the glass transition temperature of 2-MeTHF (137 K), a new vibronically structured band was observed at around 550–700 nm, which is assignable to a phosphorescence band because it matched with an emission spectrum measured with a delay time of 50 ms (Fig. 6c and d). Ultimately, cis-2a and cis-2b showed photoluminescence quantum yields, including fluorescence and phosphorescence, of 0.40 and 0.37, respectively, at 77 K.
 | ||
| Fig. 6 Temperature-dependent fluorescence spectra of (a) cis-2a and (b) cis-2b in 2-MeTHF. Phosphorescence spectra of (c) cis-2a and (d) cis-2b in 2-MeTHF at 77 K with a delay time of 50 ms. | ||
Conclusions
A series of 9,10-dihydro-9,10-diboraanthracenes with arylthiomethyl substituents on the ortho positions of the phenyl groups on the boron atom were synthesized as a new class of intramolecularly sulfur-coordinated boron-containing π-conjugated molecules. The B–S coordination bonds were sufficiently strong to allow the separation of the cis and trans isomers by silica gel column chromatography. A crystallographic analysis revealed that the strength of the B–S coordination bond was perturbed by introducing an electron-withdrawing CF3 group on the arylthio moiety. A NBO analysis complemented the experimental findings suggesting that the CF3-containing derivatives have smaller WBI values. The B–S coordination bonds were retained even in solution at room temperature, whereas dissociation partially occurred in the ground state at elevated temperature, as evidenced by their 1H NMR spectra. The introduction of the CF3 group reduced the enthalpy change in the cis–trans isomerization. The B–S bonds also dissociated in response to light irradiation, resulting in photodissociation-induced emissions from tricoordinate species with large apparent Stokes shifts. Some derivatives also showcased substantially red-shifted emission bands with ICT transition character. Thus, the arylthio-substituted DBAs exhibited multifaceted emissions from a tetracoordinate species, a tricoordinate species after photodissociation, and a tricoordinate species in the ICT state in S1, as well as phosphorescence, depending on the environment. These results demonstrate the potential utility of B–S coordination bonds for the design of unprecedented multiply emissive materials.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
H. N., M. W. and S. Y. conceived the idea. H. N. synthesized the compounds and evaluated their properties with the support of H.-W. L., H. N. performed the X-ray crystal structure analyses of cis-1a, cis-1b, and trans-2b. A. V. performed the X-ray crystal structure analysis of trans-2a. H. N. and S. Y. wrote the manuscript, and all authors discussed and commented on the manuscript. M. W. and S. Y. directed the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by KAKENHI grants 23H00295 and 22K21346 from MEXT/JSPS as well as CREST (JPMJCR21O5) from the Japan Science and Technology Agency (JST). H. N. thanks the JSPS for a Research Fellowship for Young Scientists and the “Graduate Program of Transformative Chem-Bio Research” at Nagoya University, supported by MEXT (WISE Program). ITbM is supported by the World Premier International Research Center (WPI) Initiative, Japan.Notes and references
- (a) C. D. Entwistle and T. B. Marder, Angew. Chem., Int. Ed., 2002, 41, 2927–2931 CrossRef CAS; (b) C. D. Entwistle and T. B. Marder, Chem. Mater., 2004, 16, 4574–4585 CrossRef CAS; (c) F. Jäkle, Coord. Chem. Rev., 2006, 250, 1107–1121 CrossRef; (d) S. Yamaguchi and A. Wakamiya, Pure Appl. Chem., 2006, 78, 1413–1424 CrossRef CAS; (e) Z. M. Hudson and S. Wang, Acc. Chem. Res., 2009, 42, 1584–1596 CrossRef CAS PubMed; (f) F. Jäkle, Chem. Rev., 2010, 110, 3985–4022 CrossRef PubMed; (g) A. Lorbach, A. Hübner and M. Wagner, Dalton Trans., 2012, 41, 6048–6063 RSC; (h) A. Wakamiya and S. Yamaguchi, Bull. Chem. Soc. Jpn., 2015, 88, 1357–1377 CrossRef CAS; (i) Y. Ren and F. Jäkle, Dalton Trans., 2016, 45, 13996–14007 RSC; (j) L. Ji, S. Griesbeck and T. B. Marder, Chem. Sci., 2017, 8, 846–863 RSC; (k) H. Helten, Chem.–Asian J., 2019, 14, 919–935 CrossRef CAS PubMed; (l) S. K. Mellerup and S. Wang, Chem. Soc. Rev., 2019, 48, 3537–3549 RSC; (m) X. Yin, J. Liu and F. Jäkle, Chem.–Eur. J., 2021, 27, 2973–2986 CrossRef CAS PubMed; (n) S. M. Berger, M. Ferger and T. B. Marder, Chem.–Eur. J., 2021, 27, 7043–7058 CrossRef CAS PubMed.
- (a) Z. Yuan, N. J. Taylor, T. B. Marder, I. D. Williams, S. K. Kurtz and L.-T. Cheng, J. Chem. Soc. Chem. Commun., 1990, 1489–1492 RSC; (b) Z. Yuan, J. C. Collings, N. J. Taylor, T. B. Marder, C. Jardin and J.-F. Halet, J. Solid State Chem., 2000, 154, 5–12 CrossRef CAS; (c) Z. Yuan, C. D. Entwistle, J. C. Collings, D. Albesa-Jové, A. S. Batsanov, J. A. K. Howard, N. J. Taylor, H. M. Kaiser, D. E. Kaufmann, S.-Y. Poon, W.-Y. Wong, C. Jardin, S. Fathallah, A. Boucekkine, J.-F. Halet and T. B. Marder, Chem.–Eur. J., 2006, 12, 2758–2771 CrossRef CAS PubMed.
- (a) M. Lequan, R. M. Lequan and K. C. Ching, J. Mater. Chem., 1991, 1, 997–999 RSC; (b) M. Lequan, R. M. Lequan, K. C. Ching, M. Barzoukas, A. Fort, H. Lahoucine, G. Bravic, D. Chasseau and J. Gaultier, J. Mater. Chem., 1992, 2, 719–725 RSC.
- (a) Y. Shirota, M. Kinoshita, T. Noda, K. Okumoto and T. Ohara, J. Am. Chem. Soc., 2000, 122, 11021–11022 CrossRef CAS; (b) W.-L. Jia, D.-R. Bai, T. McCormick, Q.-D. Liu, M. Motala, R.-Y. Wang, C. Seward, Y. Tao and S. Wang, Chem.–Eur. J., 2004, 10, 994–1006 CrossRef CAS PubMed; (c) H. Hirai, K. Nakajima, S. Nakatsuka, K. Shiren, J. Ni, S. Nomura, T. Ikuta and T. Hatakeyama, Angew. Chem., Int. Ed., 2015, 54, 13581–13585 CrossRef CAS PubMed; (d) K. Suzuki, S. Kubo, K. Shizu, T. Fukushima, A. Wakamiya, Y. Murata, C. Adachi and H. Kaji, Angew. Chem., Int. Ed., 2015, 54, 15231–15235 CrossRef CAS PubMed; (e) D. L. Crossley, I. A. Cade, E. R. Clark, A. Escande, M. J. Humphries, S. M. King, I. Vitorica-Yrezabal, M. J. Ingleson and M. L. Turner, Chem. Sci., 2015, 6, 5144–5151 RSC; (f) Y.-J. Shiu, Y.-C. Cheng, W.-L. Tsai, C.-C. Wu, C.-T. Chao, C.-W. Lu, Y. Chi, Y.-T. Chen, S.-H. Liu and P.-T. Chou, Angew. Chem., Int. Ed., 2016, 55, 3017–3021 CrossRef CAS PubMed.
- (a) X. Li, X. Guo, L. Cao, Z. Xun, S. Wang, S. Li, Y. Li and G. Yang, Angew. Chem., Int. Ed., 2014, 53, 7809–7813 CrossRef CAS PubMed; (b) J. Liu, X. Guo, R. Hu, X. Liu, S. Wang, S. Li, Y. Li and G. Yang, Anal. Chem., 2016, 88, 1052–1057 CrossRef CAS PubMed; (c) J. Liu, S. Zhang, C. Zhang, J. Dong, C. Shen, J. Zhu, H. Xu, M. Fu, G. Yang and X. Zhang, Chem. Commun., 2017, 53, 11476–11479 RSC.
- (a) S. Griesbeck, Z. Zhang, M. Gutmann, T. Lühmann, R. M. Edkins, G. Clermont, A. N. Lazar, M. Haehnel, K. Edkins, A. Eichhorn, M. Blanchard-Desce, L. Meinel and T. B. Marder, Chem.–Eur. J., 2016, 22, 14701–14706 CrossRef CAS PubMed; (b) S. Griesbeck, M. Ferger, C. Czernetzi, C. Wang, R. Bertermann, A. Friedrich, M. Haehnel, D. Sieh, M. Taki, S. Yamaguchi and T. B. Marder, Chem.–Eur. J., 2019, 25, 7679–7688 CrossRef CAS PubMed; (c) S. Griesbeck, E. Michail, C. Wang, H. Ogasawara, S. Lorenzen, L. Gerstner, T. Zang, J. Nitsch, Y. Sato, R. Bertermann, M. Taki, C. Lambert, S. Yamaguchi and T. B. Marder, Chem. Sci., 2019, 10, 5405–5422 RSC; (d) S. Griesbeck, E. Michail, F. Rauch, H. Ogasawara, C. Wang, Y. Sato, R. M. Edkins, Z. Zhang, M. Taki, C. Lambert, S. Yamaguchi and T. B. Marder, Chem.–Eur. J., 2019, 25, 13164–13175 CrossRef CAS PubMed; (e) S. M. Berger and T. B. Marder, Mater. Horiz., 2022, 9, 112–120 RSC.
- (a) Ž. Ban, S. Griesbeck, S. Tomić, J. Nitsch, T. B. Marder and I. Piantanida, Chem.–Eur. J., 2020, 26, 2195–2203 CrossRef PubMed; (b) H. Amini, Ž. Ban, M. Ferger, S. Lorenzen, F. Rauch, A. Friedrich, I. Crnolatac, A. Kenđel, S. Miljanić, I. Piantanida and T. B. Marder, Chem.–Eur. J., 2020, 26, 6017–6028 CrossRef CAS PubMed; (c) S. M. Berger, J. Rühe, J. Schwarzmann, A. Phillipps, A.-K. Richard, M. Ferger, I. Krummenacher, L.-M. Tumir, Ž. Ban, I. Crnolatac, D. Majhen, I. Barišić, I. Piantanida, D. Schleier, S. Griesbeck, A. Friedrich, H. Braunschweig and T. B. Marder, Chem.–Eur. J., 2021, 27, 14057–14072 CrossRef CAS PubMed.
- Y. Sugihara, N. Inai, M. Taki, T. Baumgartner, R. Kawakami, T. Saitou, T. Imamura, T. Yanai and S. Yamaguchi, Chem. Sci., 2021, 12, 6333–6341 RSC.
- (a) S. Yamaguchi, S. Akiyama and K. Tamao, J. Am. Chem. Soc., 2001, 123, 11372–11375 CrossRef CAS PubMed; (b) S. Yamaguchi, T. Shirasaka, S. Akiyama and K. Tamao, J. Am. Chem. Soc., 2002, 124, 8816–8817 CrossRef CAS PubMed; (c) C. R. Wade, A. E. J. Broomsgrove, S. Aldridge and F. P. Gabbaï, Chem. Rev., 2010, 110, 3958–3984 CrossRef CAS PubMed.
- (a) T. W. Hudnall and F. P. Gabbaï, J. Am. Chem. Soc., 2007, 129, 11978–11986 CrossRef CAS PubMed; (b) C.-W. Chiu and F. P. Gabbaï, Dalton Trans., 2008, 814–817 RSC; (c) C.-W. Chiu, Y. Kim and F. P. Gabbaï, J. Am. Chem. Soc., 2009, 131, 60–61 CrossRef CAS PubMed; (d) Y. Kim, H. Zhao and F. P. Gabbaï, Angew. Chem., Int. Ed., 2009, 48, 4957–4960 CrossRef CAS PubMed; (e) T. Matsumoto, C. R. Wade and F. P. Gabbaï, Organometallics, 2010, 29, 5490–5495 CrossRef CAS; (f) C. R. Wade and F. P. Gabbaï, Inorg. Chem., 2010, 49, 714–720 CrossRef CAS PubMed; (g) Y. Kim, H.-S. Huh, M. H. Lee, I. L. Lenov, H. Zhao and F. P. Gabbaï, Chem.–Eur. J., 2011, 17, 2057–2062 CrossRef CAS PubMed; (h) H. Zhao, L. A. Leamer and F. P. Gabbaï, Dalton Trans., 2013, 42, 8164–8178 RSC; (i) C.-H. Chen and F. P. Gabbaï, Chem. Sci., 2018, 9, 6210–6218 RSC.
- (a) K. Schickedanz, T. Trageser, M. Bolte, H.-W. Lerner and M. Wagner, Chem. Commun., 2015, 51, 15808–15810 RSC; (b) N. Ando, T. Kushida and S. Yamaguchi, Chem. Commun., 2018, 54, 5213–5216 RSC; (c) G. Meng, T. Peng, Y. Shi, H. Li, X. Wang, X. Yin, D.-T. Yang, S. Wang and N. Wang, J. Mater. Chem. C, 2020, 8, 7749–7754 RSC; (d) J. Guo, Y. Yang, C. Dou and Y. Wang, J. Am. Chem. Soc., 2021, 143, 18272–18279 CrossRef CAS PubMed; (e) J. He, F. Rauch, A. Friedrich, J. Krebs, I. Krummenacher, R. Bertermann, J. Nitsch, H. Braunschweig, M. Finze and T. B. Marder, Angew. Chem., Int. Ed., 2021, 60, 4833–4840 CrossRef CAS PubMed.
- N. Ando, T. Yamada, H. Narita, N. N. Oehlmann, M. Wagner and S. Yamaguchi, J. Am. Chem. Soc., 2021, 143, 9944–9951 CrossRef CAS PubMed.
- S. Saito, K. Matsuo and S. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 9130–9133 CrossRef CAS PubMed.
- K. Matsuo, S. Saito and S. Yamaguchi, Angew. Chem., Int. Ed., 2016, 55, 11984–11988 CrossRef CAS PubMed.
- (a) K. Matsuo, S. Saito and S. Yamaguchi, J. Am. Chem. Soc., 2014, 136, 12580–12583 CrossRef CAS PubMed; (b) Y. Kitamoto, F. Kobayashi, T. Suzuki, Y. Miyata, H. Kita, K. Funaki and S. Oi, Dalton Trans., 2019, 48, 2118–2127 RSC; (c) Z. Fan, W. Sun, Y. Yang, J. Guo, C. Dou and Y. Wang, Chin. Chem. Lett., 2023, 34, 107729 CrossRef CAS.
- (a) T. Matsumoto, K. Tanaka, K. Tanaka and Y. Chujo, Dalton Trans., 2015, 44, 8697–8707 RSC; (b) T. Matsumoto, H. Takamine, K. Tanaka and Y. Chujo, Mater. Chem. Front., 2017, 1, 2368–2375 RSC; (c) Q. Hou, L. Liu, S. K. Mellerup, N. Wang, T. Peng, P. Chen and S. Wang, Org. Lett., 2018, 20, 6467–6470 CrossRef CAS PubMed; (d) M. Gon, K. Tanaka and Y. Chujo, Bull. Chem. Soc. Jpn., 2019, 92, 7–18 CrossRef CAS; (e) Y. Shi, Y. Zeng, P. Kucheryavy, X. Yin, K. Zhang, G. Meng, J. Chen, Q. Zhu, N. Wang, X. Zheng, F. Jäkle and P. Chen, Angew. Chem., Int. Ed., 2022, 61, e202213615 CrossRef CAS PubMed.
- S. J. Cassidy, I. Brettell-Adams, L. E. McNamara, M. F. Smith, M. Bautista, H. Cao, M. Vasiliu, D. L. Gerlach, F. Qu, N. I. Hammer, D. A. Dixon and P. A. Rupar, Organometallics, 2018, 37, 3732–3741 CrossRef CAS.
- (a) Y. Shi, J. Wang, H. Li, G. Hu, X. Li, S. K. Mellerup, N. Wang, T. Peng and S. Wang, Chem. Sci., 2018, 9, 1902–1911 RSC; (b) J. Wang, N. Wang, G. Wu, S. Wang and X. Li, Angew. Chem., Int. Ed., 2019, 58, 3082–3086 CrossRef CAS PubMed.
- H. Shimogawa, O. Yoshikawa, Y. Aramaki, M. Murata, A. Wakamiya and Y. Murata, Chem.–Eur. J., 2017, 23, 3784–3791 CrossRef CAS PubMed.
- (a) J. M. Breunig, F. Lehmann, M. Bolte, H.-W. Lerner and M. Wagner, Organometallics, 2014, 33, 3163–3172 CrossRef CAS; (b) Y. Cao, J. K. Nagle, M. O. Wolf and B. O. Patrick, J. Am. Chem. Soc., 2015, 137, 4888–4891 CrossRef CAS PubMed; (c) Y. Wang, K. Liu, P. Chen, X. Yin, T. Peng, J. Iqbal and N. Wang, J. Mater. Chem. C, 2022, 10, 10981–10987 RSC; (d) M. Kawashiro, T. Mori, M. Ito, N. Ando and S. Yamaguchi, Angew. Chem., Int. Ed., 2023, 62, e202303725 CrossRef CAS PubMed.
- (a) A. Escande and M. J. Ingleson, Chem. Commun., 2015, 51, 6257–6274 RSC; (b) E. von Grotthuss, A. John, T. Kaese and M. Wagner, Asian J. Org. Chem., 2018, 7, 37–53 CrossRef CAS; (c) S. E. Prey and M. Wagner, Adv. Synth. Catal., 2021, 363, 2290–2309 CrossRef CAS; (d) T. Jin, L. Kunze, S. Breimaier, M. Bolte, H.-W. Lerner, F. Jäkle, R. F. Winter, M. Braun, J.-M. Mewes and M. Wagner, J. Am. Chem. Soc., 2022, 144, 13704–13716 CrossRef CAS PubMed; (e) Y. Guo, C. Chen and X.-Y. Wang, Chin. J. Chem., 2023, 41, 1355–1373 CrossRef CAS; (f) S. E. Prey, C. Herok, F. Fantuzzi, M. Bolte, H.-W. Lerner, B. Engels and M. Wagner, Chem. Sci., 2023, 14, 849–860 RSC; (g) M. Metzler, M. Bolte, A. Virovets, H.-W. Lerner and M. Wagner, Org. Lett., 2023, 25, 5827–5832 CrossRef CAS PubMed; (h) M. Metzler, A. Virovets, H.-W. Lerner and M. Wagner, J. Am. Chem. Soc., 2023, 145, 23824–23831 CrossRef CAS PubMed.
- (a) C. Hoffend, F. Schödel, M. Bolte, H.-W. Lerner and M. Wagner, Chem.–Eur. J., 2012, 18, 15394–15405 CrossRef CAS PubMed; (b) T.-L. Wu, M.-J. Huang, C.-C. Lin, P.-Y. Huang, T.-Y. Chou, R.-W. Chen-Cheng, H.-W. Lin, R.-S. Liu and C.-H. Cheng, Nat. Photonics, 2018, 12, 235–240 CrossRef CAS; (c) C.-M. Hsieh, T.-L. Wu, J. Jayakumar, Y.-C. Wang, C.-L. Ko, W.-Y. Hung, T.-C. Lin, H.-H. Wu, K.-H. Lin, C.-H. Lin, S. Hsieh and C.-H. Cheng, ACS Appl. Mater. Interfaces, 2020, 12, 23199–23206 CrossRef CAS.
- J. Jovaišaitė, S. Kirschner, S. Raišys, G. Kreiza, P. Baronas, S. Juršėnas and M. Wagner, Angew. Chem., Int. Ed., 2023, 62, e202215071 CrossRef.
- (a) T. Agou, M. Sekine and T. Kawashima, Tetrahedron Lett., 2010, 51, 5013–5015 CrossRef CAS; (b) Z. Zhou, A. Wakamiya, T. Kushida and S. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 4529–4532 CrossRef CAS PubMed.
- S. Osumi, S. Saito, C. Dou, K. Matsuo, K. Kume, H. Yoshikawa, K. Awaga and S. Yamaguchi, Chem. Sci., 2016, 7, 219–227 RSC.
- A. Lorbach, M. Bolte, H.-W. Lerner and M. Wagner, Chem. Commun., 2010, 46, 3592–3594 RSC.
- (a) E. Januszewski, A. Lorbach, R. Grewal, M. Bolte, J. W. Bats, H.-W. Lerner and M. Wagner, Chem.–Eur. J., 2011, 17, 12696–12705 CrossRef CAS PubMed; (b) A. John, S. Kirschner, M. K. Fengel, M. Bolte, H.-W. Lerner and M. Wagner, Dalton Trans., 2019, 48, 1871–1877 RSC.
- S. Brend'amour, J. Gilmer, M. Bolte, H.-W. Lerner and M. Wagner, Chem.–Eur. J., 2018, 24, 16910–16918 CrossRef PubMed.
- M. Mantina, A. C. Chamberlin, R. Valero, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 5806–5812 CrossRef CAS PubMed.
- (a) S. Toyota and M. Ōki, Bull. Chem. Soc. Jpn., 1992, 65, 1832–1840 CrossRef CAS; (b) H. Höpfl, J. Organomet. Chem., 1999, 581, 129–149 CrossRef.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, NBO 7.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2018 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectral data, details of the computational studies, and crystallographic data for cis-1a, cis-1b, trans-2a, and trans-2b. CCDC 2387655–2387657 and 2416563. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc01726b |
| This journal is © The Royal Society of Chemistry 2025 |