
Effect of pH on the incorporation of acrylic acid units in the core of polymer nanoparticles prepared by PISA and on their morphology†
Clément
Debrie
a,
Noémie
Coudert
b,
Jean-Michel
Guigner
c,
Fanny
Coumes
 a,
Clément
Guibert
d,
Simon
Harrisson
e,
François
Stoffelbach
a,
Olivier
Colombani
*b and
Jutta
Rieger
*a
a,
Clément
Guibert
d,
Simon
Harrisson
e,
François
Stoffelbach
a,
Olivier
Colombani
*b and
Jutta
Rieger
*a
aSorbonne Université & CNRS (UMR 8232), Institut Parisien de Chimie Moléculaire (IPCM), Polymer Chemistry Team, 4 Place Jussieu, 75252 Paris, Cedex 05, France. E-mail: jutta.rieger@sorbonne-universite.fr
bLe Mans Université & CNRS (UMR 6283), Institut des Molécules et Matériaux du Mans (IMMM), Avenue Olivier Messiaen, 72085 Le Mans, Cedex 9, France. E-mail: olivier.colombani@univ-lemans.fr
cSorbonne Université, MNHN, CNRS (UMR 7590), Institut de Minéralogie, de Physique des Matériaux et de Cosmochimie (IMPMC), IRD, 75252 Paris, Cedex 05, France
dSorbonne Université & CNRS (UMR 7197), Laboratoire de Réactivité de Surface, 4 Place Jussieu, 75252 Paris, Cedex 05, France
eUniversité de Bordeaux, Bordeaux-INP & CNRS (UMR 5629), Laboratoire de Chimie de Polymères Organiques, 16 Avenue Pey Berland, 33067 Pessac, Cedex, France
First published on 21st May 2024
Abstract
Polymerization-induced self-assembly (PISA) is a polymerization process in which amphiphilic block copolymers are simultaneously synthesized and self-assembled in a selective solvent, leading to nanoparticles of different morphologies. Copolymerization has been reported as a means to tune morphology by changing the properties of the core block. Most examples in aqueous dispersed media are however limited to the copolymerization of hydrophobic monomers, while copolymerization between hydrophobic and hydrophilic/ionisable monomers has scarcely been reported. In this work, we study the aqueous PISA copolymerization of a mildly hydrophobic monomer (methoxyethylacrylate, MEA) with a hydrophilic pH-responsive monomer, acrylic acid (AA). Kinetic studies reveal that the incorporation of AA is strongly dependent on the pH at which the polymerization is performed: quasi-random copolymers form at low pH, whereas gradient composition profiles with lower AA contents are obtained at higher pH. The incorporation of AA, its degree of ionization, as well as its distribution in the copolymer chain (determined by the pH of the polymerization medium) strongly affect the particle morphology. Moreover, the amount and degree of ionization of AA, and the composition profile of the copolymer have a strong effect on the responsiveness of the particles post-polymerization, both to pH and to temperature. This work highlights that the incorporation of ionisable units within hydrophobic blocks by a typical aqueous PISA process strongly impacts their composition profile and, at the same time, provides a powerful tool to modify the morphology of the resulting nanoparticles.
I. Introduction
Polymerization-induced self-assembly (PISA)1–3 is a powerful and straightforward technology to produce polymer nanoparticles. It combines the synthesis of amphiphilic AB block copolymers in a heterogeneous polymerization process and their simultaneous self-assembly into core–shell structures. PISA allows nanoparticles to be produced directly in water, at high concentration (typically 20 to 30 wt%) and in scalable conditions. In addition to spherical particles, vesicles, worm-like particles or long nanofibers can be prepared. The particle morphology can be tuned by changing polymerization parameters such as the monomer concentration, and the molar ratio R between the monomer and the solvophilic reactive A block which governs the degree of polymerization (DP) of the hydrophobic B block. The solvent also strongly affects the particle morphology.4 During the polymerization the solvent properties changes as the monomer is consumed, and we have recently shown that the presence of unreacted monomer favors the formation of higher order morphologies such as worms or vesicles.5 The formation of higher order morphologies can also be promoted by adding a small molecule “transformer” such as toluene.6,7Nanoparticle morphologies are also affected by the chemical structure of the polymers.2,8,9 Highly hydrophobic monomers (such as styrene10 or benzyl methacrylate4,11) generally give rise to spherical particles in water, while less hydrophobic monomers can produce all kinds of morphologies.12 Mathers, O'Reilly et al. have classified monomers according to their octanol/water partition coefficient (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) in order to predict suitable monomers for aqueous PISA.13 The morphologies formed during PISA are, however, generally out-of-equilibrium and therefore process-dependent,5 complicating their ab initio prediction.
P) in order to predict suitable monomers for aqueous PISA.13 The morphologies formed during PISA are, however, generally out-of-equilibrium and therefore process-dependent,5 complicating their ab initio prediction.
Instead of changing the structure of the B block monomer to obtain a targeted morphology, the properties of the core block can be tuned through copolymerization with small amounts of a comonomer. This strategy has less frequently been proposed in the literature. There are several reports of copolymerization with organosoluble comonomers,14–17 whereas copolymerization with hydrophilic comonomers has rarely been reported.18,19 The copolymerization of a hydrophobic monomer with a hydrophilic one in an aqueous dispersed system is challenging because unequal partitioning of the comonomers between the discrete and continuous phases can be expected.20–23 Sumerlin et al. studied the incorporation of a hydrophilic comonomer, N,N-dimethyl acrylamide, within the hydrophobic core block of poly(N,N-dimethyl acrylamide)-block-poly(diacetone acrylamide) (PDMAc-b-PDAAm) nanoparticles formed through aqueous dispersion PISA.19,24 They successfully incorporated DMAc within the core block, but the incorporation of DMAc was strongly dependent on the polymerization conditions and gradient composition profiles were generally observed.25 TEM studies demonstrated that the incorporation of DMAc favored the formation of higher order morphologies.19
The incorporation of stimuli-responsive hydrophilic comonomers in the core of polymer nanoparticles by aqueous PISA has also been reported.15,18,26 Examples include the incorporation of 2-(dimethylamino)ethyl methacrylate (DMAEMA) in the hydrophobic poly(hydroxypropyl methacrylate) (PHPMA) block of PPEGMA-b-P(HPMA-co-DMAEMA) nanoparticles (with PPEGMA: poly(poly(ethylene glycol) methacrylate)),15 and of AA in the poly(N-cyanomethyl acrylamide) (PCMAm) block of PDMAc-b-P(CMAm-co-AA) nanoparticles.26 In these examples, the presence of pH-sensitive comonomers favoured the production of lower order morphologies, typically spheres. Incorporation of hydrophilic comonomer units to form higher order stimuli-responsive morphologies during PISA has not been reported. Moreover, the effect of pH on the incorporation of a pH-sensitive monomer in PISA conditions has never been studied.
We have recently studied the aqueous dispersion polymerization of 2-methoxyethyl acrylate (MEA) in the presence of a PDMAc macroRAFT agent (DPn = 24).5 At a monomer concentration close to 20 wt%, we have shown that only spherical nano-objects were formed up to targeted DPn,PMEA ≤ 250. In a related work out of the context of PISA, we have recently shown that the aqueous dispersion copolymerization of MEA with AA in the presence of a molecular RAFT agent functionalized by a sulfonate functional group was very sensitive to the degree of ionization of AA at which the polymerization was performed (α0).20 Good control over polymerization was maintained in all conditions, and the composition profile of the resulting copolymers could be tuned from random to block-like by varying α.
In this work, we copolymerized MEA with AA in a typical aqueous PISA process using a PDMAc macroRAFT agent to generate PDMAc-b-P(MEA-co-AA) particles comprising pH-responsive AA units in their core. We studied the effect of the molar fraction of AA, fAA, and the degree of ionization of AA, α, on the copolymerization kinetics, and investigated the possibility of tuning the composition profile of the P(MEA-co-AA) core block simply by controlling α. The final objective was to investigate the effect of fAA and α on the particle morphologies. We also studied the pH- and T-sensitivity of the particle morphologies post-polymerization.
II. Results and discussion
II.1. Incorporation of neutral AA in the hydrophobic block (PISA at α ∼ 0)
To study the impact of the incorporation of AA in the PMEA block of PDMAc-b-PMEA nano-objects, we performed a series of copolymerizations in water at 40 °C using VA-044 as an initiator and solids contents close to 20 wt% (Scheme 1).5,20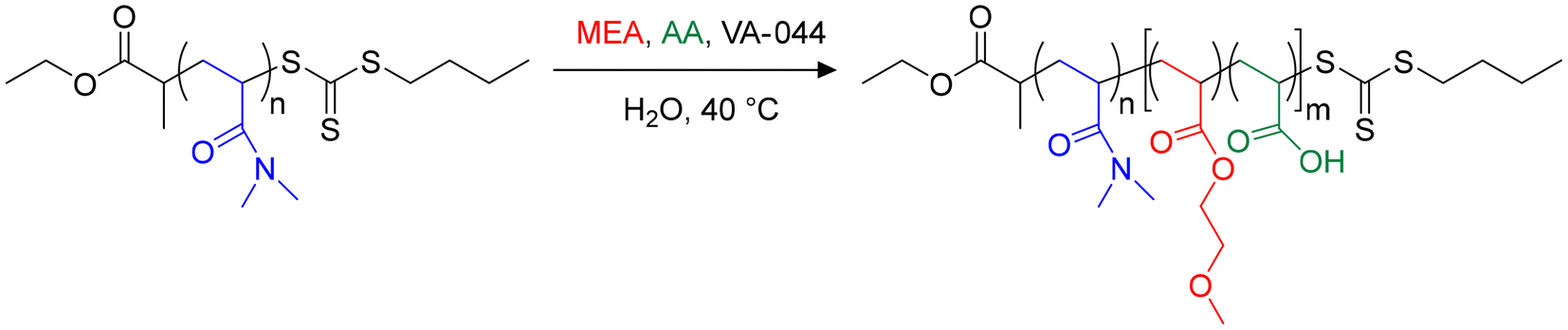 | ||
| Scheme 1 Chain extension of a PDMAc24-TTC macroRAFT agent (macroCTA-1) by MEA and AA in aqueous PISA. | ||
A trithiocarbonate (TTC) PDMAc macroRAFT agent (macroCTA-1) with a number-average degree of polymerization, DPn, of 24 was used as the chain transfer agent (Table S2†). In this series of experiments (Table S3†) no base was added to ionize AA. The molar fraction of AA in the feed, fAA,0, was varied, while the initial molar ratio of the comonomers to macroCTA-1, R = ([MEA]0 + [AA]0)/[macroCTA-1]0, which determines the number-average degree of polymerization of the core block B, DPn,B, was kept constant at 200, in order to produce hydrophobic B blocks with comparable DPn,B close to full conversion. AA is a weak acid (pKa ≈ 4.1),27 and as such is nearly fully protonated in dilute aqueous solution in the absence of added base (α ∼ 0, see ESI†). As PMEA is a weakly hydrophobic polymer, the molar fraction of AA introduced in the feed (fAA,0) was kept below 0.3 to ensure the formation of sufficiently hydrophobic P(MEA-co-AA) blocks that promote the in situ assembly of the diblock copolymers into core–shell particles, as in preliminary experiments on the dispersion copolymerization of MEA and AA, water-soluble chains were formed at fAA of 0.5 for α > 0. The copolymers were named R-fAA,0, where R was kept at 200 in this first series of experiments as mentioned above and the molar fraction of AA in the monomer feed (fAA,0) was varied. All experimental details are reported in the ESI (Table S3,† series 200-fAA,0).
For all studied AA contents, colloidally-stable, turbid dispersions were obtained. Generally, high conversions (>90%) of both monomers were achieved in less than 10 h. The kinetics of two typical polymerizations performed with 10 mol% (200-10) or 30 mol% of AA (200-30) are displayed in Fig. 1 (left). Kinetic monitoring of the individual conversions of MEA and AA by 1H NMR showed that in both polymerizations, both monomers were consumed at similar rates, suggesting quasi-random incorporation of the AA units (see Fig. 1, left). As a result, the content of AA in the produced copolymers (FAA, Table S3†) was always close to the ratio in the initial monomer feed (fAA,0). This was consistent with our previous study of the aqueous copolymerization of MEA and AA.20
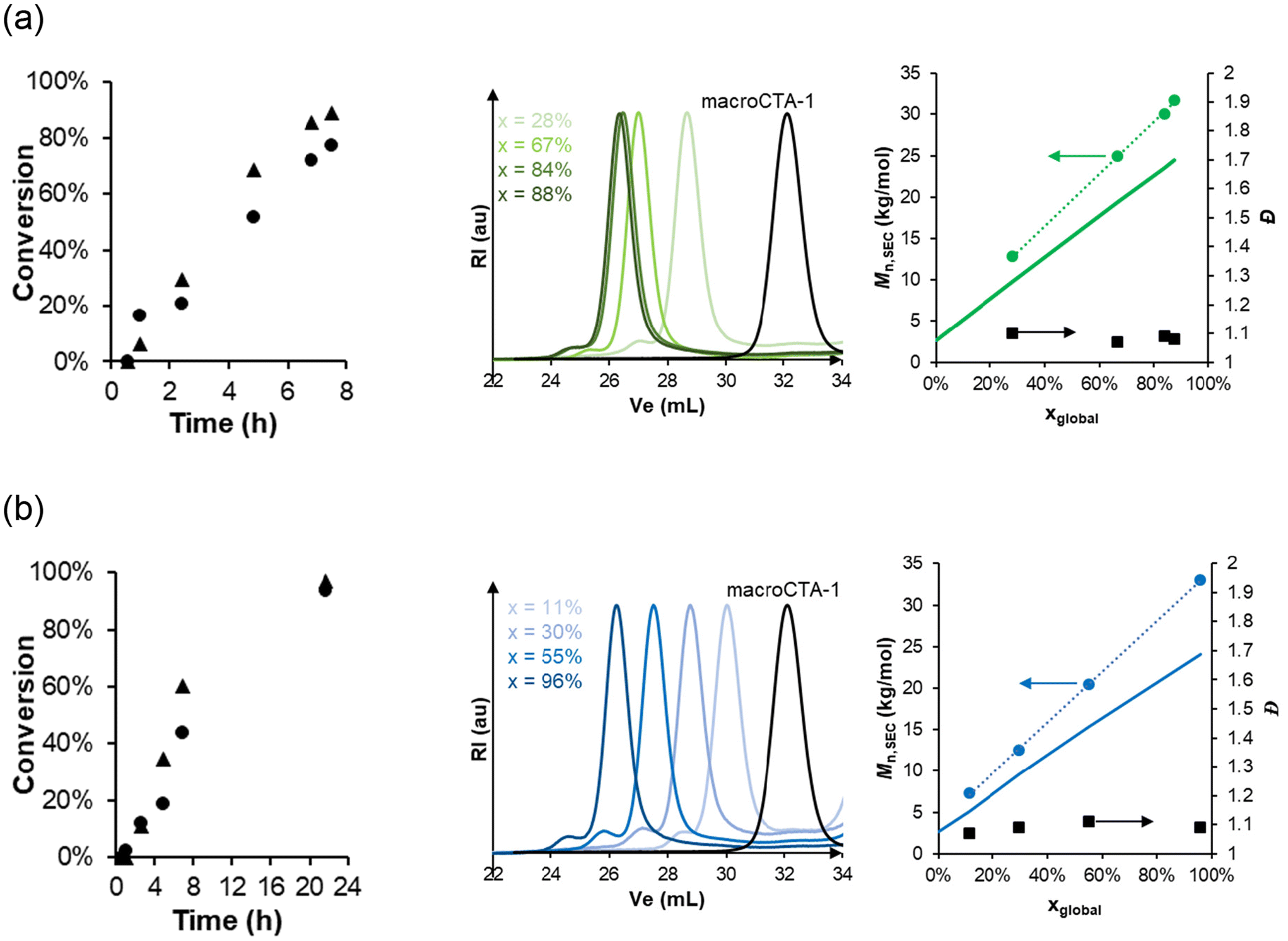 | ||
| Fig. 1 Kinetic monitoring of the aqueous dispersion copolymerization of MEA and AA in the presence of macroCTA-1 (R ≈ 200). (a) Sample 200-10 and (b) sample 200-30. (Left) Individual conversion determined by 1H NMR of MEA (▲) and AA (●). (Middle) Overlay of the normalized SEC RI traces. The global monomer conversions, x, are indicated in the insert. Ve stands for elution volume. (Right) Number-average molar mass, Mn, and dispersity, Đ, determined by SEC with PMMA calibration. The solid line shows the theoretical molar mass. | ||
Molar mass dispersities were low (Đ ∼ 1.1) throughout the polymerization (Fig. 1). In addition, the molar masses evolved linearly with conversion for both samples, even though the theoretically expected molar masses (solid lines) were quite different from the experimental ones determined by SEC, which may be explained by the use of a PMMA calibration curve. These results together with strong absorption at 309 nm indicating the presence of a trithiocarbonate end-group (see Fig. S4†) reveal a very good blocking efficiency and a good control over the polymerizations (Fig. 2a and Table S3†).
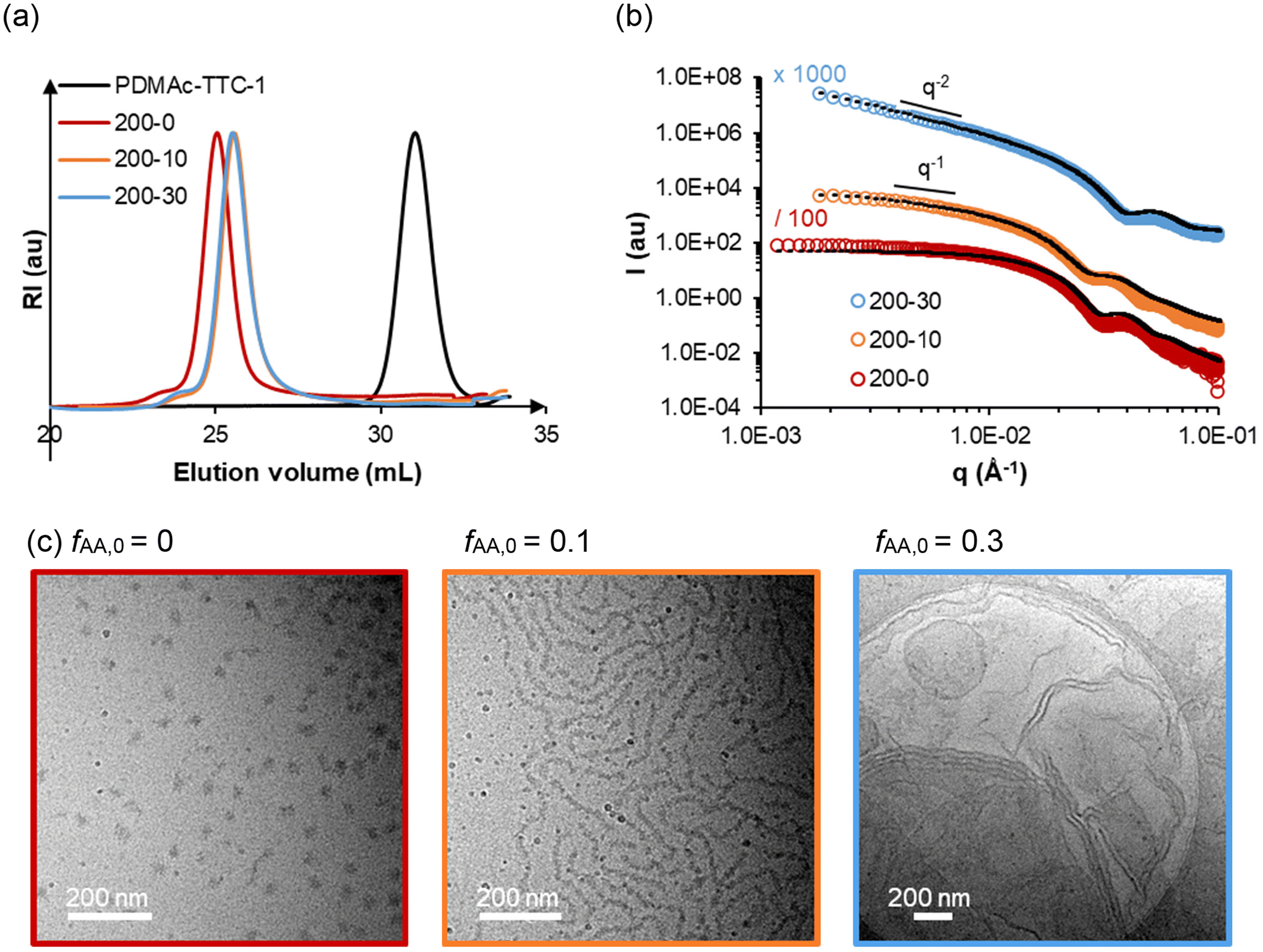 | ||
| Fig. 2 (a) SEC traces, (b) SAXS data and (c) cryo-TEM images of samples 200-0, 200-10, and 200-30 (samples synthesized with variable fAA,0, and constant R ≈ 200). The black dotted lines are models fitting the experimental data. The model used and dimensions are reported in Table S4.† | ||
In a previous study, we had shown that PDMAc-b-PMEA diblock copolymers prepared by PISA in aqueous media self-assembled into spherical nano-objects for R ≤ 250.5 Higher order morphologies (vesicles) were only obtained for R ≥ 280. To investigate a possible effect of the AA units in the PMEA block on particle morphology, we characterized three samples containing 0, 10 or 30 mol% of AA in the PMEA block (with constant R = 200) by cryo-TEM, SAXS and DLS (samples 200-0, 200-10 and 200-30, Fig. 2). Consistent with the increasing turbidity observed by the naked eye, DLS measurements showed a strong increase of the z-average diameter, Dz, from 49 to 206 to 616 nm with increasing content of AA (Table S3†). By cryo-TEM imaging, long nanoworms and lamellar objects were observed for sample 200-10 and 200-30 respectively, while the reference sample 200-0 contained spherical objects. The fitted SAXS data were consistent with the morphologies determined by cryo-TEM. The morphologies and dimensions calculated by cryo-TEM and fitted SAXS data are summarized in Table S4.†
The analyses show that the incorporation of hydrophilic AA units in the B block triggers a clear change from spheres towards higher order morphologies. One possibility to rationalize this trend would be from a thermodynamic point of view based on the packing parameter,28,29 because the introduction of AA units within the solvophobic PMEA block might alter the interaction parameters and thereby the volumes occupied by the hydrophilic and the hydrophobic segments. It is also possible that the incorporation of AA units modifies the ability of the polymer chains to reorganize by exchange of free polymer chains between the self-assemblies and/or by fusion/fission of self-assembled particles,5 thereby allowing morphological transitions which are kinetically hindered with pure PMEA blocks. Amphiphilic block copolymers indeed often form out-of-equilibrium (frozen) structures hardly able to reorganize30 in water through exchange of free unimer chains or fusion/fission; but the incorporation of AA units within their hydrophobic block may favour exchange of unimers20,31–33 and possibly fusion/fission of particles too, allowing reorganizations in PISA.
Strikingly, we have recently observed that the incorporation of AA within the core block of PDMAc-b-PCMAm nano-objects obtained by a similar synthesis strategy through aqueous dispersion PISA26 had the opposite effect: the incorporation of AA within the core block (PCMAm) prevented the formation of higher order morphologies. For instance, for DPn,PCMAm = 200, fibers of variable lengths were observed for AA contents below 13 mol% (FAA < 0.13), whereas only spherical objects where obtained for higher FAA. Explaining why these two systems behave differently when AA is introduced in the hydrophobic B block is not straightforward. One possibility might be that PMEA has a lower critical solution temperature (LCST) close to 0–5 °C,34,35 whereas PCMAm and P(CMAm-co-AA) exhibit an upper critical solution temperature, UCST-type behavior.36 Additionally, the hydration of each core block may differ, as suggested by the Armes group.37
It is well known that the molar masses of the shell and core-forming blocks affect the morphology of particles obtained by PISA.2,5,9,38 In order to establish a pseudo-phase diagram, in a second series of experiments, R was systematically varied between 150 and 400 (to vary the degree of polymerization of the core forming block, DPn,B), while fAA,0 was modified between 0.05 and 0.3 (see Table S3†). Low molar mass dispersities (Đ ≤ 1.2) were obtained in all cases, and the conversions of MEA and AA were similar (Table S3 and Fig. S5†). The morphology of the nano-objects was determined by cryo-TEM and SAXS analyses (Fig. 3 and Fig. S7†). As expected, higher order morphologies were preferentially formed for long hydrophobic blocks. For instance, at constant fAA,0 of 0.10 and nearly full conversion, spheres, worms, and vesicles were obtained as R was increased. The combined analyses of all samples synthesized at various R and fAA,0 (see additional cryo-TEM images and fitted SAXS data in Fig. S6 and S7†) allowed us to construct a pseudo-phase diagram that is shown in Fig. 4. It confirms that the introduction of AA units into the PMEA core block generally favours the formation of higher order morphologies, in particular vesicles/lamellae. Overall, vesicles and/or lamellae could be obtained over a large range of DPn,B and FAA, whereas the experimental window in which fibers can be obtained was small.
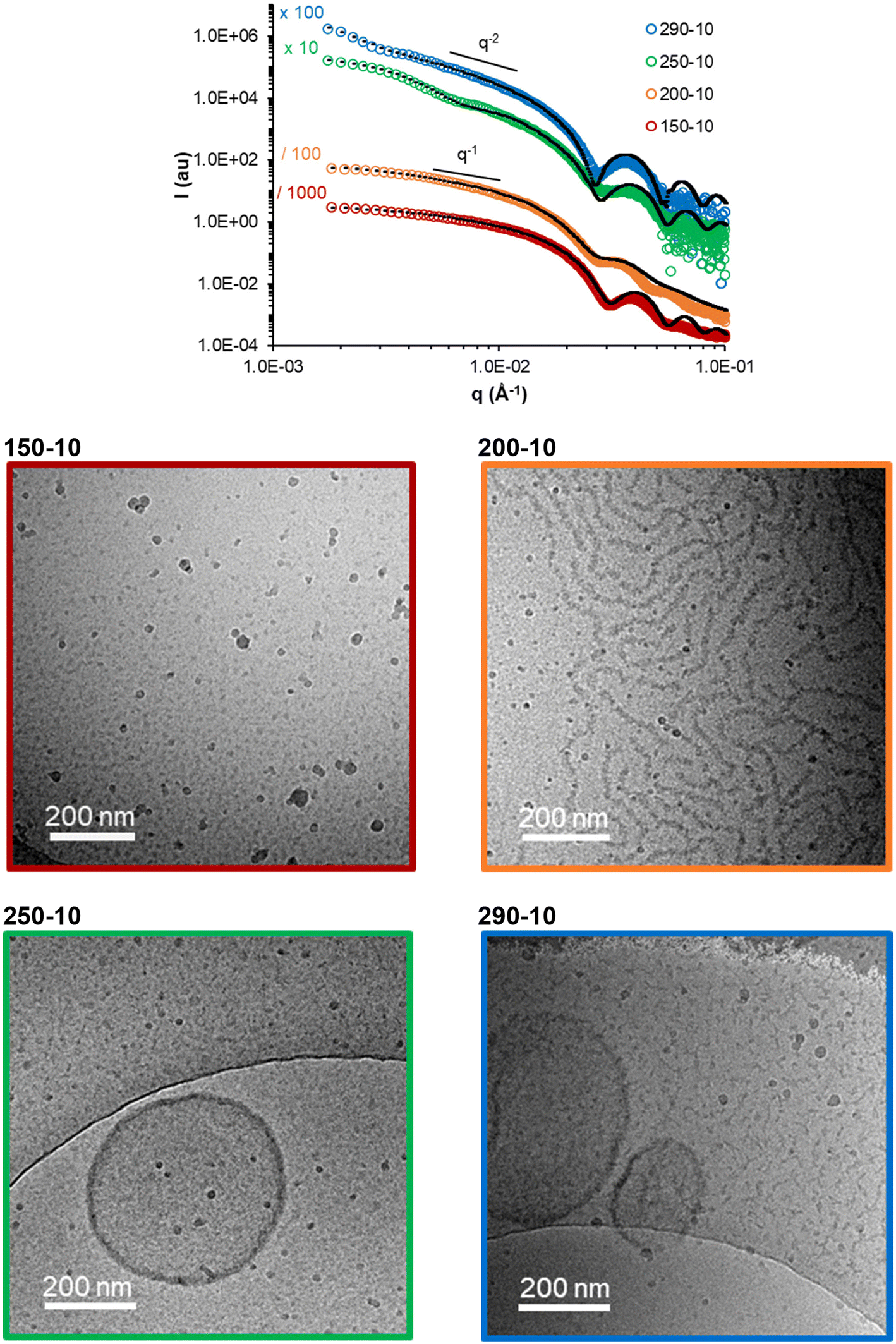 | ||
| Fig. 3 Impact of the Mn of the core block with constant AA content (fAA,0 = 0.1): (top) fitted SAXS data. The model used and dimensions are reported in Table S4.† (Bottom) Representative cryo-TEM pictures of the samples. | ||
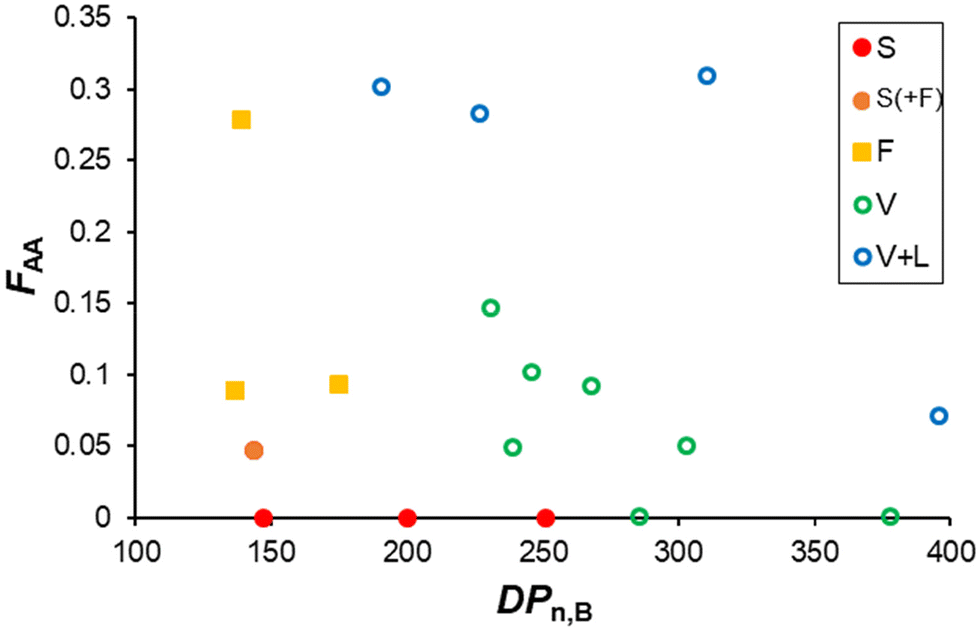 | ||
| Fig. 4 Summary of the experiments performed at natural pH (α ∼ 0). The color indicates the final morphology obtained determined by SAXS and cryo-TEM. S = spheres, F = fibers, V = vesicles and L = lamellae. | ||
In agreement with the cryo-TEM analyses, SAXS data suggested that the diameters of the vesicles were rather polydisperse and no clear effect of the molar content of AA (FAA) or the DPn,B on the vesicle size could be observed (see details in the ESI, Table S4†). In contrast, the distinct signature at intermediate q values (around 2 × 10−2 Å−1 in Fig. 5a) allowed us to determine the mean thickness e of the vesicle membranes by fitting the data with a vesicle model (Table S4†). The membrane thickness e of all vesicles and lamellae steadily increases with DPn,B as shown in Fig. 5b.
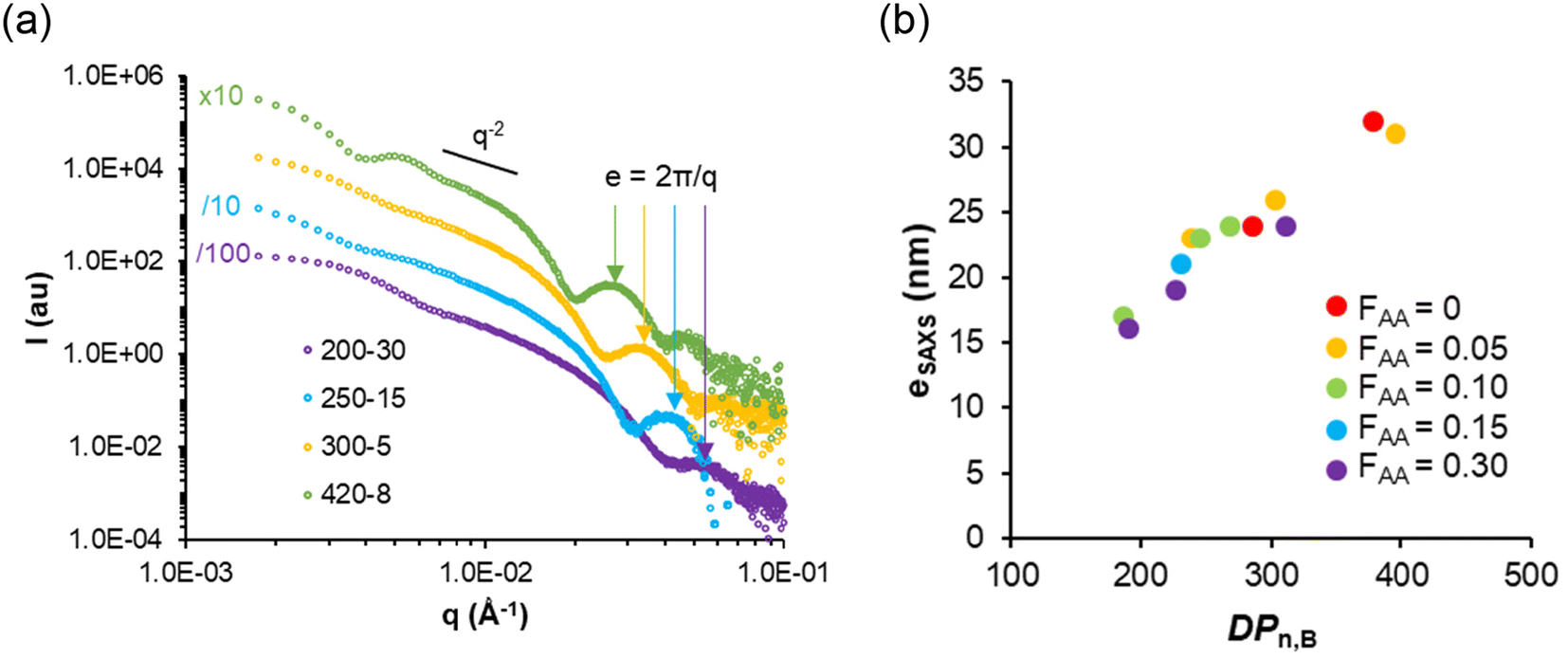 | ||
| Fig. 5 (a) SAXS data for typical lamellar objects with various DPn,B. The arrows indicate that the membrane thickness, e, varies. Values of e reported in Table S4† were determined by fitting the data with a vesicle model. (b) Variation of e with the DPn,B of the core block e was determined by fitting SAXS data with a vesicle or lamellar model (see fitted SAXS data in Fig. S6, and dimensions in Table S4†). | ||
II.2. Syntheses of PDMAc-b-P(MEA-co-AA) nano-objects at α > 0
The self-assembly of amphiphilic copolymers containing pH-sensitive monomer units, such as AA, in the hydrophilic and/or the hydrophobic block,24,26,39–41 is sensitive to their degree of ionization α. Such studies were generally performed post-polymerization on polymers that had been synthesized using homogeneous polymerization processes, purified and then assembled. As mentioned above, the polymerization conditions in PISA have a great impact on the particle morphologies, and predicting the morphology of PISA-derived particles is difficult because frozen particles, i.e. kinetically trapped and process-dependent morphologies,30,42 are generally obtained.5,43 These particles are kinetically trapped due to the extremely slow exchange of polymer chains between particles and thus unable to evolve to their thermodynamic equilibrium state. As a result, the obtained morphologies depend strongly on the process used to obtain the particles. This is also true for PDMAc-b-PMEA nanoparticles produced by PISA.5,34 Additionally, our previous study on the aqueous dispersion copolymerization of MEA and AA in the presence of an anionic molecular RAFT agent20 revealed that the composition and composition profile of the copolymers is controlled by the degree of ionization of AA (α0): while random copolymers formed at α0 ∼ 0, gradient copolymers containing fewer AA units than the initial monomer feed were produced when AA was partially ionized (α0 > 0). These great differences in reactivity of AA were explained by the difference in intrinsic reactivity44 of sodium acrylate (NaA) and acrylic acid, which is greatly amplified by the use of a heterogeneous polymerization process, where the partitioning of NaA is in favour of the continuous water phase. Thus PISA of MEA and AA performed at α0 > 0, using a macromolecular RAFT agent instead of a sulfonate-functional one, should also lead to copolymers with distinct composition and composition profiles; which may further impact the morphologies.39In all experiments, the molar fraction of AA was kept low at 5 mol% (fAA,0 = 0.05) to preserve the self-assembly of the B block. R was kept constant at 300, while α0 was varied through the addition of NaOH before polymerization (Table S5†). Theses samples were named A-x where x is related to the degree of ionization of AA (α0) in the monomer feed (e.g. A-01 corresponds to α0 = 0.1).
SEC analyses (Fig. S8†) did not reveal any significant impact of the variation of α0 on the control over the polymerizations. The incorporation of AA as a function of the degree of ionization was investigated by kinetically monitoring the individual conversions of MEA and AA by 1H NMR. As shown in Fig. 6, a strong effect of α0 on the polymerization rate of AA was observed. While the polymerization rate of AA greatly decreased when α0 was increased, little impact on MEA conversion was observed, except at the highest α0 (0.7 and 1), where the polymerization of MEA slowed down too. As in our previous study,20 we used the Jaacks method45 to determine rMEA because the AA content is negligible in the initial monomer feed. The Jaacks plot in Fig. 6 shows the experimental data and their fits. rMEA steadily increased from 1 to 12 when α0 increased from 0 to 1 (values reported in Fig. 6). These values are very close to those found in our earlier study.20 Therefore, it seems that the reactivity ratios are not impacted by the nature of the stabilizing shell. As the molar fraction in AA is low (fAA,0 = 0.05), the occurrence of AA-terminated radicals is very low and the composition profile is dominated by rMEA. Thus, the reactivity of AA can be estimated solely from the evolution of rMEA = kp MEA–MEA/kp MEA–AA and clearly decreases with increasing α0.
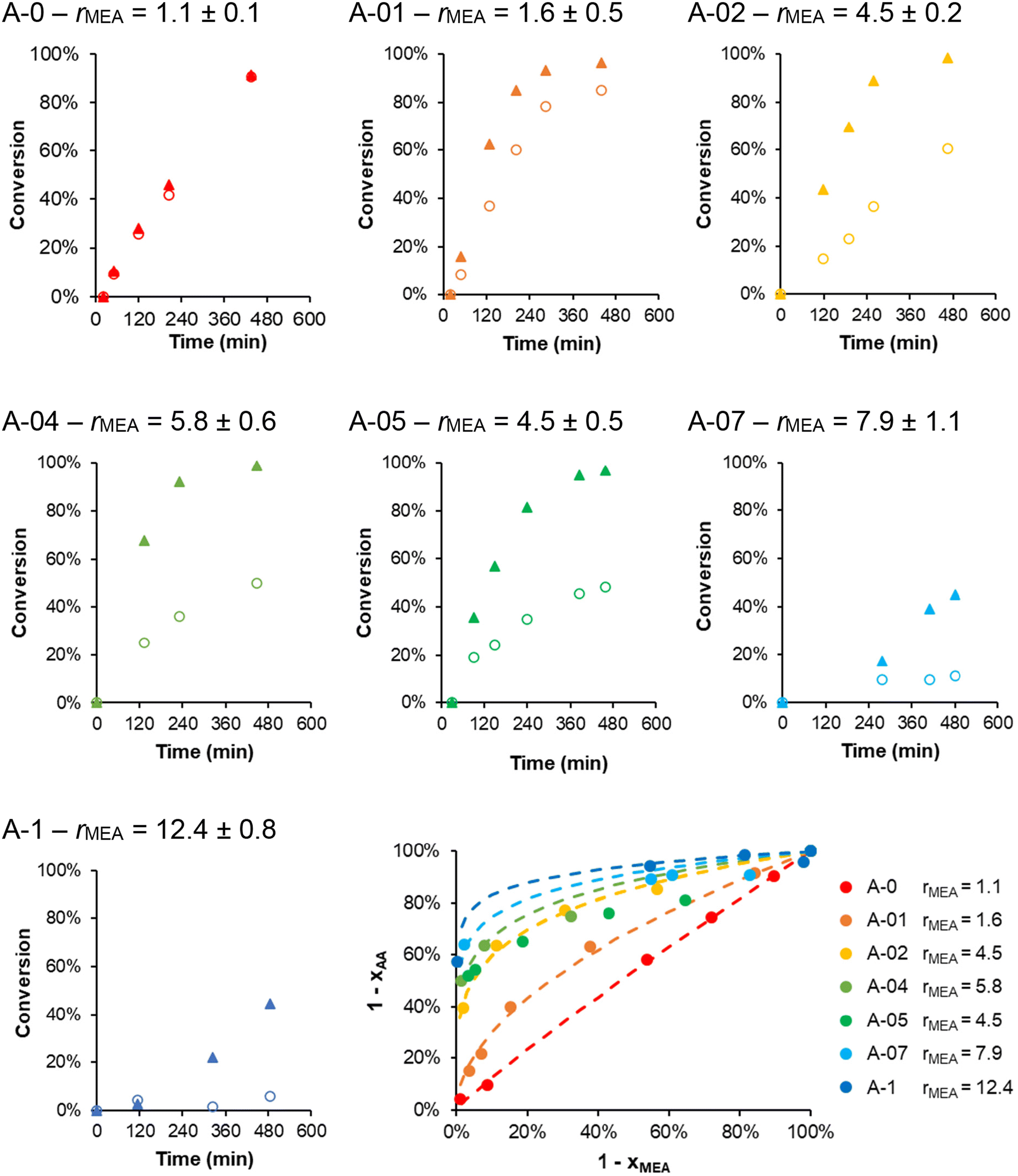 | ||
| Fig. 6 Kinetic monitoring of PISA conducted at different α0 (samples A-0 to A-1, with R = 300 and fAA,0 = 0.05) displaying the individual conversions in MEA (▲) and AA (○). Jaacks plot showing the fitting curves in dotted lines. A-0 corresponds to sample 300-5. | ||
While the MEA conversion was almost quantitative within 8 h for the polymerizations performed at α0 ≤ 0.5 (Fig. 6), the conversion of AA was incomplete when α0 > 0 (e.g. less than 50% for A-05). Consequently, the final AA molar content in the hydrophobic block (FAA) tends to decrease from 0.05 to 0.02 when α0 increases, although all polymerizations started from the same initial monomer feed, fAA,0 = 0.05 (Table S5†). The composition profile of the B block of the copolymers should also vary as a function of α0. To illustrate this, we simulated the composition profile of 100 chains using the experimentally determined apparent reactivity ratio of MEA for each α0 tested and an arbitrary constant rAA = 1. Changing this value did not strongly impact the results because the AA content was very low, decreasing strongly the probability to incorporate two consecutive AA units within the polymer chain no matter rAA. While at α0 = 0 and 0.1 the distribution of AA units in the polymer chains is close to random, at α0 > 0.2 the incorporation of the AA during copolymerization is delayed leading to gradient B blocks because of the strong differences in reactivity between MEA and AA (see Fig. 7 and Fig. S9†). The higher α0, the stronger the gradient towards the chain end and the fewer AA units are located close to the PDMAc block. Overall, the composition profile of the core-forming block, the number of AA in the chain, and the average number and distribution of negative charges per polymer chain greatly depend on α0. This must be taken into account when comparing the morphology of the samples and their thermo-responsiveness that are discussed below.
 | ||
| Fig. 7 Simulation of 100 monodisperse PDMAc-b-P(MEA-co-AA) chains, stacked horizontally. The PDMAc block is represented in green, MEA in grey and AA units in blue. Each line on a figure corresponds to a different chain. The direction of polymerization is from left to right. Experimental fAA,0, R, reactivity ratios of MEA and the final global conversion were used in the terminal model to determine the composition profiles (see ESI, Fig. S9†).46 | ||
In all conditions, stable colloidal dispersions were again obtained, and the morphologies of the nano-objects were characterized by cryo-TEM and SAXS. The cryo-TEM pictures presented in Fig. 8 reveal a strong impact of α0 on the particle morphology although the molar fraction of AA was only 5 mol%. Whereas at α0 = 0 typical vesicles were observed (A-0), deprotonating 10% of the AA units gave a mixture of objects (mainly vesicles mixed with small, mostly elongated nano-objects). When α0 was further increased, small vesicles remained the predominant morphology, but the vesicle membranes became heterogeneous and ill-defined, possibly due to a dissociation of the membrane induced through the presence of charges. Above 70% ionization, vesicles were no longer observed, and small spheres predominated. SAXS analyses confirmed that spherical objects with diameters below 40 nm were the main morphology formed in these conditions (Fig. S10 and Table S6†).
 | ||
| Fig. 8 Representative cryo-TEM images (C = 30 g L−1) of samples of series A (R = 300, fAA,0 = 0.05, prepared by PISA at different α0). | ||
Although the initial monomer feed was the same for all samples (fAA,0 = 0.05), the AA content in the polymer (FAA) progressively decreased from 0.05 to 0.02 when α0 was increased from 0 to 1. Considering a DPn,B of 300, this means that an average of only 6 AA units instead of 15 were incorporated per chain. However, the change in morphology, from vesicles (A-0) to spheres (A-1), cannot be attributed to the lower amount of AA units in A-1 compared to A-0, as pure vesicles were also obtained for the AA-free reference sample 300-0 (see Tables S3 and S4†).5 The formation of spheres (A-1) must therefore be attributed to the presence of charged AA units in the PMEA block. The presence of a few charged AA units thus seems to hinder the formation of higher order morphologies while favouring the formation of spherical aggregates.
α 0 and fAA,0 clearly impact the composition profile and FAA, which in turn have a strong effect on the morphology of the resulting particles. It is however difficult to determine whether the changes of morphology as a function of α0 and fAA,0 have a thermodynamic or a kinetic origin (or both). Indeed, a change in FAA, α and composition profile should alter the packing parameter, which may alter the morphology from a thermodynamic point of view.28,47,48 Simultaneously, addition of AA units within hydrophobic blocks and variation of their ionization should alter the exchange rate of unimers,31–33,49 which may favour morphological transitions from a kinetic point of view. At this stage, it is not possible to determine whether thermodynamics, kinetics or both explain our observations.
We also note that it has previously been proposed2,43,50,51 that the presence of charges in the corona hinders the formation of higher order morphologies because of electrostatic repulsion limiting chain aggregation. Similarly, in the studied system, spherical aggregates are formed in the early stages of PISA, and the subsequent incorporation of a few charges might prevent the fusion of primary particles (because of electrostatic repulsion), which has been suggested to be a key step in the formation of higher order morphologies.2,5,52 Alternatively, it is also possible that a reorganization of initial aggregates occurs during PISA, as the AA units are incorporated at late stages of the polymerization.
II.3. Stimuli-responsiveness of the nano-objects
All nano-objects prepared above contain AA units in the PMEA block and are therefore expected to be sensitive to pH, i.e. variations in α. Moreover, we have previously demonstrated that P(MEA-co-AA) statistical copolymers are thermoresponsive and exhibit a LCST-type temperature transition that depends on pH.20,49 We therefore studied post-polymerization the impact of α and temperature (T) on representative samples with different morphologies, molar content of AA (FAA) and DPn,B.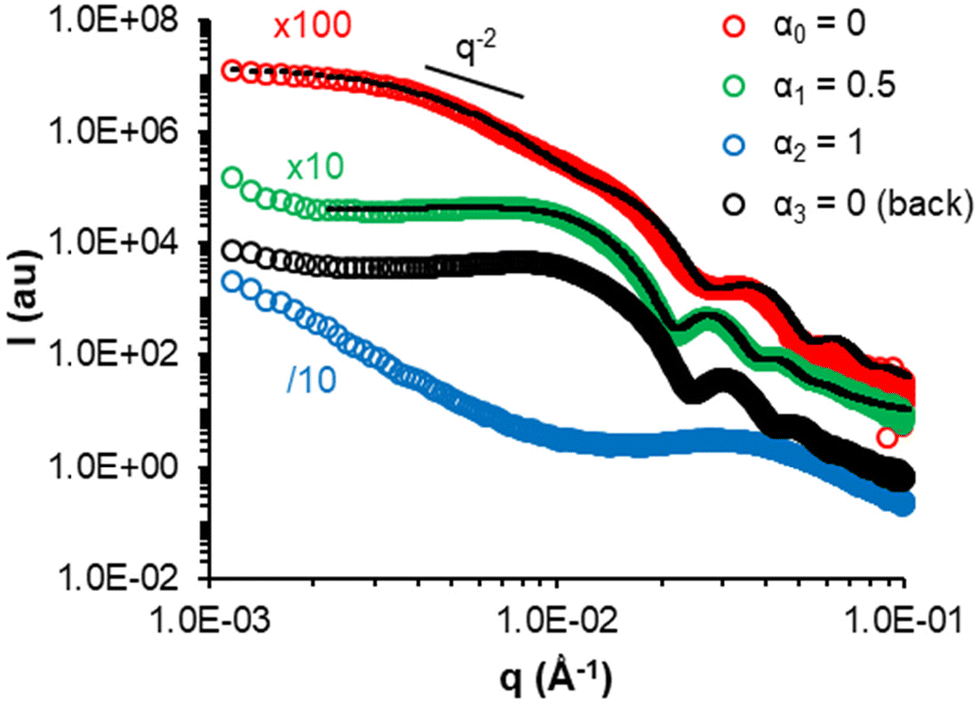 | ||
| Fig. 9 SAXS monitoring of the progressive deprotonation of the AA units of a representative sample, 300-5 (= A-0, vesicles), post-polymerization and return to α = 0 (back). The black dotted lines are models fitting the data (see Table S7†). | ||
Generally, the change in the slope of I = f(q) and data fitting suggested that higher order morphologies evolved into spherical assemblies (e.g. 300-5 at α1 = 0.5 or 310-30 at α1 = 0.1) and ultimately into unimers when the AA units were sufficiently ionized (e.g. 300-5 at α2 = 1 or 310-30 at α2 = 0.30). Decreasing α back to 0 led to re-assembly of the unimers, but into structures different from the initial ones, which is consistent with the DLS measurements performed on diluted samples (black symbols in Fig. S11†).
Samples A-05 and A-1 (Table S5†), for which the AA units are at least partially ionized, are also responsive to changes in α. While sample A-05 behaved similarly to the previously discussed samples (Fig. S14†), sample A-1 behaved differently (Fig. 10). This sample was prepared by PISA directly at α = 1. It contains only 2 mol% of AA (FAA = 0.02) with a strong gradient towards the chain end and a relatively long hydrophobic block (DPn,B ∼ 300). The low AA content combined with the strong gradient profile might explain why sample A-1 remains assembled even at α = 1 contrary to the other samples of this study (Fig. 10).53 Upon progressive protonation towards α = 0, a morphological transition from spheres to vesicles was observed by SAXS and cryo-TEM analyses (Fig. 10).
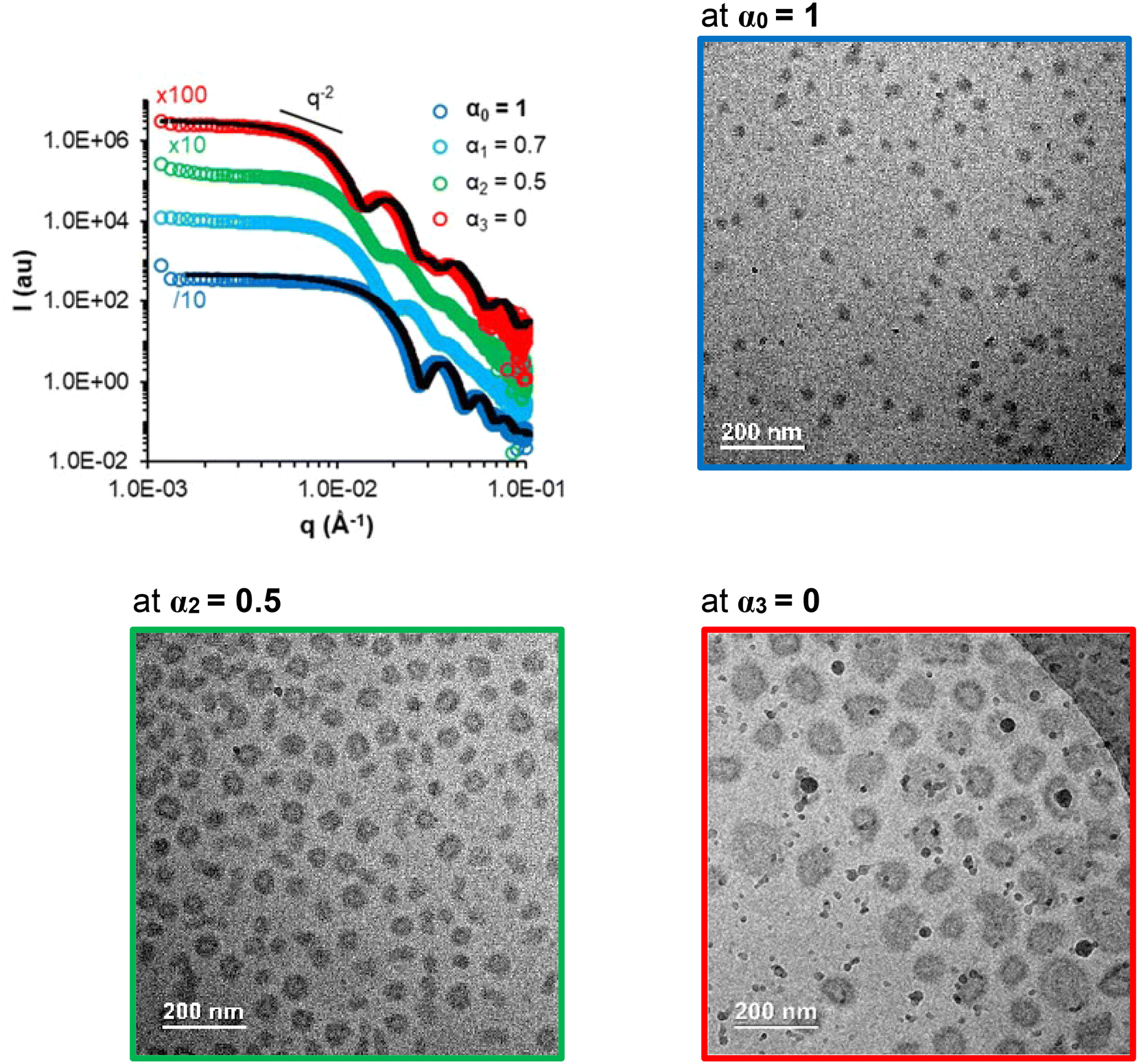 | ||
| Fig. 10 SAXS (at C = 50 g L−1) and cryo-TEM (at C = 30 g L−1) monitoring the progressive protonation of the AA units in the B block of sample A-1 (spheres, FAA = 0.02, gradient). The black dotted lines are models fitting the data (see Table S7†). | ||
Generally, the pathway dependency and non-reversibility of the morphological transitions suggest that the final nanostructures produced by PISA are out-of-equilibrium. A notable exception is sample 150-30 (short worms, Fig. 11 top), which reversibly formed elongated morphologies after ionization into unimers and reprotonation: the initial worms (formed on synthesis at α = 0) transformed into spheres when α was increased to 0.1 and dissolved completely at α = 0.3. The similar SAXS profiles and data fitting (Table S7†) suggest that worms with similar characteristics to the initial ones reformed on returning to α = 0. This quasi-reversible responsiveness might be understood by the fact that this sample has the shortest B block and the highest molar fraction of AA, which might lead to thermodynamically controlled assemblies.33,34,49
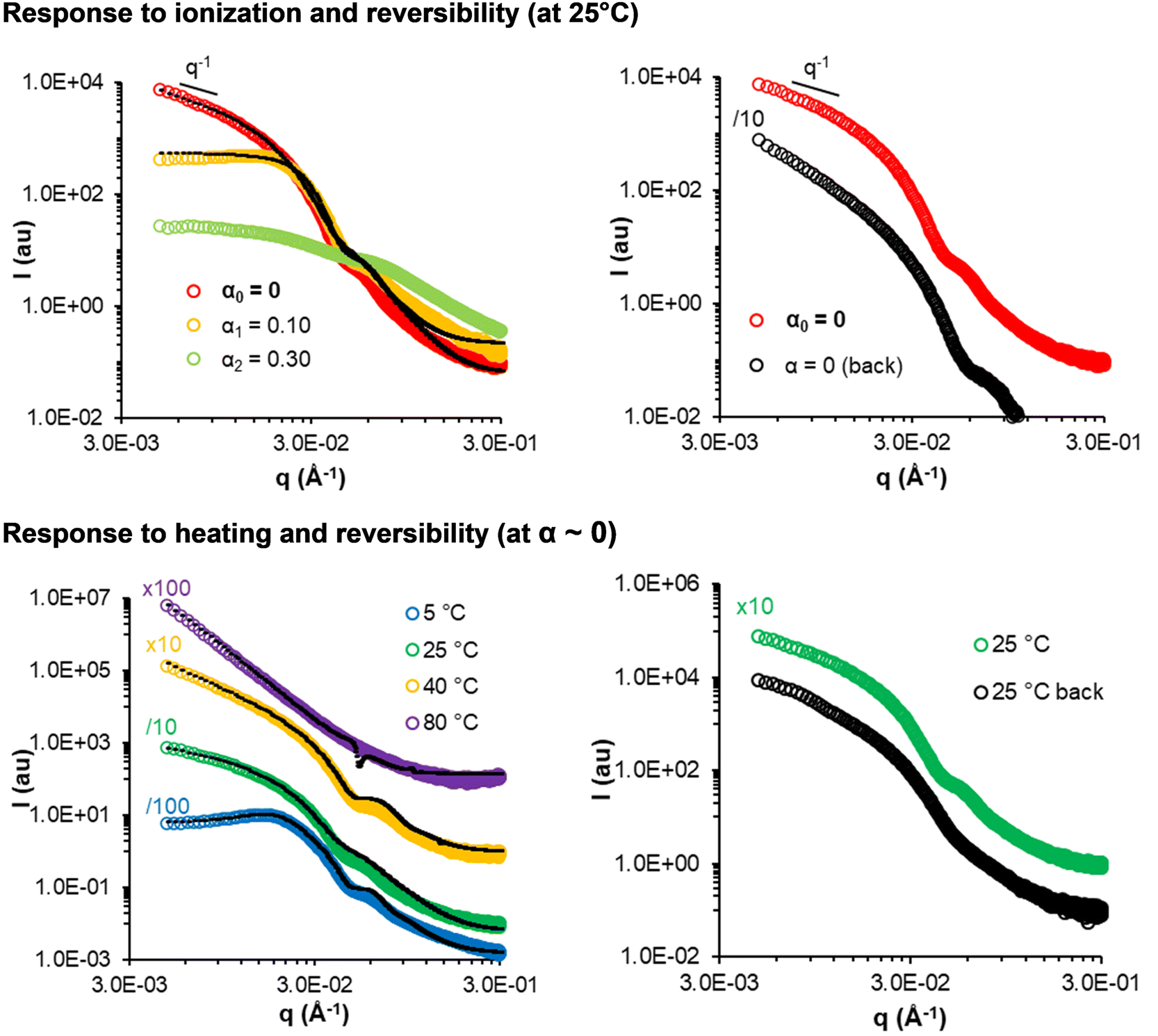 | ||
| Fig. 11 SAXS monitoring of the response of sample 150-30 (short fibers) to progressive deprotonation at 25 °C (top) and temperature (bottom, at α ∼ 0) (C = 50 g L−1). The black dotted lines are models fitting the data (see Tables S7 and S8†). | ||
Again, 150-30 is an exception. The SAXS data of sample 150-30 (short fibers) are displayed in Fig. 11, bottom. The change in the slope observed at low q indicates a morphological transition from spheres to worms to vesicles/lamellae and larger aggregates when the sample was progressively heated from 5 °C to 80 °C. This means that all main morphologies could be produced from this single copolymer composition simply by changing the temperature. The SAXS data recorded at 25 °C before and after heating to 80 °C (Fig. 11 bottom, right) were similar, suggesting that elongated objects reformed from vesicles after heating. A similar behaviour has recently been reported for other nanoparticles prepared using PISA,53–55 namely particles containing poly(2-hydroxypropyl methacrylate) and poly(4-hydroxybutyl acrylate-co-diacetone acrylamide) as the hydrophobic core.
It should be noted that PDMAc-b-PMEA samples that did not contain any AA units (see 150-0, 200-0, 400-0 in Fig. S17†) also exhibited morphology changes with the temperature. We can therefore conclude that the thermoresponsive character of the hydrophobic block is mainly due to the MEA units, although PMEA does not exhibit a classical LCST-type behaviour34 contrary to P(MEA-co-AA) copolymers.20
III. Conclusion
In this study, we investigated the impact of acrylic acid (AA) in the aqueous PISA of MEA at different ionization degrees of AA. The consequences on the kinetics of copolymerization, composition profile of the polymer chains, morphology of the self-assembled structures and post-polymerization responsiveness of the nanostructures to ionization and temperature were investigated.We showed that the incorporation of protonated AA units within the PMEA hydrophobic block favored the formation of higher order morphologies, namely worms and vesicles. While random copolymers were synthesized using uncharged AA, ionizing AA prior to polymerization led to the formation of gradient copolymers. The higher the degree of AA ionization in the feed, α0, the greater the gradient, and the lower the fraction of AA inserted in the polymer. The presence of ionized AA disfavored the formation of higher order morphologies, leading to the exclusive formation of spherical particles at α = 1, even though the polymer contained only 2% AA.
Finally, the nano-objects were sensitive to changes in temperature and pH. Small fractions of AA (2 to 5 mol% relative to MEA) triggered morphological transitions towards spherical objects when ionized. In contrast, increasing the temperature favoured the formation of higher order morphologies or large secondary aggregates. Generally, these pH- or temperature-induced transitions were irreversible. Therefore, the particles formed during PISA were generally out-of-equilibrium, implying that the morphologies obtained are probably determined both by thermodynamic aspects (DPn,B, content and ionization of the AA units affecting the packing parameter) and by kinetic aspects (rate of unimer exchange and/or of fusion/fission of particles events compared to the rate of polymerization). In one case, however, for a relatively short polymer (DPn,B = 150) with a high AA content (fAA,0 = 30 mol%), quasi reversible morphological transitions between spheres, worms and vesicles were observed, suggesting that this system was close to thermodynamic equilibrium.
These results show that PISA copolymerization of a temperature-responsive PISA monomer with a pH-responsive one is a straightforward and powerful method to prepare stimuli-responsive nano-objects in a great variety of morphologies.
Author contributions
Clément Debrie – investigation, formal analysis, visualization, data curation, writing – original draft; Noémie Coudert – formal analysis; Jean-Michel Guigner – investigation, formal analysis, resources; Fanny Coumes – investigation, formal analysis, writing – review & editing; Clément Guibert – investigation, formal analysis, writing – review & editing; Simon Harrisson – writing – review & editing, formal analysis; François Stoffelbach – investigation, formal analysis, supervision, writing – review & editing; Olivier Colombani – conceptualization, funding acquisition, project administration, supervision, writing – review & editing, methodology; Jutta Rieger – writing – original draft, conceptualization, funding acquisition, supervision, methodology, data curation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Agence Nationale de la Recherche in the framework of the DYNAMIC-PISA ANR project (ANR-19-CE06-0002-01). The authors thank Taco Nicolai for fruitful discussions and Rouguy Sognane (IPCM) for technical support in the polymerizations. We thank SOLEIL for the provision of synchrotron radiation facilities. We are grateful to Dr Thomas Bizien and Dr Javier Perez for assistance in using the SWING beamline (proposal numbers 20201382, 20220204 and 20221181) and to the SOLEIL staff for smoothly running the facility. This work benefited from the use of the SasView application, originally developed under NSF award DMR-0520547. SasView contains code developed with funding from the European Union's Horizon 2020 research and Innovation Program under the SINE2020 project, grant agreement no. 654000.References
- Y. Pei, A. B. Lowe and P. J. Roth, Macromol. Rapid Commun., 2017, 38, 1600528 CrossRef PubMed.
- F. D'Agosto, J. Rieger and M. Lansalot, Angew. Chem., Int. Ed., 2020, 59, 8368–8392 CrossRef PubMed.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS PubMed.
- X. Zhang, J. Rieger and B. Charleux, Polym. Chem., 2012, 3, 1502–1509 RSC.
- C. Debrie, N. Coudert, J.-M. Guigner, T. Nicolai, F. Stoffelbach, O. Colombani and J. Rieger, Angew. Chem., Int. Ed., 2023, 62, e202215134 CrossRef CAS PubMed.
- K. Parkatzidis, N. P. Truong, M. Rolland, V. Lutz-Bueno, E. H. Pilkington, R. Mezzenga and A. Anastasaki, Angew. Chem., Int. Ed., 2022, 61, e202113424 CrossRef CAS PubMed.
- S. Boissé, PhD Dissertation, 13/10/2020, Université Pierre et Marie Curie, https://theses.fr/027787087, 2010 Search PubMed.
- J. Rieger, Macromol. Rapid Commun., 2015, 36, 1458–1471 CrossRef CAS PubMed.
- F. Coumes, F. Stoffelbach and J. Rieger, in Macromolecular Engineering, John Wiley & Sons, Ltd, 2022, pp. 1–84 Search PubMed.
- J. Rieger, F. Stoffelbach, C. Bui, D. Alaimo, C. Jérôme and B. Charleux, Macromolecules, 2008, 41, 4065–4068 CrossRef CAS.
- V. J. Cunningham, A. M. Alswieleh, K. L. Thompson, M. Williams, G. J. Leggett, S. P. Armes and O. M. Musa, Macromolecules, 2014, 47, 5613–5623 CrossRef CAS.
- A. A. Cockram, T. J. Neal, M. J. Derry, O. O. Mykhaylyk, N. S. J. Williams, M. W. Murray, S. N. Emmett and S. P. Armes, Macromolecules, 2017, 50, 796–802 CrossRef CAS PubMed.
- J. C. Foster, S. Varlas, B. Couturaud, J. R. Jones, R. Keogh, R. T. Mathers and R. K. O'Reilly, Angew. Chem., Int. Ed., 2018, 57, 15733–15737 CrossRef CAS PubMed.
- W. Zhang, F. D'Agosto, P.-Y. Dugas, J. Rieger and B. Charleux, Polymer, 2013, 54, 2011–2019 CrossRef CAS.
- J. Tan, X. Zhang, D. Liu, Y. Bai, C. Huang, X. Li and L. Zhang, Macromol. Rapid Commun., 2017, 38, 1600508 CrossRef PubMed.
- P. Yang, O. O. Mykhaylyk, E. R. Jones and S. P. Armes, Macromolecules, 2016, 49, 6731–6742 CrossRef CAS PubMed.
- P. Chambon, A. Blanazs, G. Battaglia and S. P. Armes, Langmuir, 2012, 28, 1196–1205 CrossRef CAS PubMed.
- S. L. Canning, T. J. Neal and S. P. Armes, Macromolecules, 2017, 50, 6108–6116 CrossRef CAS PubMed.
- C. A. Figg, R. N. Carmean, K. C. Bentz, S. Mukherjee, D. A. Savin and B. S. Sumerlin, Macromolecules, 2017, 50, 935–943 CrossRef CAS.
- C. Debrie, N. Coudert, J. Abdul, S. Harrisson, O. Colombani and J. Rieger, Macromolecules, 2023, 56, 8497–8506 CrossRef CAS.
- X. Zhang, F. Boisson, O. Colombani, C. Chassenieux and B. Charleux, Macromolecules, 2014, 47, 51–60 CrossRef CAS.
- F. M. Silva, E. L. Lima and J. C. Pinto, J. Appl. Polym. Sci., 2004, 93, 1077–1088 CrossRef CAS.
- A. M. Santos, J. Guillot and T. F. McKenna, Chem. Eng. Sci., 1998, 53, 2143–2151 CrossRef CAS.
- J. I. Bowman, C. B. Eades, J. Korpanty, J. B. Garrison, G. M. Scheutz, S. L. Goodrich, N. C. Gianneschi and B. S. Sumerlin, Macromolecules, 2023, 56, 3316–3323 CrossRef CAS.
- G. M. Scheutz, J. I. Bowman, S. Mondal, J. Y. Rho, J. B. Garrison, J. Korpanty, N. C. Gianneschi and B. S. Sumerlin, ACS Macro Lett., 2023, 12, 454–461 CrossRef CAS PubMed.
- N. Audureau, F. Coumes, J.-M. Guigner, C. Guibert, F. Stoffelbach and J. Rieger, Macromolecules, 2022, 55, 10993–11005 CrossRef CAS.
- A. Rojas-Hernández, E. L. Ibarra-Montaño, N. Rodríguez-Laguna and A. Aníbal Sánchez-Hernández, J. Appl. Solution Chem. Model., 2015, 4, 7–18 CrossRef.
- J. N. Israelachvili, Intermolecular and Surface Forces, 3rd edn, 2011 Search PubMed.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969 RSC.
- T. Nicolai, O. Colombani and C. Chassenieux, Soft Matter, 2010, 6, 3111–3118 RSC.
- C. Charbonneau, C. Chassenieux, O. Colombani and T. Nicolai, Macromolecules, 2011, 44, 4487–4495 CrossRef CAS.
- C. Charbonneau, C. Chassenieux, O. Colombani and T. Nicolai, Macromolecules, 2012, 45, 1025–1030 CrossRef CAS.
- A. Shedge, O. Colombani, T. Nicolai and C. Chassenieux, Macromolecules, 2014, 47, 2439–2444 CrossRef CAS.
- N. Coudert, C. Debrie, J. Rieger, T. Nicolai and O. Colombani, Macromolecules, 2022, 55, 10502–10512 CrossRef CAS.
- R. Hoogenboom, A.-M. Zorn, H. Keul, C. Barner-Kowollik and M. Moeller, Polym. Chem., 2012, 3, 335–342 RSC.
- N. Audureau, C. Veith, F. Coumes, T. P. T. Nguyen, J. Rieger and F. Stoffelbach, Macromol. Rapid Commun., 2021, 42, 2100556 CrossRef CAS PubMed.
- S. J. Hunter and S. P. Armes, J. Colloid Interface Sci., 2023, 634, 906–920 CrossRef CAS PubMed.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS PubMed.
- J. Zhang, B. Farias-Mancilla, I. Kulai, S. Hoeppener, B. Lonetti, S. Prévost, J. Ulbrich, M. Destarac, O. Colombani, U. S. Schubert, C. Guerrero-Sanchez and S. Harrisson, Angew. Chem., Int. Ed., 2021, 60, 4925–4930 CrossRef CAS PubMed.
- E. Lejeune, M. Drechsler, J. Jestin, A. H. E. Müller, C. Chassenieux and O. Colombani, Macromolecules, 2010, 43, 2667–2671 CrossRef CAS.
- C. Charbonneau, M. M. D. S. Lima, C. Chassenieux, O. Colombani and T. Nicolai, Phys. Chem. Chem. Phys., 2013, 15, 3955–3964 RSC.
- R. C. Hayward and D. J. Pochan, Macromolecules, 2010, 43, 3577–3584 CrossRef CAS.
- J. Rieger, Macromol. Rapid Commun., 2015, 36, 1458–1471 CrossRef CAS PubMed.
- I. Lacík, S. Beuermann and M. Buback, Macromol. Chem. Phys., 2004, 205, 1080–1087 CrossRef.
- V. Jaacks, Makromol. Chem., 1972, 161, 161–172 CrossRef CAS.
- S. Harrisson, F. Ercole and B. W. Muir, Polym. Chem., 2010, 1, 326–332 RSC.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS PubMed.
- S. Y. Khor, J. F. Quinn, M. R. Whittaker, N. P. Truong and T. P. Davis, Macromol. Rapid Commun., 2019, 40, 1800438 CrossRef PubMed.
- N. Coudert, C. Debrie, S. Harrisson, J. Rieger, T. Nicolai and O. Colombani, Macromolecules, 2023, 56, 9584–9594 CrossRef CAS.
- M. Chenal, L. Bouteiller and J. Rieger, Polym. Chem., 2013, 4, 752–762 RSC.
- M. Semsarilar, V. Ladmiral, A. Blanazs and S. P. Armes, Langmuir, 2013, 29, 7416–7424 CrossRef CAS.
- C. Grazon, J. Rieger, N. Sanson and B. Charleux, Soft Matter, 2011, 7, 3482–3490 RSC.
- N. Audureau, F. Coumes, J.-M. Guigner, J. Rieger and F. Stoffelbach, Eur. Polym. J., 2024, 208, 112858 CrossRef CAS.
- S. J. Byard, C. T. O'Brien, M. J. Derry, M. Williams, O. O. Mykhaylyk, A. Blanazs and S. P. Armes, Chem. Sci., 2020, 11, 396–402 RSC.
- L. P. D. Ratcliffe, M. J. Derry, A. Ianiro, R. Tuinier and S. P. Armes, Angew. Chem., 2019, 131, 19140–19146 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00373j |
| This journal is © The Royal Society of Chemistry 2024 |