DOI:
10.1039/D4PY00804A
(Paper)
Polym. Chem., 2024,
15, 4134-4140
Sulfur-doped anthanthrenes as effective organic photocatalysts for metal-free ATRP and PET-RAFT polymerization under blue and green light†
Received
20th July 2024
, Accepted 17th September 2024
First published on 18th September 2024
Abstract
Organocatalytic atom transfer radical polymerization (O-ATRP) as a metal-free controlled polymerization method represents an appealing approach for polymer synthesis in which the organic photocatalyst is critical to the polymerization efficiency and control. Herein, we introduce sulfur-doped anthanthrene (SDA) as a new and effective organic photoredox catalyst (OPC) for O-ATRP by following the photocatalyst design logic of heteroatom-doping of polycyclic arenes (HDPA). SDA could promote the O-ATRP of methacrylate monomers with good control on both molecular weight and low dispersity at 10–100 ppm catalyst loadings under visible light, even under green light in the absence of external electron donors. Further extension of the SDA catalyst to photoinduced electron/energy transfer addition–fragmentation chain-transfer (PET-RAFT) polymerization under green light is also demonstrated.
Introduction
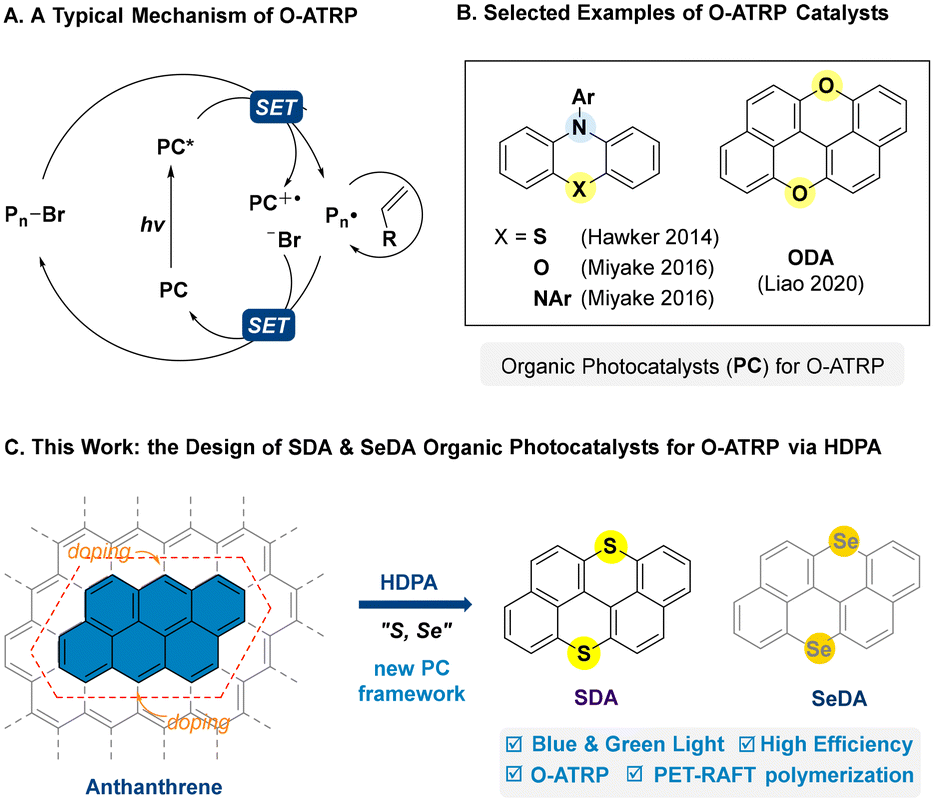
Controlled radical polymerizations (CRPs) are powerful and versatile methods for the synthesis of polymers with well-defined structures.1,2 Recently, the convergence of CRPs and photocatalysis has brought about the development of controlled radical polymerization methods with additional spatiotemporal control over chain growth,3–10 including photo-controlled atom transfer radical polymerization (ATRP)11,12 and reversible addition–fragmentation chain-transfer (RAFT) polymerization,13,14 which have significantly advanced the state-of-the-art of polymer synthesis.15 In particular, organocatalytic atom transfer radical polymerization (O-ATRP) has drawn considerable attention in the past decade by virtue of its metal-free essence, which is critical to its application in microelectronic and medical fields.16,17 Since its debut in 2014,11,18 this field has witnessed a continuous effort in the development of suitable photocatalysts for O-ATRP.17–20 However, it has proved challenging for a small organic photocatalyst to achieve an effective mediation in the photoredox processes and activation–deactivation equilibria involved in O-ATRP (Scheme 1A), and only a few catalysts could afford a good control on molecular weight and low dispersity.18–20 Furthermore, low catalytic efficiency, low chain-end fidelity, catalyst initiation, etc. were often encountered.17–20 Nonetheless, the development of O-ATRP could be undoubtedly accelerated with the evolution of organic photocatalysts, in particular, by the introduction of new photocatalyst frameworks.20,21
 |
| | Scheme 1 (A) O-ATRP: mechanism and selected organic photocatalysts for O-ATRP. (B) Selected examples of O-ATRP catalysts. (C) This work: the design of SDA and SeDA catalysts via heteroatom-doping of polycyclic arenes (HDPA). | |
In 2020, we introduced HDPA (heteroatom-doping of polycyclic arenes) as a design logic for organic photocatalysts,22–27 which allowed for the successful development of oxygen-doped anthanthrene (ODA) as a suitable class of photoredox catalysts for O-ATRP,22 as well as a class of bisphosphonium salt (BPS) photocatalysts for photo-controlled cationic polymerization from the design of phosphonium (P+)-doping of anthanthrene.25,26ODA catalysts represent a new and different photocatalyst skeleton when compared with the O-ATRP catalysts based on N-aryl phenothiazine-,11 phenoxazine-,28 and dihydrophenazine-frameworks29 that contain a characteristic triarylamine moiety and show high effectiveness in O-ATRP (Scheme 1B).28–31 According to the design logic of HDPA,22 it remains unclear whether the doping of anthanthrene with other chalcogenide elements rather than oxygen would lead to a new and effective photocatalyst for O-ATRP or not. In particular, the heavy atom effect (e.g. Se) may enhance the spin–orbit coupling (SOC) and favor the intersystem crossing (ISC) from singlet to triplet states.32,33 Recently, via oxidation/cyclization of the corresponding thioethers and selenides, we could synthesize sulfur-doped anthanthrene (SDA) and selenium-doped anthanthrene (SeDA) starting from commercially available 2,2′-dibromo-1,1′-binaphthalene.34,35 To our delight, SDA showed a strong reducing ability in excited states similar to that of ODA22 and could well mediate the atom transfer radical polymerization of methacrylate monomers with good control under low-energy blue and even green light at 10–100 ppm levels of catalyst loading. Herein, we wish to report our efforts in this study (Scheme 1C).
Experimental
Materials
Methyl methacrylate (MMA), benzyl methacrylate (BzMA), N,N-dimethylacrylamide (DMAA), 3-(trimethoxysilyl)propyl methacrylate (TMSPA) and styrene (St) were purchased from TCI Chemicals. 2,2,2-Trifluoroethyl methacrylate (TFEMA) was purchased from Adamas Chemicals and n-butyl acrylate (BA) was purchased from J&K Chemicals. MMA was first degassed and dried over CaH2 overnight, followed by vacuum distillation; then, MMA was further purified by titration with neat tri(n-octyl)aluminum (Aldrich Chemical) to a yellow endpoint and distillation under reduced pressure. The monomer was deoxygenized by the freeze–pump–thaw cycle three times, stored under a nitrogen atmosphere and sealed up. BzMA, DMAA, TMSPA, and BA were degassed and dried over CaH2, followed by vacuum distillation. St and TFEMA were purified by passing them through a plug of aluminum oxide (activated, basic) to remove the inhibitor, deoxygenized by the freeze–pump–thaw cycle three times, backfilled with argon, and sealed up. Subsequently, all of the purified monomers were stored under an inert atmosphere at −20 °C. SDA and SeDA were prepared following the reported method.33 Dichloromethane (DCM) was dried over CaH2, followed by vacuum distillation. Dimethyl sulfoxide (DMSO), N,N-dimethyl formamide (DMF), N,N-dimethyl acetamide (DMAc), ethyl acetate (EA), tetrahydrofuran (THF), anisole and toluene dried using 4 Å molecular sieves were purchased from Shanghai Chemical Reagents Co., Ltd. All other chemicals were obtained from Shanghai Chemical Reagents Co., Ltd and used as received unless mentioned.
Characterization
1H NMR, 13C NMR and 19F NMR were used to demonstrate the structures of synthesized substances, conversions for polymerization and nuclear magnetic molecular weights of polymers, and tracing reaction, which were recorded on a Bruker AVIII 400 MHz or Bruker AVANCE NEO 500 (500 MHz) nuclear magnetic resonance instrument, and CDCl3/DMSO-d6/CD2Cl2 was used as a deuterium solvent for testing samples and tetramethylsilane as the internal standard. The number-average molecular weight (Mn,GPC) and molecular weight distributions of the obtained polymers were determined by using a Waters 1515 series gel permeation chromatography (GPC) analyzer equipped with a Waters 2414 refractive-index detector, using a Styragel HR 3 THF (7.8 × 300 mm) column and a Styragel HR 4 THF (7.8 × 300 mm) column with measurable molecular weights ranging from 102 to 106 g mol−1. THF was used as an eluent at a flow rate of 1.0 mL min−1 at 40 °C. GPC samples were injected manually and Shodex Polystyrene (PS) samples and poly(methyl methacrylate) (PMMA) samples from Polymer Standards Service were used as calibration standards for the experimental number-average molecular weight (Mn,GPC) and dispersity (Đ). Mass spectra were recorded on an Agilent Q-TOF 6520 system using electrospray ionization in positive/negative ion detection (ESI+/ESI−) mode, and the significant fragments are reported in the following fashion: m/z (relative intensity). Single-crystal X-ray diffraction measurements were performed on a CCD area diffractometer (Saturn 724+, Rigaku, Japan), using graphite monochromated Mo Kα radiation (λ = 0.71073 Å). Intensity data sets were collected using ω scan techniques and corrected for Lp effects. The structures were solved by direct methods and refined by full-matrix least-squares on F2 using the Siemens SHELXTL™ Version 5 package of crystallographic software with anisotropic thermal parameters for all non-hydrogen atoms.
Synthesis and characterization of SDA catalysts
The arylated compound (0.2 mmol, 1.0 eq.) and H2O2 (30% (w/w) in H2O) (0.34 mL, 15 eq.) were dissolved in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) CHCl3:CH3COOH mixture (12 mL). The mixture was stirred at room temperature until the complete conversion of arylmethyl sulfide on TLC (within 1 hour). The reaction mixture was quenched with saturated NaOH aq. solution at 0 °C, until pH ∼ 12. The aqueous layer was extracted with DCM three times. The combined organic extract was dried over Na2SO4. After evaporation of the solvents, the mixture was purified by column chromatography (eluent: hexane/EtOAc = 1/1). The corresponding sulfoxide was placed in a Schlenk tube and DCE (3.0 mL) was added. With continuous N2 streaming into the tube, TfOH (1.5 mL) was added dropwise. After stirring for 24 hours at room temperature, 1 mL of H2O and 4 mL of pyridine were added and heated at 100 °C for 1 h. After cooling to rt, the solvent was removed in vacuo and diluted with H2O and DCM. The organic layer was separated and the aqueous layer was extracted with DCM three times. The combined organic extract was washed with H2O and brine, dried over Na2SO4, and concentrated in vacuo. The obtained crude material was purified through column chromatography (eluent: hexane/DCM = 20/1) and GPC (CHCl3) to give the cyclized product.
:1 (v/v) CHCl3:CH3COOH mixture (12 mL). The mixture was stirred at room temperature until the complete conversion of arylmethyl sulfide on TLC (within 1 hour). The reaction mixture was quenched with saturated NaOH aq. solution at 0 °C, until pH ∼ 12. The aqueous layer was extracted with DCM three times. The combined organic extract was dried over Na2SO4. After evaporation of the solvents, the mixture was purified by column chromatography (eluent: hexane/EtOAc = 1/1). The corresponding sulfoxide was placed in a Schlenk tube and DCE (3.0 mL) was added. With continuous N2 streaming into the tube, TfOH (1.5 mL) was added dropwise. After stirring for 24 hours at room temperature, 1 mL of H2O and 4 mL of pyridine were added and heated at 100 °C for 1 h. After cooling to rt, the solvent was removed in vacuo and diluted with H2O and DCM. The organic layer was separated and the aqueous layer was extracted with DCM three times. The combined organic extract was washed with H2O and brine, dried over Na2SO4, and concentrated in vacuo. The obtained crude material was purified through column chromatography (eluent: hexane/DCM = 20/1) and GPC (CHCl3) to give the cyclized product.
Typical ATRP procedure with SDA as the organocatalyst
In a glovebox, the typical O-ATRP procedure under the standard reaction conditions is as follows: MMA (0.5 ml, 4.7 mmol, 100 eq.) as the model monomer, DBMM (9 μL, 47 μmol, 1 eq.), SDA-2 (0.35 mg, 0.94 μmol, 0.02 eq.), and 0.5 mL of DCM were successively added to a Schlenk tube. Subsequently, the tube was placed under the irradiation of purple or white LEDs for a specified reaction time. Then, the tube was opened under argon, and 20 μL of the reaction mixture was syringed out and added to CDCl3 (containing 250 ppm BHT) for 1H NMR analysis to determine the monomer conversion. Another aliquot sample re-dissolved in HPLC grade, instabilized tetrahydrofuran and the Mn,GPC, and Mw/Mn were directly analyzed by GPC. Analysis of the kinetics, the polymerization procedure for chain extension from the PMMA macroinitiator, and the typical PET-RAFT polymerization procedure can be found in the ESI.†
Computational methods
All of the theoretical calculations were performed using the Gaussian 09 package. Geometry optimization calculations were carried out using a meta-GGA hybrid functional PBE0 with a 6-31G* basis set for all atoms. Vibrational frequencies were calculated analytically at the same level to obtain the thermodynamic corrections. The CPCM solvation model using the self-consistent reaction field (SCRF) method with the solvents of acetonitrile was employed to account for the solvent effect. The changes in Gibbs free energy are reported in the content. For more details, please see the ESI.†
Results and discussion
Photophysical and electrochemical properties of the catalysts
The synthesis of SDA catalysts is detailed in the ESI.† Se-doped anthanthrene was also successfully synthesized via a similar approach. The ultraviolet/visible (UV/Vis) absorption and photoluminescence properties of the SDA and SeDA catalysts were investigated first (Table 1). In contrast to ODA with the absorption maximum located in the blue region (λmax = 443 nm),22SDA-1 showed a significant redshift of maximum absorption to 468 nm, while εmax is lower (entry 1 vs. entry 2, Fig. S4†). Based on the emission wavelengths and cyclic voltammetry (CV), we could assess the excited-state reduction ability of these photocatalysts. To our delight, the excited-state reduction ability of these S- or Se-doped catalysts ranges from −1.72 to −1.86 V vs. SCE (saturated calomel electrode), which is sufficient for typical alkyl bromide initiator activation22,36 (Scheme 1A). Density functional theory (DFT) was used to estimate the reduction potentials of the triplet excited states (E0(PC˙+/3PC*)), which are in the range of −1.55 to −1.61 V vs. SCE (Fig. 1B, for more details, please see the ESI†). The CV analysis (Fig. S7†) showed good stability of the radical cations (PC˙+) of SDA catalysts, with an estimated oxidizing power of around 0.60 V vs. SCE, which is lower than that of ODA catalysts22,37,38 but higher than that of dihydrophenazine-based29 photocatalysts. These results (ODAvs.SDA) demonstrated that switching the doping heteroatoms from oxygen to sulfur or selenium39–41 could lead to significant differences in the photo-physical and redox properties of the catalysts, even though the same parental polycyclic arene was employed.
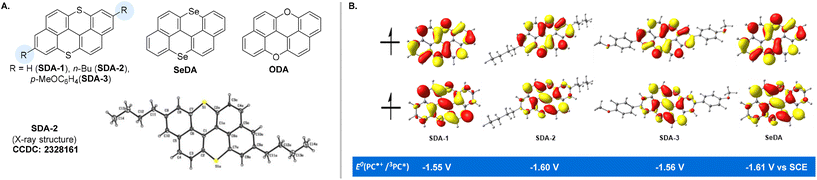 |
| | Fig. 1 (A) Structures of SDA, SeDA and ODA photocatalysts; (B) SOMOs and triplet reducing potentials of SDA and SeDA. | |
Table 1 Photophysical and electrochemical properties of HDPA
| Entry |
PC |
λ
maxa (nm) |
ε (M−1 cm−1) |
λ
em,maxb (nm) |
τ
(ns) |
Φ
|
E
s1 (eV)d |
E
1/2 (PC˙+/PC) |
E
0* (PC˙+/1PC*)e |
|
λ
max (maximum absorption wavelength) measured in DCM.
Emission wavelength and lifetime were recorded by fluorescence spectroscopy in DCM. λex = 450 nm.
Photoluminescence quantum efficiency (Φ) in solution was determined with excitation at the corresponding λmax, and the error of the quantum yield measurement is in the range of 10% (three replicas).
Energy calculated at the lowest energy intersection (λint) between normalized absorption and emission spectra, Es1 = 1240.5/λint.
Excited-state redox potentials were calculated using energies estimated from λint and the experimentally measured E1/2.
|
| 1 |
ODA
|
443 |
17450 |
481 |
5.6 |
0.347 |
2.65 |
0.82 |
−1.83 |
| 2 |
SDA-1
|
468500 |
9500 |
518556 |
5.2 |
0.012 |
2.45 |
0.64 |
−1.81 |
| 3 |
SDA-2
|
469502 |
3700 |
521560 |
8.0 |
0.025 |
2.42 |
0.56 |
−1.86 |
| 4 |
SDA-3
|
470505 |
4700 |
523563 |
6.8 |
0.022 |
2.42 |
0.62 |
−1.80 |
| 5 |
SeDA
|
489 |
7700 |
528 |
2.3 |
0.003 |
2.39 |
0.67 |
−1.72 |
Polymerization of methyl methacrylate
We conducted the initial evaluation of these S- and Se-doped catalysts in the polymerization of methyl methacrylate (MMA) by using DBMM as the initiator and dichloromethane (DCM) as the solvent under the irradiation of blue light emitting diodes (LEDs, λmax = 460 nm). To our delight, in the presence of 0.1 mol% of SDA-1, the polymerization of MMA could proceed in a controlled manner, affording PMMA with controlled molecular weight and low dispersity (Đ 1.31, Table 2, entry 1). As at this catalyst loading, SDA-1 did not completely dissolve in DCM, we thus decreased the loading to 0.01 mol%, and, to our delight, we obtained a faster polymerization and a higher initiator efficiency (entry 2). This result is similar to that of ODA,22 in which lower catalyst loading even gave a better result in terms of conversion; additional fluorescence tests at different photocatalyst concentrations also suggest that high photocatalyst loadings could lead to self-quenching (Fig. S9†) and a decrease of charge transfer to initiators.42–44 Therefore, we kept catalyst loadings at 100 ppm in the subsequent catalyst evaluation (entries 2–5) and solvent examination (entries 6–8). Pleasingly, the alkyl-modified catalyst, SDA-2, could deliver a well-controlled polymerization and a lower dispersity than SDA-1 (entry 3). The aryl-substituted catalyst, SDA-3, gave similar results (entry 4), while SeDA was found to be much less efficient and gave a relatively uncontrolled polymerization, probably due to its instability or inefficiency in deactivation41,42 under these photoirradiation conditions (entry 5). Other solvents like EA, THF, and DMAc were also examined, and PMMA with controlled MW was obtained, though with variable initiator efficiencies (entries 6–8). Notably, DMAc could afford the PMMA product with a much narrower molecular weight distribution, which might be ascribed to a better stabilizing effect of DMAc on the radical cations of the photocatalysts (entry 8). Moreover, the catalyst loading can be further reduced to 50 ppm (entry 9), 10 ppm (entry 10), and even 5 ppm (entry 11), while still maintaining the polymerization in a controlled manner, albeit with higher dispersity. In addition, controlled experiments (entries 12 & 13) suggest that both the light irradiation and catalyst were required and also critical for the good control of polymerization. Notably, the current polymerization with SDA-2 can be conducted under the irradiation of green light (516 nm) in the absence of any external electron donors,45 and polymerization with good control can also be achieved (Mn,GPC 9.9 kg mol−1, Đ 1.25, entry 14). This represents a rare example of O-ATRP catalysts that can afford good control under this low-energy irradiation.46–51 It is worth mentioning that even though ODA showed better catalytic activity than that of SDA under blue light,22ODA delivered uncontrolled polymerizations with a poor initiator efficiency (entries 15 & 16 vs. entry 14) under green light either in DCM (entry 15) or DMAc (entry 16). These results further manifest the advantage of SDA catalysts under longer wavelength, green light irradiation conditions.
Table 2
SDA-catalyzed atom-transfer radical polymerization of MMAa
Polymerization of different monomers
The monomer scope was also examined with the SDA-catalyzed ATRP method. As shown in Table 3, the polymerization of methacrylate monomers, including benzyl methacrylate (BzMA), trifluoroethyl methacrylate (TFEMA), t-butyl methacrylate (TBMA), and 3-(trimethoxysilyl)propyl methacrylate (TSPMA), can proceed in a controlled manner in terms of molecular weight and dispersity (entries 1–8). Notably, these monomers were also successfully polymerized in a controlled manner under green light conditions, although it took a longer time to reach a similar conversion (entries 2, 4, 6, and 8). For acrylates like MA and n-butyl acrylate (BA), a moderately controlled dispersity (Đ 1.47 and 1.57) was observed (entries 9 & 10). Of note, this O-ATRP system could also accommodate hydrophilic monomers like TSPMA and GMA, albeit with a higher dispersity (for more details, please see Table S7†). To our delight, the SDA catalyst was also effective for the polymerization of styrene (St). Furthermore, we also extended this method using SDA derivatives as the photoredox catalysts for the synthesis of poly(acrylonitrile) (PAN) under visible light, and a polymer product with controlled molecular weight together with a low dispersity can also be obtained (entry 12).
Table 3 Investigations on the monomer scope of O-ATRP under blue and green lighta
| Entry |
M |
PC |
Solvent |
Conv.b |
M
n,GPCc |
Đ
|
|
[M]/[DBMM]/[PC] = 100/1/0.01, [M] = 9.4 M, r.t., under the irradiation of blue LEDs (entries 1, 3, 5, 7 & 9–12) or green LEDs (entries 2, 4, 6 & 8), 7–48 h.
Determined by 1H NMR.
M
n,GPC and Đ were measured before precipitation by GPC with PMMA standards using THF as the eluent.
With 6 W green LEDs.
[M]/[DBMM] 50/1.
EBP is used as the initiator.
Polymers precipitated from methanol were used for GPC analysis.
Conv. determined gravimetrically, GPC with PS standards, using DMF as the eluent.
|
| 1 |
BzMA |
SDA-2
|
DMAc |
84% |
14.0 |
1.27 |
| 2d |
BzMA |
SDA-2
|
DMAc |
75% |
14.0 |
1.40 |
| 3 |
TFEMA |
SDA-2
|
DCM |
84% |
16.9 |
1.38 |
| 4d |
TFEMA |
SDA-2
|
DCM |
69% |
19.2 |
1.34 |
| 5 |
TBMA |
SDA-2
|
DCM |
89% |
20.8 |
1.51 |
| 6d |
TBMA |
SDA-2
|
DCM |
66% |
12.3 |
1.45 |
| 7e |
TSPMA |
SDA-3
|
Anisole |
84% |
12.0 |
1.39 |
| 8d,e |
TSPMA |
SDA-2
|
Anisole |
69% |
10.9 |
1.54 |
| 9f |
MA |
SDA-3
|
DMAc |
83% |
12.2 |
1.57 |
| 10f |
BA |
SDA-3
|
DMAc |
75% |
13.9 |
1.47 |
| 11f,g |
St |
SDA-2
|
DMAc |
57% |
13.1 |
1.67 |
| 12g,h |
AN |
SDA-2
|
DMAc |
79% |
12.8 |
1.23 |
Reaction kinetics and chain extension experiments
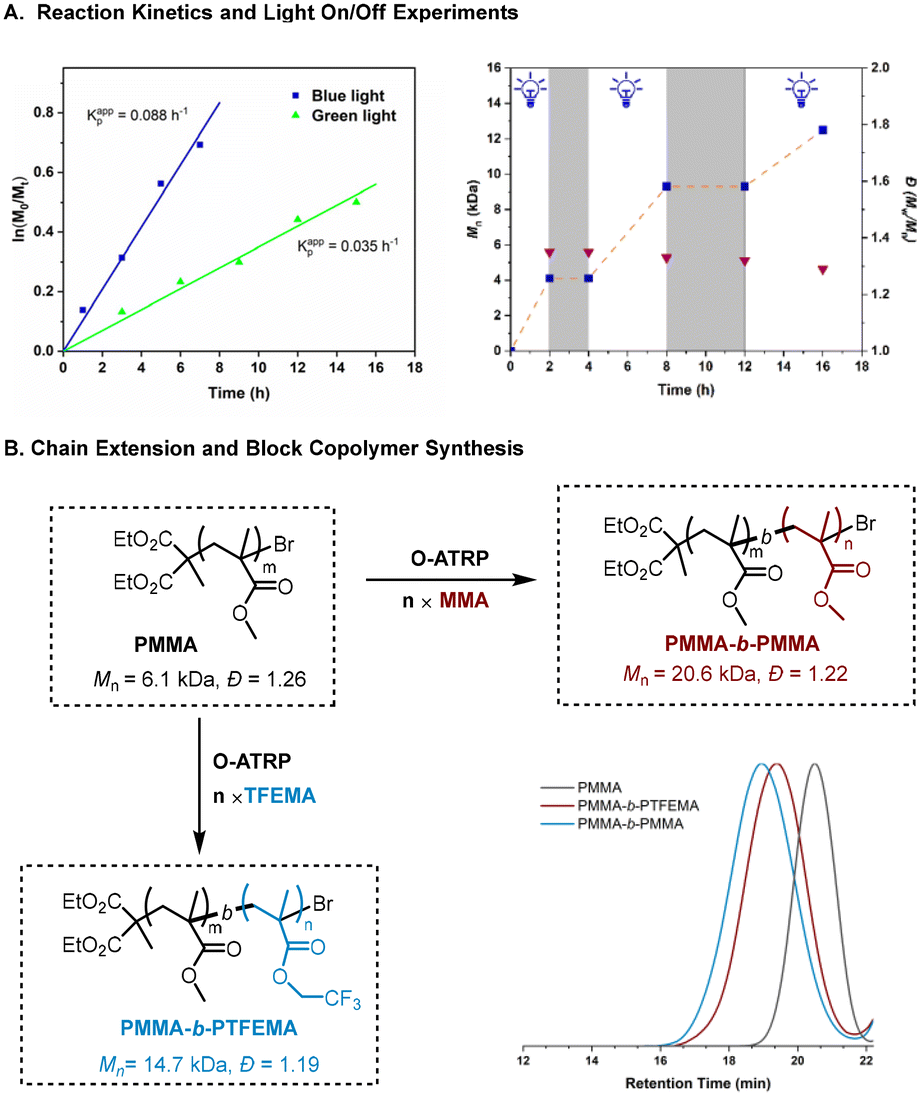
To gain some insight into the polymerization, we examined the reaction kinetics under the irradiation of blue and green light. The kinetic plots in Fig. 2A (left) appear to follow the approximate first-order kinetics under both conditions, while a faster polymerization was achieved under blue LED irradiation than green, as suggested by kappp (blue light) = 0.088 h−1 > kappp (green light) = 0.035 h−1. Furthermore, the light “on/off” experiments show the gradually increased molecular weight, which is in agreement with a “living” feature of the current polymerization. Control over the polymerization by irradiation was also demonstrated using a pulsed-irradiation on–off experiment (Fig. 2A, right). Moreover, chain extension and block copolymerization synthesis were also successfully performed with this O-ATRP method by using the PMMA macro-initiator (Fig. 2B), followed by the addition of MMA and TFEMA, respectively. The GPC traces show a clear shift to higher molecular weight regions in both cases, indicating that well-controlled polymerization and chain-end fidelity were achieved.
 |
| | Fig. 2 Temporal control and kinetic study on 50 ppm SDA-2-catalyzed ATRP of MMA. All polymerizations were performed at a ratio of [MMA]0:[DBMM]0:[SDA-2]0 = 200:1:0.005; the blue line is under blue LED irradiation (460 nm, 30 mW cm−2) and the green line is under green LED irradiation (520 nm, 30 mW cm−2). | |
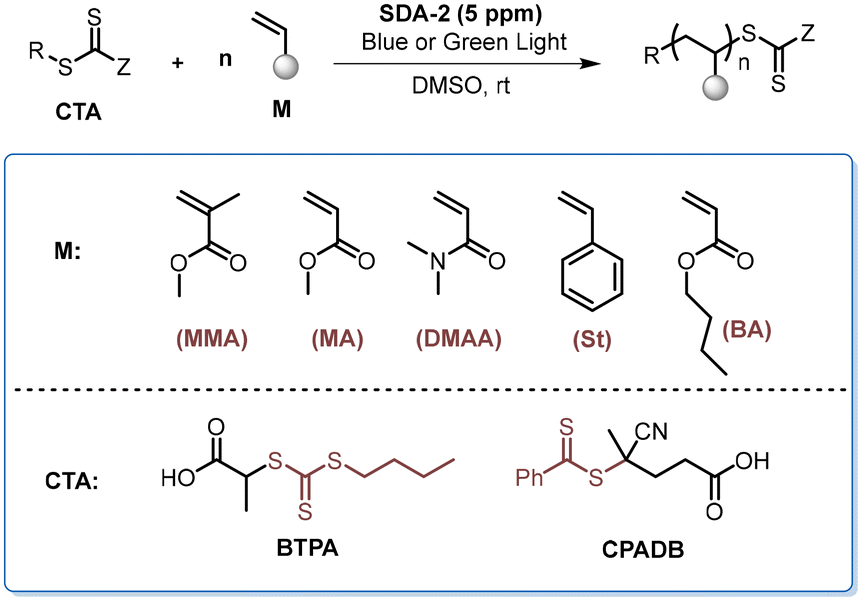
Application of the SDA catalyst in PET-RAFT polymerization
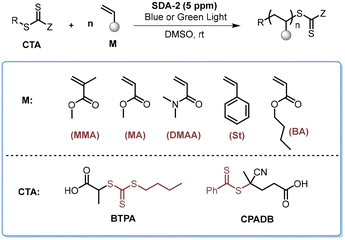
Photoinduced electron/energy transfer reversible addition–fragmentation chain transfer (PET-RAFT) polymerization provides a powerful method for the synthesis of well-defined polymers.52,53 The employment of organic photocatalysts could allow for the synthesis of metal-free polymer products under the irradiation of low-energy lights.54 Therefore, we examined the performance of the newly developed SDA catalysts in the PET-RAFT polymerization of different monomers under the irradiation of blue and green light bulbs. As shown in Table 4, the PET-RAFT polymerization of MMA and BzMA, not unexpectedly, could afford the polymer products with controlled molecular weights and low dispersity (entries 1–3). Notably, the polymerizations of MA, BA, St, and N,N-dimethylacrylamide (DMAA) all proceeded well in the presence of 5 ppm of SDA-2 as the catalyst (entries 4–11). The polymerization of St was slower, while over 50% conversion of St was attained with SDA-2 as the catalyst after 48 hours of irradiation (entries 10 & 11), with ∼100% initiator efficiency and a brilliant control over the molecular weight and molecular weight distribution (entry 11).
Table 4 Further extension of SDA photocatalysts to the PET-RAFT polymerization under blue or green lighta
|
|
| Entry |
M |
CTA |
L.S. |
Conv.b |
M
n,GPCc |
Đ
|
|
Reaction conditions: [M]0/[CTA]0/[SDA-2]0 = 100/1/0.0005, [M]0 = 9.4 M in DMSO under irradiation with a 460 nm blue bulb (3 mW cm−2), r.t., 6–48 h.
Determined by 1H NMR.
kg mol−1, determined by GPC analysis (THF, PMMA standards) before precipitation, entries 8–11 refer to the PS standard.
Measured by GPC with PS standards using DMF as the eluent.
48 h. L.S. light source. B = blue, G = green.
|
| 1 |
MMA |
CPADB |
B |
65% |
8.7 |
1.10 |
| 2 |
MMA |
CPADB |
G |
70% |
9.8 |
1.14 |
| 3 |
BzMA |
CPADB |
B |
82% |
12.0 |
1.14 |
| 4 |
MA |
BTPA |
B |
88% |
6.9 |
1.13 |
| 5 |
MA |
BTPA |
G |
97% |
10.7 |
1.09 |
| 6 |
BA |
BTPA |
B |
67% |
10.5 |
1.06 |
| 7 |
BA |
BTPA |
G |
70% |
9.7 |
1.11 |
| 8 |
DMAA |
BTPA |
B |
87% |
12.0d |
1.04d |
| 9 |
DMAA |
BTPA |
G |
84% |
12.9d |
1.07d |
| 10e |
St |
BTPA |
B |
54% |
5.9 |
1.34 |
| 11e |
St |
BTPA |
G |
59% |
6.4 |
1.15 |
Conclusions
In summary, based on the photocatalyst design logic of heteroatom-doping of polycyclic arenes (HDPA), sulfur-doped anthanthrenes (SDAs) have been successfully identified as a new family of photoredox catalysts for the development of metal-free O-ATRP and PET-RAFT polymerization. In contrast, the selenium-doped catalyst (SeDA) gave a poorly controlled polymerization with a low initiator efficiency under the same ATRP conditions, while the oxygen-doped one (ODA) failed to afford a controlled polymerization under the same conditions with green light. Of note, SDA could well promote the polymerization of various acrylate, methacrylate, and acrylonitrile monomers in a controlled manner at 10–100 ppm catalyst loadings under the irradiation of visible light, even green light. Further utilization of SDA photocatalysts in the PET-RAFT polymerization under both blue and green light, as well as block copolymer synthesis, is also demonstrated with success at low ppm of catalyst loading. Further application of SDA catalysts in photoinduced polymerizations and organic reactions in the future can be anticipated.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We gratefully acknowledge the Recruitment Program of Global Experts, the National Natural Science Foundation of China (22371240 & 22361132535), the Hundred-Talent Project of Fujian, Fuzhou University, and Xiamen University for the financial support.
References
- K. c. Matyjaszewski and N. V. Tsarevsky, J. Am. Chem. Soc., 2014, 136, 6513–6533 CrossRef CAS PubMed
 .
.
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Prog. Polym. Sci., 2020, 111, 101311 CrossRef CAS .
- F. A. Leibfarth, K. M. Mattson, B. P. Fors, H. A. Collins and C. J. Hawker, Angew. Chem., Int. Ed., 2013, 52, 199–210 CrossRef CAS PubMed .
- P. Xiao, J. Zhang, F. Dumur, M. A. Tehfe, F. Morlet-Savary, B. Graff, D. Gigmes, J. P. Fouassier and J. Lalevée, Prog. Polym. Sci., 2015, 41, 32–66 CrossRef CAS .
- X. Pan, M. A. Tasdelen, J. Laun, T. Junkers, Y. Yagci and K. Matyjaszewski, Prog. Polym. Sci., 2016, 62, 73–125 CrossRef CAS .
- M. Chen, M. Zhong and J. A. Johnson, Chem. Rev., 2016, 116, 10167–10211 CrossRef CAS PubMed .
- S. Dadashi-Silab, S. Doran and Y. Yagci, Chem. Rev., 2016, 116, 10212–10275 CrossRef CAS PubMed .
- N. Corrigan, J. Yeow, P. Judzewitsch, J. Xu and C. Boyer, Angew. Chem., Int. Ed., 2019, 58, 5170–5189 CrossRef CAS PubMed .
- P. Garra, J. P. Fouassier, S. Lakhdar, Y. Yagci and J. Lalevée, Prog. Polym. Sci., 2020, 107, 101277 CrossRef CAS .
- C. Aydogan, G. Yilmaz, A. Shegiwal, D. M. Haddleton and Y. Yagci, Angew. Chem., Int. Ed., 2022, 61, e202117377 CrossRef CAS PubMed .
- N. J. Treat, H. Sprafke, J. W. Kramer, P. G. Clark, B. E. Barton, J. Read de Alaniz, B. P. Fors and C. J. Hawker, J. Am. Chem. Soc., 2014, 136, 16096–16101 CrossRef CAS PubMed .
- J. Theriot, C.-H. Lim, H. Yang, M. Ryan, C. Musgrave and G. Miyake, Science, 2016, 352, 1082–1086 CrossRef CAS PubMed .
- J. Xu, K. Jung, A. Atme, S. Shanmugam and C. A. Boyer, J. Am. Chem. Soc., 2014, 136, 5508–5519 CrossRef CAS PubMed .
- M. L. Allegrezza and D. Konkolewicz, ACS Macro Lett., 2021, 10, 433–446 CrossRef CAS PubMed .
- K. Jung, N. Corrigan, M. Ciftci, J. Xu, S. E. Seo, C. J. Hawker and C. Boyer, Adv. Mater., 2020, 32, e1903850 CrossRef PubMed .
- J. Kreutzer and Y. Yagci, Polymers, 2018, 10, 23–30 Search PubMed .
- D. A. Corbin and G. M. Miyake, Chem. Rev., 2022, 122, 1830–1874 CrossRef CAS PubMed .
- G. M. Miyake and J. C. Theriot, Macromolecules, 2014, 47, 8255–8261 CrossRef CAS .
- N. Zivic, M. Bouzrati-Zerelli, A. Kermagoret, F. Dumur, J.-P. Fouassier, D. Gigmes and J. Lalevée, ChemCatChem, 2016, 8, 1617–1631 CrossRef CAS .
- E. H. Discekici, A. Anastasaki, J. Read De Alaniz and C. J. Hawker, Macromolecules, 2018, 51, 7421–7434 CrossRef CAS .
- C. Wu, N. Corrigan, C.-H. Lim, W. Liu, G. Miyake and C. Boyer, Chem. Rev., 2022, 122, 5476–5518 CrossRef CAS PubMed .
- Q. Ma, J. Song, X. Zhang, Y. Jiang, L. Ji and S. Liao, Nat. Commun., 2021, 12, 429 CrossRef CAS PubMed .
- J. Kreutzer, Nat. Rev. Chem., 2021, 5, 73 CrossRef CAS PubMed .
- Q. Ma, X. Zhang, Y. Jiang, J. Lin, B. Graff, S. Hu, J. Lalevée and S. Liao, Polym. Chem., 2022, 13, 209–219 RSC .
- X. Zhang, Y. Jiang, Q. Ma, S. Hu and S. Liao, J. Am. Chem. Soc., 2021, 143, 6357–6362 CrossRef CAS PubMed .
- Z. Yang, J. Chen and S. Liao, ACS Macro Lett., 2022, 11, 1073–1078 CrossRef CAS PubMed .
- G. Wang, H. Shao, J. Ma and S. Liao, Macromol. Chem. Phys., 2023, 224, 2200382 CrossRef CAS .
- R. M. Pearson, C.-H. Lim, B. G. McCarthy, C. B. Musgrave and G. M. Miyake, J. Am. Chem. Soc., 2016, 138, 11399–11407 CrossRef CAS PubMed .
- C.-H. Lim, M. D. Ryan, B. G. McCarthy, J. C. Theriot, S. M. Sartor, N. H. Damrauer, C. B. Musgrave and G. M. Miyake, J. Am. Chem. Soc., 2017, 139, 348–355 CrossRef CAS PubMed .
- J. C. Theriot, G. M. Miyake and C. A. Boyer, ACS Macro Lett., 2018, 7, 662–666 CrossRef CAS PubMed .
- V. K. Singh, C. Yu, S. Badgujar, Y. Kim, Y. Kwon, D. Kim, J. Lee, T. Akhter, G. Thangavel, L. S. Park, J. Lee, P. C. Nandajan, R. Wannemacher, B. Milián-Medina, L. Lüer, K. S. Kim, J. Gierschner and M. S. Kwon, Nat. Catal., 2018, 1, 794–804 CrossRef CAS .
- A. Köhler and H. Bässler, Mater. Sci. Eng., R, 2009, 66, 71–109 CrossRef .
- S. P. Mcglynn, F. J. Smith and G. Cilento, Photochem. Photobiol., 1964, 3, 269 CrossRef CAS .
- Z. Liu, W. Han, J. Lan, L. Sun, J. Tang, C. Zhang and J. You, Angew. Chem., Int. Ed., 2023, 62, e202211412 CrossRef CAS PubMed .
- O. Matuszewska, T. Battisti, R. R. Ferreira, N. Biot, N. Demitri, C. Mézière, M. Allain, M. Sallé, S. Mañas-Valero, E. Coronado, E. Fresta, R. D. Costa and D. Bonifazi, Chem. – Eur. J., 2023, 29, e202203115 CrossRef CAS PubMed .
- H. Roth, N. Romero and D. Nicewicz, Synlett, 2015, 714–723 Search PubMed .
- D. Stassen, N. Demitri and D. Bonifazi, Angew. Chem., Int. Ed., 2016, 55, 5947–5951 CrossRef CAS PubMed .
- C. Pezzetta, A. Folli, O. Matuszewska, D. Murphy, R. W. M. Davidson and D. Bonifazi, Adv. Synth. Catal., 2021, 363, 4740–4753 CrossRef CAS .
- S. M. Winter, S. Hill and R. T. Oakley, J. Am. Chem. Soc., 2015, 137, 3720–3730 CrossRef CAS PubMed .
- C. Huang, X. Liao, K. Gao, L. Zuo, F. Lin, X. Shi, C.-Z. Li, H. Liu, X. Li, F. Liu, Y. Chen, H. Chen and A. K.-Y. Jen, Chem. Mater., 2018, 30, 5429–5434 CrossRef CAS .
- D. R. Lee, J. Park and J. Y. Lee, Org. Electron., 2022, 106, 106534 CrossRef CAS .
- A. Allushi, S. Jockusch, G. Yilmaz and Y. Yagci, Macromolecules, 2016, 49, 7785–7792 CrossRef CAS .
- A. Breder and C. Depken, Angew. Chem., Int. Ed., 2019, 58, 17130–17147 CrossRef CAS PubMed .
- D. A. Corbin, C. Cremer, K. O. Puffer, B. S. Newell, F. W. Patureau and G. M. Miyake, ChemCatChem, 2022, 14, e202200485 CrossRef CAS PubMed .
- J. Kreutzer and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3475–3482 CrossRef CAS .
- X. Liu, L. Zhang, Z. Cheng and X. Zhu, Polym. Chem., 2016, 7, 689–700 RSC .
- C. Kutahya, F. S. Aykac, G. Yilmaz and Y. Yagci, Polym. Chem., 2016, 7, 6094–6098 RSC .
- N. El Achi, Y. Bakkour, W. Adhami, J. Molina, M. Penhoat, N. Azaroual, L. Chausset-Boissarie and C. Rolando, Front. Chem., 2020, 8, 740 CrossRef CAS PubMed .
- S. Dadashi-Silab, F. Lorandi, M. J. DiTucci, M. Sun, G. Szczepaniak, T. Liu and K. Matyjaszewski, J. Am. Chem. Soc., 2021, 143, 9630–9638 CrossRef CAS PubMed .
- W.-W. Fang, G.-Y. Yang, Z.-H. Fan, Z.-C. Chen, X.-L. Hu, Z. Zhan, I. Hussain, Y. Lu, T. He and B.-E. Tan, Nat. Commun., 2023, 14, 2891 CrossRef CAS PubMed .
- W. Wang, Z. Zhou, D. Sathe, X. Tang, S. Moran, J. Jin, F. Haeffner, J. Wang and J. Niu, Angew. Chem., Int. Ed., 2022, 61, e202113302 CrossRef CAS PubMed .
- Y. Lee, C. Boyer and M. S. Kwon, Chem. Soc. Rev., 2023, 52, 3035–3097 RSC .
- C. Wu, N. Corrigan, C.-H. Lim, K. Jung, J. Zhu, G. Miyake, J. Xu and C. Boyer, Macromolecules, 2019, 52, 236–248 CrossRef CAS PubMed .
- N. P. Truong, G. R. Jones, K. G. E. Bradford, D. Konkolewicz and A. Anastasaki, Nat. Rev. Chem., 2021, 5, 859–869 CrossRef CAS PubMed .
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 b,
Xun
Zhang
a,
Jianxu
Chen
c and
Saihu
Liao
b,
Xun
Zhang
a,
Jianxu
Chen
c and
Saihu
Liao