
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePlatinum(II) and ruthenium(II) coordination complexes equipped with an anchoring site for binding the protein kinase enzyme pockets: synthesis, molecular docking and biological assays†‡
Matthieu
Scarpi-Luttenauer
a,
Katia
Galentino
b,
Christophe
Orvain§
 c,
Audrey
Fluck
a,
Marco
Cecchini
b,
Georg
Mellitzer§
c,
Christian
Gaiddon¶
*c and
Pierre
Mobian
*a
c,
Audrey
Fluck
a,
Marco
Cecchini
b,
Georg
Mellitzer§
c,
Christian
Gaiddon¶
*c and
Pierre
Mobian
*a
aUniversité de Strasbourg, CNRS, UMR 7140, F-67000 Strasbourg, France. E-mail: mobian@unistra.fr
bUniversité de Strasbourg, CNRS, UMR 7177, F-67000 Strasbourg, France
cInserm U1113 IRFAC, Team STREINTH, Strasbourg, France
First published on 21st January 2025
Abstract
To mimic the structural aspects of staurosporine, a potent but unspecific kinase inhibitor, several coordination compounds based on two readily available diimine ligands containing hydrogen bonding donor/acceptor sites (NH–CO fragment) have been designed and synthesized. These complexes are constructed around Ru(II) and Pt(II) metal centers. A total of 9 compounds, named Ru(1)–(5) and Pt(1)–(4), were obtained through straightforward synthetic approaches. The cytotoxicity of the compounds was evaluated on AGS gastric cancer cells (GC) through standard MTT assays. All ruthenium and platinum complexes with low toxicity, i.e.Ru(3), Ru(5), Pt(3) and Pt(4), were docked in the ATP binding pocket of two protein kinases (S6K1 and MST2). The docking scores highlighted a preferred affinity of Ru(5) for the MST2 binding pocket, whereas the platinum compounds are predicted to bind stronger to the S6K1 binding site. Inhibitory activity of the metal complexes on the MST2 and S6K1 signaling pathways was evaluated by analyzing via western blot experiments the phosphorylation state of YAP, a downstream component of the Hippo pathway and the protein expression of S6 and its phosphorylated analogue p-S6. A clear difference of behavior between the Pt(II) and the Ru(II) complexes depending on the type of kinase was observed.
Introduction
Protein kinases are an essential class of enzymes which play a fundamental role in human cellular signaling pathways.1 These enzymes are crucial for the regulation of cellular metabolism, including cell growth and survival. They catalyze the addition of a phosphate group originated from the hydrolysis of ATP onto a target substrate, such as a protein, a sugar or a lipid. In some cases, overexpression or mutations in protein kinases can contribute to tumor growth.2–7 Hence, protein kinase inhibitors represent a promising class of anticancer drugs allowing the selective inhibition of a protein kinase involved in cancer development or spreading. In addition, these inhibitors are essential tools to understand the role of kinases in signaling pathways and physio-pathological processes. Currently, 80 different protein kinase inhibitors are already used for the clinical treatment of different cancer types.8 Most of them are organic compounds built around a long organic scaffold containing hydrogen-bond donor and acceptor sites for efficient binding of the drug into the ATP binding pocket of the enzyme. Thus, the interaction between the drug and the enzyme hinders the interaction of the latter with ATP. Since the structure of the ATP binding pocket of all protein kinases is highly conserved, it is a challenging task to engineer selective inhibitors.Staurosporine is a natural alkaloid acting as an extremely potent protein kinase inhibitor (Fig. 1).9–11 The structure of this molecule is composed of a flat indolocarbazole fragment responsible for the binding to the active site of the enzyme through hydrogen bonding interactions and a voluminous glycosidic unit. While being extremely potent, staurosporine is not selective, which prevented its use in clinical studies.
 | ||
| Fig. 1 (a) Staurosporine; (b) an example of an organometallic Ru(II)-complex mimicking the structural aspects of staurosporine developed by Meggers et al. as inhibitor of the MST1 kinase. | ||
Despite this lack of selectivity, the structure of this efficient kinase inhibitor has inspired the design of non-natural kinase inhibitors. In this context, Meggers et al. have developed metal complexes mimicking the structural aspects of staurosporine to prepare kinase inhibitors incorporating a metallic scaffold. Starting from a sophisticated pyridocarbazole-based bidentate nitrogen ligand, they were able to vary the coordination sphere around the metal for fine-tuning selectivity of the inhibitors, yielding a library of different metal complexes, exhibiting nM-range, in vitro IC50 towards different protein kinases with a very high selectivity.12–15 In particular, Meggers et al. identified inhibitors for MST1 (see Fig. 1b). MST1 is a kinase involved in the Hippo pathway that plays a critical role in mecanotransduction (regulation of cell and organ growth via physical force sensing) and proliferation/survival via the transcription co-factors YAP and TAZ.16,17 Alterations of this pathway leads to cancer progression by hyperactivation of YAP and/or TAZ. Inspired by the Meggers lab strategy, we have tried to develop novels and more selective inhibitors for MST1/2 kinases.
Recently, we have reported octahedral titanium(IV) complexes constructed around a TiO4N2 core where the nitrogen bidentate ligands linked to the metal center bear hydrogen bond donor and acceptor recognition units (NH–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). Docking experiments with these titanium complexes on the Hippo protein kinase MST2 showed that the titanium complexes were too voluminous to fit the binding site of the kinase. Nevertheless, the free ligands, compounds L1–2 (Fig. 2), fitted well the enzymatic pocket of this kinase.18 The phenanthroline ligand L1 functionalized with a pyridinone unit at the position 5, and the diimine ligand L2, on which two pyridine moieties are fused together by an amide group are easily accessible in a few synthetic steps. Hence, these two ligands represent good candidates to mimic the flat indolocarbazole fragment found in staurosporine.18,19 Thus, L1–2 appeared as two interesting building starting units to develop metal complexes having an optimized shape regarding the enzyme binding pocket of MST2. In addition, compounds L1–2 showed good solubility in organic media allowing their facile use for the preparation of various transition metal complexes.
O). Docking experiments with these titanium complexes on the Hippo protein kinase MST2 showed that the titanium complexes were too voluminous to fit the binding site of the kinase. Nevertheless, the free ligands, compounds L1–2 (Fig. 2), fitted well the enzymatic pocket of this kinase.18 The phenanthroline ligand L1 functionalized with a pyridinone unit at the position 5, and the diimine ligand L2, on which two pyridine moieties are fused together by an amide group are easily accessible in a few synthetic steps. Hence, these two ligands represent good candidates to mimic the flat indolocarbazole fragment found in staurosporine.18,19 Thus, L1–2 appeared as two interesting building starting units to develop metal complexes having an optimized shape regarding the enzyme binding pocket of MST2. In addition, compounds L1–2 showed good solubility in organic media allowing their facile use for the preparation of various transition metal complexes.
 | ||
| Fig. 2 Ligands L1 and L2. | ||
Here, we report the synthesis of transition metal complexes bearing the L1–2 diimine ligands. For this purpose, Ru(II) and Pt(II) were selected to generate species offering distinct geometries. Moreover, they are thoroughly investigated in therapeutic chemistry devoted to anticancer treatments. Development of novels Ru(II) and Pt(II) complexes may provide solutions to avoid the existing resistance mechanisms.20–25 Ru(II) typically displays octahedral or pseudo-octahedral coordination spheres, whereas 4-fold coordinated Pt(II) compounds adopt a square planar geometry. In addition, the carbon ruthenium bond in Ru(II) complexes confers original biological activities linked to the redox behavior of these organometallic species.26
Hence, we have prepared several Ru(II) or Pt(II) complexes based on ligand L1 or L2 displaying a diversity of shapes and geometries aiming at characterizing their potential to inhibit enzymatic activities, in particular MST1/2. The cytotoxicity of these complexes was then assayed, followed by structure–activity relationship evaluation via molecular docking in a protein kinase of the Hippo pathway, MST2. To compare the selectivity, we extended the structure–activity studies to S6K1, which is a key regulatory enzyme of the activity of the ribosomal protein S6. The docking predictions were validated experimentally with the quantification of the phosphorylation of YAP, the downstream component of the Hippo pathway,27 and S6, the phosphorylation target of S6K1.28
Results and discussion
Synthesis of Pt(1)–(4) and Ru(1)–(5) complexes and hydrolytic stability
Aiming to develop new coordination compounds mimicking the structural aspects of the staurosporine, we have prepared a series of ruthenium(II) and platinum(II) complexes incorporating the ligands L1 or L2. The synthetized complexes are depicted in Fig. 3. | ||
| Fig. 3 Pt(II) and Ru(II) complexes studied in this work. | ||
A total of 5 ruthenium(II) complexes were synthetized from either ligand L1 or L2. The synthetic protocols allowing the preparation of these complexes are given in Fig. 4. The synthesis of octahedral ruthenium(II) polypyridyl complexes Ru(1) and Ru(2) was first carried out starting from known dichloro precursor [Ru(bpy)2Cl2] (1) which reacted with ligands L1 and L2 respectively in an ethylene glycol/water 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 mixture for 6 hours as detailed in Fig. 4.29 After precipitation with the addition of a saturated aqueous KPF6 solution, purification by column chromatography of the crude products over silica gel resulted in the isolation of Ru(1) and Ru(2).
:10 mixture for 6 hours as detailed in Fig. 4.29 After precipitation with the addition of a saturated aqueous KPF6 solution, purification by column chromatography of the crude products over silica gel resulted in the isolation of Ru(1) and Ru(2).
 | ||
| Fig. 4 Synthetic pathways leading of complexes Ru(1)–(5). | ||
The two complexes were characterized by 1H and 13C NMR spectroscopy, ESI-MS and elemental analysis. Single crystals of Ru(2) were obtained when diethyl ether vapors were allowed to diffuse in an acetone solution of the complex. The solid-state structure of Ru(2) resolved by single crystal X-Ray diffraction is shown in Fig. 5.
 | ||
| Fig. 5 (a) Ball and stick representation of the solid-state structure of Ru(2), obtained from single-crystal X-ray crystallography (CCDC 2392833‡). (b) Ru(2) packing in the crystal. The hydrogen bonds are represented in blue. Ruthenium atoms are depicted in turquoise, oxygen atoms in red, carbon atoms in grey, nitrogen atoms in blue and hydrogen atoms in light grey. The hydrogen atoms, the PF6− anions and the acetone crystallization solvent molecules are omitted for clarity. | ||
The compound Ru(2) displays a slightly distorted octahedral geometry. The N–Ru–N angles between the nitrogen atoms in trans are of 172.3°, 173.6° and 173.7°. The N–Ru bond lengths between the bipyridine ligands and the metal are comprised between 2.061 and 2.071 Å, whereas the N–Ru bond lengths between the ligand L2 and the metal are slightly longer (2.076 Å and 2.078 Å). The N–Ru–N angles displayed by the two bipyridine ligands are similar (78.6° and 78.7°), whereas the one displayed by L2 is slightly wider (79.7°). Surprisingly, the solid-state arrangement of Ru(2) shows that the amide fragment of L2 is involved in a hydrogen bond interaction with the oxygen atom of an acetone molecule, which is characterized by a ![[N with combining low line]](https://www.rsc.org/images/entities/char_004e_0332.gif) –H⋯
–H⋯![[O with combining low line]](https://www.rsc.org/images/entities/char_004f_0332.gif) length of 2.840 Å (Fig. 5b) which contrasts with the dimerization observed for the previous reported Ti(IV)-complex bearing the same nitrogen ligand L2.16
length of 2.840 Å (Fig. 5b) which contrasts with the dimerization observed for the previous reported Ti(IV)-complex bearing the same nitrogen ligand L2.16
Cycloruthenated complexes Ru(3) and Ru(4) were prepared by adapting procedures described by Pfeffer et al.30,31Ru(3) was prepared by reacting precursor 2 with ligand L2 at 45 °C for 3 days. After purification by column chromatography over silica gel, the desired compound was obtained in a moderate yield of 20%. The 1H NMR spectrum of Ru(3) is complex due to the dissymmetry of the ligand L2. The 1H NMR spectrum of Ru(3) displays 20 signals which were attributed according 2D 1H and 13C NMR analysis to the two different isomers shown Fig. 6a. In particular, the 2D ROESY spectrum proved the presence of these two isomers since two demonstrative correlations are observed as shown in Fig. 6b. The first correlation was found between the H9 proton belonging to L2 and the benzylamine Ha proton of one stereoisomer. The second correlation was assigned to the L2 H2′ proton and the benzylamine Ha′ proton of the second stereoisomer. This unique spatial proximity between the benzylamine proton and one L2 proton (H2′ or H9) per isomer was verified by constructing molecular models of each isomer and analyzing the crystal structure of an analogous complex where the ligand L2 is replaced by a 2,2′-bipyridine ligand.32 DOSY NMR analysis confirmed that the two species were stereoisomers as only one diffusion value of D = 7.00 × 10−10 m2 s−1 was obtained. Additionally, integration of the signals on the 1H NMR spectrum revealed that the two stereoisomers are found in a 1:1 ratio in solution.
 | ||
| Fig. 6 (a) Representation of the two stereoisomers corresponding to Ru(3). The arrows indicate the correlation in space between the two protons seen on the ROESY spectrum, for each isomer. (b) Region of the 2D ROESY spectra of Ru(3) highlighting two strong correlations; one between the protons Ha′ and H2′ and a second between Ha and H2. | ||
Ru(4) was synthesized by reacting L2 with precursor 333 in methanol at reflux overnight. The desired complex was isolated by column chromatography over alumina. NMR analysis of the compound showed a similar situation as described for Ru(3), as the spectrum showed more signals related to the presence of a single stereoisomer in solution as two sets of signals are observed by 1H NMR. Furthermore, the 2D COSY spectrum allowed the identification of the signals for L2 for each stereoisomer. Additionally, Ru(3) and Ru(4) were also characterized by ESI-MS and elemental analysis.
Piano-stool complex, Ru(5), was synthesized according to a procedure described by Queyriaux et al.34 The one-step reaction of commercially available dimeric species 4 with L2 in methanol at reflux during 4 h allowed after precipitation with saturated aqueous KPF6 the isolation of the desired complex which was characterized by 1H and 13C NMR spectroscopy, ESI-MS spectrometry and elemental analysis.
The synthetic pathways leading to the formation of square-planar Pt(II) complexes containing either ligand L1 or L2 are described in Fig. 7. Robust square-planar PtN4 complexes incorporating diimine ligands for DNA intercalation have recently been described by Aldrich-Wright et al.35–37 This scaffold appeared of great interest for the synthesis of protein kinase inhibitors constructed around a square-planar Pt(II) core incorporating ligands L1 and L2 (Fig. 7).
 | ||
| Fig. 7 Synthetic pathways leading to complexes Pt(1)–(4). | ||
Pt(1) and Pt(2) were prepared by reacting known 1,2-diaminocyclohexane (1,2-DACH) precursor [Pt(1,2-DACH)Cl2] (5) with ligands L1 and L2 respectively in water at reflux.35 The desired complexes were then isolated after purification over reverse phase resin.
Pt(3) was synthesized in a one-pot two steps procedure.36 First, the chloro ligands were eliminated with the addition of AgNO3 to the kiteplatin precursor 6 in DMF. In a second step, the AgCl precipitate was centrifuged, and L2 was added to the supernatant. The resulting mixture was stirred first at room temperature for 16 hours and next at 50 °C for one day. The desired complex Pt(3) was isolated after purification over reverse phase resin.
Finally, complex Pt(4a) incorporating (S,S)-diphenylethylenediamine (9) was prepared in a two-step procedure. First, the dichloro precursor 8 was obtained from the substitution of the DMSO ligands of [Pt(DMSO)2Cl2] (7) by L2.37 The complex was then refluxed without further purification with the corresponding nitrogenated ligands to yield the targeted complex. The solid-state structure of Pt(4) obtained after single crystal XRD analysis is shown in Fig. 8a. The crystalline packing shows that the complex units are stacked together through the diimine ligand (Fig. 8b). The mean distance between the closest carbon atoms is of 3.424 ± 0.049 Å. Surprisingly, in the crystal, the amide fragments of ligand L2 are deprotonated, which leads to hydrogen bonding interactions (N(L2)⋯HN(9)) between this deprotonated nitrogen and NH2 proton of the diamino ligand 9 (N⋯HN length = 3.222 Å).
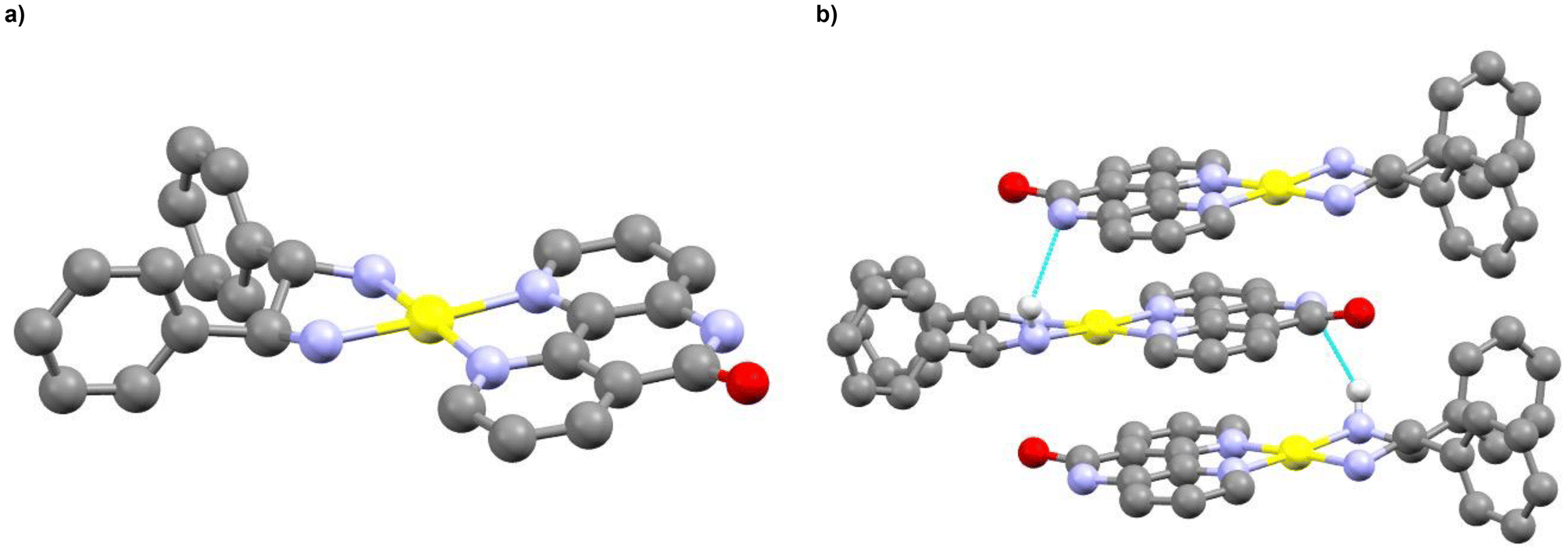 | ||
| Fig. 8 (a) Ball and stick representation of the solid-state structure of Pt(4), obtained from single-crystal X-ray crystallography (CCDC 2392834‡). (b) Pt(4) packing in the crystal. The hydrogen bonds are represented in blue. Platinum atoms are depicted in yellow, oxygen atoms in red, carbon atoms in grey, nitrogen atoms in blue and hydrogen atoms in light grey. The hydrogen atoms (excepted the H atoms involved in the hydrogen bonding), the PF6− anion and the DMF crystallization solvent molecules are omitted for clarity. | ||
In order to evaluate the hydrolytic stability of the Ru(1)–(5) and Pt(1)–(4) complexes, the evolution of UV-visible absorption spectra of these complexes were recorded in Phosphate Buffer Saline (PBS). Stock solutions of the complexes in DMSO (c = 50 mM) were diluted in an aqueous PBS buffer solution to reach a concentration of c = 50 μM and a spectrum was recorded every hour. A slight drop in intensity was observed for the analyzed complexes, which was explained by the medium evaporation (see the ESI‡).
Overall, no changes were noticed, meaning that the complexes are stable in a biologically relevant condition. The only exception concerned complex Ru(3). The recorded spectra are displayed Fig. 9. Over time, a hypsochromic shift of the visible absorption band was observed with an isosbestic point at λ = 455 nm. This observation indicated that the initial complex Ru(3) was slowly converted into a new species. Since Pfeffer et al. have previously demonstrated that the acetonitrile ligands within analogous complexes can be substituted by water molecules, it was assumed that the observed transformation for Ru(3) was linked to ligands exchange.38 To confirm this hypothesis, the solution of Ru(3) in PBS obtained after 24 hours was analyzed by HPLC-MS.
 | ||
| Fig. 9 UV-visible hydrolytic stability assay for Ru(3) in PBS buffer (c = 50 μM). A spectrum was recorded every hour over 24 h. | ||
Table 1 summarizes the major signals observed, and the m/z values associated with them. An adduct corresponding to the product on which one acetonitrile ligand has been exchanged by a DMSO molecule can be found at 3.87 min, meaning that the acetonitrile ligands have already been exchanged in the stock solution in DMSO. Finally, the evolution of a Ru(3) solution in CD2Cl2 in which D2O (50 μL) were added was monitored by 1H NMR analysis. Fig. 10 highlights the presence of an increasing amount of free acetonitrile at 1.97 ppm over time (24 h). After 48 h, the presence of free CH3CN is even accentuated. Altogether, these analysis indicated that the acetonitrile ligands are labile ligands within Ru(3) and can be substituted by coordinating species such as DMSO or water.
 | ||
| Fig. 10 Close-up on the acetonitrile CH3 protons from the 1H NMR spectrum of Ru(3) in a mixture CD2Cl2:D2O 8:1. The coordinated acetonitrile signals are labelled by a red dot and the free acetonitrile signals are signaled by a green star. The spectra were recorded: (a) before the addition of D2O, (b) just after the addition of D2O, (c) 24 h after the addition of D2O and (d) 48 h after the addition of D2O. | ||
| Retention time (min) | m/z (z) | Adduct |
|---|---|---|
| 3.16 | 217.0335 (2) | [C20H20N4ORu + H]2+ |
| 258.0600 (2) | [C24H25N6ORu + H]2+ | |
| 3.87 | 552.1039 (1) | [C24H28N5O2RuS]+ |
| 4.15 | 515.1166 (1) | [C24H26N6ORu]+ |
Overall, several Ru(II) and Pt(II) complexes bearing either ligand L1 or L2 were synthetized using straightforward approaches. These species were fully characterized and their stability in a medium compatible with the biological assays evaluated. We noticed that Ru(3) was unstable under these conditions due to the substitution of labile acetonitrile ligands. Nevertheless, after this ligand exchange, the rest of the Ru(3) backbone remained unaltered.
In vitro cytotoxic activity of the Ru(II) and Pt(II) complexes
Having in hands this series of metal complexes designed for kinase inhibition, the impact of the Ru(II) and Pt(II) complexes on cell survival was evaluated. For instance, inhibition of MST1/2 should favors YAP/TAZ function, leading to increase survival and proliferation. Thus, the cytotoxicity of the compounds was evaluated through standard MTT assays. We choose the AGS GC cells as GC is a particularly aggressive cancer with limited therapeutic options that has been shown to have deregulation in the YAP/TAZ pathway.39 The Ru(II) and Pt(II) complexes and oxaliplatin (oxa), reference chemotherapy for the treatment of gastric cancer patients,40 were incubated for 48 h with AGS cells. Cell viability was then determined by addition of MTT and absorption measurements and IC50 values were calculated (Table 2).| IC50 (μM) | |
|---|---|
| Ru(1) | 104 ± 23 |
| Ru(2) | 164 ± 27 |
| Ru(3) | >200 |
| Ru(4) | 13 ± 2.0 |
| Ru(5) | >200 |
| Pt(1) | 16 ± 3.5 |
| Pt(2) | 5.5 ± 2.6 |
| Pt(3) | >200 |
| Pt(4) | >200 |
| Oxa | 1.7 ± 0.8 |
Ru(1) to Ru(3) and Ru(5) are considered with a very low or moderate toxicity since their IC50 values are over 100 μM. Despite being really close to Ru(1) from a structural view point, complex Ru(4) exhibited a much lower IC50 (13 ± 2.0 μM). The cytotoxic properties of Ru(4) analogues were studied by Pfeffer et al.41 They reported that the cytotoxicity of these cyclometallated Ru-complexes arise from the redox properties of such complexes containing a Ru–C linkage. Hence, the toxicity displayed by Ru(4) is assumed to be a consequence of the Ru–C bond within this species.
Concerning the Pt(II) species, only Pt(1) and Pt(2) showed cytotoxicity when incubated with AGS cells (IC50 = 16 ± 3.5 and 5.5 ± 2.6 μM respectively). The biological activity of similar analogues was first reported by Aldrich-Wright et al.42 The cytotoxic effect of these complexes were attributed to their capacity to intercalate double-stranded DNA, due to their planar structure. Therefore, it is not surprising to see that Pt(3) and Pt(4), bearing more bulky 1,4-DACH or diphenylethylenediamine ligands, respectively, were non-toxic.
Docking studies of the Ru(II) and Pt(II) complexes in MST2 and S6K1
Based on the cytotoxicity of the synthesized complexes, only the compounds with low toxicity (IC50 > 200 μM) were selected for docking studies on two protein kinases, i.e. S6K1 (4rlp.pdb) and MST2 (5dh3.pdb). These proteins have two distinct cellular functions. MST2 is part of the Hippo signaling pathway, which contributes to the regulation of organ growth and apoptosis activation,16 and is a relevant target for studying the effects of downregulation of the Hippo pathway on cellular growth.17 S6K1 is a protein kinase that controls the activity of the 40S ribosomal protein S6 promoting protein synthesis.43 Both the 40S ribosomal protein S6 and S6K1 have been implicated in morbidity such as diabetes, aging, obesity or even cancer.44–46Eight docking experiments were carried out using the software PLANTS with the CHEMPLP scoring function,47 which includes energy contributions accounting for hydrogen bonding, protein–ligand shape complementarity, and intra-ligand torsion and clash penalties (see Methods for details). To validate the docking protocol, we re-docked the two ligands co-crystallized with the MST2 and S6K1 kinases and compared results with the X-ray structures. The RMSD between the predicted lowest-energy binding pose and the experimentally determined X-ray binding pose was 0.5 Å for XMU-MP-1 in MST217 and 0.89 Å for FL772 in S6K1.48 The re-docked compounds show significant overlap with the crystal coordinates establishing the same interactions with the protein (see ESI, Fig. S31‡).
All non-toxic ruthenium and platinum complexes, i.e.Ru(3), Ru(5), Pt(3) and Pt(4), were docked in the ATP binding pocket of the two protein kinases. The docking results indicate a different propensity of the ligands to bind S6K1 or MST2 and predict that the platinum compounds Pt(3) and Pt(4) have a better docking score for S6K1, whereas the ruthenium compound Ru(5) interacts stronger with MST2 (Table 3). Concerning Ru(3), the docking calculations predict an unfavorable interaction at MST2 likely due to steric clashes with the protein, as indicated by the positive score in Table 3.
| Compound | 4rlp (S6K1) | 5dh3 (MST2) | Δ(S6K1-MST2) |
|---|---|---|---|
| Ru(3) | −61.3 | +13.7 | −75 |
| Ru(3)–H2O | — | −57.8 | — |
| Ru(3)–DMSO | — | +12.7 | — |
| Ru(5) | −68.9 | −74.6 | +5.7 |
| Pt(3) | −79.5 | −67.5 | −11.9 |
| Pt(4) | −91.5 | −68.6 | −22.8 |
Fig. 11 shows an overview of the key protein–ligand interactions that each compound establishes in the binding site of the kinase for which it has the best docking score. Analysis of the protein–ligand interactions using the software PLIP49 provides insights into the binding selectivity. The comparison with the interaction patterns established by the reference compounds in the X-ray structures (i.e. FL772 in S6K1 and XMU-MP-1 in MST2) reveals that the ruthenium compounds fit reasonably well the ATP-binding pocket at S6K1 but they are unable to establish the critical H-bonds that steer protein–ligand recognition likely due to their shape or bulkiness of the substituents (Table S1‡). By contrast, the platinum compounds show a suboptimal fit of the ATP-binding pocket in MST2 albeit establishing similar hydrogen bonding interactions as the reference compound (Table S2‡). We conclude that the binding selectivity predicted by docking originates from an interplay of shape complementarity and directional interactions steering the recognition of rigid and bulky ligands at the nucleotide binding site of the two kinases.
 | ||
| Fig. 11 Protein–ligand interaction profiles of Ru(3) and Ru(5) compounds in MST2 (PDB code: 5dh3) and of Pt(3) and Pt(4) compounds in S6K1 (PDB code: 4rlp). Hydrogen bonding and hydrophobic interactions are highlighted in yellow and green, respectively. | ||
Inhibitory activity of the metal complexes in MST2 and S6K1
Having docked both Ru(II) and Pt(II) complexes in MST2 and S6K1, we next aimed to evaluate their inhibitory activity in vitro using the GC cell line AGS. GC is a public health problem due to high aggressiveness with a 5-year survival rate of less than 25% and a median survival of about 11 months.50 The reasons for this high mortality are multiple, including the variability in response to first treatment at equivalent tumor stage and that about 75% of cancers become resistant. Patient management is still based on surgery combined with oxaliplatin-based chemotherapies (oxaliplatin + 5-fluoro-uracyl, 5-FU), and only in a few cases anti-HER2 or anti-PD1 targeted therapies are applied.51,52 Consequently, there is urgent need for the development of new therapeutic solution to improve the management of gastric cancer patients. In this respect, components of the Hippo pathway, like MST2, or the S6K1 signaling pathway have been reported to be frequently altered in GC and are subjects to the development of targeted therapies.53–55 To investigate the inhibitory effect of our compounds on the protein kinase MST2, we chose to analyze the phosphorylation state of YAP, a downstream in the Hippo pathway. For this AGS cell were treated for 24 h with the indicated compounds at 2 or 25 μM concentrations, and the expression of YAP and phosphorylated YAP was analyzed by western blot (Fig. 12a). | ||
| Fig. 12 (a) AGS cells were treated for 24 h with 2 or 25 μM of the indicated drugs and the expression of YAP and YAP p-S127 was analyzed by western blot. The expression of actin served as a loading control. Western blot analysis of YAP. The same blot was first probed with an antibody detecting phosphorylated YAP (mouse monoclonal antibody) and then re-probed with an antibody detecting total YAP (rabbit-monoclonal antibody), ensuring accurate comparison while eliminating variability due to loading and migration differences. (b) Relative pYAP and YAP expression levels were normalized to the corresponding actin expression levels and are represented as ratio pYAP/YAP. The results are given ± SEM from three distinct experiments. Statistical difference was established by a two-value's t test. *p < 0.05. | ||
The results show that among the four compounds tested only Ru(3) at 25 μM significantly diminished the phosphorylation of YAP, suggesting inhibition of the upstream protein kinase MST2. By contrast, Ru(5) and Pt(3) showed only a very slight effect, which did not reach statistical significance, and Pt(4) showed the opposite effect (Fig. 12). This result is in contradiction with the positive docking score obtained from the modeling of Ru(3) in the binding pocket of MST2. However, since the acetonitrile ligands responsible for steric clashing with the protein are effectively exchanged with less bulky substituents such as water in a biologically relevant medium (see above), it is possible that the product of such ligand exchange is a good fit in the protein binding pocket. This was confirmed by new docking experiments where the acetonitrile ligands were exchanged from water or DMSO ligands. These modeled complexes are named Ru(3)–H2O and Ru(3)–DMSO and the docking scores obtained for these two complexes with the binding pocket of MST2 are found in Table 3. The negative docking score for Ru(3)–H2O (−57.8) demonstrates that this aqua adduct well fits the binding pocket of the enzyme and has significant affinity. By contrast, the DMSO adduct (Ru-DMSO) has an unfavorable docking score (+12.7) similar as the Ru(3) species. Clearly, this investigation demonstrates that the efficacy of Ru(3) is enhanced by water and not DMSO substitution. Additionally, the binding mode of Ru–H2O predicted by docking resembles that of staurosporine with two H-bonding interactions with the same protein loop.
Next, we evaluated the inhibitory activity of the four complexes on S6K1 by analyzing the protein expression of S6 and its phosphorylated analogue p-S6. For this purpose, AGS cells were treated for 24 h with 2 or 25 μM of Ru(3), Ru(5), Pt(3) and Pt(4) and the expression of S6 and p-S6 analyzed by western blot (Fig. 13a).
 | ||
| Fig. 13 (a) AGS cells were treated for 24 h with 2 or 25 μM of the indicated drugs and the expression of S6 and p-S6 was analyzed by western blot. The expression of actine served as a loading control. The nitrocellulose blot was successively probed and revealed for S6 p-S235/236, S6 and actin (b) relative pS6 and S6 expression levels where normalized with the corresponding actin expression level and are represented as ratio pS6/S6. The results are given ± SEM from three distinct experiments. Statistical difference was established by a two-value's t test. *p < 0.05, **p < 0.01, ***p < 0.005, ****p < 0.001. | ||
The results show that, in contrast to the YAP phosphorylation, all compounds significantly decrease the phosphorylation of S6, suggesting S6K1 inhibition at 25 μM concentration (Fig. 13). Interestingly and in contrast to the previous results on YAP phosphorylation, Pt(3) showed to be much more potent than Ru(3) to inhibit S6K1. Overall, the Pt(II) complexes seemed to be more efficient in inhibiting the activity of the protein kinase S6K1 than the Ru(II) complexes. These observations are consistent with conclusions drawn from the docking experiments, as the Pt(II) complexes have lower docking scores than the Ru(II) complexes in the S6K1 binding pocket (Table 3). Since the S6K1 binding pocket is bigger than the MST2 binding pocket, these data confirm that bulkier complexes such as the Pt(II) complexes, in comparison to the Ru(II) complexes, will have stronger interactions with larger protein binding pockets.
Conclusion
Five Ru(II) complexes, i.e.Ru(1)–(5), and four Pt(II) complexes, i.e.Pt(1)–(4), were generated from ligands L1 and L2 that incorporate a NH–CO fragment. All of these species are stable in aqueous media with the exception of Ru(3) where the labile acetonitrile ligands are substituted. Complexes Ru(1), Ru(2), Ru(4), Pt(1) and Pt(2) are toxic molecules for AGS cells, whereas compounds Ru(3), Ru(5), Pt(3) and Pt(4) displayed IC50 > 200 μM. Hence, the non-toxic species (Ru(3), Ru(5), Pt(3) and Pt(4)) were selected for docking studies in two protein kinases, i.e. MST2 and S6K1. From this investigation, it appeared that all complexes could fit the ATP-binding pocket of these enzymes. Nevertheless, docking results predicted that platinum compounds have stronger affinities for the ATP-binding pocket of S6K1. Finally, inhibitory activities in vitro using the gastric cancer cell line AGS were evaluated. It was found that Ru(3) at 25 μM significantly diminished the phosphorylation of YAP, suggesting that it inhibited the activity of the upstream protein kinase MST2. Albeit in contradiction with the docking results, which indicated that this compound would not fit the ATP-binding pocket of MST2, this observation along with the detected lability of the acetonitrile ligands within Ru(3) in water suggest that the active form of this compound may be water or DMSO coordinated. Concerning the inhibition of S6K1, the two platinum species showed the best activities, consistent with the docking predictions.Overall, albeit the structure of the ATP binding pockets of protein kinases are similar, this investigation has highlighted a preferred interaction for a particular type of kinase depending on the architecture and the substitution of the metal complex inhibitor. These results open to the development of selective metal complex kinase inhibitors based on the diimine ligand L2.
Experimental part
Materials and methods
Antibodies list: ESI.‡Synthetic procedures
All reagents and products were purchased from Sigma Aldrich, Alfa Aesar or TCI and used as received. Ultrapure water used was purified by a Milli-Q UV purification system (Sartorius Stedim Biotech SA). Gibco® Versene solution, Gibco® Trypsin/EDTA solution, Gibco® MEM Non-Essential Amino Acids solution (NEAA), 10% SDS solution, PenicillinStreptomycin (10000 U mL−1), Dulbecco's Phosphate-Buffered Saline (10×). Hyclone™ RPMI 1640, DMEM medium and fetal bovine serum (FBS) were purchased from Thermo Fisher Scientific Inc. Bio-rad Protein Assay Dye Reagent Concentrate, 40% acrylamide/bis solution, 10× Tris/glycine buffer, TEMED, 4× Laemmli Sample Buffer, nitrocellulose membrane, 0.2 μm and 0.45 μm were purchased from Bio-rad Laboratories. Complete™, mini Protease Inhibitor Cocktail Tablets. Luminata™ Classico and Luminata™ Crescendo Western HRP Substrate were purchased from Merck Millipore Corporation.
Bruker Avance-500 and Avance-600 spectrometers were used for solution NMR analyses performed at 25 °C. Deuterated solvents for 1H NMR analysis were dried over molecular sieves before use. 1H NMR spectra were recorded at 500.13 MHz and referenced to SiMe4. 13C{1H} NMR spectra (broadband decoupled) were recorded at 125.77 MHz and referenced to SiMe4. Chemical shifts are reported in ppm and coupling constants in Hz; the latter are proton–proton coupling constants. Multiplicity: s = singlet, d = doublet, t = apparent triplet, m = multiplet. The 13C{1H} signals are singlets. DOSY measurements were performed at 600.13 MHz with a 5 mm 1H/X z-gradient BBI probe and applying a PFGSTE pulse sequence using bipolar gradients. Electrospray analyses were performed on a MicroTOF (Bruker) apparatus equipped with an electrospray (ES) source. The elemental analyses were performed using a Flash 2000 apparatus (Thermo Fisher Scientific) for C, H, and N elements. UV-vis liquid and solid spectra were recorded with a PerkinElmer Lambda650s spectrometer. The X-ray diffraction data were collected by two different means. Mean 1: the data were collected at and 173 K on a Bruker SMART CCD diffractometer with MoKα radiation (λ = 0.71073 Å). The diffraction data were corrected for absorption using the SADABS program.56 The structures were solved using SHELXS9757 and refined by full matrix least-squares on F2 using SHELXL-201458 in the anisotropic approximation for all non-hydrogen atoms. The hydrogen atoms were introduced at calculated positions and not refined (riding model). z with a 5 mm 1H/X z-gradient BBI probe and applying a PFGSTE pulse sequence using bipolar gradients. Mean 2: the X-ray diffraction data were collected on a Bruker PHOTON-III DUO Kappa CPAD diffractometer equipped with an Oxford Cryosystem liquid N2 device, using Mo-Kα radiation (λ = 0.71073 Å). The crystal-detector distance was 37 mm. The cell parameters were determined (APEX3 software)59 from reflections taken from two sets of 6 frames, each at 10 s exposure. The structure was solved by Direct methods using the program SHELXT-2014. The refinement and all further calculations were carried out using SHELXL-2014. The H-atoms were included in calculated positions and treated as riding atoms using SHELXL default parameters. The non-H atoms were refined anisotropically, using weighted full-matrix least-squares on F2. A semi-empirical absorption correction was applied using SADABS in APEX3. The residual electron density was assigned to one molecule of the chloroform solvent. Absorbance and fluorescence on 96-well plates were measured using Tristar2 Multimode Reader LB942 from Berthold Technologies.
Cell culture and cell survival
AGS (CRL-1739™), and KATOIII (HTB-103TM) cells were obtained from ATCC. 0.5 × 104 cells were seeded per well in 96-well microplates (Falcon Multiwell), 24 h prior to any treatment, drugs were added in fresh medium for 48 h. Then the medium was replaced by fresh medium supplemented with 0.5 mg mL−1 MTT (Sigma) for 1 h. Subsequently, the medium was aspirated, and the purple formazan crystals were dissolved in DMSO (100 μL). Absorbance was measurements at 590 nm with the LB942 Tristar2 Multimode Reader (Berthold Technologies). The IC50 and IC75 was calculated with Prism 9.5.0 using non-linear regression. The experiments were performed in 4 replicates for each drug concentration and were carried out at least three times independently.Western blot protocol
AGS cells were grown at 500000 cells per well (2 mL) on Cellstar® 6-well plates (Greiner Bio-One) for 24 h or 17 h cells were lysed with NP40 Lysis Buffer (125 mM TrisHCl pH 7.5, NaCl 150 mM, NP40 0.5%, 10% Glycerol). A total of 50 μg of proteins were resolved by 10% or 15% SDS-PAGE (depending on protein molecular weight) according to standard methods. Proteins were visualized with selected antibodies (see ESI Methods‡) and enhanced chemiluminescence using the Luminata™ Classico and Luminata™ Crescendo western blotting Substrate Millipore reagent, and according to the manufacturer instructions. To evaluate the phosphorylation of YAP at S127 and S6 at S235/236, the same blot was first probed with an antibody detecting the phosphorylated protein and then re-probed with an antibody detecting the total protein, ensuring accurate comparison while eliminating variability due to loading and migration differences. For instance, for YAP, the blot was probed first with the mouse monoclonal antibody directed against the phosphorylated protein (YAP, anti-rabbit, S127-D9W2I Cell Signaling; in 1/1000 in TBS 1×, Tween 20 0.1%, BSA 1%) overnight, then it was washed (TBS 1×, Tween 20 0.1%, BSA 1%) 3 times 5 min and probed with the anti-mouse secondary antibody (sc-101190 Santa Cruz, 1/5000 in TBS 1×, Tween 20 0.1%, BSA 1%) before revelation with ECL. The same blot was thoroughly washed (TBS 1×, Tween 20 0.1%, BSA 1%) and then probed for the whole protein with an antibody that have been produced in rabbit to avoid any cross-reaction with the mouse phospho-antibody previously used. The use of two antibodies produced in different species avoid the cross-reaction and the necessity of stripping that may lead to a protein loss on the blot. The ECL signals were acquired on a Pxi Imager (Syngene®). Expression intensities of the protein of interest were measured using ImageJ software and normalized to their respective loading control (Actine). Finally, the protein of interest to loading control ratios were further normalized by setting the value of this ratio to 1 in the negative control.
Molecular docking
The initial coordinates of the MST2 protein were taken from the crystal structure of MST2 solved in complex with the inhibitor 4-[(5,10-dimethyl-6-oxo-6,10-dihydro-5H-pyrimido[5,4-b]thieno[3,2-e][1,4]diazepin-2-yl)amino]benzenesulfonamide, a.k.a. XMU-MP-1 or 5BS (PDB code: 5DH3). The initial coordinates of the S6K1 protein were taken from the crystal structure of S6K1 solved in complex with the ruthenium complex FL772 (PDB code: 4RLP). Starting from 2D chemical structures, PrepFlow provides 3D molecular coordinates, after tautomer, stereoisomer and conformer enumeration at a given pH, here 7.0. All docking experiments were performed using the program PLANTS in combination with the scoring function CHEMPLP, which includes energy contributions accounting for hydrogen bonding, protein–ligand shape complementarity, and intra-ligand torsion and clash penalties. The searching space for docking was defined based on the center of mass of the co-crystallized ligand and a sphere radius of 8.0 Å. For each compound, 10 binding poses were generated. The best binding mode according to the docking score was selected and analyzed as the most representative binding pose.Ru(1)
In a two-necked round-bottom flask, a mixture of ethylene glycol and water (9:1, 3 mL) was degassed for 15 min by Ar bubbling. 1 (50 mg, 103 μmol) and L1 (28 mg, 103 μmol) were added and the mixture was stirred at 120 °C for 6 h with light excluded. The orange mixture was cooled to r.t. and water (4.5 mL) was added. An aqueous saturated solution of KPF6 was added drop by drop until no precipitate formed anymore. The orange solid was filtered, washed with cold water and diethyl ether. The crude product was purified by column chromatography (SiO2, MeCN:water:KNO3, 100:10:1). The fractions containing product were combined and the solvent was removed in vacuo. The residue was dissolved in MeCN, the excess KNO3 was filtered off and an aqueous saturated solution of KPF6 was added. The solution was concentrated until a precipitate formed. The precipitate was filtered, washed with cold water and diethyl ether to isolate Ru(1) (56 mg, 56%). 1H NMR (CD3CN, 500 MHz): δ (ppm) = 8.58–8.49 (m, 7H), 8.15 (s, 1H), 8.12–8.08 (m, 4H), 8.02–7.98 (m, 2H), 7.86–7.84 (m, 2H), 7.75–7.65 (m, 3H), 7.59–7.58 (m, 2H), 7.55–7.53 (m, 1H), 7.47–7.44 (m, 2H), 6.61 (d, 1H). 13C NMR (126 MHz, CD2Cl2): δ = 162.45, 157.80, 157.76, 157.57, 157.56, 152.95, 152.92, 152.56, 152.53, 152.48, 152.38, 148.68, 147.62, 143.04, 138.41, 138.40, 138.31, 138.29, 137.08, 136.81, 136.26, 135.64, 131.00, 130.93, 128.10, 128.05, 127.97, 127.93, 126.92, 126.42, 124.83, 124.80, 124.75, 121.20 ppm. MS (ESI) calcd for C37H27N7ORu2+ 343.57; found 343.57. Anal. calcd for C37H27F12N7OP2Ru·H2O: C, 44.68; H, 2.94, N, 9.86; found C, 44.17; H, 2.93; N, 9.65.
Ru(2)
In a two-necked round-bottom flask, a mixture of ethylene glycol and water (9:1, 3 mL) was degassed for 15 min by Ar bubbling. 1 (50 mg, 103 μmol) and L2 (20 mg, 103 μmol) were added and the mixture was stirred at 120 °C for 6 h with light excluded. The orange mixture was cooled to r.t. and water (4.5 mL) was added. An aqueous saturated solution of KPF6 was added drop by drop until no precipitate formed anymore. The orange solid was filtered, washed with cold water and diethyl ether. The crude product was purified by column chromatography (SiO2, MeCN:water:KNO3 100:10:1). The fractions containing product were combined and the solvent was removed in vacuo. The residue was dissolved in MeCN, the excess KNO3 was filtered off and an aqueous saturated solution of KPF6 was added. The solution was concentrated until a precipitate formed. The precipitate was filtered, washed with cold water and diethyl ether to isolate Ru(2) (51 mg, 81%). Crystals were obtained from acetone/Et2O. T = 296(2) K; monoclinic; space group P21/c; a = 13.4931(5) Å, b = 13.8886(4) Å, c = 19.9737(8) Å; β = 94.6980(10); V = 3730.5(3) Å3; Z = 4; Dcalcd = 1.707 g cm−3; reflections collected: 35303; Rint = 0.0671; R1(F) (I > 2σ(I)) = 0.0637, wR2(F2) (all data) = 0.1935; GOF(F2) = 1049. CCDC: 2392833. 1H NMR (CD3CN, 500 MHz): δ (ppm) = 8.68 (dd, 1H, 3J = 8.1 Hz, 4J = 1.2 Hz, H4), 8.52–8.48 (m, 4H, H2 + Hbpy), 8.10–8.03 (m, 5H), 7.95 (dd, 1H, 3J = 8.3 Hz, 4J = 1.2 Hz), 7.80–7.77 (m, 2H), 7.74–7.71 (m, 2H), 7.55–7.50 (m, 2H), 7.44–7.41 (m, 2H), 7.37–7.33 (m, 2H). 13C NMR (126 MHz, CD3CN): δ (ppm) = 160.58, 158.20, 157.87, 157.85, 156.98, 155.16, 153.31, 153.26, 153.08, 152.96, 147.63, 139.27, 138.93, 138.90, 137.58, 136.99, 129.55, 128.55, 128.50, 128.49, 128.47, 128.24, 127.36, 125.25, 125.19, 125.15, 124.84. MS (ESI) calcd for C31H23N7ORu2+ 305.55; found 305.55. Anal. calcd for C31H23F12N7OP2Ti: C, 41.34 H, 2.57, N, 10.89; found C, 40.28; H, 2.72; N, 10.25.
Ru(3)
In a two-necked round-bottom flask, MeCN (5 mL) was degassed by argon bubbling for 15 min. 2 (50 mg, 100 μmol) and L2 (20 mg, 100 μmol) were added and the mixture was stirred at 30 °C for 48 h, then 40 °C for 24 h and finally 45 °C for 48 h. The mixture was filtered, and the purple solution was purified by column chromatography (SiO2, MeCN:water:KNO3 100:3:1). The red band was recovered, the fractions were combined, and the solvent was removed in vacuo. The residue was redissolved in a minimum of MeCN, and an aqueous saturated KPF6 was added (2 mL). MeCN was removed to obtain a dark precipitate which was filtered and washed with ether to isolate Ru(3) (12 mg, 18%). 1H NMR (CDCl3, 500 MHz): δ (ppm) = 11.33–11.24 (mbr, 2H, N–H), 9.79 (d, 1H, 3J = 5 Hz, H2), 9.27 (d, 1H, 3J = 4.7 Hz, H2′), 8.87 (d, 1H, 3J = 5.4 Hz, H9′), 8.83 (d, 1H, 3J = 8 Hz, H4), 8.62 (d, 1H, 3J = 7.9 Hz, H7′), 8.40 (d, 1H, 3J = 5.3 Hz, H9), 8.13 (dd, 1H, 3J1 = 7.8 Hz, 3J2 = 5.3 Hz H3), 8.05 (d, 1H, 3J1 = 8.2 Hz, H4′), 7.96 (dd, 1H, 3J1 = 8.1 Hz, 3J2 = 4.9 Hz H3′), 7.86–7.81 (m, 3H, H7,a,a′), 7.46 (dd, 1H, 3J1 = 7.7 Hz, 3J2 = 5.7 Hz H8′), 7.35 (dd, 1H, 3J1 = 8.2 Hz, 3J2 = 5.4 Hz H8), 7.26–7.21 (m, 2H, Hb,b′), 7.09–7.07 (m, 2H, Hd,d′), 6.99–6.94 (m, 2H, Hc,c′), 3.94 (m, 2H, Hf,f′), 3.34 (d, 1H, 3J = 5 Hz, Hg), 3.31 (d, 1H, 3J = 5 Hz, Hg′), 2.55 (m, 6H, MeCN), 2.31 (s, 6H, Hh,h′), 2.11 (s, 3H, MeCN), 2.10 (s, 3H, MeCN), 1.42 (s, 3H, Hi), 1.40 (s, 3H, Hi′). 13C NMR (CDCl3, 126 MHz): δ (ppm) = 161.83, 161.28, 157.51, 156.64, 156.11, 152.38, 148.95, 147.68, 146.91, 141.01, 137.41, 136.85, 135.13, 135.12, 134.62, 133.71, 128.87, 128.08, 127.54, 126.63, 125.21, 125.10, 124.63, 123.54, 122.56, 120.73, 120.60, 73.30, 52.85, 52.78, 51.41, 51.09, 4.83, 4.21, 4.20. MS (ESI) calcd for C24H25N6ORu+ 515.11, found 515.11. Anal. calcd for C24H25F6N6OPRu·2 CH2Cl2: C, 37.65; H, 3.52; N, 10.13; found C, 35.94; H, 3.50; N, 10.17.
Ru(4)
In a two-necked round-bottom flask, MeOH (10 mL) was degassed by argon bubbling for 15 min. 3 (95 mg, 149 μmol) and L2 (29 mg, 149 μmol) were added and the mixture was stirred at 70 °C overnight. The mixture was cooled down at room temperature and the solvent was removed in vacuo. The residue was purified by column chromatography (Al2O3, MeCN:water 99:1). The red band was recovered, the fractions were combined, and the solvent was removed in vaccuo to isolate a purple crystalline powder (20 mg, 18%). 1H NMR (CDCl3, 500 MHz): δ (ppm) = 10.07 (sbr, 2H, N–H + N–H′), 8.64 (dd, 1H, 3J = 8.1 Hz, 4J = 1.3 Hz, H57), 8.46 (dd, 3J = 8 Hz, 4J = 1.4 Hz, H57′), 8.32–8.30 (m, 5H), 8.26 (dd, 1H, 3J = 5.6 Hz, 4J = 1.4 Hz, H57′), 8.22 (dd, 3J = 5.3 Hz, 4J = 1.4 Hz, H57), 8.02–7.99 (m, 2H), 7.87–7.78 (m, 13H), 7.75 (dd, 1H, 3J = 5.2 Hz, 4J = 1.1 Hz), 7.70–7.66 (m, 6H), 7.62 (dd, 1H, 3J = 8.4 Hz, 4J = 1 Hz), 7.58–7.55 (m, 4H), 7.48 (dd, 1H, 3J1 = 5.6 Hz, 3J2 = 8.1 Hz, Hphen′), 7.36 (dd, 1H, 3J1 = 5.5 Hz, 3J2 = 8.4 Hz), 7.18–7.14 (m, 2H), 6.92–6.83 (m, 7H), 6.47–6.43 (m, 2H). 13C NMR (CD3CN, 126 MHz): δ (ppm) = 167.67, 160.71, 160.63, 158.50, 157.42, 157.38, 157.33, 155.58, 153.98, 152.97, 151.53, 151.48, 151.15, 151.04, 150.95, 149.50, 144.57, 137.52, 136.80, 136.60, 136.43, 136.39, 136.09, 135.09, 134.68, 134.55, 133.56, 128.99, 128.75, 128.02, 127.49, 126.89, 126.87, 126.86, 126.67, 126.64, 126.61, 126.28, 124.65, 123.57, 123.53, 123.48, 122.89, 122.83, 122.82, 121.45, 119.37, 119.32. MS (ESI) calcd for C32H23N6ORu+ 609.10, found 609.10. Anal. Calcd for C32H23F6N6OPRu·2 CH2Cl2: C, 44.22; H, 2.95, N, 9.10; found C, 43.50; H, 2.93; N, 9.31.
Ru(5)
A solution of 4 (61 mg, 122 μmol) and L2 (50 mg, 254 μmol) was heated to reflux in MeOH (21 mL) for 4 h. Solvent was removed in vacuo and the residue was redissolved in water (10 mL). The unsoluble material was filtered off and a saturated aqueous solution of KPF6 was added until no precipate formed anymore. The precipitate was filtered on a fritted funnel, washed with water (3 × 10 mL), with Et2O (3 × 10 mL) to obtain a yellow powder. 1H NMR (CD3CN, 500 MHz): δ (ppm) = 9.95 (dd, 1H, 3J = 5.4 Hz, 4J = 1.3 Hz, H2), 9.18 (dd, 1H, 3J = 5.3 Hz, 4J = 0.9 Hz, H9), 8.81 (dd, 1H, 3J = 8.1 Hz, 4J = 1.31 Hz, H4), 7.97–7.93 (m, 2H, H3,7), 7.80 (dd, 1H, 3J = 8.1 Hz, 4J = 1.31 Hz, H8), 6.07 (s, 6H, Hbenz). 13C NMR (CD3CN, 126 MHz): δ (ppm) = 159.59, 159.38, 152.83, 150.39, 139.04, 137.12, 136.53, 129.10, 127.87, 126.48, 126.42, 87.16. MS (ESI) calcd for C17H13ClN3ORu+ 411.98, found 411.98. Anal. calcd for C17H13ClF6N3OPRu: C, 36.67; H, 2.35; N, 7.55; found C, 36.51; H, 2.48; N, 7.53.Pt(1)
5 (48 mg, 126 μmol) and L1 (35 mg, 126 μmol) were suspended in distilled water (12 mL) and the mixture was stirred at reflux for three days. The clear yellow solution was concentrated in vacuo until the volume reached around 2 mL. The product was purified on a Porapak Rxn RP 20cc column with water as eluent. The fractions containing product were combined, concentrated in vacuo until the volume reached around 3 mL. Saturated aqueous KPF6 was added to the solution until no precipate formed anymore. The yellow residue was filtered on a fritted funnel, and air-dried. Yield: 20%. 1H NMR (CD3CN, 500 MHz): δ (ppm) = 8.94–8.89 (m, 4H, H2,4,7,9), 8.15 (s, 1H, H6), 8.08 (dd, 1H, 3J1 = 8.3 Hz, 3J2 = 5.4 Hz, H3 or 8), 8.06 (dd, 1H, 3J1 = 8.6 Hz, 3J2 = 5.4 Hz, H3 or 8), 7.63 (dd, 1H, 3J = 9.4 Hz, 4J = 2.7 Hz, Hpyr), 7.58 (d, 1H, 4J = 2.6 Hz, Hpyr), 6.63 (d, 1H, 3J = 9.5 Hz, Hpyr), 5.82 (sbr, 2H, NH2), 5.17 (sbr, 2H, NH2), 2.78 (m, 2H, HDACH), 2.25 (m, 2H, HDACH) 1.71 (m, 2H, HDACH), 1.53 (m, 2H, HDACH), 1.31 (m, 2H, HDACH). 13C NMR (CD3CN, 126 MHz): δ (ppm) = 162.89, 152.63, 152.47, 149.04, 147.85, 143.28, 142.09, 140.64, 137.57, 137.15, 131.68, 128.39, 127.78, 127.39, 121.81, 114.91, 62.88, 62.84, 33.15, 24.87. HRMS (ESI) calcd for C23H24N5OPt2+ 291.0849, found 291.0862.Pt(2)
5 (77 mg, 203 μmol) and L2 (40 mg, 203 μmol) were suspended in distilled water (18 mL) and the mixture was stirred at reflux overnight. The clear yellow solution was concentrated in vacuo until the volume reached around 2 mL. The product was purified on a Porapak Rxn RP 20cc column with water as eluent. The fractions containing product were combined and dried in vacuo to obtain a yellow solid. Yield: 39%. 1H NMR (D2O, 500 MHz): δ (ppm) = 9.01 (d, 1H, 3J = 8.1 Hz, H4), 8.95 (d, 1H, 3J = 5.6 Hz, H2), 8.50 (d, 1H, 3J = 5.3 Hz, H9), 8.20 (d, 1H, 3J = 8.6 Hz, H7), 8.01 (dd,1H, 3J1 = 5.7 Hz, 3J2 = 8.06 Hz, H3), 7.87 (dd, 1H, 3J1 = 5.4 Hz, 3J2 = 8.5 Hz, H8), 2.71–2.69 (m, 2H, HDACH), 2.21 (d, 2H, 3J = 13 Hz, HDACH), 1.66 (d, 2H, 3J = 8.8 Hz, HDACH), 1.47 (d, 2H, 3J = 9.2 Hz, HDACH), 1.29–1.20 (q, 2H, 3J = 13 Hz, HDACH) ppm. 13C NMR (D2O, 125 MHz): δ (ppm) = 161.08, 154.52, 154.50, 145.86, 139.87, 138.88, 135.44, 129.08, 128.56, 127.77, 125.51, 61.67, 61.64, 32.01, 31.97, 23.75 ppm. MS (ESI) calcd for C17H21N5OPt2+ 253.07; found 253.07. Anal. calcd for C17H21Cl2N5OPt·2 H2O: C, 33.29; H, 4.11; N, 11.42; found C, 32.22; H, 4.10; N, 11.05.Pt(3)
6 (60 mg, 158 μmol) was dissolved in DMF (4 mL). AgNO3 (59 mg, 347 μmol) was added and the mixture was stirred for 16 h with light excluded. The mixture was centrifuged for 5 min at 4000 rpm. To the supernatant was added L2 (37 mg, 189 μmol) and the mixture was stirred for 16 h and then for 24 h at 50 °C. The yellow mixture was cooled to room temperature and centrifuged for 5 min at 4000 rpm. The yellow precipitate was washed with CH2Cl2 (3 × 5 mL) and air-dried. The residue was dissolved in water (10 mL), filtered and a saturated aqueous solution of KPF6 was added. The solution was concentrated in vacuo until a precipate appeared. The mixture was allowed to rest O.N. with light excluded. The solid was centrifuged 5 min at 4000 rpm, the solid was washed with a 50:50 mixture of iPrOH:Et2O and air-dried. Yield: 16%. 1H NMR (DMSO-d6, 500 MHz): δ (ppm) = 8.83 (d, 1H, 3J = 8.01 Hz, H4), 8.64 (d, 1H, 3J = 5.2 Hz, H2), 7.99 (dd, 3J1 = 7.8 Hz, 3J2 = 5.8 Hz, H3), 7.93–7.88 (m, 2H, H7/9), 7.69 (dd, 3J1 = 8.6 Hz, 3J2 = 5.2 Hz, H8), 6.52 (s, 4H, NH2), 3.44 (sbr, 2H, CH–NH2), 1.8–1.72 (m, 8H). 13C NMR (DMSO-d6, 126 MHz): δ (ppm) = 167.11, 154.11, 149.51, 140.46, 138.55, 137.86, 135.41, 126.65, 126.28, 125.11, 46.93, 46.83, 21.75, 21.70. MS (ESI) calcd for C17H20N5OPt+ [M − H]+ 505.13, found 505.13.
Pt(4)
8 (50 mg, 108 μmol) and (1S,2S)-9 (46 mg, 216 μmol) were suspended in water (100 mL) and stirred for 24 h at reflux. The mixture was allowed to cool at room temperature and was concentrated in vacuo until 50 mL solvent remained. A saturated aqueous solution of KPF6 was added until no precipate formed anymore. The mixture was filtered on a fritted funnel, washed with water and Et2O to obtain a yellow powder. Yield: 38%. Crystals were obtained from MeCN/Et2O. T = 296(2) K; orthorhombic; space group P212121; a = 6.6613(4) Å, b = 16.7503(10) Å, c = 32.2265(18) Å; α = β = γ = 90°; V = 3595.8(4) Å3; Z = 4; Dcalcd = 1.518 g cm−3; reflections collected: 106407; Rint = 0.0807; R1(F) (I > 2σ(I)) = 0.0556, wR2(F2) (all data) = 0.1337; GOF(F2) = 1.118. CCDC: 2392834.‡1H NMR (CD3CN, 500 MHz): δ (ppm) = 8.75 (d, 1H, 3J = 8 Hz, H4), 8.13 (d, 1H, 3J = 5.2 Hz, H2), 7.73 (d, 1H, 3J = 8.6 Hz, H7), 7.67–7.66 (m, 5H, H9, HPh), 7.39–7.33 (m, 7H, H3, HPh), 7.13 (dd, 1H, 3J1 = 8.4 Hz, 3J2 = 5.2 Hz, H8), 6.43–6.09 (mbr, 4H, NH2), 4.86 (s, 2H, CH–NH2). 13C NMR (CD3CN, 126 MHz): δ (ppm) = 168.89, 152.96, 152.09, 147.05, 142.17, 140.29, 137.70, 136.24, 136.16, 135.23, 130.11, 130.09, 129.74, 129.72, 129.02, 128.95, 127.56, 127.24, 126.30, 66.76, 66.47. [α]D = 5.8° dm−1 cm3 g−1 (c = 0.67 mM, DMSO, 20 °C). MS (ESI) calcd for C25H23N5OPt2+ 302.08, found 302.08. Anal. calcd for C25H23F12N5OP2Pt·2MeCN·C5H12: C, 38.31; H, 3.80; N, 9.48; found C, 39.13; H, 3.41; N, 9.12.
Data availability
The data supporting this article have been included as part of the ESI.‡Crystallographic data for Ru(2) and Pt(4) has been deposited at the CCDC 2392833 and 2392834‡ respectively.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Strasbourg and the CNRS for financial support. This research has been supported by the University of Strasbourg's IdEx program (Idex-CNRS interdisciplinary project 2017). M. Scarpi-Luttenauer thanks the French Ministry of Research for his PhD fellowship. CG, GM and CO are supported by ARC, Ligue contre le Cancer, ITMO cancer, INCa and the ITI Innovec.References
- G. Manning, D. B. Whyte, R. Martinez, T. Hunter and S. Sudarsanam, The Protein Kinase Complement of the Human Genome, Science, 2002, 298(5600), 1912–1934, DOI:10.1126/science.1075762.
- H. J. Su Huang, M. Nagane, C. K. Klingbeil, H. Lin, R. Nishikawa, X. D. Ji, C. M. Huang, G. N. Gill, H. S. Wiley and W. K. Cavenee, The Enhanced Tumorigenic Activity of a Mutant Epidermal Growth Factor Receptor Common in Human Cancers Is Mediated by Threshold Levels of Constitutive Tyrosine Phosphorylation and Unattenuated Signaling, J. Biol. Chem., 1997, 272(5), 2927–2935, DOI:10.1074/jbc.272.5.2927.
- A. J. Wong, S. H. Bigner, D. D. Bigner, K. W. Kinzler, S. R. Hamilton and B. Vogelstein, Increased Expression of the Epidermal Growth Factor Receptor Gene in Malignant Gliomas Is Invariably Associated with Gene Amplification, Proc. Natl. Acad. Sci. U. S. A., 1987, 84(19), 6899–6903, DOI:10.1073/pnas.84.19.6899.
- D. J. Hicklin and L. M. Ellis, Role of the Vascular Endothelial Growth Factor Pathway in Tumor Growth and Angiogenesis, J. Clin. Oncol., 2005, 23(5), 1011–1027, DOI:10.1200/JCO.2005.06.081.
- M. Malumbres and M. Barbacid, Cell Cycle, CDKs and Cancer: A Changing Paradigm, Nat. Rev. Cancer, 2009, 9(3), 153–166, DOI:10.1038/nrc2602.
- J. D. Benson, Y.-N. P. Chen, S. A. Cornell-Kennon, M. Dorsch, S. Kim, M. Leszczyniecka, W. R. Sellers and C. Lengauer, Validating Cancer Drug Targets, Nature, 2006, 441, 451–456, DOI:10.1038/nature04873.
- J. Downward, Targeting RAS Signalling Pathways in Cancer Therapy, Nat. Rev. Cancer, 2003, 3(1), 11–22, DOI:10.1038/nrc969.
- R. Roskoski Jr. , FDA-approved small molecule protein kinase inhibitorshttps://www.brimr.org/PKI/PKIs.htm (accessed Oct 25, 2021).
- S. Omura, H. Tanaka, R. Oiwa, J. Awaya, R. Masuma and K. Tanaka, New Antitumor Antibiotics, OS-4742 A1, A2, B1 and B2 Produced by a Strain of Streptomyces, J. Antibiot., 1977, 30(11), 908–916 CrossRef CAS PubMed.
- L. M. Toledo and N. B. Lydon, Structures of Staurosporine Bound to CDK2 and CAPK - New Tools for Structure-Based Design of Protein Kinase Inhibitors, Structure, 1997, 5(12), 1551–1556, DOI:10.1016/S0969-2126(97)00304-3.
- J. Berger, Staurosporine, a Potent Inhibitor of Phospholipid/Ca++ Dependent Protein Kinase, Biochem. Biophys. Res. Commun., 1986, 135(2), 397–402 CrossRef PubMed.
- G. E. Atilla-Gokcumen, D. S. Williams, H. Bregman, N. Pagano and E. Meggers, Organometallic Compounds with Biological Activity: A Very Selective and Highly Potent Cellular Inhibitor for Glycogen Synthase Kinase 3, ChemBioChem, 2006, 7(9), 1443–1450, DOI:10.1002/cbic.200600117.
- J.É Debreczeni, A. N. Bullock, G. E. Atilla, D. S. Williams, H. Bregman, S. Knapp and E. Meggers, Ruthenium Half-Sandwich Complexes Bound to Protein Kinase Pim-1, Angew. Chem., Int. Ed., 2006, 45(10), 1580–1585, DOI:10.1002/anie.200503468.
- R. Anand, J. Maksimoska, N. Pagano, E. Y. Wong, P. A. Gimotty, S. L. Diamond, E. Meggers and R. Marmorstein, Toward the Development of a Potent and Selective Organoruthenium Mammalian Sterile 20 Kinase Inhibitor, J. Med. Chem., 2009, 52(6), 1602–1611, DOI:10.1021/jm8005806.
- H. Bregman and E. Meggers, Ruthenium Half-Sandwich Complexes as Protein Kinase Inhibitors: An N-Succinimidyl Ester for Rapid Derivatizations of the Cyclopentadienyl Moiety, Org. Lett., 2006, 8(24), 5465–5468, DOI:10.1021/ol0620646.
- Y. Zheng and D. Pan, The Hippo Signaling Pathway in Development and Disease, Dev. Cell, 2019, 50(3), 264–282, DOI:10.1016/j.devcel.2019.06.003.
- F. Fan, Z. He, L. L. Kong, Q. Chen, Q. Yuan, S. Zhang, J. Ye, H. Liu, X. Sun, J. Geng, L. Yuan, L. Hong, C. Xiao, W. Zhang, X. Sun, Y. Li, P. Wang, L. Huang, X. Wu, Z. Ji, Q. Wu, N. S. Xia, N. S. Gray, L. Chen, C. H. Yun, X. Deng and D. Zhou, Pharmacological Targeting of Kinases MST1 and MST2 Augments Tissue Repair and Regeneration, Sci. Transl. Med., 2016, 8(352), 1–14, DOI:10.1126/scitranslmed.aaf2304.
- M. Scarpi-Luttenauer, K. Galentino, C. Orvain, M. Cecchini, C. Gaiddon and P. Mobian, TiO4N2 Complexes Formed with 1,10-Phenanthroline Ligands Containing a Donor-Acceptor Hydrogen Bond Site: Synthesis, Cytotoxicity and Docking Experiments, Inorg. Chim. Acta, 2022, 540, 121036, DOI:10.1016/j.ica.2022.121036.
- S. Arribas, O. F. Wendt, S. Siegel and W. Kenneth, 4,5,9-Triazaphenanthren-10-One. 2005.
- K. M. Deo, D. L. Ang, B. McGhie, A. Rajamanickam, A. Dhiman, A. Khoury, J. Holland, A. Bjelosevic, B. Pages, C. Gordon and J. R. Aldrich-Wright, Platinum Coordination Compounds with Potent Anticancer Activity, Coord. Chem. Rev., 2018, 375, 148–163, DOI:10.1016/j.ccr.2017.11.014.
- H. Yin, J. Roque, P. Konda, S. Monro, K. L. Colo, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. Mcfarland, Transition Metal Complexes and Photodynamic Therapy from a Tumor-Centered Approach : Challenges, Opportunities, and Highlights from the Development of TLD1433, Chem. Rev., 2019, 119(2), 797–828, DOI:10.1021/acs.chemrev.8b00211.
- Pragti, B. K. Kundu and S. Mukhopadhyay, Target Based Chemotherapeutic Advancement of Ruthenium Complexes, Coord. Chem. Rev., 2021, 448, 214169–214209, DOI:10.1016/j.ccr.2021.214169.
- Y. Lu, D. Zhu, Q. Le, Y. Wang and W. Wang, Ruthenium-Based Antitumor Drugs and Delivery Systems from Monotherapy to Combination Therapy, Nanoscale, 2022, 14(44), 16339–16375, 10.1039/d2nr02994d.
- L. Zeng, P. Gupta, Y. Chen, E. Wang, L. Ji, H. Chao and Z. S. Chen, The Development of Anticancer Ruthenium(II) Complexes: From Single Molecule Compounds to Nanomaterials, Chem. Soc. Rev., 2017, 46(19), 5771–5804, 10.1039/c7cs00195a.
- C. Licona, J. B. Delhorme, G. Riegel, V. Vidimar, R. Cerón-Camacho, B. Boff, A. Venkatasamy, C. Tomasetto, P. Da Silva Figueiredo Celestino Gomes, D. Rognan, J. N. Freund, R. Le Lagadec, M. Pfeffer, I. Gross, G. Mellitzer and C. Gaiddon, Anticancer Activity of Ruthenium and Osmium Cyclometalated Compounds: Identification of ABCB1 and EGFR as Resistance Mechanisms, Inorg. Chem. Front., 2020, 7(3), 678–688, 10.1039/c9qi01148j.
- C. Gaiddon and M. Pfeffer, The Fate of Cycloruthenated Compounds: From C–H Activation to Innovative Anticancer Therapy, Eur. J. Inorg. Chem., 2017, 2017(12), 1639–1654, DOI:10.1002/ejic.201601216.
- S. Ma, Z. Meng, R. Chen and K. L. Guan, The Hippo Pathway: Biology and Pathophysiology, Annu. Rev. Biochem., 2019, 88, 577–604, DOI:10.1146/annurev-biochem-013118-111829.
- E. B. Wright and D. A. Lannigan, Therapeutic Targeting of P90 Ribosomal S6 Kinase, Front. Cell Dev. Biol., 2023, 11, 1–15, DOI:10.3389/fcell.2023.1297292.
- A. P. Halverson, T. A. Elmaaty and L. W. Castle, Complexes of (Bpy) 2Ru(II) and (Ph 2bpy) 2Ru(II) with a Series of Thienophenanthroline Ligands: Synthesis, Characterization, and Electronic Spectra, J. Coord. Chem., 2011, 64(21), 3693–3699, DOI:10.1080/00958972.2011.629296.
- R. Le Lagadec, L. Rubio, L. Alexandrova, R. A. Toscano, E. V. Ivanova, R. Meškys, V. Laurinavičius, M. Pfeffer and A. D. Ryabov, Cyclometalated N,N-Dimethylbenzylamine Ruthenium(II) Complexes [Ru(C6HR1R2R3-o-CH2NMe2)(Bpy)(RCN)2]PF6 for Bioapplications: Synthesis, Characterization, Crystal Structures, Redox Properties, and Reactivity toward PQQ-Dependent Glucose Dehydrogenase, J. Organomet. Chem., 2004, 689(25), 4820–4832, DOI:10.1016/j.jorganchem.2004.09.056.
- B. Boff, M. Ali, L. Alexandrova, N. A. Espinosa-Jalapa, R. O. Saavedra-Díaz, R. Le Lagadec and M. Pfeffer, Rational Synthesis of Heteroleptic Tris(Chelate) Ruthenium Complexes [Ru, Organometallics, 2013, 32(18), 5092–5097, DOI:10.1021/om400611t.
- A. D. Ryabov, V. S. Sukharev, L. Alexandrova, R. Le Lagadec and M. Pfeffer, New Synthesis and New Bio-Application of Cyclometalated Ruthenium(II) Complexes for Fast Mediated Electron Transfer with Peroxidase and Glucose Oxidase, Inorg. Chem., 2001, 40(25), 6529–6532, DOI:10.1021/ic010423h.
- A. D. Ryabov, R. Le Lagadec, H. Le; Estevez, R. A. Toscano, S. Hernandez, L. Alexandrova, V. S. Kurova, A. Fischer, C. Sirlin and M. Pfeffer, Synthesis, Characterization, and Electrochemistry of Biorelevant Photosensitive Low-Potential Orthometalated Ruthenium Complexes, Inorg. Chem., 2005, 44(5), 1626–1634, DOI:10.1021/ic048270w.
- N. Queyriaux, E. Giannoudis, J. F. Lefebvre, V. Artero and M. Chavarot-Kerlidou, Synthesis of Ruthenium Tris-Diimine Photosensitizers Substituted by Four Methylphosphonate Anchoring Groups for Dye-Sensitized Photoelectrochemical Cell Applications, Eur. J. Inorg. Chem., 2019, 2019(15), 2154–2161, DOI:10.1002/ejic.201900151.
- N. S. Ng, P. Leverett, D. E. Hibbs, Q. Yang, J. C. Bulanadi, M. Jie Wu and J. R. Aldrich-Wright, The Antimicrobial Properties of Some Copper(II) and Platinum(II) 1,10-Phenanthroline Complexes, Dalton Trans., 2013, 42(9), 3196–3209, 10.1039/c2dt32392c.
- B. J. Pages, F. Li, P. Wormell, D. L. Ang, J. K. Clegg, C. J. Kepert, L. K. Spare, S. Danchaiwijit and J. R. Aldrich-Wright, Synthesis and Analysis of the Anticancer Activity of Platinum(II) Complexes Incorporating Dipyridoquinoxaline Variants, Dalton Trans., 2014, 43(41), 15566–15575, 10.1039/c4dt02133a.
- A. M. Krause-Heuer, R. Grunert, S. Kuhne, M. Buczkowska, N. J. Wheate, D. D. Le Pevelen, L. R. Boag, D. M. Fisher, J. Kasparkova, J. Malina, P. J. Bednarski, V. Brabec and J. R. Aldrich-Wright, Studies of the Mechanism of Action of Platinum(II) Complexes with Potent Cytotoxicity in Human Cancer Cells, J. Med. Chem., 2009, 52(17), 5474–5484, DOI:10.1021/jm9007104.
- L. Leyva, C. Sirlin, L. Rubio, C. Franco, R. Le Lagadec, J. Spencer, P. Bischoff, C. Gaiddon, J. P. Loeffler and M. Pfeffer, Synthesis of Cycloruthenated Compounds as Potential Anticancer Agents, Eur. J. Inorg. Chem., 2007,(19), 3055–3066, DOI:10.1002/ejic.200601149.
- A. Blanchet, A. Bourgmayer, J. E. Kurtz, G. Mellitzer and C. Gaiddon, Isoforms of the P53 Family and Gastric Cancer: A Ménage à Trois for an Unfinished Affair, Cancers, 2021, 13(4), 1–45, DOI:10.3390/cancers13040916.
- L. Depotte, J. Palle, C. Rasola, C. Broudin, V.-A. Afrăsânie, A. Mariani and A. Zaanan, New Developments and Standard of Care in the Management of Advanced Gastric Cancer, Clin. Res. Hepatol. Gastroenterol., 2024, 48(1), 102245, DOI:10.1016/j.clinre.2023.102245.
- C. Gaiddon, I. Gross, X. Meng, M. Sidhoum, G. Mellitzer, B. Romain, J.-B. Delhorme, A. Venkatasamy, A. C. Jung and M. Pfeffer, Bypassing the Resistance Mechanisms of the Tumor Ecosystem by Targeting the Endoplasmic Reticulum Stress Pathway Using Ruthenium- and Osmium-Based Organometallic Compounds: An Exciting Long-Term Collaboration with Dr. Michel Pfeffer, Molecules, 2021, 26(17), 5386, DOI:10.3390/molecules26175386.
- N. J. Wheate, R. I. Taleb, A. M. Krause-Heuer, R. L. Cook, S. Wang, V. J. Higgins and J. R. Aldrich-Wright, Novel Platinum(II)-Based Anticancer Complexes and Molecular Hosts as Their Drug Delivery Vehicles, Dalton Trans., 2007,(43), 5055–5064, 10.1039/b704973k.
- H. B. J. Jefferies, S. Fumagalli, P. B. Dennis, C. Reinhard, R. B. Pearson and G. Thomas, Rapamycin Suppresses 5′TOP MRNA Translation through Inhibition of P70(S6k), EMBO J., 1997, 16(12), 3693–3704, DOI:10.1093/emboj/16.12.3693.
- C. Selman, J. M. A. Tullet, D. Wieser, E. Irvine, S. J. Lingard, A. I. Choudhury, M. Claret, H. Al-qassab, D. Carmignac, A. Woods, I. C. A. Robinson, E. Schuster, R. L. Batterham, L. Partridge, D. Gems and D. J. Withers, Ribosomal Protein S6 Kinase 1 Signaling Regulates Mammalian Lifespan, Science, 2009, 326(5949), 140–144, DOI:10.1126/science.1177221.Ribosomal.
- S. H. Um, F. Frigerio, M. Watanabe, F. Picard, M. Joaquin, M. Sticker, S. Fumagalli, P. R. Allegrini, S. C. Kozma, J. Auwerx and G. Thomas, Absence of S6K1 Protects against Age- and Diet-Induced Obesity While Enhancing Insulin Sensitivity, Nature, 2004, 431(7005), 200–205, DOI:10.1038/nature02866.
- M. Bärlund, F. Forozan, J. Kononen, L. Bubendorf, Y. Chen, M. L. Bittner, J. Torhorst, P. Haas, C. Bucher, G. Sauter and O. Kallioniemi, Detecting Activation of Ribosomal Protein S6 Kinase by Complementary DNA and Tissue Microarray Analysis, J. Natl. Cancer Inst., 2000, 92(15), 1252–1259 CrossRef PubMed.
- O. Korb, T. Stützle and T. E. Exner, An Ant Colony Optimization Approach to Flexible Protein–Ligand Docking, Swarm Intell., 2007, 1(2), 115–134, DOI:10.1007/s11721-007-0006-9.
- J. Qin, R. Rajaratnam, L. Feng, J. Salami, J. S. Barber-Rotenberg, J. Domsic, P. Reyes-Uribe, H. Liu, W. Dang, S. L. Berger, J. Villanueva, E. Meggers and R. Marmorstein, Development of Organometallic S6K1 Inhibitors, J. Med. Chem., 2015, 58(1), 305–314, DOI:10.1021/jm5011868.
- M. F. Adasme, K. L. Linnemann, S. N. Bolz, F. Kaiser, S. Salentin, V. J. Haupt and M. Schroeder, PLIP 2021: Expanding the Scope of the Protein-Ligand Interaction Profiler to DNA and RNA, Nucleic Acids Res., 2021, 49(W1), W530–W534, DOI:10.1093/nar/gkab294.
- M. Mihmanli, E. Ilhan, U. O. Idiz, A. Alemdar and U. Demir, Recent Developments and Innovations in Gastric Cancer, World J. Gastroenterol., 2016, 22(17), 4307–4320, DOI:10.3748/wjg.v22.i17.4307.
- H. Wong and T. Yau, Targeted Therapy in the Management of Advanced Gastric Cancer: Are We Making Progress in the Era of Personalized Medicine?, Oncologist, 2012, 17(3), 346–358, DOI:10.1634/theoncologist.2011-0311.
- R. J. Kelly, Immunotherapy for Esophageal and Gastric Cancer, Am. Soc. Clin. Oncol. Educ. B, 2017, 37, 292–300, DOI:10.1200/EDBK_175231.
- Z. Zheng, Y. Zheng, M. Zhang, J. Wang, G. Yu and W. Fang, Reciprocal Expression of P-AMPKa and p-S6 Is Strongly Associated with the Prognosis of Gastric Cancer, Tumor Biol., 2016, 37(4), 4803–4811, DOI:10.1007/s13277-015-4193-5.
- L. Seeneevassen, P. Dubus, C. Gronnier and C. Varon, Hippo in Gastric Cancer: From Signalling to Therapy, Cancers, 2022, 14(9), 2282–2309, DOI:10.3390/cancers14092282.
- S. Yoshida, K. Matsumoto, T. Arao, H. Taniguchi, I. Goto, T. Hanafusa, K. Nishio and Y. Yamada, Gene Amplification of Ribosomal Protein S6 Kinase-1 and -2 in Gastric Cancer, Anticancer Res., 2013, 33(2), 469–476 CAS.
- Inc., B. A. SADABS. Madison, WI 2001.
- G. M. Sheldrick, A Short History of SHELX, Acta Crystallogr., Sect. A:Found. Crystallogr., 2008, 64(1), 112–122, DOI:10.1107/S0108767307043930.
- G. M. Sheldrick, Crystal Structure Refinement with SHELXL, Acta Crystallogr., Sect. C:Struct. Chem., 2015, 71, 3–8, DOI:10.1107/S2053229614024218.
- Bruker, SADABS. Bruker AXS Inc., Madison, Wisconsin, USA, 2001 Search PubMed.
Footnotes |
| † This article is dedicated to the memory of Pr. Marc Henry. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2392833 and 2392834. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02984d |
| § Present address: INSERM, UMR 1260, CRBS, Regenerative Nanomedicine, “GP_SMIT” Laboratory, CRBS; 1 Rue Eugène Boeckel, 67085 Strasbourg, France. |
| ¶ Present address: UMR7242, Biotechnology et Signalisation Cellulaire, group STREINTH, 300 Bld S. Brant, FR-67412 Illkirch Cedex, France. |
| This journal is © The Royal Society of Chemistry 2025 |