
Revealing the synergistic effect of LiF and Li3N in solid electrolyte interphases for stable lithium metal batteries using in situ electrochemical atomic force microscopy†
Shuwei
Wang
a,
Jianxun
Zhang
a,
Lihan
Zhang
*b,
Xiaojing
Li
c,
Rui
Zhao
c,
Yuanming
Liu
c,
Zile
Wang
a,
Xuewei
Lu
a,
Yan
Xin
a,
Huajun
Tian
*a,
Feiyu
Kang
c and
Baohua
Li
 *c
*c
aKey Laboratory of Power Station Energy Transfer Conversion and Systems of Ministry of Education, North China Electric Power University, Beijing 102206, China. E-mail: Huajun.Tian@ncepu.edu.cn
bBeijing Key Laboratory of Microstructure and Properties of Solids Institute of Microstructure and Properties of Advanced Materials, Beijing University of Technology, Beijing 100124, China. E-mail: zhanglh06@bjut.edu.cn
cShenzhen Key Laboratory on Power Battery Safety Research and Shenzhen Geim Graphene Center, Tsinghua Shenzhen International Graduate School, Shenzhen 518055, China. E-mail: libh@sz.tsinghua.edu.cn
First published on 24th January 2024
Abstract
Inorganic components in the solid electrolyte interphases (SEIs) on lithium (Li) metal play a critical role in Li metal batteries (LMBs) since the presence of numerous inorganic constituents may promote ion diffusion and mechanical strength. However, manipulating inorganic components and acquiring an in-depth understanding of their roles in LMBs remain challenging. Herein, we deliberately introduce different kinds of inorganic components (e.g., LiF, Li3N, and their complexes) into SEIs on Li metal in LMBs by electrolyte engineering. Through in situ atomic force microscopy and electrochemical experiments, the roles of the specific inorganic component in SEIs played in the process of Li nucleation and evolution are revealed, and the detailed synergistic effect of LiF and Li3N is clarified. We demonstrate that the simultaneous presence of LiF and Li3N in SEIs enables homogeneous and dense lithium metal nucleation and growth, improving both the chemical stability and mechanical strength of SEIs for highly stable LMBs. In contrast, individual LiF and Li3N in SEIs induce needle morphology of Li metal and discrete Li deposition, respectively. These findings can provide rational guidelines for optimizing SEIs through electrolyte engineering for stable LMBs.
1 Introduction
Lithium (Li) metal has been considered a promising anode material for decades due to its desirable properties, such as a high theoretical specific capacity (3860 mA h g−1), low redox potential (−3.04 V vs. standard hydrogen electrode), and low density (0.534 g cm−3),1–3 making it ideal for developing high energy density lithium metal battery (LMB) systems (e.g., Li–O2, Li–air, and Li–S). Nevertheless, several challenges need to be addressed before LBMs can be put into practical applications, including the uncontrolled dendrite growth and continuous consumption of fresh Li source and electrolyte components, which are responsible for the short cycle lives and safety hazards of LMBs, respectively.4–7 The key issue is the high reactivity of Li metal making almost all organic liquid electrolytes thermodynamically unstable, further resulting in the formation of heterogeneous electrolyte decomposition species that coat the surface of anodes, which is referred to as the solid electrolyte interphase (SEI).8,9 An ideal SEI layer can act as a physical barrier by passivating further detrimental reactions, but in fact, uncontrolled and heterogeneous SEI growth results in irreversible electrode and electrolyte degradation reactions. Therefore, it is believed that the properties of SEI species contribute significantly to the abovementioned issues.10–13The properties of SEI species are dependent upon their chemical compositions,14 which can be classified into two main categories: (i) organic constituents (e.g., ROLi and ROCO2Li, R representing –CH2CH3, –CH3, –H, etc.) and (ii) often poor ion-conducting inorganic components (e.g., LiF, Li3N, Li2O, and Li2CO3). The organic constituents usually can increase the mechanical flexibility of the SEI layer to accommodate infinite volume variation during the Li plating/stripping process. However, the intrinsic lithiophilicity of organic species leads to strong bonding between organic-rich SEI and Li metal, limiting the Li+ diffusion along the SEI/Li metal interface and promoting vertical Li penetration into the SEI layer directly by forming Li dendrites.15–17 In contrast, a large amount of inorganic compounds have weak bonding with Li metal, these inorganic species can facilitate the Li+ lateral diffusion along the SEI/Li metal interface to suppress the growth of Li dendrites.18,19 Thus, the inorganic components in SEIs play a critical role in suppressing Li dendrites and the cracking of the SEI layer, which benefits reversible Li plating/stripping and hence the LMBs' cycle life.20–22
Among numerous inorganic species, LiF has been considered one of the most effective inorganic components in SEI layers owing to its following three superiorities: (i) sufficiently high Young's modulus of 64.9 GPa (ref. 21) to enhance the mechanical strength, (ii) extremely low electronic conductivity of 10−31 S cm−1 to reduce SEI thickness,23 and (iii) high surface energy towards Li metal to suppress the growth of Li dendrites.24 When used for Li–S batteries, the LiF-rich SEIs can efficiently suppress the polysulfide attack against the Li metal anode.25–27 Despite these promising merits, the concern is the poor ionic conductivity of the inorganic LiF that will increase the interface impedance at the Li metal anode.28 On the other hand, inorganic Li3N has been utilized to stabilize Li metal anode due to its excellent thermodynamic stability and ultrahigh ionic conductivity of 10−3 S cm−1 for the faster Li+ diffusion, which usually originates from the reduction of LiNO3 in the conventional electrolyte.29 However, LiNO3 has low solubility in the common electrolyte, limiting the Li3N content in SEIs and thus increasing the difficulty in understanding the functional mechanism of Li3N towards SEIs' properties. Although LiF- and Li3N-rich SEIs have been investigated for decades,30–35 challenges still remain, motivating further fundamental studies towards the understanding of the individual and synergistic effects of LiF and Li3N and their relation to Li nucleation and growth. The key difficulties focus on the following two aspects: (i) the heterogeneous and complex compositions of SEIs make it hard to obtain pure LiF- or Li3N-dominated SEI layers, (ii) the SEI structure is sensitive to radiation, air, and moisture,36,37 making it difficult to study the detailed mechanism through traditional characterization techniques.
Herein, we deliberately selected a lithium bis(oxalate) borate (LiBOB)-based electrolyte, which does not contain any fluoride and nitrogen species, in order to obtain pure LiF-, Li3N- and LiF/Li3N-dominated SEI layers by adding fluoroethylene carbonate (FEC), LiNO3, and FEC/LiNO3 additives into the LiBOB-based electrolyte, respectively. Then, we conducted real-time observations of Li nucleation and evolution behaviour influenced by LiF-, Li3N- and LiF/Li3N-dominated SEI layers using in situ atomic force microscope (AFM). In combination with X-ray photoelectron spectroscopy (XPS) and scanning electron microscopy (SEM), our direct in situ AFM investigation demonstrates the detailed synergistic effect of LiF and Li3N. The simultaneous presence of LiF and Li3N in SEIs enables homogeneous and dense lithium metal nucleation and growth, improving both the chemical stability and mechanical strength of SEIs for reversible Li plating/striping and hence stable LBMs. In contrast, individual LiF and Li3N in SEIs induce needle morphology of Li metal and discrete Li deposition, respectively, resulting in poor cycling stability of LBMs. These results indicate the importance of the synergistic effect of LiF and Li3N inorganic components in SEIs, which can be achieved by electrolyte engineering for higher reversibility for LMBs.
2 Results and discussion
Regulating the SEI composition requires that the preferred pure LiF and Li3N species should come from a sole electrolyte component. Among a large number of electrolyte additives, FEC is the most effective for carbonate electrolytes because the fluorine in FEC can be removed from cyclic carbonate and react with Li to form LiF (Fig. 1a). As another strong oxidant additive, LiNO3 can accept a few electrons to break the N–O bonds and react with Li to produce Li3N (Fig. 1b). Considering the Li salt, LiPF6 has the advantages of excellent thermal stability and high compatibility with the anode and cathode. However, FEC is thermally unstable in the LiPF6-based electrolytes, which will trigger the formation of unwanted HF and other various acids. These acids will induce severe dissolution of transition metal ions into the electrolyte and finally result in the serious degradation of the cathode material.38 Besides, the reduction of PF6− can also lead to the generation of LiF, which increases the difficulty in analysing the function mechanism of LiF. According to the above discussion, we selected LiBOB as the ideal lithium salt, which has the same advantages as LiPF6. Besides, LiBOB cannot generate LiF and Li3N inorganic components (Fig. 1c), which is helpful to explore the function mechanism of LiF and Li3N in lithium metal anodes, separately. | ||
| Fig. 1 Generalized decomposition reactions of (a) FEC, (b) LiNO3, (c) LiBOB, (d) voltage–time curves of symmetric cells in pure, FEC, LiNO3, and FEC–LiNO3 electrolytes at various currents of 20, 40, 60, 80, 100, and 120 μA for extracting the overpotential at each current. (e) Exchange current density (i0) for Li plating/stripping using Li‖Cu cells in pure, FEC, LiNO3, and FEC–LiNO3 electrolytes. (f) Coulombic efficiency of the repeated Li plating/stripping at 1.0 mA cm−2 for 2.0 mA h cm−2 using the Li‖Cu cells in pure, FEC, LiNO3, and FEC–LiNO3 electrolytes. | ||
To study the effect of LiF- and Li3N-rich SEIs on the Li plating/stripping kinetics in lithium metal batteries, Li–Cu half cells were assembled (see more details in the experimental section in the ESI†). The Cu electrode was pre-deposited with 3.0 mA h cm−2 Li metal, followed by galvanostatic plating/stripping at various currents from 20 to 120 μA, as shown in Fig. 1d. The hysteresis in FEC–LiNO3 electrolyte is the smallest during the entire galvanostatic plating/stripping process, indicating the minimal interface reaction.39,40Fig. 1e was obtained by extracting the overpotential at each current to plot the overpotential versus current curves shown in Fig. 1d. The exchange current density (i0) in Fig. 1d was measured using eqn (1)
 | (1) |
Scanning electron microscopy (SEM) was further used to study the morphology of the deposited Li metal on a Cu foil in FEC-, LiNO3- and FEC/LiNO3-based electrolytes after plating at a current density of 1.0 mA cm−2 to an area capacity of 2.0 mA h cm−2. It was observed that the deposited Li in the electrolyte without any additives exhibited a needle-like structure (Fig. S2†). The needle morphology of Li metal would easily lead to preferential deposition of Li+ during Li metal plating and Li residues after the stripping of Li metal. The deposited Li in the FEC electrolyte exhibited a similar up-growing mechanism, whereas the as-deposited Li was more compact than that in the pure electrolyte (Fig. 2a, d, and S3†). For the LiNO3 electrolyte, the larger diameter of the Li deposits was observed from the top-view SEM image (Fig. 2b and S4†). The cross-section SEM image shows the inhomogeneous morphology of Li deposits with both mushroom- and spindle-like structures (Fig. 2e), which results in the larger diameter of deposited metallic Li, as observed from the top-view SEM image. In contrast, a columnar morphology of the deposited Li was formed in the FEC–LiNO3 electrolyte with the size of several micrometers (Fig. 2c, f, and S5†), representing a different growth mechanism of Li metal as illustrated in Fig. 2i. The columnar Li deposits with high deposit density and small exposed surface are favorable for improving high Li metal plating/stripping reversibility by minimizing the side reactions between metallic Li with the electrolyte.8
 | ||
| Fig. 2 Top view (a–c) and cross-sectional view (d–f) SEM images of Li metal deposited at 1.0 mA cm−2 for 2 h (2.0 mA h cm−2) in the FEC electrolyte (a and d), LiNO3 electrolyte (b and e) and FEC–LiNO3 electrolyte (c and f) on a Cu foil. Schematic illustration of Li metal deposited for 2.0 mA h cm−2 in the FEC electrolyte (g), LiNO3 electrolyte (h), and FEC–LiNO3 electrolyte (i). | ||
Then, we studied the spatial chemical properties of SEIs using XPS depth profiling. Fig. 3 and S6–S8† show the Li 1s, C 1s, and F 1s on Li metal after the 10th plating/stripping in different electrolytes. In pure electrolyte, the as-formed SEI is composed of inorganic components (e.g. Li2CO3 at 55.5 eV) and organic components (CO2 at 292.2 eV, ROCO2Li at 290.2 eV, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 287.7 eV, etc.), as shown in Fig. S6 and S7.† Besides, CO2 was detected in the SEI, which was derived from the solvent and salt reduction process. CO2 gas residue would further result in the formation of porous SEI. It should be mentioned that the loose and porous SEI is in agreement with the lowest modulus as shown in Fig. S9.† In the FEC electrolyte, the LiF (56.6 eV) and Li2CO3 are the dominant inorganic components in SEI (Fig. 3a and d). The LiF and Li2CO3 content ratio remained stable from the outer layer to the inner layer with an increase in the sputtering time from 0 to 360 s. For LiNO3 and FEC–LiNO3 electrolytes, Li3N (55.0 eV) is the reduction product of LiNO3. Moreover, Li3N and LiF appear as the dominant inorganic components of SEI in the FEC–LiNO3 electrolyte (Fig. 3c and f). Li3N has a high Li+ conductivity of 10−3 cm−1 and LiF is a well-known high mechanical strength component with a high Young's modulus of 64.9 GPa. Upon the etching process, Li3N/LiF decreased slowly in the SEI on the plated Li, denoting the formation of a gradient heterostructure SEI layer with high Li+ conductivity Li3N from outside and mechanically stable LiF inside. It is worth noting that the ROCO2Li diminishes in SEI (Fig. S7†), which stems from that the chemically stable SEI does not react with Li metal. In combination with the minimum exchange current density in the FEC–LiNO3 electrolyte (Fig. 1d), the mixed LiF and Li3N components contribute to facilitated rapid Li+ migration and optimize the SEI's chemical and mechanical stabilities. Also, the linear sweep voltammetry (LSV) curves corresponding to the stripping process appear at the lower redox peak in the FEC–LiNO3 electrolyte, which means a smaller hysteresis during the Li stripping process (Fig. S10†).
O at 287.7 eV, etc.), as shown in Fig. S6 and S7.† Besides, CO2 was detected in the SEI, which was derived from the solvent and salt reduction process. CO2 gas residue would further result in the formation of porous SEI. It should be mentioned that the loose and porous SEI is in agreement with the lowest modulus as shown in Fig. S9.† In the FEC electrolyte, the LiF (56.6 eV) and Li2CO3 are the dominant inorganic components in SEI (Fig. 3a and d). The LiF and Li2CO3 content ratio remained stable from the outer layer to the inner layer with an increase in the sputtering time from 0 to 360 s. For LiNO3 and FEC–LiNO3 electrolytes, Li3N (55.0 eV) is the reduction product of LiNO3. Moreover, Li3N and LiF appear as the dominant inorganic components of SEI in the FEC–LiNO3 electrolyte (Fig. 3c and f). Li3N has a high Li+ conductivity of 10−3 cm−1 and LiF is a well-known high mechanical strength component with a high Young's modulus of 64.9 GPa. Upon the etching process, Li3N/LiF decreased slowly in the SEI on the plated Li, denoting the formation of a gradient heterostructure SEI layer with high Li+ conductivity Li3N from outside and mechanically stable LiF inside. It is worth noting that the ROCO2Li diminishes in SEI (Fig. S7†), which stems from that the chemically stable SEI does not react with Li metal. In combination with the minimum exchange current density in the FEC–LiNO3 electrolyte (Fig. 1d), the mixed LiF and Li3N components contribute to facilitated rapid Li+ migration and optimize the SEI's chemical and mechanical stabilities. Also, the linear sweep voltammetry (LSV) curves corresponding to the stripping process appear at the lower redox peak in the FEC–LiNO3 electrolyte, which means a smaller hysteresis during the Li stripping process (Fig. S10†).
 | ||
| Fig. 3 XPS depth profiles of Li 1s of the SEIs after Li plating and stripping process in the FEC electrolyte (a and d), LiNO3 electrolyte (b and e), and FEC–LiNO3 electrolyte (c and f) with different depths of SEIs due to Ar+ sputtering (0 s, 30 s, 60 s, 180 s, and 360 s). | ||
In addition to the chemical properties of SEIs, the mechanical properties of SEIs in different electrolytes were also investigated by atomic force microscope (AFM). The topography images, modulus distribution, and the average Young's modulus of SEIs formed on Li deposits and after Li metal stripping off are shown in Fig. 4. In FEC electrolytes, Li deposits are composed of small-sized grains and the valley between the grains. The SEI formed on the surface of Li deposits exhibits a large average modulus of 6334.34 MPa (Fig. 4a), attributed to the LiF-rich SEIs formed in the FEC electrolyte. After the stripping process, the fine-grained residual Li metal and deeper valley have a lower surface modulus (Fig. 4b), suggesting that the SEI collapse was caused by uneven stripping. The modulus of SEI on Li deposits is significantly higher than that after Li stripping due to the voids between SEI and the residual Li metal. The tip of the residual Li metal increases electric fields and accelerates additional tip growth, resulting in the formation of Li dendrites in the subsequent plating/stripping process. As shown in Fig. 4c, varying-sized Li deposits can be observed under the same view, while the potholes on the surface cause inhomogeneous surface modulus distribution in the LiNO3 electrolyte. The Young's modulus of the Li3N-rich SEIs formed in the LiNO3 electrolyte is 1477.12 MPa, which is significantly lower than that of the LiF-rich SEIs in the FEC electrolyte. The following stripping brings about the sink on the surface, leading to the inhomogeneous modulus of the SEI surface (Fig. 4d). Whereas, the smooth SEI containing LiF and Li3N components in FEC–LiNO3 electrolyte reaches large-scale consistency of topography and modulus distribution in the area. Such an SEI has the highest average modulus of 8726.34 MPa (Fig. 4e). These led to homogeneous Li deposition and inhibited electrolyte side reactions. The wrinkle on SEI appeared and kept its integrity to confine Li metal after Li stripping. As expected, the modulus of SEI after Li stripping reaches 2158.86 MPa (Fig. 4f), which is much higher than that in the FEC (1685.68 MPa) and LiNO3 electrolytes (1078.87 MPa). The mechanical uniformity and strength of the SEIs at Li plating and stripping in different electrolytes confirmed the synthetic effect of Li3N and LiF components on the interface arrangement.
 | ||
| Fig. 4 AFM and modulus images of SEI on Li metal after plating at 1.0 mA cm−2 for 2 h (2.0 mA h cm−2) and stripping to 1 V vs. Li+/Li in the FEC electrolyte (a and b), LiNO3 electrolyte (c and d), and FEC–LiNO3 electrolyte (e and f). | ||
Furthermore, the Li metal deposits on the Cu foil were investigated by in situ EC-AFM to gain more insights into the Li nucleation and growth process. Note that a high negative potential of Cu foil causes the reductive electrolyte to generate an SEI layer on the electrode at the beginning of the electroplating. Li+ goes through SEI and is then deposited on the electrode, meaning that the SEI on the Cu foil has a significant effect on the overpotential of the Li metal nucleation. It is worth noting that the plated Li particle sizes in the in situ AFM three-electrode cell are smaller than those in the conventional coin cells due to their different cell pressures.41,42Fig. 5a–c and S11† show the AFM images before the Li electroplating, and the plating process with increasing deposition capacity in pure, FEC, LiNO3, and FEC–LiNO3 electrolytes. Before Li electroplating, the even surface of the Cu foil at the open circuit potential (OCP) can be observed (OCP in Fig. 5a–c and S11a†). As shown in Fig. S11b–e,† the Li metal is difficult to nucleate and grow uniformly without FEC and LiNO3 in the electrolyte. In the FEC electrolyte, dispersive particles of ∼200 nm in diameter accumulated on the Cu foil (Fig. 5e), marking the initial nuclei formation (0.005 mA h cm−2 in Fig. 5a). Corresponding to the overpotential profile (Fig. 5d), the high overpotential is favorable to form independent Li metal nuclei. After 0.02 mA h cm−2, a layer of sub-micrometer-sized Li nuclei (∼400 nm) emerged above the layer of the initial nuclei. As the plating process continued, the continuous upward nucleation of Li metal (at a capacity of 0.05 mA h cm−2) and inhomogeneous Li protrusions (at a capacity of 0.3 mA h cm−2) were observed. In the LiNO3 electrolyte, two distinctive features were observed: one is the larger initial nucleus size (Fig. 5e) with broader size distribution after plating Li metal at 0.005 mA h cm−2, as shown in Fig. 5b, and the other is that the continuous nucleation of Li metal in a longer time scale can be observed in 0.02 mA h cm−2 and 0.05 mA h cm−2, as shown in Fig. 5b. Up to 0.3 mA h cm−2, the difference in the particle size increases, and non-uniform protrusions can be observed. In contrast, in the FEC–LiNO3 electrolyte, initial nuclei are sparsely scattered on the Cu foil and the nucleus size is similar to that formed in the FEC electrolyte (0.005 mA h cm−2 in Fig. 5c). The nucleus size is inversely proportional to the overpotential, while the nucleation overpotential in the FEC–LiNO3 electrolyte is lower (Fig. 5d). Moreover, the obvious decrease in subsequent Li metal deposition overpotential,41 resulting in no obvious cuspidal protrusion can be observed at 0.3 mA h cm−2, as shown in Fig. 5c. Combining the above SEM results of the high capacity of Li deposition, the Li deposition process is fully presented in Fig. 6. These indicate that the SEI's properties should be the decisive factors in the Li metal nucleation and growth. Also, it is worth demonstrating that LiF and Li3N components are conducive to uniform Li metal deposition. Thus, for electrolytes, the addition of electrolyte additives, such as LiFSI, LiNO3, LiDFOB, and FEC can improve the LiF and Li3N content in SEI. For artificial SEI, the homogenously distributed LiF and Li3N can be introduced to the current collector's surface or the functional groups containing nitrogen and fluorine can be modified on the current collector's surface to form LiF and Li3N.
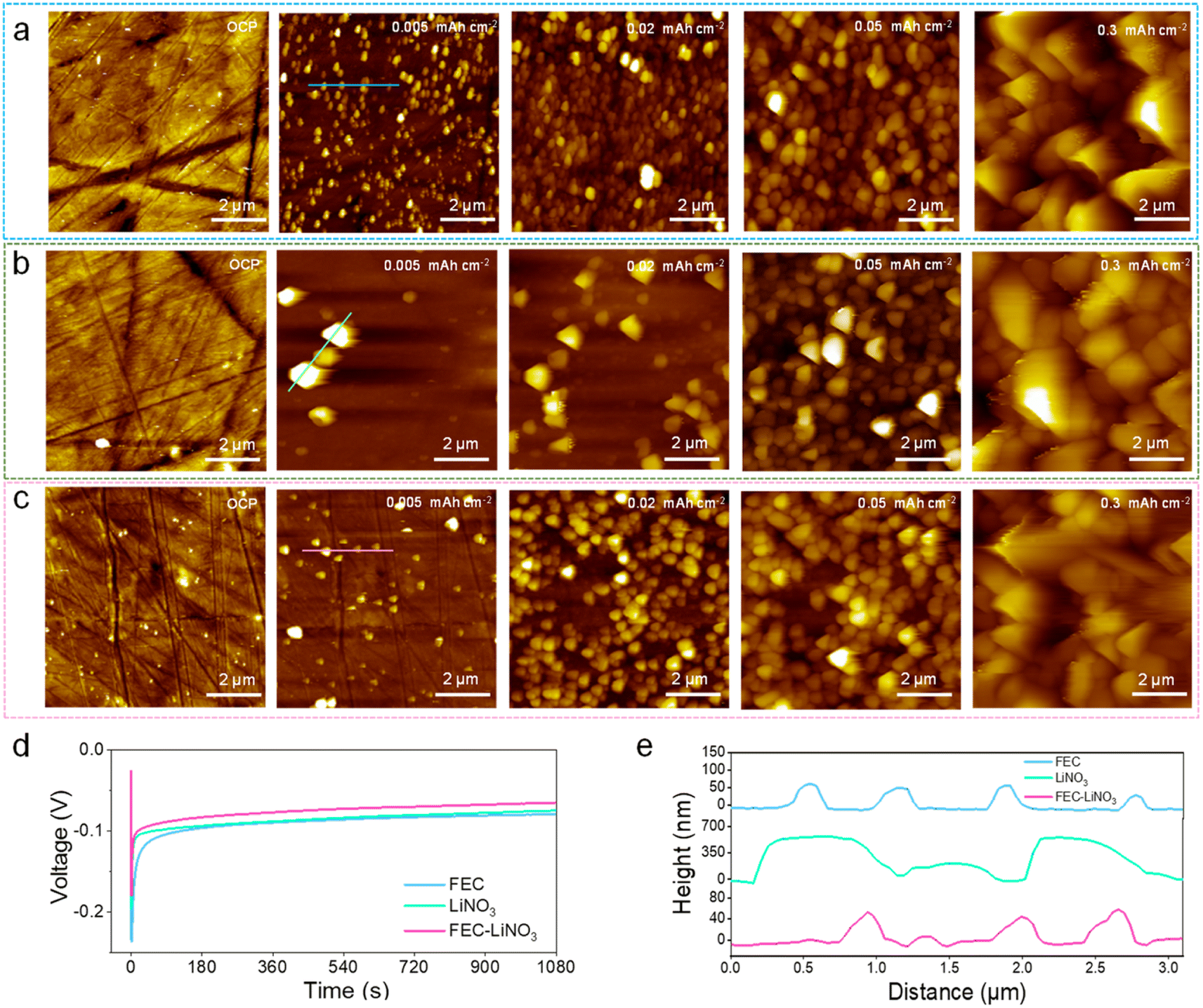 | ||
| Fig. 5 In situ AFM images of Li metal nucleation and growth. Morphology of the Cu foil during galvanostatic discharge at 1.0 mA cm−2 for 18 s (0.005 mA h cm−2), 72 s (0.02 mA h cm−2), 180 s (0.05 mA h cm−2), and 1080 s (0.3 mA h cm−2) in the FEC electrolyte (a), LiNO3 electrolyte (b) and FEC–LiNO3 electrolyte (c). (d) Overpotential profiles of the Li deposition in different electrolytes. (e) The corresponding height curves correspond to the marked regions in the AFM images in different electrolytes. | ||
 | ||
| Fig. 6 Schematic of the Li deposition mechanism with different inorganic components in SEIs. | ||
3 Conclusions
In summary, various SEI layers with individual or combined inorganic LiF and Li3N components were obtained through electrolyte engineering. Based on that, we further systematically investigated the functional mechanism of LiF and Li3N in SEIs during the process of Li nucleation and evolution by using in situ and ex situ experimental techniques and revealed the detailed synergistic effect of LiF and Li3N. Specifically, LiF/Li3N can synergistically promote the SEI's chemical and mechanical stability and enable the Li metal to nucleate and grow homogeneously. In contrast, the LiF and Li2CO3 in SEI induce upward nucleation and needle morphology of Li deposits, resulting in poor cycling stability of Li plating/striping. On the other hand, Li3N in SEI induces inhomogeneous nucleation and discrete Li deposits. These results imply the detailed synergistic effect of LiF and Li3N inorganic components on the Li plating process, which provides a promising research direction for practical applications of the SEI design.4 Experimental methods
4.1 AFM characterization
The in situ AFM measurement (Bruker Corp., Dimension Icon) was carried out using a three-electrode cell powered by an electrochemical workstation (CHI760E) in an argon-filled glove box (MBRAUN, H2O <0.1 ppm, O2 <0.1 ppm). A polished Cu foil was used as the working electrode and lithium strips were used as the counter and reference electrodes. The battery-grade lithium bis(oxalate)borate (LiBOB), lithium nitrate (LiNO3), 1,4-butyrolactone (γ-GBL), and fluoroethylene carbonate (FEC) were obtained from Shenzhen Capchem Technology Corporation. The pure electrolyte (0.5 M LiBOB in γ-GBL), FEC electrolyte (0.5 M LiBOB in γ-GBL with 5 wt% FEC), LiNO3 electrolyte (0.5 M LiBOB in γ-GBL with 5 wt% LiNO3) and FEC–LiNO3 electrolyte (0.5 M LiBOB in γ-GBL with 2.5 wt% FEC and 2.5 wt% LiNO3) were prepared in an argon-filled glove box (MBRAUN, H2O <0.1 ppm, O2 <0.1 ppm). AFM topography images were collected simultaneously during the galvanostatic deposition process using a ScanAsyst-Fluid+ tip (k = 0.7 N m−1, Bruker Corporation) in the Peakforce tapping mode. The initial Li deposition on Cu substrate AFM images of the Cu foil were collected during the galvanostatic deposition process using a ScanAsyst-Fluid+ tip (k = 0.7 N m−1, Bruker Corporation) in the Peakforce tapping mode. This mode causes minimal damage to the sample surface, due to the superior force control with a pN-level force between the tip and sample. Quantitative nanomechanics mode (QNM) was applied to analyse Young's modulus of the SEI formed on Li metal. The QNM mode performs a force curve at every pixel in the image, providing a two-dimensional image of the mechanical properties that have the same resolution as the height profile. The elastic deformation of the sample is related to its Young's modulus. To measure Young's modulus, we use the contact part of the force curves to determine the loading force (F) and the tip-sample distance (d). By fitting the force curve using the Derjaguin–Muller–Toropov (DMT) model, Young's modulus of each point can be obtained.4.2 Material characterization
The morphology of deposited Li metal on the Cu current collector was measured on a cold-field scanning electron microscope (HITACHI, SU8010) at 5 kV. The chemical composition after lithium deposition was analysed by XPS (PHI 5000 VersaProbe II) with monochromatic Al Kα radiation. The sample was rinsed with DMC in the glove box to remove surface residuals and then transferred to the XPS vacuum chamber with a special transfer device to avoid air exposure. Depth profile measurements were carried out using an Ar+ cluster source and sputtered for 30 s, 60 s, 180 s, and 360 s. The etching rate was confirmed using a SiO2 standard sample and it is 8 nm min−1.4.3 Electrochemical testing
For the Li‖Cu cell measurements, standard coin cells (CR2032) were assembled with 70 μL of the electrolyte in the glove box. The coulombic efficiency (CE) was tested on a LAND (Wuhan LAND Electronics Co., Ltd) cell test system at room temperature. The galvanostatic cycling was performed by first depositing Li metal on a Cu current collector at 4.0 mA h cm−2 and then charging/discharging at a certain current density with a certain cut-off capacity.Conflicts of interest
There are no confilicts to declare.Acknowledgements
We would like to acknowledge the support provided by the National Natural Science Foundation of China (No. 52302249, 12304003, 52072208, 52261160384 and 22379085), Chinese Postdoctoral Science Foundation (No. 2023M741154 and 2022M720331), Fundamental Research Funds for the Central Universities (2023MS018), Postdoctoral Science Foundation of Beijing, China (2023-zz-57) and Key-Area Research and Development Program of Guangdong Province (No. 2020B090919003). The authors thank the Materials and Devices Testing Center of Tsinghua University Shenzhen International Graduate School (Tsinghua SIGS).References
- M. Winter, B. Barnett and K. Xu, Chem. Rev., 2018, 118, 11433–11456 CrossRef CAS PubMed.
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS PubMed.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- X. Sun, X. Zhang, Q. Ma, X. Guan, W. Wang and J. Luo, Angew. Chem., Int. Ed., 2020, 59, 6665–6674 CrossRef CAS PubMed.
- H. Chen, M. Li, C. Li, X. Li, Y. Wu, X. Chen, J. Wu, X. Li and Y. Chen, Chin. Chem. Lett., 2022, 33, 141–152 CrossRef CAS.
- S. Ni, J. Sheng, C. Zhang, X. Wu, C. Yang, S. Pei, R. Gao, W. Liu, L. Qiu and G. Zhou, Adv. Funct. Mater., 2022, 32, 2200682 CrossRef CAS.
- Y. Xiao, R. Xu, L. Xu, Y. X. Zhan, J. F. Ding, S. Zhang, Z. H. Li, C. Yan and J. Q. Huang, Adv. Energy Mater., 2023, 13, 2300959 CrossRef CAS.
- Q. Wang, C. Zhao, S. Wang, J. Wang, M. Liu, S. Ganapathy, X. Bai, B. Li and M. Wagemaker, J. Am. Chem. Soc., 2022, 144, 21961–21971 CrossRef CAS PubMed.
- Y. Wang, M. Li, F. Yang, J. Mao and Z. Guo, Energy Materials and Devices, 2023, vol. 1, p. 9370005 Search PubMed.
- M. Zhou, Y. Liao, L. Li, R. Xiong, G. Shen, Y. Chen, T. Huang, M. Li, H. Zhou and Y. Zhang, J. Energy Chem., 2023, 85, 181–190 CrossRef CAS.
- Q. Liu, W. Jiang, J. Xu, Y. Xu, Z. Yang, D. J. Yoo, K. Z. Pupek, C. Wang, C. Liu, K. Xu and Z. Zhang, Nat. Commun., 2023, 14, 3678 CrossRef CAS PubMed.
- H. T. Kim, Nat. Energy, 2023, 8, 911–912 CrossRef CAS.
- O. Tamwattana, H. Park, J. Kim, I. Hwang, G. Yoon, T. Hwang, Y. Kang, J. Park, N. Meethong and K. Kang, ACS Energy Lett., 2021, 6, 4416–4425 CrossRef CAS.
- Z. Zhang, Y. Li, R. Xu, W. Zhou, Y. Li, S. T. Oyakhire, Y. Wu, J. Xu, H. Wang, Z. Yu, D. T. Boyle, W. Huang, Y. Ye, H. Chen, J. Wan, Z. Bao, W. Chiu and Y. Cui, Science, 2022, 375, 66–70 CrossRef CAS PubMed.
- S. Liu, X. Ji, N. Piao, J. Chen, N. Eidson, J. Xu, P. Wang, L. Chen, J. Zhang, T. Deng, S. Hou, T. Jin, H. Wan, J. Li, J. Tu and C. Wang, Angew. Chem., Int. Ed., 2021, 60, 3661–3671 CrossRef CAS PubMed.
- C. Fang, J. Li, M. Zhang, Y. Zhang, F. Yang, J. Z. Lee, M. Lee, J. Alvarado, M. A. Schroeder, Y. Yang, B. Lu, N. Williams, M. Ceja, L. Yang, M. Cai, J. Gu, K. Xu, X. Wang and Y. S. Meng, Nature, 2019, 572, 511–515 CrossRef CAS PubMed.
- B. Sun, Z. Sun, Y. Yang, X. L. Huang, S. C. Jun, C. Zhao, J. Xue, S. Liu, H. K. Liu and S. X. Dou, ACS Nano, 2024, 18, 28–66 CrossRef CAS PubMed.
- S. Liu, X. Ji, J. Yue, S. Hou, P. Wang, C. Cui, J. Chen, B. Shao, J. Li, F. Han, J. Tu and C. Wang, J. Am. Chem. Soc., 2020, 142, 2438–2447 CrossRef CAS PubMed.
- X. Fan, X. Ji, F. Han, J. Yue, J. Chen, L. Chen, T. Deng, J. Jiang and C. Wang, Sci. Adv., 2018, 4, eaau9245 CrossRef CAS PubMed.
- J. Chen, Q. Li, T. P. Pollard, X. Fan, O. Borodin and C. Wang, Mater. Today, 2020, 39, 118–126 CrossRef.
- P. Shi, F. Liu, Y. Feng, J. Zhou, X. Rui and Y. Yu, Small, 2020, 16, 2001989 CrossRef CAS PubMed.
- S. Ni, M. Zhang, C. Li, R. Gao, J. Sheng, X. Wu and G. Zhou, Adv. Mater., 2023, 35, 2209028 CrossRef CAS PubMed.
- J. Pan, Y. T. Cheng and Y. Qi, Phys. Rev. B, 2015, 91, 134116 CrossRef.
- S. Yan, F. Liu, Y. Ou, H. Zhou, Y. Lu, W. Hou, Q. Cao, H. Liu, P. Zhou and K. Liu, ACS Nano, 2023, 17, 19398–19409 CrossRef CAS PubMed.
- H. Duan, J. Zhang, X. Chen, X. Zhang, J. Li, L. Huang, X. Zhang, J. Shi, Y. Yin, Q. Zhang, Y. Guo, L. Jiang and L. Wan, J. Am. Chem. Soc., 2018, 140, 18051–18057 CrossRef CAS PubMed.
- Q. K. Zhang, X. Q. Zhang, J. Wan, N. Yao, T. L. Song, J. Xie, L. P. Hou, M. Y. Zhou, X. Chen, B. Q. Li, R. Wen, H. J. Peng, Q. Zhang and J. Q. Huang, Nat. Energy, 2023, 8, 725–735 CrossRef CAS.
- X. Zhang, Q. Su, G. Du, B. Xu, S. Wang, Z. Chen, L. Wang, W. Huang and H. Pang, Angew. Chem., Int. Ed., 2023, 62, e202304947 CrossRef CAS PubMed.
- Q. Wang, Z. Yao, C. Zhao, T. Verhallen, D. P. Tabor, M. Liu, F. Ooms, F. Kang, A. Aspuru Guzik, Y. Hu, M. Wagemaker and B. Li, Nat. Commun., 2020, 11, 4188 CrossRef PubMed.
- M. Liu, Z. Cheng, K. Qian, T. Verhallen, C. Wang and M. Wagemaker, Chem. Mater., 2019, 31, 4564–4574 CrossRef CAS.
- S. Wang, W. Zhang, Y. Chen, Z. Dai, C. Zhao, D. Wang and C. Shen, Appl. Surf. Sci., 2017, 426, 217–223 CrossRef CAS.
- R. Zhao, S. Wang, D. Liu, Y. Liu, X. Lv, X. Zeng and B. Li, ACS Appl. Energy Mater., 2021, 4, 492–499 CrossRef CAS.
- S. Huang, S. Wang, G. Hu, L. Cheong and C. Shen, Appl. Surf. Sci., 2018, 441, 265–271 CrossRef CAS.
- J. Ding, R. Xu, X. Ma, Y. Xiao, Y. Yao, C. Yan and J. Huang, Angew. Chem., Int. Ed., 2022, 61, e202115602 CrossRef CAS PubMed.
- S. Wang, X. Yin, D. Liu, Y. Liu, X. Qin, W. Wang, R. Zhao, X. Zeng and B. Li, J. Mater. Chem. A, 2020, 8, 18348–18357 RSC.
- S. Wang, D. Liu, X. Cai, L. Zhang, Y. Liu, X. Qin, R. Zhao, X. Zeng, C. Han, C. Zhan, F. Kang and B. Li, Nano Energy, 2021, 90, 106510 CrossRef CAS.
- S. Wang, Q. Liu, C. Zhao, F. Lv, X. Qin, H. Du, F. Kang and B. Li, Energy Environ. Mater., 2018, 1, 28–40 CrossRef.
- M. Yousaf, U. Naseer, A. Imran, Y. Li, W. Aftab, A. Mahmood, N. Mahmood, X. Zhang, P. Gao, Y. Lu, S. Guo, H. Pan and Y. Jiang, Mater. Today, 2022, 58, 238–274 CrossRef CAS.
- T. Teufl, D. Pritzl, L. Hartmann, S. Solchenbach, M. A. Mendez and H. A. Gasteiger, J. Electrochem. Soc., 2023, 170, 020531 CAS.
- Y. Guo, P. Niu, Y. Liu, Y. Ouyang, D. Li, T. Zhai, H. Li and Y. Cui, Adv. Mater., 2019, 31, 1900342 CrossRef PubMed.
- X. Wang, S. Wang, H. Wang, W. Tu, Y. Zhao, S. Li, Q. Liu, J. Wu, Y. Fu, C. Han, F. Kang and B. Li, Adv. Mater., 2021, 33, 2007945 CrossRef CAS PubMed.
- A. Pei, G. Zheng, F. Shi, Y. Li and Y. Cui, Nano Lett., 2017, 17, 1132–1139 CrossRef CAS PubMed.
- K. Yan, J. Wang, S. Zhao, D. Zhou, B. Sun, Y. Cui and G. Wang, Angew. Chem., Int. Ed., 2019, 58, 11364–11368 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta08019f |
| This journal is © The Royal Society of Chemistry 2024 |