DOI:
10.1039/D4DT03379E
(Paper)
Dalton Trans., 2025,
54, 6386-6401
Benzimidazole-based mononuclear polypyridyl Cu(II) complexes: DNA binding, cleavage, and in vitro antiproliferative studies†
Received
4th December 2024
, Accepted 23rd February 2025
First published on 26th February 2025
Abstract
This paper addresses the synthesis, characterization, DNA binding, cleavage, and in vitro antiproliferative activity studies of a series of heteroleptic mononuclear copper(II) complexes [Cu(L)(bpy)](ClO4)2, {1}; [Cu(L)(phen)](ClO4)2, {2}; and [Cu(L)(Mephen)](ClO4)2, {3} derived from different polypyridyl ligands, where in the complex architecture, one 2,6-bis(1-methyl-1H-benzo[d]imidazol-2-yl)pyridine(Mebzimpy) (L) moiety is connected to the central Cu metal in a tridentate fashion and the bidentate co-ligands are 2,2′-bipyridine (bpy), 1,10-phenanthroline (phen) and 2,9-dimethyl-1,10-phenanthroline (Mephen). All the synthesized complexes were characterized using various spectroscopic and analytical methods, along with the single-crystal X-ray diffraction (SCXRD) technique. The complexes crystallize in a penta-coordinated distorted square pyramidal geometry. The redox properties of the complexes were also studied by using cyclic voltammetry (CV) and differential pulse voltammetry (DPV). The DNA binding nature of the complexes was investigated utilizing absorbance spectral measurement and fluorescence quenching experiments with ethidium bromide (EB) as a DNA intercalator, employing double-stranded salmon sperm DNA (ss-DNA). Both the binding constant (Kb) and the Stern–Volmer constant (KSV) were found to be in the order of 104. In silico molecular docking analysis confirmed that all the complexes could interact with the minor groove of duplex DNA. The DNA cleaving ability of the complexes was studied by gel electrophoresis using supercoiled plasmid DNA; however, no DNA cleavage was found. DNA-binding polypyridyl complexes are well known to disrupt DNA metabolic pathways and cause cytotoxicity to rapidly growing cancer cells. Hence, cell viability analysis was also carried out with complexes 1–3. It was observed that complexes 2 and 3 prevented the proliferation of the human osteosarcoma cell line U2OS and the triple-negative breast cancer cell line MDA-MB-231. Overall, these findings could be beneficial in the design and development of future antitumor agents.
Introduction
Since the discovery of cisplatin, it has been used worldwide as a clinical anticancer agent for the cure of various types of cancers (prostate, lung, brain, colon cancer, etc.).1–4 It can readily bind to DNA which helps to stop the process of DNA replication, ultimately resulting in DNA damage by inducing cancer cell apoptosis.5,6 Despite its several successful usages in the field of cancer therapy, there are some issues like drug resistance, nephrotoxicity, nausea, hair loss, neurotoxicity, low water solubility, etc. that limit the application of cisplatin and other platinum-based drugs (e.g. carboplatin and oxaliplatin) in the medicinal world.7,8 In search of better alternatives, transition metal-based polypyridyl complexes have been utilised for DNA binding, cleavage, and anticancer therapeutic aspects. Several transition metals (Co, Ni, Fe, Cu, Zn, Ru, etc.) with suitable redox-active ligand backbones that contain heteroatoms in the coordination site are reported in the literature.9–15 Among them, Cu complexes are found to be less toxic and more effective.16,17 Cu is a biologically essential metal that plays crucial roles in many biological events like neurotransmission and cellular respiration, and catechol oxidase, superoxide dismutase, amine oxidase, and tyrosinase activities.18–22 These complexes are also effective for inducing oxidative DNA cleavage, with greater affinity among all divalent transition metals.23–25 It is now recognized that metal complexes interact with DNA majorly in non-covalent ways, i.e. via groove binding, DNA intercalation, and external binding.26–28 The extent of interaction majorly depends on important factors like the type of metal ion, flexibility in the coordination valency of the central metal, the type of coordinating donor-atom in the ligands, the steric bulk of the ligand core, the planarity of the coordinating ligand framework, etc.29,30 The electrostatic mode of interaction is generally very weak, which is why metal complexes that can interact with DNA either by groove binding or through intercalation are the prime focus of modern-day research.31 Copper(II) complexes coordinated with different redox-active polypyridyl ligand cores such as 2,2′-bipyridine (bpy), 1,10-phenanthroline (phen), etc. are effective for this purpose.32,33 The aromatic nature of the polypyridyl systems and their high coordination ability with the Cu(II) metal cause better interaction with DNA, particularly in the groove binding and/or intercalation mode.22,34,35 [Cu(phen)2]2+ is a classical example of a copper(II) based metallodrug that shows promising activity in DNA binding and cleavage and also acts as an effective chemical nuclease in the presence of H2O2 as a co-oxidant.36 Since then, investigation on Cu(II) based molecular systems and their interaction with DNA have gained much attention.37–40
Alternatively, benzimidazole-based systems and their derivatives are also well documented as a vital class of heteroaromatic compounds with diverse biological importance, for example, anticancer, antimicrobial, antifungal, antiviral, antiulcer, and anticoagulant properties.31–46 These systems have also functioned as a crucial intermediate in different transformation reactions and the synthesis of several valuable products on an industrial scale.47 Because of the above-mentioned applications, the benzimidazole moiety and its derivatives are gaining much attention for synthetic and bio-investigation in recent times. In addition to that, benzimidazole incorporated tridentate systems upon complexation with different transition metals (Zn, Cu, Fe, etc.) showed promising activity in numerous research fields such as catalysis, spin crossover, sensing and biological anticancer therapeutics.48–51 Thus, it is very important and interesting to synthesize benzimidazole-based ligand systems and their metal complexes in combination with different polypyridyl ligand frameworks as well as study their interaction with DNA. Also, these complexes are useful for DNA cleavage and anticancer in vitro therapeutic applications with different cancer cell lines. Kose and co-workers reported substituted benzimidazole-based transition metal complexes with Cu, Mn, and Zn of type [M(bzimpy)Cl2] (where bzimpy = 2,6-bis(1H-benzo[d]imidazol-2-yl)pyridine) and studied their DNA binding interaction which revealed a groove binding mode of interaction in all cases.52 As an extension to their previous work, Kose's group prepared the N-butylbenzimidazole derivative of the ligand bzimpy and investigated its in vitro antifungal and antibacterial activities towards different Gram (+) and Gram (−) bacteria and yeasts.53 Later on, several benzimidazole-based derivatives were documented by different research groups just by replacing the N–H proton of the benzimidazole moiety.54,55 In recent years, noticeable attention has been paid to synthesizing biocompatible Cu(II) complexes based on tridentate–bidentate mixed ligand systems to perform biological studies on DNA binding, cleavage, and therapeutic applications.56–59 In this connection, we have synthesised three mononuclear Cu(II) complexes 1–3 with 2,6-bis(1-methyl-1H-benzo[d]imidazol-2-yl)pyridine (Mebzimpy) (L) as a tridentate ligand and different bidentate polypyridyl ligands such as 2,2′-bipyridine (bpy) for 1, 1,10-phenanthroline (phen) for 2 and 2,9-dimethyl-1,10-phenanthroline (Mephen) for 3. The complexes were comprehensively characterised by UV-vis, FT-IR, mass spectrometry, EPR, and elemental analyses. The molecular integrity of the complexes was established by using the single-crystal X-ray diffraction technique. All the complexes were tested for DNA binding activity using salmon sperm DNA (ss-DNA) and in silico molecular docking analysis was also performed to gain more insight into the binding. Furthermore, the DNA cleaving ability of complexes 1–3 has been studied by gel electrophoresis using supercoiled plasmid DNA. An in vitro cell viability assay was performed on the osteosarcoma cell line U2OS and the triple-negative breast cancer cell line MDA-MB-231 to evaluate their growth inhibitory potential.
Results and discussion
Synthesis
The tridentate ligand 2,6-bis(1-methyl-1H-benzo[d]imidazol-2-yl)pyridine (Mebzimpy)[L] was prepared in a two-step manner using a previously reported procedure.60 In the first step, pyridine-2,6-dicarboxylic acid was mixed with 10 mL of o-phosphoric acid and then o-phenylene diamine was added to the solution. The reaction mixture was then refluxed for 4 h, which after neutralization with 10% Na2CO3 resulted in the formation of the 2,6-bis(1H-benzo[d]imidazol-2-yl)pyridine (bzimpy) ligand. In the next step, the bzimpy ligand was methylated using methyl iodide in the presence of sodium hydride in dry dimethylformamide (DMF) solvent which led to the formation of our desired Mebzimpy (L) ligand in good yield (Scheme 1). Ligand L was then well characterised using several spectroscopic methods. The metal complexes were synthesized by taking an equimolar quantity of Cu(ClO4)2·6H2O and ligand L in methanol solvent and refluxed for 2 h. Then, methanolic solutions of the respective bidentate ligands were added dropwise to the reaction mixture while hot and refluxed for 4 h to obtain the desired respective metal complexes (Scheme 2). The complexes were further purified by recrystallizing in an acetonitrile–methanol (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) solvent mixture which results in green colour crystals suitable for single-crystal X-ray diffraction analysis. All the complexes were extensively characterised using UV-vis, FT-IR, mass spectrometry, EPR spectroscopy, etc.
:1 v/v) solvent mixture which results in green colour crystals suitable for single-crystal X-ray diffraction analysis. All the complexes were extensively characterised using UV-vis, FT-IR, mass spectrometry, EPR spectroscopy, etc.
 |
| | Scheme 1 Synthesis of ligand L. | |
 |
| | Scheme 2 Synthesis of complexes 1–3. | |
Caution: Perchlorate salts are generally explosive and should be handled carefully and used in small quantities.
Spectral studies
In the 1H NMR spectra, L showed a sharp singlet at 4.24 ppm for the six of two equivalent methyl (–CH3) groups bonded to the nitrogen center of the two equivalent benzimidazole rings situated at the ortho positions of the central pyridine moiety. A sharp doublet at 8.42 ppm is observed for the 2 equivalent protons situated at the meta position to the central pyridine unit; a clear triplet for a single proton at 8.02 ppm is also observed for the para hydrogen of the central pyridine moiety. Other NMR signals for the remaining protons of the benzene ring of the two equivalent benzimidazole situated ortho to the central pyridine are also found in the desired aromatic range of 7.88 to 7.33 ppm (Fig. S1†). The FT-IR spectrum of L showed an aromatic ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) peak at 1427 cm−1, a ν(CN) peak at 1566 cm−1, an sp3ν(C–H) peak at 2921 cm−1, and ν(C–H) out of plane bending at 732 cm−1 (Fig. S2†).53 For all complexes, the sharp peaks at ∼1080 cm−1 and ∼623 cm−1 indicate the perchlorate (ClO4−) counterions (Fig. S3†).6 Other characteristic bands for the complexes such as ν(CC) andν(CN) are found to be somewhat shifted compared to the free ligand (L).57,61 The ESI mass spectrum of L was taken in methanol (Fig. S4†). A molecular ion peak corresponding to ligand L was found at m/z = 340.1559 [L + H]+ (calc.: 340.1562). The ESI mass spectra of 1–3 were taken in acetonitrile solvent (Fig. S4†). Molecular ion peaks for the complexes at m/z = 657.0945 [1 − ClO4]+ (calcd 657.0953), m/z = 681.0931 [2 − ClO4]+ (calcd 681.0953) and m/z = 709.1300 [3 − ClO4]+ (calcd: 709.1266) validate their respective complex formation. The electronic absorption (UV-vis) spectra of L and the complexes were recorded in Tris-HCl buffer (5 mM, pH 7.4) at room temperature (Fig. 1 and Table 1). The ligand (L) shows characteristic bands in the region of 200 nm to 400 nm, and the band at 239 nm can be ascribed as the π–π* transition of the aromatic ring, and the higher-intensity band at 319 nm represents the n–π* transition of the azomethine (CN) groups. The UV-vis spectra of complexes 1–3 closely resemble the free ligand L where the transition bands are found to be somewhat shifted (either bathochromic or hypsochromic) compared to the free ligand L. This shift of electronic transition bands in their UV-vis spectra indicated successful complex formation with the ligand (L). The higher energy bands in the range of 210–330 nm can be assigned to the ligand-based π–π* and n–π* transitions, and the lower energy bands in the range of 340–400 nm can be ascribed to the ligand to metal charge transfer transition.52,53 All the complexes were investigated for their stability using UV-vis spectra in 5 mM Tris-HCl buffer. The findings of the stability study suggest that the complexes were stable for a couple of days (Fig. S5†).
C) peak at 1427 cm−1, a ν(CN) peak at 1566 cm−1, an sp3ν(C–H) peak at 2921 cm−1, and ν(C–H) out of plane bending at 732 cm−1 (Fig. S2†).53 For all complexes, the sharp peaks at ∼1080 cm−1 and ∼623 cm−1 indicate the perchlorate (ClO4−) counterions (Fig. S3†).6 Other characteristic bands for the complexes such as ν(CC) andν(CN) are found to be somewhat shifted compared to the free ligand (L).57,61 The ESI mass spectrum of L was taken in methanol (Fig. S4†). A molecular ion peak corresponding to ligand L was found at m/z = 340.1559 [L + H]+ (calc.: 340.1562). The ESI mass spectra of 1–3 were taken in acetonitrile solvent (Fig. S4†). Molecular ion peaks for the complexes at m/z = 657.0945 [1 − ClO4]+ (calcd 657.0953), m/z = 681.0931 [2 − ClO4]+ (calcd 681.0953) and m/z = 709.1300 [3 − ClO4]+ (calcd: 709.1266) validate their respective complex formation. The electronic absorption (UV-vis) spectra of L and the complexes were recorded in Tris-HCl buffer (5 mM, pH 7.4) at room temperature (Fig. 1 and Table 1). The ligand (L) shows characteristic bands in the region of 200 nm to 400 nm, and the band at 239 nm can be ascribed as the π–π* transition of the aromatic ring, and the higher-intensity band at 319 nm represents the n–π* transition of the azomethine (CN) groups. The UV-vis spectra of complexes 1–3 closely resemble the free ligand L where the transition bands are found to be somewhat shifted (either bathochromic or hypsochromic) compared to the free ligand L. This shift of electronic transition bands in their UV-vis spectra indicated successful complex formation with the ligand (L). The higher energy bands in the range of 210–330 nm can be assigned to the ligand-based π–π* and n–π* transitions, and the lower energy bands in the range of 340–400 nm can be ascribed to the ligand to metal charge transfer transition.52,53 All the complexes were investigated for their stability using UV-vis spectra in 5 mM Tris-HCl buffer. The findings of the stability study suggest that the complexes were stable for a couple of days (Fig. S5†).
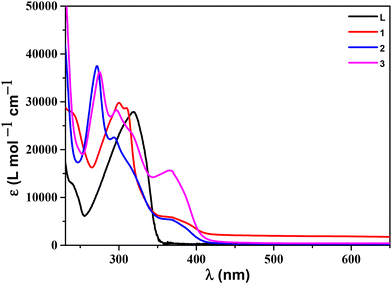 |
| | Fig. 1 Absorption spectra of ligand L and complexes 1–3 (all 20 μM) in Tris–HCl buffer (pH 7.4) at room temperature. | |
Table 1 Physiochemical characterization data
| Complex |
UV-vis λ/nm (ε, M−1 cm−1) Tris–HCl buffer |
EPR data/g values |
Redox data |
| Cu(II)/Cu(I)a |
Cu(I)/Cu(0)a |
Ligand-baseda |
Potentials  vs. SCE in CH3CN/0.1 M TBAP. vs. SCE in CH3CN/0.1 M TBAP.
|
|
1
|
242 (27345), 300 (29776), 310 (28704), 369 (5821) |
2.063 |
0.07 V |
−0.62 V |
−1.17 V |
|
2
|
272 (37469), 294 (22549), 313 (16907), 370 (5268) |
2.058 |
0.06 V |
−1.37 V |
−1.58 V |
|
3
|
275 (36211), 297 (28261), 317 (23315), 368 (14011) |
2.051 |
0.04 V |
−0.64 V |
−1.01 V, −1.71 V |
EPR studies
The EPR spectra for all the complexes were taken in the solid state at room temperature (Fig. 2 and Table 1). The employed conditions of EPR spectra for the complexes are: microwave frequency = 9.450 GHz, center field = 336 mT, and width +/− = 250 mT. The observed EPR activity of 1–3 is a consequence of unpaired electrons in the dx2−y2 orbital of the paramagnetic copper(II) metal center in a square pyramidal geometry.
 |
| | Fig. 2 The solid state EPR spectra of (a) 1, (b) 2 and (c) 3 at room temperature. | |
The solid-state EPR spectra of complexes 1, 2, and 3 showed isotropic signals corresponding to the g values of 2.063, 2.058, and 2.051, respectively, which is in accordance with the literature values.62,63 Unfortunately, no hyperfine splitting (four-line) was observed for 1 and 2 as expected for 63Cu (I = 3/2). This kind of similar observation for the square pyramidal complexes was reported in the literature.64,65 On the other hand, for complex 3, we get the desired four-line hyperfine splitting in their respective EPR spectrum.
Crystallographic description
The single crystals of the mononuclear complexes were grown using a (3:1, v/v) acetonitrile and methanol solvent mixture at room temperature. The molecular integrity of the synthesized complexes was identified by single-crystal X-ray diffraction investigation. The ORTEP representations of the complexes are shown in Fig. 3. A polyhedral view of the complexes is shown in Fig. S6.† Complexes 1 and 2 both crystallise as triclinic systems with the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. Also, for complex 1 in the asymmetric unit, two non-equivalent Cu(II) centers are found to be present in the crystal symmetry (Fig. S7†). Complex 3 crystallises in a monoclinic fashion with the space group P21/c. The selected crystallographic information and the other significant parameters related to the refinement methods are shown in Table S1.† Important bond distances (Å) and bond angles (°) are summarized in Table S2.† In all the three complexes, the copper(II) center is penta-coordinated with five distinct nitrogen atoms, where three nitrogens (N1, N2, and N3) are from tridentate ligand L and the other two (N4 and N5) are from the bidentate ligand (L′) composed of bpy (for 1), phen (for 2) and Mephen (for 3), respectively. Similarly, the penta coordination around the Cu(2) center in the asymmetric unit of 1 is fulfilled by the three nitrogens (N8, N9, and N10) of the tridentate ligand (L), and the remaining two are fulfilled by the N11 and N12 atoms of the bpy ligand. The trigonal index value τ5 = 0.21 and 0.33 (for 1), 0.27 (for 2) and 0.14 (for 3) [τ5 = (β − α)/60], where β is the largest angle (170.91° and 176.88° (for 1), 174.03° (for 2) and 166.68° (for 3)) and α is the second-largest angle (157.74° and 156.56° (for 1), 157.68° (for 2) and 157.94° (for 3)) between L–M–L′ indicates the distorted square pyramidal geometry around the Cu(II) center (0.0 < τ5 < 0.5).66,67 In the asymmetric unit of 1, nitrogen centers N1, N2, N3 and N4 {for Cu1} and N8, N9, N10 and N12 {for Cu2} are present in the basal plane and N5 {for Cu1} and N11 (for Cu2) atoms are located at the axial position. In the case of 2 and 3, the basal plane is occupied by the N1, N2, N3, and N4 atoms and N5 is present in the apical position. All the Cu–N in-plane bonds are quite equivalent, but the Cu1–N5 and Cu2–N11 apical bonds are found to be longer as compared to the basal Cu–N bonds which suggests a pseudo-Jahn–Teller elongation in all 1–3 complexes.66 For complex 1, N1–Cu1–N4, N3–Cu1–N2, N8–Cu2–N12 and N10–Cu2–N9 bonds are not linear [N1–Cu1–N4: 170.91°(14), N3–Cu1–N2: 157.74°(14), N8–Cu2–N12: 176.88°(14) and N10–Cu2–N9: 156.56°(14)] and also, the bond angle around the central Cu(II) unit (N–Cu1–N and N–Cu2–N) within the basal plane is not a perfect right angle (90°). A similar observation was found for complexes 2 and 3 as well [N1–Cu1–N4: 174.03°(11) and N3–Cu1–N2: 157.68°(12) for 2 and N1–Cu1–N4: 166.68°(18) and N3–Cu1–N2: 157.94°(18) for 3 and also N–Cu1–N ≠ 90 °C for both 2 and 3]. This validates a distorted square pyramidal geometry around the copper(II) center in all 1–3 complexes.
space group. Also, for complex 1 in the asymmetric unit, two non-equivalent Cu(II) centers are found to be present in the crystal symmetry (Fig. S7†). Complex 3 crystallises in a monoclinic fashion with the space group P21/c. The selected crystallographic information and the other significant parameters related to the refinement methods are shown in Table S1.† Important bond distances (Å) and bond angles (°) are summarized in Table S2.† In all the three complexes, the copper(II) center is penta-coordinated with five distinct nitrogen atoms, where three nitrogens (N1, N2, and N3) are from tridentate ligand L and the other two (N4 and N5) are from the bidentate ligand (L′) composed of bpy (for 1), phen (for 2) and Mephen (for 3), respectively. Similarly, the penta coordination around the Cu(2) center in the asymmetric unit of 1 is fulfilled by the three nitrogens (N8, N9, and N10) of the tridentate ligand (L), and the remaining two are fulfilled by the N11 and N12 atoms of the bpy ligand. The trigonal index value τ5 = 0.21 and 0.33 (for 1), 0.27 (for 2) and 0.14 (for 3) [τ5 = (β − α)/60], where β is the largest angle (170.91° and 176.88° (for 1), 174.03° (for 2) and 166.68° (for 3)) and α is the second-largest angle (157.74° and 156.56° (for 1), 157.68° (for 2) and 157.94° (for 3)) between L–M–L′ indicates the distorted square pyramidal geometry around the Cu(II) center (0.0 < τ5 < 0.5).66,67 In the asymmetric unit of 1, nitrogen centers N1, N2, N3 and N4 {for Cu1} and N8, N9, N10 and N12 {for Cu2} are present in the basal plane and N5 {for Cu1} and N11 (for Cu2) atoms are located at the axial position. In the case of 2 and 3, the basal plane is occupied by the N1, N2, N3, and N4 atoms and N5 is present in the apical position. All the Cu–N in-plane bonds are quite equivalent, but the Cu1–N5 and Cu2–N11 apical bonds are found to be longer as compared to the basal Cu–N bonds which suggests a pseudo-Jahn–Teller elongation in all 1–3 complexes.66 For complex 1, N1–Cu1–N4, N3–Cu1–N2, N8–Cu2–N12 and N10–Cu2–N9 bonds are not linear [N1–Cu1–N4: 170.91°(14), N3–Cu1–N2: 157.74°(14), N8–Cu2–N12: 176.88°(14) and N10–Cu2–N9: 156.56°(14)] and also, the bond angle around the central Cu(II) unit (N–Cu1–N and N–Cu2–N) within the basal plane is not a perfect right angle (90°). A similar observation was found for complexes 2 and 3 as well [N1–Cu1–N4: 174.03°(11) and N3–Cu1–N2: 157.68°(12) for 2 and N1–Cu1–N4: 166.68°(18) and N3–Cu1–N2: 157.94°(18) for 3 and also N–Cu1–N ≠ 90 °C for both 2 and 3]. This validates a distorted square pyramidal geometry around the copper(II) center in all 1–3 complexes.
 |
| | Fig. 3 ORTEP representations of (a) 1, (b) 2 and (c) 3. The H atoms are excluded for clarity. Thermal ellipsoids are shown at 30% probability. | |
The packing diagram of 1 shows two distinct displaced π–π stacking interactions between the terminally coordinated benzimidazole pyridine rings of the two neighbouring metal units. Among the three stacking interactions, the one with a distance of 3.555 Å can be considered as a strong interaction; the second interaction with a distance of 4.024 Å is a very weak interaction and the third interaction (distance 4.212 Å) is not a true π–π stacking interaction as its distance is much higher than the upper limit (∼4.0 Å) of a true π–π stacking interaction.68 Furthermore, such kinds of stacking interactions were found to be absent in the case of complexes 2 and 3 as suggested by the stacking distance values of 4.365 Å (for 2) and 4.519 Å and 4.399 Å (for 3), respectively (Fig. S8†).
All three complexes also exhibit several intermolecular H-bonding interactions. In complex 1, the oxygen (O3) atom of the perchlorate counter anion is found to be H-bonded with the hydrogen (H31) atom of the bipyridine unit coordinated to the Cu(1) center in the asymmetric unit (distance, H31–O3 = 2.428 Å). Another similar H-bonding is also present between the O12 of the perchlorate counter anion and the H25 of the bipyridine moiety (H25–O12 = 2.475 Å) coordinated to the Cu(1) center. On the other hand, the O13 atom of the perchlorate anion which is connected to the Cu(2) center in the asymmetric unit forms an H-bonding with the H17 atom of the tridentate ligand L coordinated to the Cu(1) center with an interaction distance H17–O13 = 2.625 Å. In complex 2, the O3, O4, and O5 atoms of the perchlorate anion form intermolecular H-bonding with the H22, H31, and H26 atoms of the Cu1 coordinated phenanthroline moiety, respectively (H22–O3 = 2.646 Å, H31–O4 = 2.581 Å and H26–O5 = 2.606 Å). Likewise, the O1 and O6 atoms of the perchlorate counter anion form H-bonding with the H21C (methyl hydrogen) and H17 (terminal benzimidazole hydrogen) atoms of the tridentate ligand (L), respectively (H21C–O1 = 2.456 Å and H17–O6 = 2.539 Å). The methyl (–CH3) proton H20A is also found to be H-bonded with the O7 and O8 atoms of the perchlorate counter anion with a bonding distance of H20A–O7 = 2.551 Å and H20A–O8 = 2.643 Å, respectively. Similar to the first two complexes, the third complex also forms several H-bonding interactions. In complex 3, the methyl (–CH3) protons H7A, H7B, and H15B from the ligand (L) form intermolecular H-bonding interactions with the perchlorate oxygen atoms O4, O1, and O2, respectively (H7A–O4 = 2.555 Å, H7B–O1 = 2.392 Å and H15B–O2 = 2.635 Å). Similarly, the methyl (–CH3) proton H22A from the bidentate (Mephen) moiety is found to be H-bonded with the O6 and O8 atoms of the perchlorate anion, respectively (H22A–O6 = 2.673 Å and H22A–O8 = 2.660 Å). The central pyridine protons H1 and H2 were also involved in H-bonding with the perchlorate O7 and O8 atoms, respectively, with an interaction distance of H1–O7 = 2.457 Å and H2–H8 = 2.500 Å. Similar to the central pyridine protons, the terminal benzimidazole proton H9 also forms an H-bonding with the O1 atom of the perchlorate counter anion (H9–O1 = 2.544 Å). A pictorial representation of the H-bonding interactions for 1–3 is shown in Fig. S9.†
Redox properties
Electrochemical studies of the ligands as well as the copper complexes were performed using cyclic voltammetry (CV) and differential pulse voltammetry (DPV) techniques. All the studies were carried out under inert conditions using dry acetonitrile as a solvent and in the presence of 0.1 M Bu4N(ClO4) [TBAP] as a supporting electrolyte and a glassy carbon electrode as the working electrode. Redox potential values are given with reference to the saturated calomel electrode (SCE). Cyclic voltammograms showing the reversible Cu(II)/Cu(I) couple for complexes 1–3 are presented in Fig. 4 and Table 1.
 |
| | Fig. 4 Electrochemical behaviour of (a) 1, (b) 2 and (c) 3 in dry CH3CN solvent using 0.1 M TBAP as a supporting electrolyte vs. SCE at a scan rate of 100 mV s−1. | |
Ligand L showed two irreversible oxidation peaks at 1.56 V and 1.82 V. (Fig. S10†) while it was found to be redox silent in the negative potential range. The cyclic voltammogram of 1 showed one reversible and one irreversible redox event at 0.07 V and −0.62 V which can be assigned to Cu(II)/Cu(I) and Cu(I)/Cu(0), respectively.21,69 The expected peak for copper deposition at the surface of the working electrode was observed at −0.21 V.70,71 Also, complex 1 shows an irreversible reduction due to the bpy-based reduction observed at −1.17 V.72 The CV diagram of 2 shows one reversible and one irreversible peak at 0.06 V and −1.37 V which could be ascribed to Cu(II)/Cu(I) and Cu(I)/Cu(0) events, respectively (Fig. 5, Table 1 and Fig. S11†). Likewise, the copper deposition at the electrode was confirmed by a sharp peak at −0.31 V. The irreversible peak at −1.58 V can be ascribed to phen-based reduction. Electrochemical studies of complex 3 show one reversible and one quasi-reversible redox couple at 0.04 V and −0.64 V which could be assigned to Cu(II)/Cu(I) and Cu(I)/Cu(0) reductions, respectively (Fig. 5 and S10†). Similarly, the peak for deposition of copper was observed at −0.18 V. Other irreversible peaks at −1.01 V and −1.71 V are observed for Mephen-based reductions.
 |
| | Fig. 5 UV-vis spectra of (a) 1, (b) 2 and (c) 3 (50 μM) in the absence (black) and presence (colour) of ss-DNA with a gradual increase in the concentrations (10–80 μM). Inset: plot of A0/A − A0vs. 1/[DNA] for the determination of the binding constant of complexes 1–3. | |
DNA binding studies
For the investigation of the binding properties of the metal complexes with DNA, the electronic absorption spectroscopic (UV-vis) studies are well known for a long time.73 A Tris–HCl buffer (5 mM, pH = 7.4) solution was employed for all the UV-vis spectral measurements to evaluate the DNA binding properties of all complexes. Complexes can interact with DNA via different non-covalent ways, like groove (minor or major) binding, electrostatic interactions via the phosphate-rich backbone and intercalation to the base pairs.74,75 The specific binding pattern defines the strength, cytotoxic effects, reversible nature, etc. Generally, binding leads to the shift of λmax in their subsequent UV-vis spectra, which can be either bathochromic or hypsochromic, with an increase (hyperchromicity) or decrease (hypochromicity) in the absorbance value. The intercalative mode of binding generally induces a bathochromic shift with a successive increase in absorbance as a result of effective stacking interactions between the ligand backbone of the metal complex and DNA base pairs.6
The DNA binding studies of 1, 2, and 3 were carried out using ss-DNA and are shown in Fig. 5. Upon successive increase in the concentration of DNA, hypochromism was observed in the intra-ligand transition bands with very minor or no bathochromic shifts for complexes 1, 2, and 3. These changes in the spectral behaviour of the complexes indicate that there are some interactions taking place between the complex and the ss-DNA. The isosbestic points with hyperchromism were also observed at around 281 nm for 1 which indicates the homogeneity as well as the reversible nature towards the DNA binding process. The intrinsic binding constant (Kb) was calculated to compare the binding strength of the complexes with ss-DNA. Eqn (1) was employed to evaluate the Kb value using the spectrophotometric method.23,61
| |  | (1) |
where the terms
A0 and
A denote the absorbance of the metal complex in the absence and presence of the ss-DNA. The corresponding molar extinction coefficient values are denoted by the terms
εG and
εH–G, respectively. The 1
:
1 complex and DNA adduct formation leads to the linear fitting plot between
A0/(
A −
A0) and 1/[DNA].
22 The
Kb values were calculated according to
eqn (1) from the ratio between the intercept to the slope. The
Kb values were calculated as 1.21 × 10
4 M
−1, 1.53 × 10
4 M
−1, and 1.44 × 10
4 M
−1 for
1–3 respectively (
Table 2), suggesting that all three complexes are moderate DNA intercalators compared to the classical DNA intercalator ethidium bromide whose binding constant value is found to be 1.40 × 10
6 M
−1.
52 The calculated
Kb values of all the complexes are found to be quite comparable to the analogous reported benzimidazole based complexes [Cr(Bzimpy)
2]
3+, [Cu(Bzimpy)(bpy)H
2O]
2+, [Ru(Bzimpy)
2]
2+, [Ru(Bzimpy)(phen)(H
2O)]
3+ and [Ru(Bzimpy)(bpy)(H
2O)]
3+ with values of 1.21 × 10
4 M
−1, 1.41 × 10
4 M
−1, 1.80 × 10
4 M
−1, 2.87 × 10
4 M
−1 and 3.58 × 10
4 M
−1, respectively.
57,76 Among all complexes, complex
2 shows the best binding ability with the highest binding constant of 1.53 × 10
4 M
−1. This observation might be due to the better stability and planarity of the phenanthroline (phen) coordinated complex compared to the bipyridine (bpy) coordinated complex
1. Complex
3 also showed decent binding interaction with ss-DNA but owing to the presence of two methyl groups in the bidentate ligand (Mephen), it might cause some sort of hindrance towards interaction with the ss-DNA and thus its
Kb value was found to be somewhat less than that of complex
2.
Table 2 Comparison table of the DNA binding activities of 1–3 with the reported literature
| Complex |
Binding constant (Kb)/M−1 (×104) |
Stern–Volmer constant (KSV)/M−1 (×104) |
Ref. |
| [CuLa(Bpy)](ClO4)2 |
1.90 |
1.50 |
82
|
| [CuLb(Bpy)](ClO4)2 |
2.80 |
3.60 |
| [CuLcCl2] |
3.46 |
1.27 |
52
|
| [CuLdCl2] |
2.44 |
4.88 |
| [CuLeCl2] |
5.00 |
1.02 |
| [Cu(Lf)(bpy)H2O]SO4 |
1.41 |
0.21 |
57
|
| [Cu(Lf)(en)H2O]SO4 |
1.32 |
0.20 |
| [Cu(Lg)(bpy)](ClO4)2 |
0.60 |
17.00 |
9
|
| [Cu(Lg)(phen)](ClO4)2 |
2.80 |
36.00 |
| [Cu(Lg)(5,6-dmp)](ClO4)2 |
10.90 |
108.00 |
| [Cu(Lg)(dpq)](ClO4)2 |
22.00 |
51.00 |
| [Cu(Lg)(crotonate)]ClO4 |
0.14 |
2.63 |
83
|
| [Cu(Lg)(methacrylate)]ClO4 |
1.34 |
3.06 |
| [Cu(Lg)(acrylate)(MeOH)]NO3H2O |
1.90 |
1.50 |
| [Cu (Lh)(pyimphen)](ClO4)2 |
1.80 |
— |
84
|
| [Cu (Li)(pyimphen)](ClO4)2 |
12.30 |
— |
| [Cu (Lj)(pyimphen)](ClO4)2 |
14.30 |
— |
| [Cu(Lk)(nap)Cl] |
23.80 |
— |
77
|
| [Cu(Ll)(nap)Cl] |
42.70 |
— |
| [Cu(Lm)(nap)Cl] |
22.40 |
— |
|
1
|
1.21 |
2.71 |
This work |
|
2
|
1.53 |
4.8 |
|
3
|
1.44 |
2.70 |
Viscosity measurement studies
Further clarification regarding the binding nature of our synthesized complexes towards ss-DNA was verified by performing viscosity studies on ss-DNA with increasing concentrations of the complexes. This investigation consists of measuring the flow time of a DNA solution via the capillary of a viscometer. Generally, an intercalative nature of interaction results in an overall rise in the viscosity of the DNA solution as a result of the elongation of the DNA helix. Conversely, molecules that bind to the DNA exclusively through grooves result in minor or no change in the overall viscosity of the solution.77 The relative viscosity coefficients for ss-DNA both in the absence (η0) and presence (η) of the complexes were calculated according to eqn (2).where the flow time for the DNA solution in the presence of added complexes is denoted by t, while t0 represents the flow time for the buffer solution only. The graph of (η/η0)1/3vs. [complex/DNA] results in the viscosity changes shown in Fig. 6. From the plot it is visible that the relative viscosity of the DNA solution increases as the concentration of the complex increases, indicating that all complexes bind in an intercalative fashion.78 The trend follows the order of 2 > 1 ≥ 3, which is also consistent with the findings of binding experiments using the UV-vis spectroscopic method.
 |
| | Fig. 6 Change in the viscosity of ss-DNA with complexes 1–3. Relative specific viscosity vs. [Complex]/[DNA], ([DNA] = 200 μM, [Complex] = 10–80 μM). | |
The ethidium bromide (EB) displacement assay
In this assay, fluorescence spectroscopy was employed to investigate the competitive binding of EB and the complexes with the ss-DNA. The lack of significant emission of EB in buffer solution is due to the quenching by solvent molecules. Nevertheless, the fluorescence intensity increases rapidly upon the addition of DNA owing to its extensive intercalative binding with the adjacent DNA base pairs.79 Upon consecutive addition of the complex, it started displacing the EB from the EB bound DNA solution (EB-DNA) due to its ability to bind with DNA effectively in comparison with EB, which leads to quenching of the intensity of the emission spectra of the bound EB-DNA solution.80 Initially, this decreased gradually for all complexes, but later the complexes reached the saturation point at different concentrations. The observations of this assay are displayed in Fig. 7 where 2 showed efficient binding with DNA as it displaces the EB from DNA more rapidly and the intensity of the emission spectra decreases gradually as the displaced or free EB will not show any emission spectra. As EB is known as a classical DNA intercalator, this assay confirms the intercalative binding nature of the synthesized complexes as they substitute the bound EB from ss-DNA. The classical Stern–Volmer equation is utilized to determine the Stern–Volmer quenching constant (KSV), according to eqn (3).81| |  | (3) |
where I0 and I represent the emission intensity of EB-DNA solution in the absence and presence of the complexes, respectively. [Q] denotes the quencher (complex) concentration.
 |
| | Fig. 7 Emission quenching profile of EB-DNA as a function of increasing concentration of (a) 1, (b) 2 and (c) 3. [DNA] = 100 μM and [EB] = 10 μM. [Complex] (μM) for 1 (1–61 μM), 2 (1–52 μM) and 3 (1–24 μM). Inset: graph of I0/I vs. [Complex]. | |
Plotting I0/I against [Q] yields a linear plot, and the slope/intercept gives the values of KSV, which are found to be 2.71 × 104 M−1, 4.80 × 104 M−1, and 2.70 × 104 M−1 for 1–3, respectively. The findings of this study support the most intercalative binding nature of 2 among all synthesized complexes. All the binding parameters of the above investigations along with a detailed comparison with similar reported complexes are listed in Chart 1 and Table 2.
 |
| | Chart 1 Tridentate ligands used for the DNA binding activity with Cu(II) complexes. | |
Circular dichroism (CD) spectral studies
CD spectral investigation has been carried out to investigate the DNA secondary structure perturbation by the influence of our synthesized complexes. The right-handed B form of ss-DNA in its CD spectrum exhibits a positive band near 275 nm corresponding to base stacking whereas another band in the negative region near 244 nm is a consequence of DNA helicity.85 Generally, small molecules upon interacting with DNA through grooves or electrostatic interactions display a minor alteration on both positive (∼275 nm) and negative bands (∼244 nm) of ss-DNA; conversely, intercalation can significantly alter the intensities of both bands to a great extent.23Fig. 8 presents the CD spectra of ss-DNA both in the presence and absence of the complexes. In all the cases, noticeable alterations of CD spectral bands were seen with the addition of the metal complexes. Spectral investigation shows that the intensity of the positive band was considerably decreased, whereas the intensity of the negative band was increased for all complexes. The investigation indicates that the binding of the complexes with DNA is significant in causing conformational changes in the ss-DNA secondary structure. All of these findings are consistent with an intercalative nature of binding as stated in the literature reports.86,87
 |
| | Fig. 8 CD spectra of ss-DNA (100 μM) in the absence (black line) and presence (red, blue, and green lines) of 1–3 (50 μM). | |
Molecular docking studies
To understand the possible interaction sites of DNA binding by complexes 1–3 and to find the preferred sterically acceptable orientation of the complexes inside the DNA groove, in silico binding analysis was performed (Fig. 9). The analysis of the docked structure revealed the identity of DNA residues that interacted with complexes 1–3via π–σ, π–alkyl, π–anion, carbon–hydrogen bond, π–anion, π–alkyl, carbon–hydrogen, π–σ, π–anion, carbon–hydrogen and van der Waals interactions. It was observed that all three complexes partly fit into the minor groove comfortably involving outside edge interactions without disrupting the double helical structure of DNA. The theoretical binding free energy of complexes 1–3 was predicted to be −8.70, −7.59 and −9.27 kcal mol−1, respectively. Notably, the relative affinities of binding obtained from the spectroscopic study and docking analysis were not the same. This could be due to the small size of limited sequence variation of the model DNA considered for the docking studies. Overall, the molecular docking model substantiated our spectroscopic results and provided further evidence of duplex DNA binding.
 |
| | Fig. 9 Molecular docking of copper containing metals (a) 1, (b) 2 and (c) 3 on the 3D crystal structure of DNA. In the docked DNA complex, the metal complexes (1–3) are highlighted in the dark grey stick model, whereas nucleotides (represented as rings) are coloured as follows: pink, green, red, and blue represent cytosine, guanine, adenine, and thymine, respectively. The backbone is represented by grey arrows. Interaction of the nucleotides with the ligands and the bonds are represented by dotted lines (π–alkyl: blue, π–σ: violet, π–anion: yellow and carbon–hydrogen bond: green). | |
DNA cleavage analysis
The DNA cleaving ability of the complexes was studied by agarose gel electrophoresis using supercoiled (SC) plasmid DNA. It was observed that 1–3 were not able to bring about the conversion of the SC form of DNA to the nicked circular (NC) form in the presence or absence of light. This shows that all complexes lack the ability to induce hydrolytic cleavage of DNA (Fig. 10). The high kinetic stability of our Cu(II) complexes in buffer solution perhaps led to electrochemical redox silencing of the Cu(II)/Cu(I) couple which results in complete loss of the ability to generate ROS.88,89 This might be the probable reason for the inability of our complexes towards DNA cleavage.
 |
| | Fig. 10 Assessment of the DNA cleavage properties of 1–3 (200 μM) by agarose gel electrophoresis in the presence and absence of light (SC and NC represent supercoiled and nicked circular, respectively). | |
Cytotoxicity analysis
The cytotoxicity of complexes 1–3 towards the human osteosarcoma cell line U2OS and the triple-negative breast cancer cell line MDA-MB-231 was studied using the XTT assay. Cell viability was represented as a dose–response curve (Fig. 11a and b). Limited cytotoxicity of complex 1 prevented its IC50 determination. However, the IC50 values of complex 2 were found to be 10.34 ± 0.07 μM and 13.87 ± 0.08 μM for MDA-MB-231 and U2OS, respectively. The IC50 values of 3 were found to be 4.87 ± 0.05 μM and 3.87 ± 0.04 μM for MDA-MB-231 and U2OS, respectively. Staining cells with a dual fluorescent dye helps to identify viable metabolically active cells from dead cells and offers a qualitative evaluation of cell death. We studied the effect of complexes 1–3 towards U2OS and MDA-MB-231 using the dual fluorescent dye Calcein-AM (green) and Hoechst (blue). Compared to the negative control, complex 1 showed little cytotoxicity (Fig. 11c). The dose dependent cell death inducing ability of complexes 2 and 3 was clearly visible at a concentration of 2 μM (Fig. 11d). In the presence of a higher concentration (5 μM) of complex 3, almost no viable cells remained but little nuclear degradation was observed (Fig. 11e). This result also supports our previous result on DNA cleavage (Fig. 10). Taken together, these results suggest that complex 3 is very efficient at inhibiting cell proliferation among the three complexes. A moderate level of inhibition is also observed with 2. Given the absence of DNA cleavage activity, it is possible that DNA binding by complex 3 could inhibit important DNA metabolic pathways in cancer cells causing inhibition of cell proliferation.
 |
| | Fig. 11 Effect of 1–3 on cancer cell survival. The XTT-based cell viability assay was performed to determine the specific cytotoxicity. The osteosarcoma cell line (U2OS) and triple negative breast cancer cell line (MDA-MB231) were incubated with (a) 1 (b) 2, and (c) 3. (d) U2OS and (e) MDA-MB231 cell lines treated with 1–3 were observed under a fluorescence microscope using DAPI and Calcein-AM dye. | |
Conclusions
This present study offers the synthesis of three novel mononuclear heteroleptic Cu(II) complexes where the tridentate ligand core consists of a biologically relevant benzimidazole-based tridentate ligand (L). The ancillary bidentate ligands are composed of bpy (for 1), phen (for 2), and Mephen (for 3). All the complexes are well characterised using different spectroscopic and analytical tools. The molecular integrity of the complexes was determined by employing SCXRD analysis. UV-vis spectroscopic analysis was performed to investigate the DNA binding ability of the synthesized complexes. The DNA intercalation ability of the complexes was studied by ethidium bromide displacement analysis. The relevant binding parameters such as Kb and Ksv were determined for all the complexes and the results suggest greater Kb and Ksv values for 2 among all complexes, whereas complexes 1 and 3 show comparable values. The greater binding ability of 2 is because of the more planar aromatic bidentate (phen) ligand moiety, compared to bpy and Mephen for 1 and 3, respectively. Molecular docking analysis confirmed that all the synthesized complexes can bind with DNA. Although the DNA cleaving ability was not observed, complexes 2 and 3 caused cytotoxicity to rapidly growing cancer cells. Analysis of cell viability revealed that compound 3 could be a potential cytotoxic compound to human cancer cells. Overall, these findings could be beneficial for the advancement of new antitumor agents.
Experimental section
Materials
Low molecular weight double-stranded salmon sperm DNA (ss-DNA), ethidium bromide (EB), 2,2′-bipyridine, 1,10-phenanthroline, 2,9-dimethyl-1,10-phenanthroline, XTT cell proliferation kit, fetal bovine serum (FBS) and Cu(ClO4)2·6H2O were bought from Sigma Aldrich India. Dulbecco's modified Eagle's medium (DMEM), trypsin–EDTA solution, antibiotic solution, Calcein-AM, and NucBlue™ Reagent (Hoechst 33342) were purchased from Thermo Fisher Scientific. Dimethyl sulfoxide (DMSO) was procured from SRL Laboratories. MDA-MB-231 (human triple-negative breast adenocarcinoma) and U2OS (human osteosarcoma) cell lines were obtained from the National Centre for Cell Science, India. Dry DMF used for the synthesis of ligand L was obtained by distillation over calcium hydride. All the solvents were of reagent grade and used directly without any further refinement.
Physical measurements
The 1H NMR spectrum of the ligand (L) was recorded using a BRUKER AVANCE III-400 MHz spectrometer in (CD3)2SO solvent. The coupling constants (Hz) along with all the chemical shift values (δ, ppm) are assigned in an accustomed manner with tetramethylsilane (TMS) (δ(H) 0.00 ppm) as a reference standard. The abbreviations of singlet = s, doublet = d, triplet = t and multiplet = m are used for all the 1H NMR spectra. A JASCO V-730 spectrophotometer was utilised to record the UV-vis electronic absorption spectra of the reported ligand L and complexes 1–3 at room temperature. The FT-IR spectral measurements were performed using a Fourier transform Bruker Alpha-P spectrometer. An Agilent Technologies (Model: HRMS Q-TOF 6538) mass spectrometer was used for obtaining high-resolution mass spectra in the electrospray ionisation (ESI) mode. Fluorescence spectral data were recorded using a Horiba Flouromax-4 fluorescence spectrophotometer at room temperature. Elemental analysis was recorded using a BRUKER EURO EA. The electrochemical investigations, i.e. cyclic voltammetry (CV) and differential pulse voltammetry (DPV), were performed using a CHI-660 potentiostat device. A one-pot three-electrode setup was used, which included a glassy carbon electrode as the working electrode, a platinum electrode as a counter electrode, and a saturated calomel electrode (SCE) as a reference electrode under an inert atmosphere. During the electrochemical experiments, 0.1 M TBAP (tetrabutylammonium perchlorate) was used as a supporting electrolyte. EPR spectra of the complexes were recorded using a JOEL EPR spectrometer. Circular dichroism spectra were recorded using a JASCO J-815 CD spectrophotometer in the range of 190 to 500 nm.
Synthesis
Synthesis of 2,6-bis(1-methyl-1H-benzo[d]imidazol-2-yl)pyridine (Mebzimpy)[L].
The meridional tridentate ligand L was synthesized in a two-step manner by following the previously reported literature with slight modification. Initially, the 2,6-bis(1H-benzo[d]imidazol-2-yl)pyridine (bzimpy) precursor ligand was synthesized by taking pyridine-2,6-dicarboxylic acid (1.67 g, 10 mM) in 10 mL of o-phosphoric acid (H3PO4) and mixing with another 10 mL H3PO4 solution of o-phenylene diamine (2.13 g, 20 mM) followed by heating the resulting reaction mixture at 220 °C for 5 h. Then, the resulting solution was poured into 1 L of vigorously stirred distilled water which results in a blue-green precipitate, which was collected by filtration. Then the solid precipitate was stirred in 500 mL of hot 10% Na2CO3 solution for 1 h. The resulting solution was then filtered and finally recrystallized from ethanol (2.5 g, 7.37 mmol, yield ∼ 74%). 1H NMR (400 MHz, CDCl3): δ (ppm) = 4.24 (s, 6H, –NCH3), 7.42–7.33 (m, 4H), 7.47 (dd, J = 7.0, 1.8 Hz, 2H), 7.36 (m, 4H), 7.88 (dd, J = 6.7, 1.8 Hz, 2H), 8.05 (t, J = 7.9, 1H), 8.42 (d, J = 7.9, 2H). ESI-MS (CH3OH): m/z = 340.1559 (calc.: 339.1484) [L + H]+. FT-IR (solid, ν, cm−1): 2900–3200 (sp3, alkyl C–H), 1576 (CN stretching of the imidazole ring), 1481 (CC stretching) UV-vis (Tris-HCl buffer) λmax (nm) [ε (L mol−1 cm−1)]: 239 [12963], 319 [27824]. Anal. calcd for C21H17N5: C, 74.32; H, 5.05; N, 20.63; found: C, 73.83; H, 5.15; N, 20.36.
General procedure for the synthesis of copper(II) complexes 1–3.
In a 10 mL methanolic solution of ligand L (0.15 mmol), another 10 mL methanolic solution of Cu(ClO4)2·6H2O (0.15 mmol) was added dropwise with constant stirring and the resulting green solution was refluxed for 2 h; then the corresponding bidentate ligand (0.15 mmol) (bpy for 1, phen for 2 and Mephen for 3) was added dropwise and the resulting reaction mixture was refluxed for another 4 h. The solution was cooled to room temperature and the resulting green precipitate was filtered and washed several times with methanol (10 mL) and diethyl ether (10 mL) and finally dried using a rotary evaporator to obtain pure products of complexes 1–3. All the complexes were recrystallised by dissolving them in a (3:1 v/v) acetonitrile and methanol solvent mixture under slow evaporation. The resulting single crystals were found to be suitable for single-crystal X-ray diffraction analysis.
[Cu(L)(bpy)](ClO4)2 (1).
Light green crystals. Yield 75 mg (67%). Anal. calcd for C31H25Cl2CuN7O8: C, 49.12; H, 3.32; N, 12.93; found: C, 49.05; H, 3.38; N, 12.86. ESI-MS (CH3CN): m/z = 657.0945 [1 − ClO4]+ (calcd: 657.0953). FT-IR (solid, ν, cm−1): 752 (aromatic C–H out of plane bending), 1081, 623 (Cl–O), 1481 (CC stretching), 1597 (CN stretching of the imidazole ring). UV-vis (Tris-HCl buffer) λmax (nm) [ε (L mol−1 cm−1)]: 242 [27345], 300 [29776], 310 [28704], 369 [5821].
[Cu(L)(phen)](ClO4)2 (2).
Dark green crystals. Yield 71 mg (60%). Anal. calcd for C33H25Cl2CuN7O8: C, 50.68; H, 3.22; N, 12.54; found: C, 50.61; H, 3.28; N, 12.08. ESI-MS (CH3CN): m/z = 681.0931 [2 − ClO4]+ (calcd: 681.0953). FT-IR (solid, ν, cm−1): 751 (aromatic C–H out of plane bending), 1079, 621 (Cl–O), 1488 (CC stretching), 1589 (CN stretching of the imidazole ring). UV-vis (Tris-HCl buffer) λmax (nm) [ε (L mol−1 cm−1)]: 272 [37469], 294 [22549], 313 [16907], 370 [5268].
[Cu(L)(Mephen)](ClO4)2 (3).
Green crystals. Yield 77 mg (63%). Anal. calcd for C35H29Cl2CuN7O8: C, 51.89; H, 3.61; N, 12.10; found: C, 51.81; H, 3.66; N, 11.04. ESI-MS (CH3CN): m/z = 709.1300 [3 − ClO4]+ (calcd: 709.1266). FT-IR (solid, ν, cm−1): 741 (aromatic C–H out of plane bending), 1080, 620 (Cl–O), 1481 (CC stretching), 1594 (CN stretching of the imidazole ring). UV-vis (Tris-HCl buffer) λmax (nm) [ε (L mol−1 cm−1)]: 275 [36211], 297 [28261], 317 [23315], 368 [14011].
Crystallography
Single crystals of complexes 1–3 were grown from a 3:1 acetonitrile and methanol solvent mixture. The crystallographic data for all the complexes were recorded using a Bruker D8 Venture instrument with a Photon III mixed mode detector using graphite-monochromatic Mo-Kα (λ = 0.71073 Å) radiation. Suitable single crystals for all the complexes were mounted using a CryoLoop (Hampton Research Corp.) by employing a layer of mineral oil. The crystal structures of the complexes were solved by direct methods using SHELXT90 and all the non-hydrogen atoms were refined on F2 by employing full-matrix least-squares methods using SHELXL.91 Hydrogen atoms were placed at the appropriate geometrically perfect positions and all the non-hydrogen atoms along with all other atoms were refined anisotropically. The crystallographic software program Olex2 was used to perform all the calculations.92 The Mercury 2022.3.0 software program was used to present the ORTEP representation crystal structures of all the complexes.93 The synthesised complexes 1–3 in the present manuscript belong to CCDC numbers 2407715–2407717.†
DNA binding studies
Absorption studies.
The stock solution of ss-DNA was prepared by using Tris–HCl buffer (5 mM, pH 7.4). To confirm the absence of proteins in the DNA preparation, the absorbance ratio (A260/A280) of ss-DNA was maintained in the range of 1.8–1.9. The ss-DNA concentration was determined spectrophotometrically at room temperature using the molar absorption coefficient value ε = 6600 L mol−1 cm−1.94 For the preparation of a stock solution of 1–3, an appropriate amount was taken and dissolved in buffer solution. The interaction between the complexes and ss-DNA was examined by recording electronic spectra at room temperature in 5 mM Tris–HCl buffer. Absorption titration spectra of all complexes were recorded keeping the metal complex concentration fixed (both 50 μM) with the successive addition of DNA concentration ranging from 10 μM to 80 μM. To eliminate the absorbance caused by DNA itself, the same quantity of ss-DNA was added to the sample as well as in the reference solution. The resulting mixture of the metal complex and DNA was equilibrated for 10 min before recording the data.
Viscosity measurement.
The viscosity measurement studies were carried out within a water bath, using an Ostwald viscometer of capacity 10 mL, maintaining a steady temperature of 298 K. The DNA concentration was kept fixed at 200 μM and the concentration of the metal complexes varied from 10 μM to 80 μM. The flow time for the DNA solutions was recorded using a digital stopwatch and for better accuracy, each sample was measured 3 times, and an average flow time was taken for the calculation of relative specific viscosity (η).
Ethidium bromide displacement assay.
The ethidium bromide (EB) displacement assay was carried out to illustrate the mode of binding between the potent compounds and DNA. For the blank measurement of the fluorescence intensity, 5 mM Tris–HCl buffer (pH = 7.4) was taken. The fluorescence quenching experiment was started after taking the initial intensity value of EB bound ss-DNA. For this experiment, the ratio of [DNA] to [EB] was maintained as [DNA]/[EB] = 10 for 10 h at room temperature. The interaction binding between the DNA and added metal complex was determined by the extent of fluorescence quenching upon addition of the complex.95 The fluorescence intensity was found to decrease with an increase in the concentration of the complexes. Thus, it can be said that the complexes displaced EB from the ss-DNA and the complexes themselves were bound to the DNA base pairs. This EB displacement assay using fluorescence quenching experiments was performed by keeping [DNA]/[EB] (100 μM/10 μM) fixed and gradually increasing the concentration of complexes 1 (1–61 μM), 2 (1–52 μM) and 3 (1–24 μM). The fluorescence intensity was recorded with an excitation wavelength of 500 nm and the emission limit was fixed at 515 nm to 780 nm.
CD spectra analysis.
CD spectra were recorded using Tris–HCl (pH 7.4) buffer at room temperature (298 K). CD spectra of ss-DNA (100 μM) were recorded in the absence and as well as the presence of all the three complexes (50 μM) in the wavelength range of 190 nm to 500 nm with a bandwidth of 10 nm and a time per point of 1 s. The scanning rate was kept at 200 nm s−1.
Molecular docking.
The molecular docking studies were performed using AutoDock 4.274 to investigate the interactions between 1–3 and DNA. For this, the structure of DNA containing the 12-mer oligonucleotide d(CpGpCpGpApApTpTpCpGpCpG) was downloaded from the Protein Data Bank (PDB: 1BNA), water molecules were removed, and polar hydrogen bonds were added using AutoDock MGL tools. Then, the DNA was converted to PDBQT format by adding formal Kohlmann charges and exported for docking. The DNA molecule was centered and a grid box was generated with dimensions of 66 × 66 × 124 Å, which covered the entire DNA molecule. The CIF file of the crystals was converted to PDB using Open Babel software. The complexes were auto-optimized for Gasteiger charges and rotatable bonds and exported in PDBQT format using AutoDock MGL tools. Docking was run using the Lamarckian genetic algorithm. Interaction analysis was done using BIOVIA Discovery Studio software.
DNA cleavage.
DNA cleavage was analysed by incubating plasmid DNA (pBS) with 1–3 (200 μM) in the presence of the reaction buffer (50 mM Tris-HCl, pH 7.4) at 37 °C for 6 h. DMSO was used for the control reaction. A number of metal complexes have been shown to exhibit DNA photocleaving ability. Therefore, each compound was incubated with DNA in the presence and absence of light. The product of the reaction was analysed by agarose gel (0.7%) electrophoresis and observed using ethidium bromide staining. The images were captured using Geldoc G-BOX (Syngene).
Cytotoxicity studies.
The cytotoxicity of complexes 1–3 was evaluated against MDA-MB-231 and U2OS cell lines through the XTT assay.96 Initially, the cells were cultured in T-25 culture flasks in complete DMEM (with 10% FBS and 1% antibiotic solution) at 37 °C under 5% CO2. The culture conditions were the same for all the successive cellular studies. The cells were harvested using trypsin-EDTA and seeded at a density of 104 cells per well in a 96-well tissue culture plate. After overnight incubation, the cells were supplemented with the complexes at different concentrations (0.25, 0.5, 1, 2.5, and 5 μM), from a stock solution of 2.5 mM (in DMSO). It was ensured that the final concentration of DMSO in the culture medium did not exceed 0.2%, to minimize the toxic effects arising from the solvent. The cells were incubated with the complexes for 48 h. Later, the cell viability was measured using XTT reagent. The protocol was followed using the manufacturer's instructions. The absorbance values were normalized with respect to that of control untreated cells.
Furthermore, the live/dead cell population was determined using co-staining with Calcein-AM/Hoechst. For this, 6 × 104 cells were seeded onto a 24-well culture plate and cultured in DMEM without phenol red, along with the complexes (1 and 5 μM) for 48 h. Then the cell population was co-stained with Calcein-AM and Hoechst at the recommended dosage and time by the supplier. The live cell population was differentially stained with Calcein-AM and the total cell nuclei were stained with Hoechst. The images were captured using a fluorescence microscope (Life Technologies-EVOS FL AUTO).
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support received from the Science and Engineering Research Board (SERB), Department of Science and Technology, India: project no. CRG/2023/003259; the Ministry of Education (MoE) (fellowship to I. R.), the University Grants Commission (fellowship to S. M.), SERB project no. CRG/2023/003259 (fellowship to K. K.), the Institute Post-Doctoral Fellowship (IPDF) IIT Hyderabad (fellowship to S. D.) and the Indian Institute of Technology Hyderabad is gratefully acknowledged.
References
- E. Wong and C. M. Giandomenico, Chem. Rev., 1999, 99, 2451–2466 CrossRef CAS PubMed.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
- T. Boulikas and M. Vougiouka, Oncol. Rep., 2003, 10, 1663–1682 CAS.
- A. M. Angeles-Boza, P. M. Bradley, P. K.-L. Fu, S. E. Wicke, J. Bacsa, K. R. Dunbar and C. Turro, Inorg. Chem., 2004, 43, 8510–8519 CrossRef CAS PubMed.
- M. Frezza, S. Hindo, D. Chen, A. Davenport, S. Schmitt, D. Tomco and Q. P. Dou, Curr. Pharm. Des., 2010, 16, 1813–1825 CrossRef CAS PubMed.
- K. S. Karumban, A. Muley, R. Raut, P. Gupta, B. Giri, S. Kumbhakar, A. Misra and S. Maji, Dalton Trans., 2022, 51, 7084–7099 RSC.
- B. Rosenberg, L. Vancamp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385–386 CrossRef CAS PubMed.
- P. C. A. Bruijnincx and P. J. Sadler, Curr. Opin. Chem. Biol., 2008, 12, 197–206 CrossRef CAS PubMed.
- R. Loganathan, S. Ramakrishnan, E. Suresh, A. Riyasdeen, M. A. Akbarsha and M. Palaniandavar, Inorg. Chem., 2012, 51, 5512–5532 CrossRef CAS PubMed.
- F. H. Haghighi, H. Hadadzadeh, F. Darabi, Z. Jannesari, M. Ebrahimi, T. Khayamian, M. Salimi and H. A. Rudbari, Polyhedron, 2013, 65, 16–30 CrossRef CAS.
- E. R. Milaeva, D. B. Shpakovsky, Y. A. Gracheva, S. I. Orlova, V. V. Maduar, B. N. Tarasevich, N. N. Meleshonkova, L. G. Dubova and E. F. Shevtsova, Dalton Trans., 2013, 42, 6817–6828 RSC.
- V. A. Kawade, A. A. Kumbhar, A. S. Kumbhar, C. Näther, A. Erxleben, U. B. Sonawane and R. R. Joshi, Dalton Trans., 2010, 40, 639–650 RSC.
- B. Giri, T. Saini, S. Kumbhakar, K. S. Karumban, A. Muley, A. Misra and S. Maji, Dalton Trans., 2020, 49, 10772–10785 RSC.
- A. Saha, I. Mondal, A. Kumari, A. Kumar Sonkar, R. Mishra, R. Kulshreshtha and A. K. Patra, Dalton Trans., 2024, 53, 1551–1567 RSC.
- J. Sayala, E. Srivastava, P. Kumar, N. Shukla, A. Kumar and A. K. Patra, Dalton Trans., 2024, 53, 4580–4597 RSC.
- C. Marzano, M. Pellei, F. Tisato and C. Santini, Anticancer Agents Med. Chem., 2009, 9, 185–211 CrossRef CAS PubMed.
- S. Adsule, V. Barve, D. Chen, F. Ahmed, Q. P. Dou, S. Padhye and F. H. Sarkar, J. Med. Chem., 2006, 49, 7242–7246 CrossRef CAS PubMed.
- N. Berthet, V. Martel-Frachet, F. Michel, C. Philouze, S. Hamman, X. Ronot and F. Thomas, Dalton Trans., 2013, 42, 8468–8483 RSC.
- C. M. Chang, V. J. Klema, B. J. Johnson, M. Mure, J. P. Klinman and C. M. Wilmot, Biochemistry, 2010, 49, 2540–2550 CrossRef CAS PubMed.
- E. D. Getzoff, J. A. Tainer, P. K. Weiner, P. A. Kollman, J. S. Richardson and D. C. Richardson, Nature, 1983, 306, 287–290 CrossRef CAS PubMed.
- Saswati, A. Chakraborty, S. P. Dash, A. K. Panda, R. Acharyya, A. Biswas, S. Mukhopadhyay, S. K. Bhutia, A. Crochet, Y. P. Patil, M. Nethaji and R. Dinda, Dalton Trans., 2015, 44, 6140–6157 RSC.
- C. Santini, M. Pellei, V. Gandin, M. Porchia, F. Tisato and C. Marzano, Chem. Rev., 2014, 114, 815–862 CrossRef CAS PubMed.
- P. Sureshbabu, A. A. J. S. Tjakraatmadja, C. Hanmandlu, K. Elavarasan, N. Kulak and S. Sabiah, RSC Adv., 2015, 5, 22405–22418 RSC.
- J. Hormann, C. Perera, N. Deibel, D. Lentz, B. Sarkar and N. Kulak, Dalton Trans., 2013, 42, 4357–4360 RSC.
- Y. Lu, Chem. – Eur. J., 2002, 8, 4588–4596 CrossRef CAS PubMed.
- E. C. Long and J. K. Barton, Acc. Chem. Res., 1990, 23, 271–273 CrossRef CAS.
- A. A. Adeleke, S. J. Zamisa, Md. S. Islam, K. Olofinsan, V. F. Salau, C. Mocktar and B. Omondi, Molecules, 2021, 26, 1205 CrossRef CAS PubMed.
- H. Xu, K.-C. Zheng, Y. Chen, Y.-Z. Li, L.-J. Lin, H. Li, P.-X. Zhang and L.-N. Ji, Dalton Trans., 2003, 2260–2268 RSC.
- K. F. Konidaris, R. Papi, E. Katsoulakou, C. P. Raptopoulou, D. A. Kyriakidis and E. Manessi-Zoupa, Bioinorg. Chem. Appl., 2010, 2010, 803424 CrossRef PubMed.
- J.-B. Ye, G. Ren, W.-Y. Li, G.-Y. Zhong, M. Zhang, J.-B. Yuan and T. Lu, Molecules, 2019, 24, 4591 CrossRef CAS PubMed.
- P. Kumar, S. Gorai, M. K. Santra, B. Mondal and D. Manna, Dalton Trans., 2012, 41, 7573–7581 RSC.
- M. A. Hoque, A. D. Chowdhury, S. Maji, J. Benet-Buchholz, M. Z. Ertem, C. Gimbert-Suriñach, G. K. Lahiri and A. Llobet, Inorg. Chem., 2021, 60, 5791–5803 CrossRef CAS PubMed.
- X.-Q. Zhou, Q. Sun, L. Jiang, S.-T. Li, W. Gu, J.-L. Tian, X. Liu and S.-P. Yan, Dalton Trans., 2015, 44, 9516–9527 RSC.
- F. Arjmand and M. Aziz, Eur. J. Med. Chem., 2009, 44, 834–844 CrossRef CAS PubMed.
- R. Ruiz, B. García, J. Garcia-Tojal, N. Busto, S. Ibeas, J. M. Leal, C. Martins, J. Gaspar, J. Borrás, R. Gil-García and M. González-Álvarez, J. Biol. Inorg. Chem., 2010, 15, 515–532 CrossRef CAS PubMed.
- C. Lüdtke, S. Sobottka, J. Heinrich, P. Liebing, S. Wedepohl, B. Sarkar and N. Kulak, Chem. – Eur. J., 2021, 27, 3273–3277 CrossRef PubMed.
- S. P. Foxon, T. Phillips, M. R. Gill, M. Towrie, A. W. Parker, M. Webb and J. A. Thomas, Angew. Chem., Int. Ed., 2007, 46, 3686–3688 CrossRef CAS PubMed.
- C. Metcalfe and J. A. Thomas, Chem. Soc. Rev., 2003, 32, 215–224 RSC.
- D. R. Graham, L. E. Marshall, K. A. Reich and D. S. Sigman, J. Am. Chem. Soc., 1980, 102, 5419–5421 CrossRef CAS.
- B. M. Zeglis, V. C. Pierre and J. K. Barton, Chem. Commun., 2007, 4565–4579 RSC.
- P. K. Mudi, R. K. Mahato, M. Joshi, M. Shit, A. R. Choudhury, H. S. Das and B. Biswas, Appl. Organomet. Chem., 2021, 35, e6211 CrossRef CAS.
- D.-S. Son, E.-S. Lee and S. E. Adunyah, Immune Netw., 2020, 20, 1–20 CrossRef PubMed.
- A. Kamal, P. K. Pogula, M. N. A. Khan, B. N. Seshadri and K. Sreekanth, Med. Chem., 2013, 9, 651–659 CrossRef CAS PubMed.
- B. Pathare and T. Bansode, Results Chem., 2021, 3, 100200 CrossRef CAS.
- A. Noor, N. G. Qazi, H. Nadeem, A. Khan, R. Z. Paracha, F. Ali and A. Saeed, Chem. Cent. J., 2017, 11, 85 CrossRef PubMed.
- O. Ebenezer, F. Oyetunde-Joshua, O. D. Omotoso and M. Shapi, Results Chem., 2023, 5, 100925 CrossRef CAS.
- P. Ghosh and A. Mandal, Tetrahedron Lett., 2012, 53, 6483–6488 CrossRef CAS.
- T. Liu, L. Chen and D. Chao, Dalton Trans., 2022, 51, 4052–4057 RSC.
- D. Maheshwaran, S. Priyanga and R. Mayilmurugan, Dalton Trans., 2017, 46, 11408–11417 RSC.
- H. Wu, X. Huang, J. Yuan, F. Kou, F. Jia, B. Liu and K. Wang, Eur. J. Med. Chem., 2010, 45, 5324–5330 CrossRef CAS PubMed.
- J. V. Geden, A. K. Pancholi and M. Shipman, J. Org. Chem., 2013, 78, 4158–4164 CrossRef CAS PubMed.
- E. Turgut, O. Gungor, H. Kirpik, A. Kose, S. A. Gungor and M. Kose, Appl. Organomet. Chem., 2021, 35, e6323 CrossRef CAS.
- M. Kose, J. Coord. Chem., 2014, 67, 2377–2392 Search PubMed.
- G. Muller, J.-C. G. Bünzli, K. J. Schenk, C. Piguet and G. Hopfgartner, Inorg. Chem., 2001, 40, 2642–2651 CrossRef CAS PubMed.
- X. Xu, Z. Xi, W. Chen and D. Wang, J. Coord. Chem., 2007, 60, 2297–2308 CrossRef CAS.
- K. Paliwal, P. Haldar, P. K. S. Antharjanam and M. Kumar, ACS Omega, 2022, 7, 21961–21977 CrossRef CAS PubMed.
- M. Sunita, B. Anupama, B. Ushaiah and C. Gyana Kumari, Arabian J. Chem., 2017, 10, S3367–S3374 Search PubMed.
- E. A. Ermakova, Y. A. Golubeva, K. S. Smirnova, L. S. Klyushova, A. S. Berezin, L. N. Fetisov, A. E. Svyatogorova, N. O. Andros, A. A. Zubenko and E. V. Lider, New J. Chem., 2023, 47, 9472–9482 RSC.
- S. Kathiresan, S. Mugesh, M. Murugan, F. Ahamed and J. Annaraj, RSC Adv., 2015, 6, 1810–1825 Search PubMed.
- K. Tenza, M. J. Hanton and A. M. Z. Slawin, Organometallic, 2009, 28, 4852–4867 CrossRef CAS.
- P. R. Reddy and P. Manjula, C&B, 2007, 4, 468–480 Search PubMed.
- J. L. Viegas and S. N. Dhuri, J. Mol. Struct., 2023, 1288, 135719 CrossRef CAS.
- M. M. Damaceno, C. B. P. Ligiero, J. D. P. Serna, O. C. Alves, L. A. S. Costa, D. S. Cukierman and N. A. Rey, J. Mol. Struct., 2023, 1288, 135827 Search PubMed.
- J. A. R. Hartman, A. L. Kammier, R. J. Spracklin, W. H. Pearson, M. Y. Combariza and R. W. Vachet, Inorg. Chim. Acta, 2004, 357, 1141–1151 CrossRef CAS.
- C. M. Buta, M. M. Radu, A. Mischie, C. M. Zălaru, G. Ioniţă and M. Ferbinteanu, Polyhedron, 2019, 170, 771–782 CrossRef CAS.
- I. Roy, A. Muley, S. Mathur, A. Verma, M. K. Kumawat and S. Maji, New J. Chem., 2024, 48, 11647–11661 Search PubMed.
- S. Mathur, K. S. Karumban, A. Muley, N. Tuti, U. P. Shaji, I. Roy, A. Verma, M. K. Kumawat, A. Roy and S. Maji, Dalton Trans., 2024, 53, 1163–1177 RSC.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885–3896 RSC.
- S. Mukherjee, S. Roy, S. Mukherjee and B. Biswas, J. Mol. Struct., 2020, 1217, 128348 CrossRef CAS.
- H. H. Hammud, G. J. McManus, M. J. Zaworotko, R. N. Tabesh, H. I. M. Ibrahim, K. Ayub and R. Ludwig, Monatsh. Chem., 2021, 152, 323–336 CrossRef CAS.
- K. Jana, T. Maity, T. S. Mahapatra, P. K. Das Mohapatra, S. C. Debnath, S. Das, M. Hossain and B. C. Samanta, Transition Met. Chem., 2017, 42, 69–78 CrossRef CAS.
- B. A. Johnson, S. Maji, H. Agarwala, T. A. White, E. Mijangos and S. Ott, Angew. Chem., Int. Ed., 2016, 55, 1825–1829 Search PubMed.
- K. S. Karumban, R. Raut, P. Gupta, A. Muley, B. Giri, S. Kumbhakar, A. Misra and S. Maji, J. Inorg. Biochem., 2022, 233, 111866 Search PubMed.
- L. Cerasino, M. J. Hannon and E. Sletten, Inorg. Chem., 2007, 46, 6245–6251 CrossRef PubMed.
- B.-L. Fei, W.-S. Xu, W.-L. Gao, J. Zhang, Y. Zhao, J.-Y. Long, C. E. Anson and A. K. Powell, J. Photochem. Photobiol., B, 2015, 142, 77–85 Search PubMed.
- B. Anupama, M. Sunita, D. Shiva Leela, B. Ushaiah and C. Gyana Kumari, J. Fluoresc., 2014, 24, 1067–1076 CrossRef CAS PubMed.
- D. Mahendiran, P. Gurumoorthy, K. Gunasekaran, R. S. Kumar and A. Kalilur Rahiman, New J. Chem., 2015, 39, 7895–7911 RSC.
- D. Mahendiran, R. Senthil Kumar, V. Viswanathan, D. Velmurugan and A. Kalilur Rahiman, Dalton Trans., 2016, 45, 7794–7814 RSC.
- A. K. Patra, T. Bhowmick, S. Ramakumar, M. Nethaji and A. R. Chakravarty, Dalton Trans., 2008, 6966–6976 RSC.
- S. Tabassum, A. Asim, F. Arjmand, M. Afzal and V. Bagchi, Eur. J. Med. Chem., 2012, 58, 308–316 CrossRef CAS PubMed.
- M. R. Eftink and C. A. Ghiron, Anal. Biochem., 1981, 114, 199–227 CrossRef CAS PubMed.
- I. A. H. A. Sherwani, A. Köse, Ö. Güngör, H. Kırpık, S. A. Güngör and M. Köse, Appl. Organomet. Chem., 2022, 36, e6585 CrossRef.
- H. Wu, H. Wang, X. Wang, G. Pan, F. Shi, Y. Zhang, Y. Bai and J. Kong, New J. Chem., 2014, 38, 1052–1061 RSC.
- V. Singh, K. Sharma, B. Shankar, S. K. Awasthi and R. D. Gupta, New J. Chem., 2016, 40, 5906–5913 RSC.
- A. Muley, S. Kumbhakar, R. Raut, S. Mathur, I. Roy, T. Saini, A. Misra and S. Maji, Dalton Trans., 2024, 53, 11697–11712 RSC.
- N. Shahabadi, M. Falsafi and N. H. Moghadam, J. Photochem. Photobiol., B, 2013, 122, 45–51 CrossRef CAS PubMed.
- S. Zhang, Y. Zhu, C. Tu, H. Wei, Z. Yang, L. Lin, J. Ding, J. Zhang and Z. Guo, J. Inorg. Biochem., 2004, 98, 2099–2106 CrossRef CAS PubMed.
- J. Heinrich, K. Bossak-Ahmad, M. Riisom, H. H. Haeri, T. R. Steel, V. Hergl, A. Langhans, C. Schattschneider, J. Barrera, S. M. F. Jamieson, M. Stein, D. Hinderberger, C. G. Hartinger, W. Bal and N. Kulak, Chem. – Eur. J., 2021, 27, 18093–18102 CrossRef CAS PubMed.
- A. Santoro, G. Walke, B. Vileno, P. P. Kulkarni, L. Raibaut and P. Faller, Chem. Commun., 2018, 54, 11945–11948 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- S. Mathur, K. S. Karumban, A. Muley, N. Tuti, U. P. Shaji, I. Roy, A. Verma, M. K. Kumawat, A. Roy and S. Maji, Dalton Trans., 2024, 53, 1163–1177 RSC.
- M. Sirajuddin, S. Ali and A. Badshah, J. Photochem. Photobiol., B, 2013, 124, 1–19 CrossRef CAS PubMed.
- D. Akula, T. R. O'Connor and R. Anindya, Biochem. Biophys. Res. Commun., 2021, 534, 114–120 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Shobhit
Mathur
a,
Shobhit
Mathur



 vs. SCE in CH3CN/0.1 M TBAP.
vs. SCE in CH3CN/0.1 M TBAP.












