
Photoinduced morphology change in ionic supramolecular block copolymer†
Kseniia M.
Karnaukh‡
 ab,
Shuyi
Xie‡
bc,
Kai-Chieh
Yang
d,
Komal
Komal
a,
Rachel A.
Segalman
*abcd and
Javier
Read de Alaniz
*a
ab,
Shuyi
Xie‡
bc,
Kai-Chieh
Yang
d,
Komal
Komal
a,
Rachel A.
Segalman
*abcd and
Javier
Read de Alaniz
*a
aDepartment of Chemistry and Biochemistry, University of California, Santa Barbara, California 93106, USA. E-mail: javier@chem.ucsb.edu; segalman@ucsb.edu
bMaterials Research Laboratory, University of California, Santa Barbara, California 93106, USA
cMitsubishi Chemical Center for Advanced Materials, University of California, Santa Barbara, California 93106, USA
dDepartment of Chemical Engineering, University of California, Santa Barbara, California 93106, USA
First published on 2nd September 2024
Abstract
Physically mixing two dissimilar polymers often results in a macroscopically segregated blend with poor optical clarity and mechanical properties. Previously, we demonstrated that chain end functionalization of immiscible polymer blends with oppositely paired acid and base groups leads to an ionic supramolecular block copolymer with electrostatically stabilized microdomains that suppress macroscopic phase separation. In this work, a functionalized polydimethylsiloxane with a photochromic diarylethene (DAE) end-group (PDMS-ω-DAE) is blended with a sulfonic acid end-functionalized polystyrene (PS-ω-SO3H), forming an ionic supramolecular block copolymer with tunable morphology by light. When the DAE is irradiated with UV light, it triggers an isomerization from the ring-opened (DAE–O) to the ring-closed (DAE–C) form. The light-induced conformational change in the chain-end group chemistry substantially alters the ionic junctions, leading to a phase structure difference in the solid-state from hexagonally packed cylinders (HEX) to lamellae (LAM). After UV radiation, the more localized positive charge on DAE–C led to stronger ionic bonds between the two dissimilar blocks, while the repulsion between adjacent DAE–C groups increased. The stronger electrostatic repulsion along the interface resulted in increased interfacial area per polymer chain and thus induced such phase transition. The light-induced phase transition of this system demonstrates that the ionic interactions can be tuned on-demand to create different morphologies from a single polymer blend.
Introduction
Physically mixing A and B homopolymers typically results in an incompatible blend that macroscopically phase separates due to the small entropy of mixing that scales with 1/N, where N is the degree of polymerization.1–3 Linking A and B chains end-to-end with covalent bonds forms block copolymers and prevents macrophase separation. Strongly segregated diblock copolymers may self-assemble into nanostructures where the covalent junctions reside along the A/B microdomain interfaces, and the morphology is determined by the architecture of the copolymer and the volume fraction of each block.4–6 Contrary to conventional block copolymers, supramolecular copolymers form in situ upon blending the two homopolymer “building blocks”.7,8 This approach allows for easy modification of the chain architecture and molecular weight by tuning the identity and blending ratio of the building blocks while bypassing the synthetic challenges in conventional copolymers.Ionic interactions between positive and negative charges that are tethered on the A and B homopolymer chain ends offer promising potential in constructing copolymers that can self-assemble into nanostructures due to the tunable bond energy and hetero-complementarity (only cation/anion binding is possible). Previous experimental and theoretical work,9–20 including ours,21 have demonstrated that blending chain-end functionalized A and B polymers with opposite charges leads to block copolymers that self-assemble into lamellar,14 double gyroid,15 and cylindrical morphologies.17 Moreover, the dynamic nature of the ionic bond offers a convenient tool to manipulate the ionic supramolecular copolymer self-assembly, as it is dictated by the competition between the effective Coulomb attraction (h) and segregation strength of the blend (χN).16 Strongly bonded blends (high h/χN) resemble conventional block copolymers. Increasing temperature or adding ionic species can disrupt the ionic interactions (decrease in h), resulting in weakly bonded blends (low h/χN) where swollen microphases and a potential phase transition were observed.19,21
Strategies that involve chemical modification to the end group chemistry of each building block have been used to adjust the thermodynamics of mixing, leading to richer phase behavior compared to conventional block copolymers. For example, Luo and coworkers found that when the neutral junction of a polydimethylsiloxane-block-poly(ethylene oxide) (PDMS-b-PEO) or polydimethylsiloxane-block-poly(methyl methacrylate) (PDMS-b-PMMA) diblock copolymer is replaced with a 1,2,3-triazolium ionic junction, the segregation strength increases, leading to an increase in the order–disorder transition temperature.22 Park and coworkers demonstrated that block copolymer assembly is sensitive to the end group and junction chemistry, and by judicious control of the secondary interactions between the functional groups, phase transition can be induced. Notably, this approach enabled access to the rare A15 phase and the recently discovered Im![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m structure (plumber's nightmare), which does not exist for any unfunctionalized block copolymers due to the large packing frustration.23–26
m structure (plumber's nightmare), which does not exist for any unfunctionalized block copolymers due to the large packing frustration.23–26
These approaches enable access to a rich range of morphologies, but controlling microstructures with an external stimulus in supramolecular polymers has not been extensively explored until recently, Yue et al. reported a far-from-equilibrium self-assembly system where a diblock copolymer is blended with a photoexcitation-induced aggregation molecule that selectively attaches to one of the blocks via supramolecular interactions. Upon UV radiation followed by solvent annealing, the conformational change of the aggregation molecule leads to a shift in the phase-volume, resulting in continuous morphology change from lamellae to gyroids, cylinders, and finally to spheres.27
Here, we propose a facile methodology to construct microstructured binary polymer blends (without any small molecule additives or a covalent diblock copolymer) where dissimilar homopolymer chains are linked by ionic bonds. Controlling the dynamic nature of the ionic junctions with light enables a photoexcitation-induced phase transition. Such a swift and non-invasive approach for tuning the phase behavior of simple polymer blends has the potential to apply to many polymeric systems where microstructures can be tuned on demand. We achieve this by introducing a photochromic diarylethene end-group (DAE) to polydimethylsiloxane (PDMS). The functionalized polymer (PDMS-ω-DAE, Mn = 7.3 kDa, Đ = 1.10) acts as a “photobase” that can react with a sulfonic acid end-functionalized polystyrene (PS-ω-SO3H, Mn = 4.1 kDa, Đ = 1.14), resulting in a PDMS-b-PS like copolymer with an ionic junction that self-assembles into hexagonally packed cylinders. Irradiation of the blend with UV light triggers isomerization from the ring-opened (DAE–O) to the ring-closed (DAE–C) form that disrupts the aromaticity of the imidazolium ring (4n + 2π electrons).28–30 The more localized positive charge on DAE–C leads to stronger ionic bonds at the block junctions, inducing a phase transition from hexagonal (HEX) to lamellae (LAM). This example of a bulk microphase order–order transition in polymer blends, induced by light stimuli, exemplifies a novel and versatile approach for constructing adjustable polymer morphologies, obviating the need for synthesizing new copolymers.
Results and discussion
PDMS-ω-DAE was synthesized from the vinyl-terminated PDMS (Mn = 6.7 kDa, Đ = 1.09), as shown in Fig. 1. In the first step, PDMS was functionalized with 3-mercaptopropanyl-N-hydroxysuccinimide ester through thiol–ene click reaction, followed by coupling with diarylethene molecule bearing primary amine group (DAE-NH2). The synthesis of the DAE-NH2 was modified from previous works and obtained in five steps.31,32 Detailed synthetic procedures are provided in ESI.† To form an ionic copolymer with an SO3−/DAE-H+ junction point, desired amounts of PDMS-ω-DAE (10 wt% in THF) and PS-ω-SO3H (10 wt% in THF) were mixed at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio between DAE and SO3H end groups for two hours at room temperature. The described blending process facilitated a proton transfer from the acid to the base group, forming the PS-ω-SO3−/PDMS-ω-DAE-H+ ionic block copolymer. The formation of DAE-H+ is confirmed by 1H NMR spectroscopy (Fig. S7†).
:1 molar ratio between DAE and SO3H end groups for two hours at room temperature. The described blending process facilitated a proton transfer from the acid to the base group, forming the PS-ω-SO3−/PDMS-ω-DAE-H+ ionic block copolymer. The formation of DAE-H+ is confirmed by 1H NMR spectroscopy (Fig. S7†).
 | ||
| Fig. 1 Synthesis of PS-ω-SO3−/PDMS-ω-DAE-H+ ionic block copolymer. Reagents and conditions: (i) 3-mercaptopropanyl-N-hydroxysuccinimide ester, 2,2-dimethoxy-2-phenylacetophenone (cat.), 365 nm, THF, rt, N2, 5 h (ii) DAE-NH2, triethylamine, DCM, rt, 36 h. | ||
The PS-ω-SO3−/PDMS-ω-DAE-H+ ionic copolymer was characterized by UV-Vis spectroscopy to monitor the photoswitching properties of the ionic blend. Upon irradiation with UV light, the solution quickly turned dark blue due to the isomerization of DAE-H+–O to the DAE-H+–C form (Fig. 2a). The absorption peaks around 380 nm and 660 nm are characteristic of the ring-closed form (Fig. 2b).28,30 The isomerization kinetics was studied by time-dependent UV-Vis spectroscopy, as demonstrated in Fig. 2c. The maximum absorption at 660 nm was achieved after 5 min radiation using 300 nm low-intensity light, and the intensity decayed slowly in a dark environment due to thermal recovery of the DAE molecule (≈10 h).
 | ||
| Fig. 2 (a) Photochromism of PS-ω-SO3−/PDMS-ω-DAE-H+ blend in 0.025 mM THF solution. (b) Time-dependent UV-Vis spectroscopy of PS-ω-SO3−/PDMS-ω-DAE-H+ blend in THF solution. Kinetic traces of blend during initial exposure to 300 nm light (≈1.32 mW cm−2) for 5 min, followed by subsequent thermal recovery in the dark (≈10 hours). | ||
Next, the blend solutions (10 wt% in THF), before and after irradiation, were cast on PTFE films (McMaster-Carr) and allowed to dry in a dark environment for two hours. Note that most THF was evaporated and the PS domain was vitrified within 20 minutes, freezing the morphology. The samples were then transferred to a high vacuum oven equipped with a turbo pump (∼10−8 torr) and dried for 12 hours at room temperature, followed by another 12 hours at 60 °C. The samples were slowly cooled to room temperature and stored in an argon-filled glovebox before solid-state characterization.
The solid-state blends were analyzed by small-angle X-ray scattering (SAXS) and transmission electron microscopy (TEM) techniques to access the associated microstructure before and after irradiation. Due to the incompatibility between the PS and PDMS blocks (Flory–Huggins parameter χ = 0.095 at 25 °C and χN = 18, with a reference volume Vref = 0.1 nm3),33 after drying, the solid-state copolymer segregated into microstructures. Transmission electron microscopy revealed a vertical cylindrical morphology of the pristine blend (the darker areas correspond to PDMS-rich domains), and the corresponding small-angle X-ray scattering profile demonstrated identifiable Bragg reflections for HEX, with relative peak positions at q/q* = √3, √4 (Fig. 3a). However, higher-order peaks are broad, possibly due to the non-ideal ordering of the cylinders. After UV treatment and casting on a PTFE film, the blend self-assembled into a LAM morphology, and typical Bragg peaks at q/q* = 2, 3, 4 were observed in the SAXS profile (Fig. 3b). Note that the 2q* peak intensity is lower than the 3q* peak, which is unusual in conventional compositionally asymmetric diblock copolymers but similar to the SAXS patterns of lyotropic lamellar phases in hydrated diblock copolymers34 and diblock copolymer/ionic liquid mixtures.35 In fact, the near absence of the 2q* peak suggests that the ionic junctions (SO3−/DAE-H+) may form a thin layer with a higher electron density compared to the PS and PDMS blocks, offering X-ray contrasts with respect to the two domains. Such thin layer results in a three-layer lamellar structure (LAM-3) similar to what has been reported in ABC terpolymers.36,37
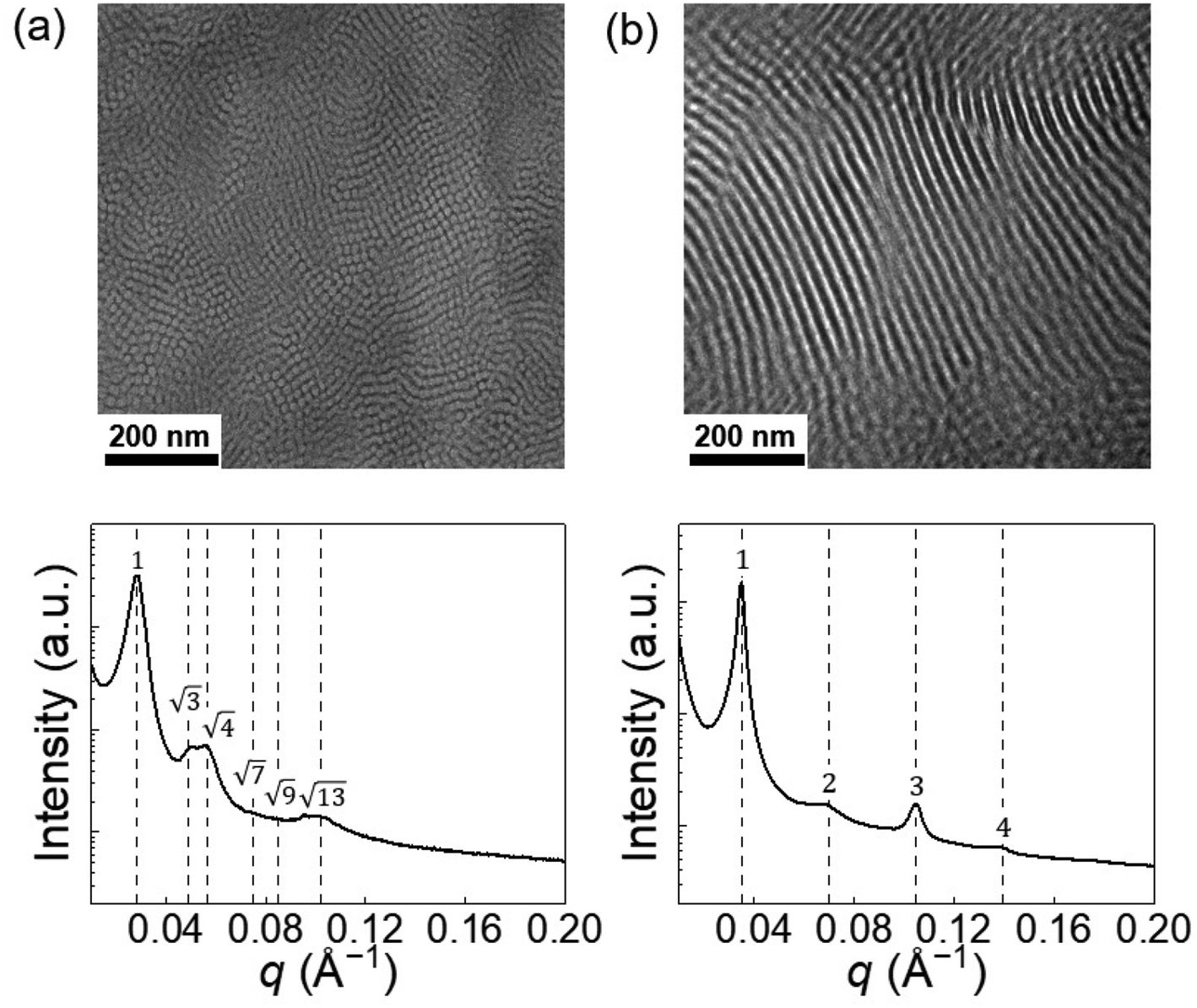 | ||
| Fig. 3 The PS-ω-SO3−/PDMS-ω-DAE-H+–O blend melt (a) self-assembles into HEX microstructure while the UV-radiated blend PS-ω-SO3−/PDMS-ω-DAE-H+–C (b) segregates into LAM microstructure. | ||
The HEX to LAM phase transition is intriguing since there is no change in block composition (fPS = 0.34 for both cases), and the change in χN can be neglected. Thus, we speculate that the order–order transition is related to the change in the ionic junction, which we attribute to the bulky SO3−/DAE-H+ functional group that takes up ca. 4 vol% of the supramolecular diblock copolymer. For direct comparison, we synthesized a PS-b-PDMS copolymer (Fig. S8†) by clicking a thiol-terminated PS (PS-ω-SH, Mn = 3.9 kDa) onto a vinyl-terminated PDMS (PDMS-ω-vinyl, Mn = 6.7 kDa). The total molar mass is slightly lower than the ionic blend, but the volume fraction of the PS block is comparable between the two cases (Table 1). The conventional copolymer demonstrates a LAM morphology (Fig. S13†) with a domain size D = 2π/q* = 16.7 nm. When the covalent bond between PS and PDMS (in this case, the –S– bond) is replaced by the bulky SO3−/DAE-H+ groups, the interface becomes more crowded, resulting in a cylindrical morphology and a curve toward the PS domain (Fig. 4). The interfacial areas per copolymer chain (∑chain) can be calculated by assuming the densities of the copolymer and ionic blend are the same and all chains pack into perfectly ordered LAM and HEX lattices. Using the lattice parameters determined by SAXS, the interfacial areas per copolymer chain (∑chain) can be calculated (Fig. S14 and S15†). The ∑chain of the cylindrical ionic blend (1.76 nm2 per chain) is 17% lower than that of the conventional lamellar copolymer (2.11 nm2 per chain). After isomerization, the electrostatic attraction between SO3−/DAE-H+–C increases, while the repulsion between two adjacent DAE-H+–C groups also increases, leading to a HEX to LAM transition accompanied by a recovery in ∑chain to 2.11 nm2 per chain, close to the value of conventional PS-b-PDMS copolymer.
 | ||
| Fig. 4 The stronger cation/cation and anion/anion repulsion on the PS-ω-SO3−/PDMS-ω-DAE-H+–C blend result in increased interfacial area per chain that leads to a phase transition from (a) a HEX phase (non-UV treated) to (b) a LAM phase (after UV treatment). | ||
| Component | M n of PS (kg mol−1) | M n of PDMS (kg mol−1) | M n of ionic junction (kg mol−1) | N | χN |
f
PSc |
|---|---|---|---|---|---|---|
| a Total volumetric degree of polymerization based on a reference volume Vref = 0.1 nm3. b Calculated at 25 °C where χ = 0.095 (Vref = 0.1 nm3) [ref. 33]. c Calculated assuming the density of PS and PDMS is 1.04 and 0.965 g cm−3, respectively. | ||||||
| PS-b-PDMS | 3.9 | 6.7 | — | 178 | 16.9 | 0.35 |
| PS-ω-SO3−/PDMS-ω-DAE-H+ | 4.1 | 6.9 | 0.4 | 191 | 18.1 | 0.34 |
Unlike covalent junctions in conventional block copolymers, the phase behaviors of ionic blend junctions are governed by the strength of the ionic bonds, which is temperature-dependent.19 Similar to hydrogen bonds, the effective ionic bond energy between the acid and base groups decreases as temperature increases, revealed by an upfield shift of the imidazole proton in 1H NMR.21,38 Such acid/base interactions are not permanent and sometimes are referred to as ionic hydrogen bonds.39 At elevated temperatures, reverse proton transfer may occur, producing the two charge-neutral homopolymers (acid and base forms).19 These neutral species will detach from the block copolymer interfaces and substantially change the microstructure of the blend, resulting in macroscopic phase separation. As shown in Fig. 5a, heating the conventional PS-b-PDMS copolymer resulted in a slight increase in the domain size (D = 2π/q*) from 16.7 to 18.4 nm, from 25 to 140 °C. Note that χ is not strongly temperature dependent for PS-b-PDMS, and the increase in D can be ascribed to thermal expansion. In comparison, for a non-photoswitchable blend, PS-ω-SO3−/PDMS-ω-imidazolium, where the PDMS was end-functionalized with an imidazole group, D substantially increased before diverging at around 100 °C, resulting in macroscopic phase separation. As discussed in our previous work, the increase in D is due to the reduced bond energy (h) and acid/base bond breaking.21
 | ||
| Fig. 5 (a) Self-assembled ionic PS/PDMS blends and PS-b-PDMS diblock copolymer microstructure domain sizes (D) as a function of temperature. (b) Photoswitchable PS-ω-SO3−/PDMS-ω-DAE-H+–(O and C) and PS-ω-SO3−/PDMS-ω-Im-H+ blends domain sizes are normalized by that of the PS-b-PDMS (D/D0). Accounting for the slight difference in molecular weight of the PS-b-PDMS diblock, D0 has been adjusted to that of the same molecular weight of the photoswitchable blends. | ||
To quantify the influence of temperature on the stability of the ionic junctions, we normalize the domain size of the blends (D) by that of the conventional block copolymer (D0) (Fig. 5b). Note that D0 has been adjusted to account for the difference in molecular weights between the PS-b-PDMS diblock and the photoswitchable blends, assuming D ∼ N2/3. The DAE group can be regarded as a functionalized imidazole group, and the PS-ω-SO3−/PDMS-ω-DAE-H+ blend demonstrates better thermal stability than the corresponding PS-ω-SO3−/PDMS-ω-imidazolium blend. We attribute this to the additional sterics of the DAE-H+ group compared to non-substituted imidazolium molecule. Recently, Čorić and coworkers emphasized that improving the directionality of ionic bonds can influence their strength, which is associated with more linear geometry and shorter distances.40 We speculate that the additional bulkiness of DAE groups associated with phenyl-substituted thiophenes rings improves the directionality of ionic bonds and, as a result, their bond strength and thermal stability. At 140 °C, the reduced domain size D/D0 of the photoswitchable blend increases by less than 25%, while the non-photoswitchable blend is already macroscopically phase separated. Besides temperature, the domain size of the PS-ω-SO3−/PDMS-ω-DAE-H+ blend can also be adjusted by light. Previously, our group confirmed that this change in electron density in photoresponsive polymeric ionic liquids leads to a stronger association of counter anions in the closed form, which was demonstrated by the modulation of conductivity.31,41 After UV radiation, the blend's domain size is systematically smaller than that of the non-treated blend. This temperature/light dual tunability may be leveraged to construct microphases with a wide characteristic dimension range, bypassing synthesizing block copolymers with various molar masses.
Conclusion
In this study, we reported a facile strategy to construct a supramolecular diblock copolymer by reacting a PS homopolymer end-functionalized with a sulfonic acid group and a PDMS homopolymer end-functionalized with a photoresponsive diarylethene (DAE) group. After proton transfer from the SO3H to the DAE group, the two dissimilar homopolymers are joined by the SO3−/DAE-H+ ion pair and self-assemble into a cylindrical microstructure. Such ionic supramolecular system has substantial dynamic features: (1) the ionic bond strength and, thus, the interfacial strength are subject to the end group/junction chemistry, which can be readily controlled by UV light irradiation; (2) the ionic attraction competes with the repulsion due to the inherent incompatibility of the two homopolymers to reach a dynamic phase equilibrium that is sensitive to temperature. Thus, the system has dual tunability by light and heat. First, after UV radiation, a cylinder-to-lamellae phase transition was observed due to the change in the interfacial area. Second, upon heating, the microstructure domains are swollen due to the dissociation of the ionic block copolymer and the release of the homopolymers from the interfaces. Leveraging controllable end-group photochemistry, we anticipate that such methodology can be applied to construct microstructured functional polymeric materials with a wide tunability window.Data availability
Data for this article entitled “Photoinduced Morphology Change in Ionic Supramolecular Block Copolymer” will be made available at Dryad.Conflicts of interest
All authors declare that they have no conflicts of interest.Acknowledgements
This work was supported by the MRSEC Program of the National Science Foundation under award no. DMR-2308708 (IRG-1). The research reported here made use of the shared facilities of the Materials Research Science and Engineering Center (MRSEC) at UC Santa Barbara: NSF DMR-2308708. The UC Santa Barbara MRSEC is a member of the Materials Research Facilities Network (https://www.mrfn.org). X-ray scattering experiments were conducted at the National Synchrotron Light Source II (NSLS-II, beamline 11-BM, Brookhaven National Laboratory). We thank our beamline scientist, Dr Ruipeng Li, for help with the experiment setup and discussion. K. M. K. and S. X. thank Elizabeth Murphy for assistance in PS-b-PDMS purification and Jie Xu (Texas A&M University) for assistance in MALDI-TOF characterization.References
- R. W. Clarke, T. Sandmeier, K. A. Franklin, D. Reich, X. Zhang, N. Vengallur, T. K. Patra, R. J. Tannenbaum, S. Adhikari, S. K. Kumar, T. Rovis and E. Y. Chen, Dynamic crosslinking compatibilizes immiscible mixed plastics, Nature, 2023, 616, 731–739 CrossRef CAS PubMed.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y. Chen, F. A. Leibfarth and H. Sardon, Critical advances and future opportunities in upcycling commodity polymers, Nature, 2022, 603, 803–814 CrossRef CAS PubMed.
- G. H. Fredrickson, S. Y. Xie, J. Edmund, M. L. Le, D. Sun, D. J. Grzetic, D. L. Vigil, K. T. Delaney, M. L. Chabinyc and R. A. Segalman, Ionic compatibilization of polymers, ACS Polym. Au, 2022, 2, 299–312 CrossRef CAS PubMed.
- F. S. Bates, Polymer-polymer phase behavior, Science, 1991, 251, 898–905 CrossRef CAS PubMed.
- F. S. Bates and G. H. Fredrickson, Block copolymers—designer soft materials, Phys. Today, 1999, 52, 32 CrossRef CAS.
- L. Leibler, Theory of microphase separation in block copolymers, Macromolecules, 1980, 13, 1602–1617 CrossRef CAS.
- L. Brunsveld, B. J. Folmer, E. W. Meijer and R. P. Sijbesma, Supramolecular polymers, Chem. Rev., 2001, 101, 4071–4098 CrossRef CAS PubMed.
- O. Ikkala and G. ten Brinke, Functional materials based on self-assembly of polymeric supramolecules, Science, 2002, 295, 2407–2409 CrossRef CAS PubMed.
- D. J. Grzetic, K. T. Delaney and G. H. Fredrickson, Electrostatic manipulation of phase behavior in immiscible charged polymer blends, Macromolecules, 2021, 54, 2604–2616 CrossRef CAS.
- A. M. Rumyantsev and J. J. de Pablo, Microphase separation in polyelectrolyte blends: Weak segregation theory and relation to nuclear “pasta”, Macromolecules, 2020, 53, 1281–1292 CrossRef CAS.
- S. Srivastava, A. E. Levi, D. J. Goldfeld and M. V. Tirrell, Structure, morphology, and rheology of polyelectrolyte complex hydrogels formed by self-assembly of oppositely charged triblock polyelectrolytes, Macromolecules, 2020, 53, 5763–5774 CrossRef CAS.
- F. Vidal, J. Gomezcoello, R. A. Lalancette and F. Jakle, Lewis pairs as highly tunable dynamic cross-links in transient polymer networks, J. Am. Chem. Soc., 2019, 141, 15963–15971 CrossRef CAS PubMed.
- A. M. Rumyantsev, A. A. Gavrilov and E. Y. Kramarenko, Electrostatically stabilized microphase separation in blends of oppositely charged polyelectrolytes, Macromolecules, 2019, 52, 7167–7174 CrossRef CAS.
- L. Zhang, L. R. Kucera, S. Ummadisetty, J. R. Nykaza, Y. A. Elabd, R. F. Storey, K. A. Cavicchi and R. A. Weiss, Supramolecular multiblock polystyrene–polyisobutylene copolymers via ionic interactions, Macromolecules, 2014, 47, 4387–4396 CrossRef CAS.
- J. Huh, J. Y. Jung, J. U. Lee, H. Cho, S. Park, C. Park and W. H. Jo, Supramolecular assembly of end-functionalized polymer mixtures confined in nanospheres, ACS Nano, 2011, 5, 115–122 CrossRef CAS PubMed.
- R. Elliott and G. H. Fredrickson, Supramolecular assembly in telechelic polymer blends, J. Chem. Phys., 2009, 131, 144906 CrossRef CAS PubMed.
- A. Noro, A. Tamura, S. Wakao, A. Takano and Y. Matsushita, Stoichiometric effects on nanostructures of block- and graft-type supramacromolecules via acid–base complexation, Macromolecules, 2008, 41, 9277–9283 CrossRef CAS.
- E. H. Feng, W. B. Lee and G. H. Fredrickson, Supramolecular diblock copolymers: a field-theoretic model and mean-field solution, Macromolecules, 2007, 40, 693–702 CrossRef CAS.
- J. Huh, H. J. Park, K. H. Kim, K. H. Kim, C. Park and W. H. Jo, Giant thermal tunability of the lamellar spacing in block-copolymer-like supramolecules formed from binary-end-functionalized polymer blends, Adv. Mater., 2006, 18, 624–629 CrossRef CAS.
- D. L. Vigil, A. Zhang, K. T. Delaney and G. H. Fredrickson, Phase separation, reaction equilibrium, and self-assembly in binary telechelic homopolymer blends, Macromolecules, 2023, 56, 9994–10005 CrossRef CAS PubMed.
- S. Xie, K. M. Karnaukh, K.-C. Yang, D. Sun, K. T. Delaney, J. Read de Alaniz, G. H. Fredrickson and R. A. Segalman, Compatibilization of polymer blends by ionic bonding, Macromolecules, 2023, 56, 3617–3630 CrossRef CAS.
- Y. Luo, D. Montarnal, N. J. Treat, P. D. Hustad, M. D. Christianson, E. J. Kramer, G. H. Fredrickson and C. J. Hawker, Enhanced block copolymer phase separation using click chemistry and ionic junctions, ACS Macro Lett., 2015, 4, 1332–1336 CrossRef CAS PubMed.
- J. Kim, H. Y. Jung and M. J. Park, End-group chemistry and junction chemistry in polymer science: Past, present, and future, Macromolecules, 2020, 53, 746–763 CrossRef CAS.
- H. Lee, S. Kwon, J. Min, S. M. Jin, J. H. Hwang, E. Lee, W. B. Lee and M. J. Park, Thermodynamically stable plumber's nightmare structures in block copolymers, Science, 2024, 383, 70–76 CrossRef CAS PubMed.
- J. Kim, K.-J. Jeong, K. Kim, C. Y. Son and M. J. Park, Enhanced electrochemical properties of block copolymer electrolytes with blended end-functionalized homopolymers, Macromolecules, 2022, 55, 2028–2040 CrossRef CAS.
- J. Min, H. Y. Jung, S. Jeong, B. Lee, C. Y. Son and M. J. Park, Enhancing ion transport in charged block copolymers by stabilizing low symmetry morphology: Electrostatic control of interfaces, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2107987118 CrossRef CAS PubMed.
- B. Yue, X. Jia, G. V. Baryshnikov, X. Jin, X. Feng, Y. Lu, M. Luo, M. Zhang, S. Shen, H. Agren and L. Zhu, Photoexcitation-based supramolecular access to full-scale phase-diagram structures through in situ phase-volume ratio phototuning, Angew. Chem., Int. Ed., 2022, 61, e202209777 CrossRef CAS PubMed.
- G. Duan, N. Zhu and V. W. W. Yam, Syntheses and photochromic studies of dithienylethene–containing imidazolium derivatives and their reactivity towards nucleophiles, Chem. – Eur. J., 2010, 16, 13199–13209 CrossRef CAS PubMed.
- T. Nakashima, M. Goto, S. Kawai and T. Kawai, Photomodulation of ionic interaction and reactivity: Reversible photoconversion between imidazolium and imidazolinium, J. Am. Chem. Soc., 2008, 130, 14570–14575 CrossRef CAS PubMed.
- B. M. Neilson and C. W. Bielawski, Photoswitchable organocatalysis: Using light to modulate the catalytic activities of n-heterocyclic carbenes, J. Am. Chem. Soc., 2012, 134, 12693–12699 CrossRef CAS PubMed.
- H. Nie, N. S. Schauser, N. D. Dolinski, J. Hu, C. J. Hawker, R. A. Segalman and J. Read de Alaniz, Light-controllable ionic conductivity in a polymeric ionic liquid, Angew. Chem., Int. Ed., 2020, 59, 5123–5128 CrossRef CAS PubMed.
- L. F. B. Wilm, M. Das, D. Janssen-Muller, C. Muck-Lichtenfeld, F. Glorius and F. Dielmann, Photoswitchable nitrogen superbases: Using light for reversible carbon dioxide capture, Angew. Chem., Int. Ed., 2022, 61, e202112344 CrossRef CAS PubMed.
- Y. Luo, D. Montarnal, S. Kim, W. Shi, K. P. Barteau, C. W. Pester, P. D. Hustad, M. D. Christianson, G. H. Fredrickson, E. J. Kramer and C. J. Hawker, Poly(dimethylsiloxane-b-methyl methacrylate): A promising candidate for sub-10 nm patterning, Macromolecules, 2015, 48, 3422–3430 CrossRef CAS.
- S. Jain, X. Gong, L. E. Scriven and F. S. Bates, Disordered network state in hydrated block-copolymer surfactants, Phys. Rev. Lett., 2006, 96, 138304 CrossRef PubMed.
- P. M. Simone and T. P. Lodge, Lyotropic phase behavior of polybutadiene–poly(ethylene oxide) diblock copolymers in ionic liquids, Macromolecules, 2008, 41, 1753–1759 CrossRef CAS.
- H. Kim, M. M. L. Arras, J. P. Mahalik, W. Wang, D. M. Yu, S. Chernyy, M. Goswami, R. Kumar, B. G. Sumpter, K. Hong, G. S. Smith and T. P. Russell, Studies on the 3-lamellar morphology of miktoarm terpolymers, Macromolecules, 2018, 51, 7491–7499 CrossRef CAS.
- Y. Tanaka, H. Hasegawa, T. Hashimoto, A. Ribbe, K. Sugiyama, A. Hirao and S. Nakahama, A study of three-phase structures in abc triblock copolymers, Polym. J., 1999, 31, 989–994 CrossRef CAS.
- S. Bhaumik, W. Shan, E. L. Thomas and N. Hadjichristidis, Synthesis and characterization of asymmetric a1ba2 supramolecular triblock copolymers via noncovalent interactions: A solution and solid-state study, Macromolecules, 2021, 54, 10730–10739 CrossRef CAS.
- M. Meot-Ner, The ionic hydrogen bond, Chem. Rev., 2005, 105, 213–284 CrossRef CAS PubMed.
- I. Hutskalov, A. Linden and I. Čorić, Directional ionic bonds, J. Am. Chem. Soc., 2023, 145, 8291–8298 CrossRef CAS PubMed.
- H. Nie, N. S. Schauser, N. D. Dolinski, Z. Geng, S. Oh, M. L. Chabinyc, C. J. Hawker, R. A. Segalman and J. Read de Alaniz, The role of anions in light-driven conductivity in diarylethene-containing polymeric ionic liquids, Polym. Chem., 2021, 12, 719–724 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR spectra, SEC, additional SAXS profiles (PDF). See DOI: https://doi.org/10.1039/d4py00682h |
| ‡ K. M. K. and S. X. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |