
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An in situ DRIFTS-MS study on elucidating the role of V in the selective oxidation of methacrolein to methacrylic acid over heteropolyacid compounds†
Sarayute
Chansai
 *a,
Yuki
Kato
b,
Wataru
Ninomiya
b and
Christopher
Hardacre
*a
*a,
Yuki
Kato
b,
Wataru
Ninomiya
b and
Christopher
Hardacre
*a
aDepartment of Chemical Engineering, Engineering Building A, The University of Manchester, Manchester M13 9PL, UK. E-mail: sarayute.chansai@manchester.ac.uk; c.hardacre@manchester.ac.uk
bMMA Group, Monomers & Catalysts Laboratory, Mitsubishi Chemical Corporation, 20-1 Miyuki-cho, Otake-shi, Hiroshima 739-0693, Japan
First published on 16th July 2024
Abstract
The role of vanadium in 11-molybdo-1-vanadophosphoric acid (H4PMo11VO40, HPMoV) has been investigated and compared with 12-modybdophosphoric acid (H3PMo12O40, HPMo) using in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) coupled with mass spectrometry (MS). It has been found for the first time that there are differences in the DRIFT spectra of carbonyl- and carboxylate-type species recorded in the region of 1900–1200 cm−1 between the two catalysts under methacrolein (MAL), MAL + O2, and MAL + O2 + H2O reaction conditions at 320 °C and the MS results show faster formation of methacrylic acid over HPMoV than over HPMo catalysts. A number of hydrocarbon species adsorbed on the surface have been identified in the DRIFTS analysis including a 5-membered ring lactone-type species, a 6-membered ring lactone-type species, bidentate methacrylate species, monodentate methacrylate species, and π-adsorbed species, with the presence of V in the heteropolyacid Keggin structure favouring the formation of monodentate methacrylate surface species compared with lactone-type surface species under MAL + O2 + H2O reaction conditions at 320 °C. The monodentate methacrylate surface species is proposed to be the intermediate in the final step for the formation of MAA. It is likely that the same mechanism of selective oxidation of MAL occurs with and without the substitution of Mo with V, with the presence of V resulting in a faster formation of active monodentate methacrylate intermediates.
1. Introduction
Heteropolyacids (HPAs) used as oxidation catalysts have been extensively investigated for many decades1–39 due to their acid and redox properties, high thermal stability, simple preparation procedure, and simple physical property modification such as solubility and surface areas.3,5,7,12–16,18,20,21,26,40,41 In general, the clusters of HPAs are made up of metal–oxygen anions with different molecular sizes, compositions, and structures.38,39 One of the HPAs that has received much attention is the Keggin-type structure with the formula [XM12O40]n−, typically composed of a central XO4 tetrahedral unit surrounded by twelve octahedrons of MO6 units, linked by corner- and edge-sharing oxygen atoms. [PMo12O40]3− is a member of the Keggin-type HPA family, which exhibits two defining characteristics (Brønsted acidity and redox behaviour). H3PMo12O40 (HPMo) and its salts have been reported to show excellent catalytic oxidation of various types of hydrocarbons.11,14,17,19,23–26,29–32,34,38,39 The selective oxidation of methacrolein (MAL) to methacrylic acid (MAA) over heteropolyacids has been extensively studied1,4,15,26–28,30–32,35–37 since MAA is an important chemical in the production of methyl methacrylate and other derivatives including polymers.Previous research4,29,30,32 has shown that partially substituting Mo with V in the primary Keggin structure enhances the catalytic activity of MAL oxidation to the selective form MAA. The vanadyl species in a range of HPA catalysts were investigated for the oxidation and dehydration of methanol,25 oxidation of isobutane14,24,38 and isobutyric acid,7,8,11,17 and partial oxidation of propane,23 for example, ascertaining the role and promotional effect of V on the catalytic activity and product selectivity.
Deußer and co-workers4 investigated the effect of V on the kinetics of catalytic MAL oxidation to MAA over various CsxH3+y–xPMo12−yVyO40 catalysts. It was reported that the presence of V moderately influenced the rate of MAL oxidation but substantially decreased the rate of MAA oxidation, hence the significant decrease in a consecutive oxidation of MAA and resulting in an increase in the selectivity of MAA.
Song et al.13 studied the redox properties of several Keggin-type HPA compounds as a function of the counter-cation, heteroatom, and polyatom substitution, in which their reduction potentials were electrochemically measured. They reported that the V-substituted H3+yPMo12−yVyO40 samples demonstrated an increase in the reduction potential in comparison with the H3PMo12O40 compound, indicative of different redox properties, which can be tuned and designed for the selective oxidation transformations.
Several studies5,16–23,25,26 have investigated the nature of the active vanadyl species in the Keggin structure that could promote the selective oxidation reactions. It was reported22 that heteropoly anions, [PMo11VO40]4−, in the H4PMo11VO40 catalyst were not stable during calcination and reaction, resulting in a partial decomposition and a defect of the Keggin structure. This behaviour facilitated the migration of V atoms from primary to secondary structures.
Recently, Zhou et al.30 reported an in-depth study on vanadyl species for the oxidation of MAL to MAA over three HPA catalysts containing different vanadyl species in primary and secondary structures, i.e. H4PMo11VO40 (V in the primary structure), HVOPMo12O40 (V in the form of VO2+ in the secondary structure), and V2O5/H3PMo12O40 (V in the form of V2O5 before calcination under air). It was found that after calcination of all V-containing HPA catalysts, the vanadyl species were detected in the form of ions, resulting in a breaking of the symmetry of the Keggin structure. However, during the oxidation reaction of MAL to MAA, these independent vanadyl ions were transformed into a defective Keggin structure or a squashed square pyramidal form of the Keggin structure. They also reported that the V2O5/H3PMo12O40 catalyst showed the best catalytic performance due to several active vanadyl species formed under calcination and reaction conditions. It was concluded that the vanadyl ions located in the secondary structure were transformed and interacted with the primary Keggin structure during the oxidation of MAL to MAA. Those vanadyl ions were identified as V4+ ions, whose amount was correlated with the selectivity of MAA.
Although the effect and properties of V in HPA catalysts4,5,16,26,29,30 in promoting MAL conversion and MAA selectivity have been reported, the mechanistic understanding of the effect of V on the surface reaction remains unclear. In particular, infrared studies on the hydrocarbon-derived surface species under reaction conditions are very limited. In the present work, DRIFTS-MS has been used to gain a better understanding of and an insight into the role of V in the reaction mechanism during the oxidation of MAL to MAA over Keggin-type H3PMo12O40 and H4PMo11V1O40 catalysts.
2. Experimental
In this study, both H3PMo12O40 (HPMo) and H4PMo11VO40 (HPMoV) catalysts were obtained from Mitsubishi Chemical Corporation. The catalyst was formed from hydrous 12-molybdophosphoric acid (H3PMo12O40·xH2O, Nippon Inorganic Colour and Chemical Co., Ltd.) extracted from an aqueous solution of H3PMo12O40 and diethyl ether. H3PMo12O40·xH2O was then recrystallised from an aqueous solution and the solid dried at 60 °C overnight.36In situ DRIFTS measurements were performed using a Bruker Tensor 27 FTIR spectrometer equipped with a liquid N2-cooled detector. Approximately 40 mg of catalyst sample was placed in a ceramic crucible in the in situ DRIFTS cell. The exit lines were connected to a Hiden Analytical HPR-20 mass spectrometer in order to monitor the gas phase species: H2O (m/z = 18), MAL (m/z = 70), and MAA (m/z = 86).
Prior to the DRIFTS experiments, the catalyst was pretreated by heating in Ar at a total flow rate of 100 cm3 min−1 ramping the temperature to 320 °C at a rate of 10 °C min−1. Then the catalyst was held at this temperature for 60 min. Subsequently, the IR spectrum of the treated HPA catalyst was taken as a background.
The reactant gases used were MAL (95%, Sigma-Aldrich), O2 (99.99% BOC), and Ar (99.99% BOC). O2 and Ar were fed from independent Aera™ PC-7700C mass flow controllers. MAL and water vapour were introduced to the system by means of separate saturators with Ar as the carrier gas. The MAL saturator was placed in an ice water bath and the temperature of the H2O saturator was controlled by Grant™ GD120 thermostatic baths. All the gas lines following the water and MAL saturators were trace heated to prevent condensation. The concentrations of the reactants used were 3500 ppm MAL, 7000 ppm O2, 7000 ppm H2O (when added), and Ar balance. The total flow rate was 100 cm3 min−1. Gas flows were carefully equilibrated using micrometer needle valves to adjust the pressure between the gas flows on each side of the four-way valve with a high-sensitivity differential pressure detector. This avoided the production of spikes on the MS signal when switching from one mixture to another.
Two different types of in-situ DRIFTS-MS experiments were carried out. The first set of experiments was designed to investigate the effect of V on the changes of surface and gas phase species under different gas feeds, i.e. MAL + Ar, MAL + O2 + Ar and MAL + O2 + H2O + Ar feeds. After Ar pretreatment, the HPMo catalyst was exposed to each gas feed for 30 min at 320 °C. Our previous work36,37 reported the important role of water in the formation of MAA, therefore, the second set of experiments was performed using H2O switches in and out of the MAL + O2 feed to probe the role of V. After Ar pretreatment, the HPMo catalyst was exposed to the MAL + O2 gas feed for only 1 min at 320 °C to allow surface species to be formed and then the cycling switches of H2O in (5 min) and out (5 min) of the MAL + O2 + Ar feed were performed for 4 cycles.
All in situ DRIFT spectra were recorded with a resolution of 4 cm−1 with an accumulation of 64 scans every 30 s and the DRIFT spectra were analysed using OPUS software. The IR data are reported as log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1/R (absorbance), with R = I/I0, where R is the sample reflectance, I is the intensity measured under reaction conditions, and I0 is the intensity measured on the pure HPMo sample under a flow of argon at 320 °C.
1/R (absorbance), with R = I/I0, where R is the sample reflectance, I is the intensity measured under reaction conditions, and I0 is the intensity measured on the pure HPMo sample under a flow of argon at 320 °C.
3. Results and discussion
3.1 In situ DRIFTS-MS and IR assignments
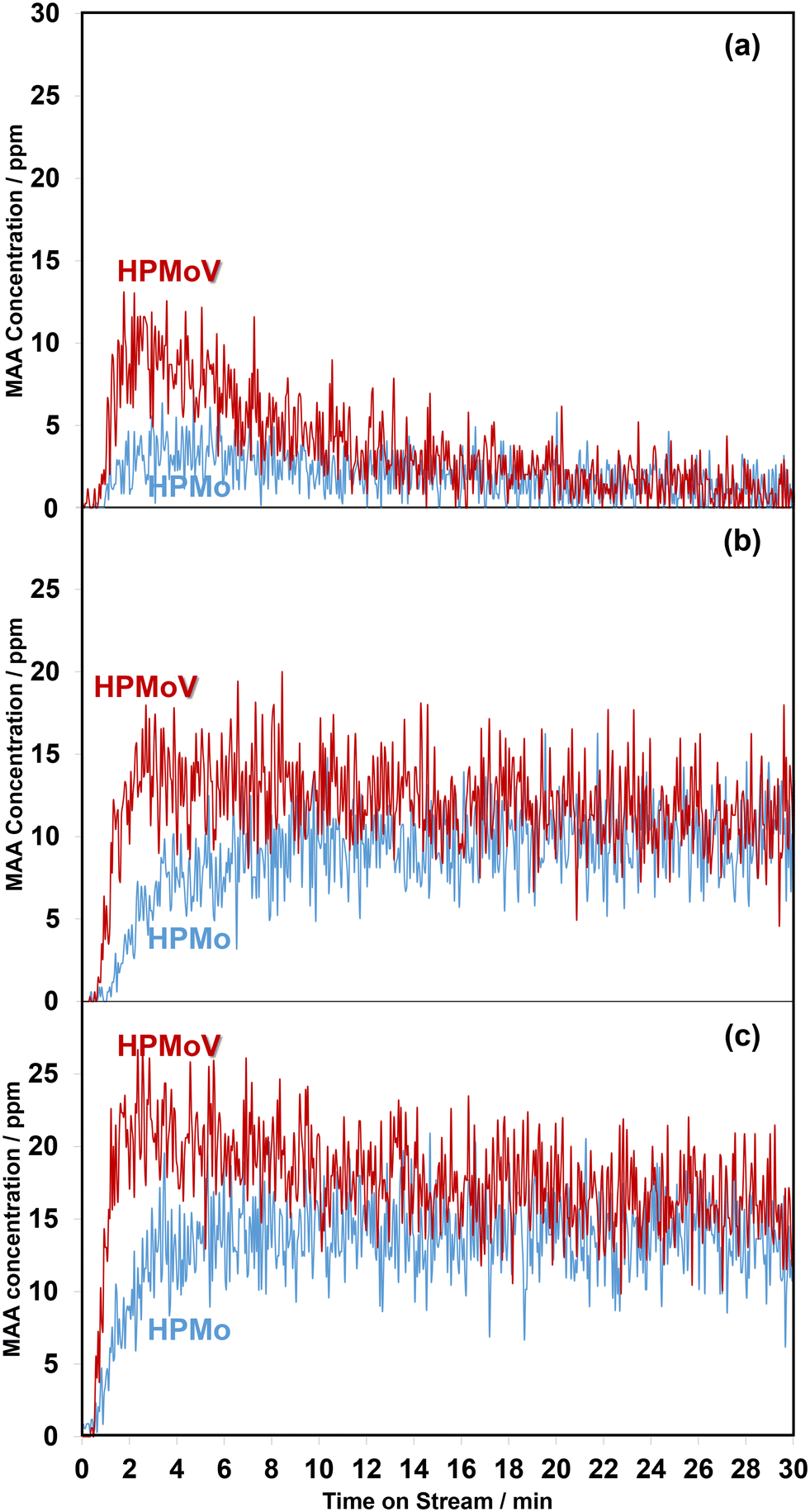 | ||
| Fig. 1 Comparison of the evolution of MAA formation at 320 °C as a function of time on stream over H3PMoO12O40 and H4PMoO11VO40 under MAL (a), MAL + O2 (b), and MAL + O2 + H2O (c). The gas feed is composed of 3500 ppm MAL (when added), 7000 ppm O2 (when added), 7000 ppm H2O (when added), and Ar balance and the total flow rate is 100 cm3 min−1. | ||
Overall, the current work shows that there is a difference in the release of desirable gas phase MAA product from the HPMo catalyst under all three different reaction conditions compared with over the HPMoV catalyst at 320 °C. A comparison of in situ DRIFT spectra, recorded at 2 min under different reaction conditions and corresponding to the gaseous MAA profiles displayed in Fig. 1, is shown in Fig. 2.
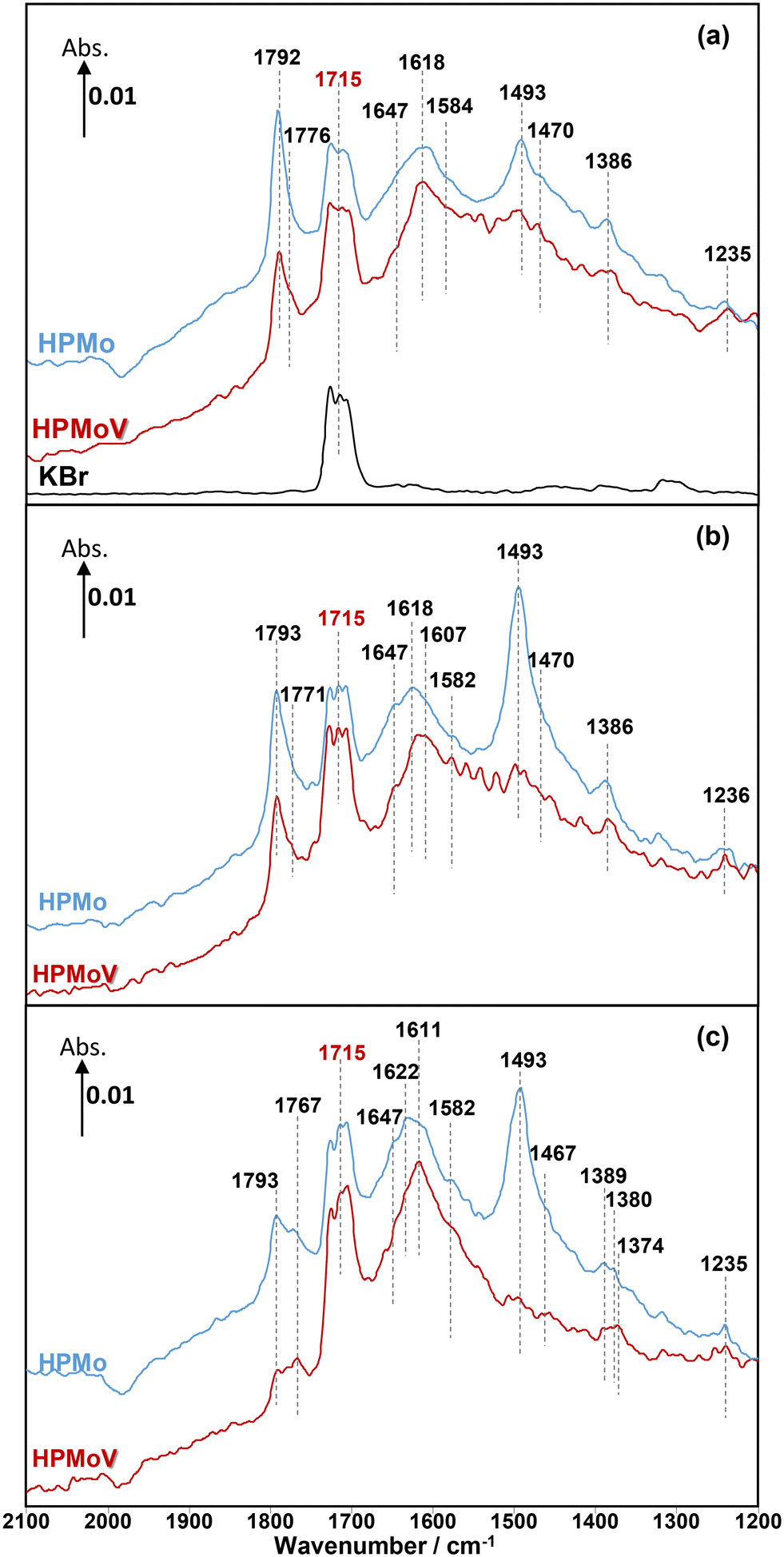 | ||
| Fig. 2 Comparison of in situ DRIFT spectra recorded at 320 °C over H3PMoO12O40 and H4PMoO11VO40 at 2 min under MAL (a), MAL + O2 (b), and MAL + O2 + H2O (c). The gas feed is composed of 3500 ppm MAL (when added), 7000 ppm O2 (when added), 7000 ppm H2O (when added), and Ar balance and the total flow rate is 100 cm3 min−1. | ||
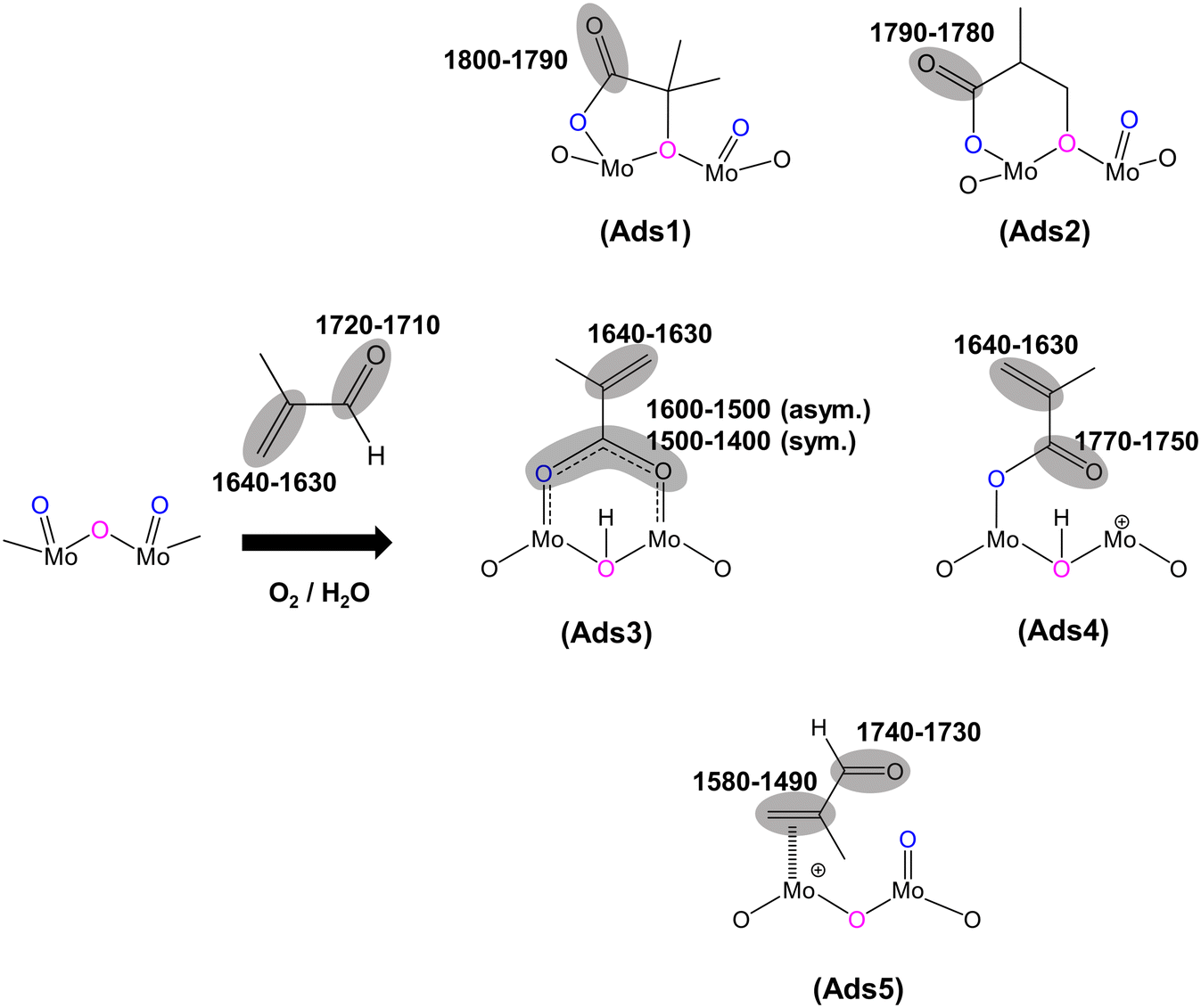 | ||
| Fig. 3 Proposed surface species under MAL reaction conditions at 320 °C over HPA catalysts: 5-membered ring lactone-type species (Ads1), 6-membered ring lactone-type species (Ads2), bidentate methacrylate species (Ads3), monodentate methacrylate species (Ads4), and π-adsorbed species [Ads5]. | ||
Under MAL adsorption conditions at 320 °C (Fig. 2a), KBr was initially used as an inert and reference material. It is clearly observed that the carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) group of gas-phase MAL at 1715 cm−1 is the only significant IR feature, which can also be seen over both HPA catalysts. In contrast to KBr powder, several IR bands were also observed over both HPMo and HPMoV catalysts (Fig. S1 and S2,† respectively).
O) group of gas-phase MAL at 1715 cm−1 is the only significant IR feature, which can also be seen over both HPA catalysts. In contrast to KBr powder, several IR bands were also observed over both HPMo and HPMoV catalysts (Fig. S1 and S2,† respectively).
Considering the IR bands above 1700 cm−1, in comparison with the gas phase MAL (free ν(CO), 1715 cm−1), the IR band observed at 1793 cm−1 would generally be associated with an anhydride species;43 however, anhydrides typically consist of two IR bands due to the asymmetric and symmetric stretch of OCO. In the present spectra, the additional higher wavenumber IR band/shoulder was not observed; this suggests that the surface species responsible for the band at 1793 cm−1 is associated with the formation of cyclic lactone-type adsorbed species, for example, Ads1 and Ads2 in Fig. 3. This feature is at a higher wavenumber than typically found for lactone carbonyl groups (1750–1700 cm−1) and the shift is likely to be due to the cyclic nature of the lactone formed over the HPA catalysts. However, Schaidle and co-workers45 recently carried out DRIFTS work on the deoxygenation of acetic acid over molybdenum carbides and reported that the double IR bands at 1800–1780 cm−1 are associated with symmetric and asymmetric ν(CO) in monodentate acetates, derived from acetic acid. Our DRIFTS work has shown that the peak at 1793 cm−1 appears as a single and strong IR band and is tentatively associated and assigned to the ν(CO) group in cyclic lactone.
Fig. 2 shows IR bands at 1647 and 1618 cm−1 which are attributed to the stretching vibration of unreacted carbon double bonds ν(CC), possibly with different adsorption configurations as bidentate methacrylate adsorbed species (Ads3), monodentate methacrylate adsorbed species (Ads4), and π-adsorbed species (Ads5). The shoulders at 1584 and 1470 cm−1 may be associated with the asymmetric and symmetric stretching vibration of carboxylate-type surface species (νasOCO and νsOCO), respectively. In this case, the formation of bidentate methacrylate adsorbed species (Ads3) shifted the vibration of the unreacted CC bond to 1647 cm−1 due to unconjugated configuration, while the carbonyl group of monodentate methacrylate adsorbed species (Ads4) is conjugated with the CC bond, resulting in a slightly lower wavenumber of the CC bond at 1618 cm−1. It is important to note that the bending vibration of adsorbed and gas phase H2O, δ(HOH), is commonly observed in the region of 1670–1570 cm−1.6,37,44 The presence of this band overlaps with the IR signal of the CC bond at 1618 cm−1 and its observation becomes increasingly challenging with H2O accumulation on the catalyst surfaces over the course of the reaction.
Furthermore, a significantly more intense peak at 1493 cm−1 was observed over HPMo compared with HPMoV. Krauß et al.9 carried out DRIFTS measurements on MAL and MAA adsorptions over Mo9V3W12Ox mixed oxides as well as the Cs2H2PMo11VO40 heteropolyacid compound. The IR band observed at 1520–1490 cm−1 was assigned to π-complex adsorption (Ads5) over partially reduced MoOx. This adsorption can be facilitated by the availability of partially reduced Mo (IV and V) species. However, it is expected that fresh HPA catalysts initially possess the terminal group of (MoOt) in the Keggin structure, meaning that π-complex adsorption (Ads5) should not be substantially formed over HPA compounds. Therefore, it is likely that the IR band at 1493 cm−1 is related to a carboxylate species,42–46 for example, the bidentate methacrylate adsorbed species (Ads3).
In addition to MAL adsorption, the adsorption of MAA on HPMo, HPMoV, and KBr at 320 °C was carried out to help identify the surface species. As shown in Fig. 4, on KBr, a strong carbonyl vibration associated with gas phase MAA was observed, which is split into bands at 1768 and 1751 cm−1 due to vibrational stretching of ν(CO) and ν(C–O−). At 320 °C, there is an absence of the ν(CC) stretching band at 1632 cm−1 and δ(CH3) bending at 1386 cm−1, which is consistent with the work by Krauß et al.9 In contrast, over HPMo and HPMoV significant differences in the surface species compared with that over KBr were observed when MAA was adsorbed. Strong IR bands at 1795–1785 cm−1, assigned to lactone-type adsorbed species (Ads1 and Ads2), were detected with the stretching vibration of unreacted ν(CC) at 1632 cm−1. This is also accompanied by the appearance of the IR bands at 1581, 1491, and 1470 cm−1, which are associated with carboxylate-type surface species.42–46 Previously, on adsorption of acetic acid,42,45,46 it was also reported that the functional group (–COOH) of acetic acid readily formed the carboxylate species as acetates (H3CCOO−) on catalyst surfaces with the IR region of 1560–1550 cm−1 for νas(COO−) and with the IR region of 1500–1450 cm−1 for νs(COO−).42–46 Therefore, it is likely that the strong IR band at 1493–1491 cm−1 is associated with a bidentate methacrylate species (Ads3).
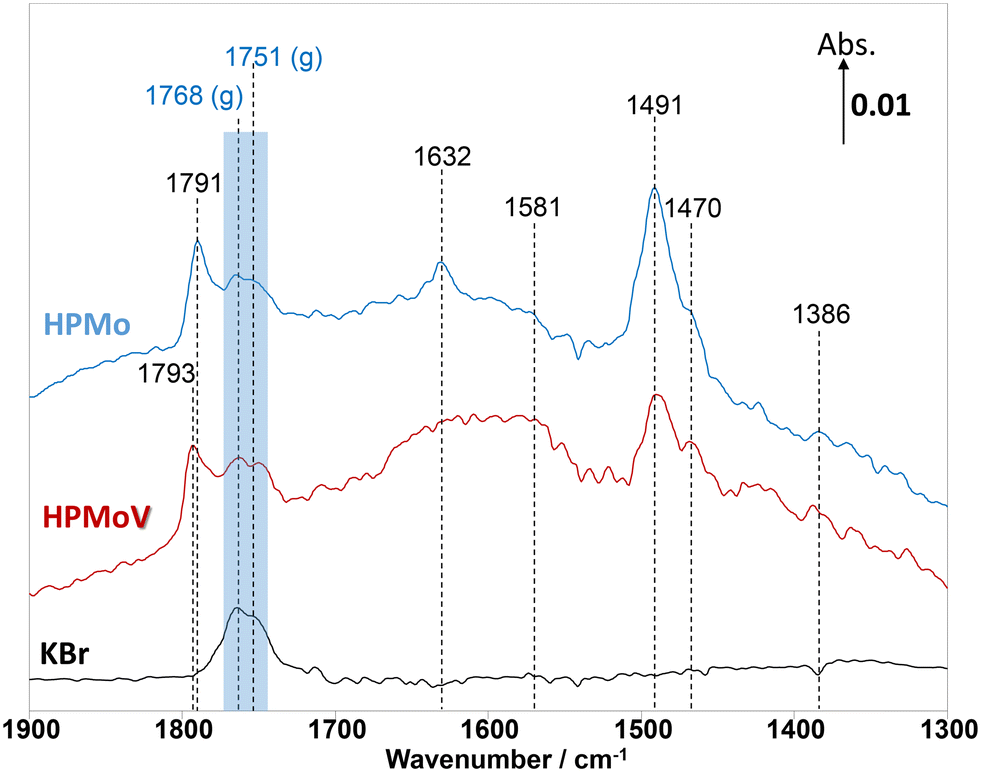 | ||
| Fig. 4 Comparison of in situ DRIFT spectra recorded at 2 min during the adsorption of methacrylic acid (MAA) at 320 °C over H3PMoO12O40 (HPMo), H4PMoO11VO40 (HPMoV), The gas feed is composed of 3500 ppm MAA and Ar balance and the total flow rate is 100 cm3 min−1. | ||
As well as examining MAL adsorption (Fig. 2a) at 320 °C over HPAs, MAL + O2 (Fig. 2b) and MAL + O2 + H2O (Fig. 2c) reactions were also carried out under steady-state conditions. The similarity in IR features and adsorbed species was observed under all reaction conditions. However, the presence of H2O in the reaction feed has a direct impact on the formation of monodentate methacrylate adsorbed species (Ads4) at around 1767 cm−1, and the occurrence of a lactone-type adsorbed species (Ads1 and Ads2) in the IR region of 1795–1785 cm−1. It is important to point out that the IR band at 1793 cm−1 under MAL + O2 + H2O reaction conditions is less pronounced than those under MAL and MAL + O2 reaction feeds, i.e. the formation of cyclic lactone-type surface species is hindered or converted by adding H2O to another adsorbed species. It is also worth noting that both Fig. 2 and 4 show similar adsorbed surface species in the IR region of 1770–1750 cm−1 under MAA adsorption, MAL adsorption, and MAL + O2 reaction with and without adding H2O at 320 °C. Therefore, it is proposed that the observation of the IR bands between 1770 and 1750 cm−1 is associated with the stretching vibration of the ν(CO) group of gas phase MAA or a weakly bound adsorbed MAA, possibly as monodentate methacrylate surface species (Ads4), which can be readily reacted and converted to form gas phase MAA.27,32,36,37
One important feature to be pointed out in both Fig. 2 and 4 is that bidentate methacrylate species (Ads3) at 1493–1491 cm−1 on the HPMoV catalyst is less significant than over HPMo. The presence of V in the HPMoV catalyst is thought to minimise the formation of bidentate methacrylate species (Ads3) but promote the formation of monodentate methacrylate adsorption (Ads4). Zhou et al.30 proposed that V4+/VO2+ species in the secondary structure rather than the primary structure has an influence on the formation and selectivity to MAA during the oxidation reaction of MAL to MAA due to the presence of accessible active vanadyl species. Bayer et al.17 also reported higher catalytic oxidative dehydrogenation of isobutyric acid to MAA on H4PMo11VO40 than on H3PMo12O40 because H4PMo11VO40 was reduced and partially decomposed, resulting in more active V4+ sites during the reaction.
Additional DRIFTS analysis by integrating the spectra between 1800 and 1760 cm−1 over the 30 min reaction period has been used to quantify the changes observed. Fig. S3† shows the comparison of changes in the ratio of monodentate methacrylate species (Ads4) in the IR range of 1770–1760 cm−1 to lactone-type adsorbed species (Ads1 and Ads2) between 1800 and 1780 cm−1. It is found that all ratios of monodentate methacrylate species (Ads4) to lactone-type surface species (Ads1 and Ads2) under three different reaction conditions at 320 °C over the HPMoV catalyst change more rapidly than those over the HPMo catalyst, indicative of faster and more significant formation of monodentate methacrylate surface species (Ads4) on the HPMoV catalyst. Therefore, in the present work, it is likely that the reducibility of the H4PMo11VO40 catalyst leads to the formation of these active V4+/VO2+ species,30 which facilitates the conversion of bidentate methacrylate species (Ads3) to and/or the direct formation of monodentate methacrylate surface species (Ads4). As a result, more MAA is formed over HPMoV than over HPMo catalysts, as shown in Fig. 1.
Recently, Tian et al.48 reported a density functional theory (DFT) study and proposed a reaction mechanism which operated via the abstraction of proton from MAL and the formation of monodentate methacrylate species over the bridge oxygen of HPA compounds as a final step to form MAA. The DFT calculations48 revealed the formation of monodentate methacrylate species with the lowest reaction energy for the HPMoV compound before producing MAA. In addition, the presence of V in the HPA compound was found to result in the highest reaction rate constant and lowest activation energy for the change from monodentate methacrylate surface species to gas phase MAA product. Interestingly, the in situ DRIFTS analysis reported, herein, is consistent with this simulation, indicating the importance of the formation of monodentate methacrylate adsorbed species (Ads4) and the vital role of V present in the HPMoV catalyst in enhancing the formation of desirable MAA product.
In addition to gaseous MAA formation detected, gas phase CO and CO2 were also observed, as shown in Fig. S4–S6.† HPMo was found to produce more CO and CO2 than HPMoV; however, both catalysts show an initial transient CO and CO2 formation within 5 min of exposure to the gas feeds. Fig. 5 shows the proposed mechanism for the formation of both CO and CO2 under MAL adsorption (without co-fed O2) at 320 °C. During MAL adsorption, the HPA surfaces are occupied by bidentate methacrylate adsorbed species (Ads3), and that the release of gaseous CO2 from the surface leads to a partially reduced Mo species, which in turn forms the π-adsorbed species (Ads5).
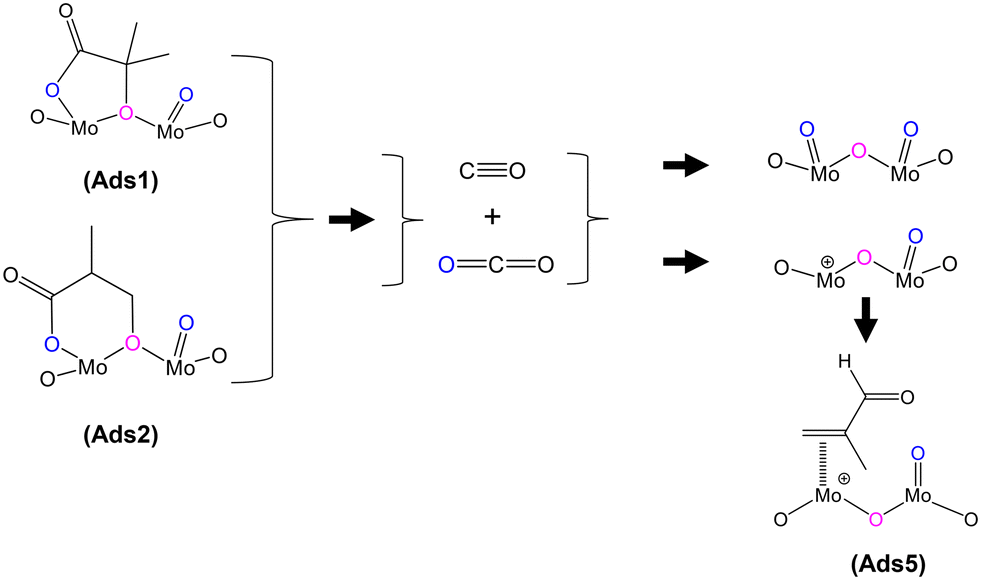 | ||
| Fig. 5 Proposed formation of by-products CO and CO2 derived from lactone-type adsorbed species during MAL adsorption at 320 °C. | ||
In addition to the steady-state reaction under different reaction conditions at 320 °C, transient experiments were carried out by performing the switches of H2O in and out of MAL + O2 to probe the influence of V on the surface-adsorbed species.
3.2 H2O transient switches
The influence of H2O on the activity of MAL oxidation and the selectivity of MAA has been reported over HPA2,27,28,36,37 and mixed oxide49,50 catalysts. Recently, Yasuda et al.36 and Chansai et al.37 reported the role of water and its involvement in the reaction mechanism over HPMo, where the proton from water is used to form an –OH group in the MAA product. Fig. 6 shows the changes in gas phase MAA, while Fig. 7 demonstrates the evolution of IR bands in the region of 2100–1200 cm−1, corresponding to Fig. 6 during the first cycles of H2O switching in and out of the MAL + O2 feed at 320 °C.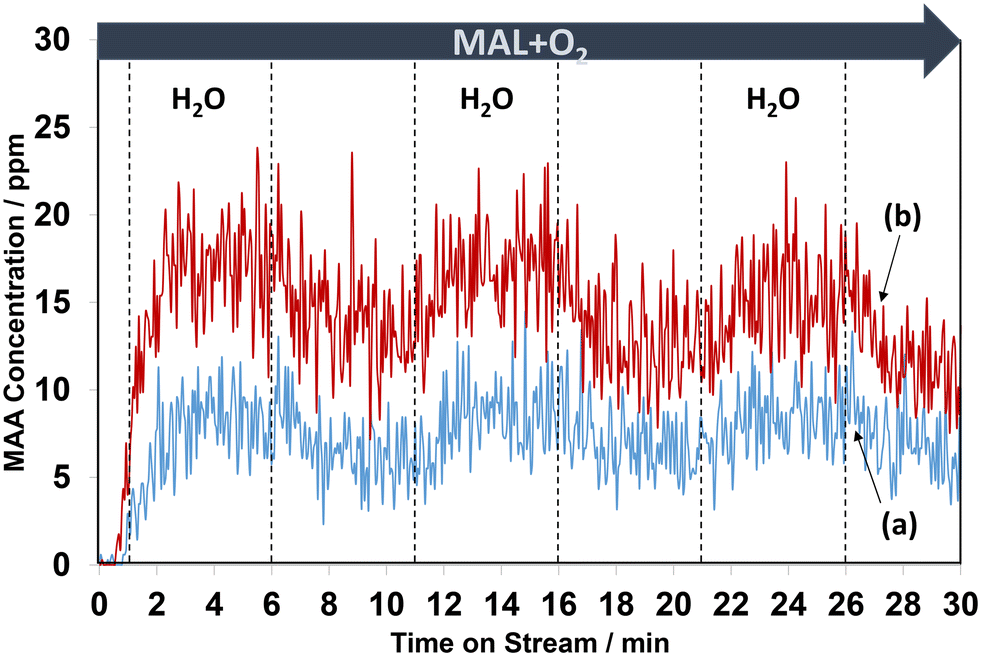 | ||
| Fig. 6 Changes in gaseous MAA product as a function of time on stream at 320 °C over HPMo (a) and HPMoV (b) samples during the switches of H2O in and out of the MAL + O2 gas feed. The gas feed is composed of 3500 ppm MAL, 7000 ppm O2, 7000 ppm H2O (when added), and Ar balance and the total flow rate is 100 cm3 min−1. | ||
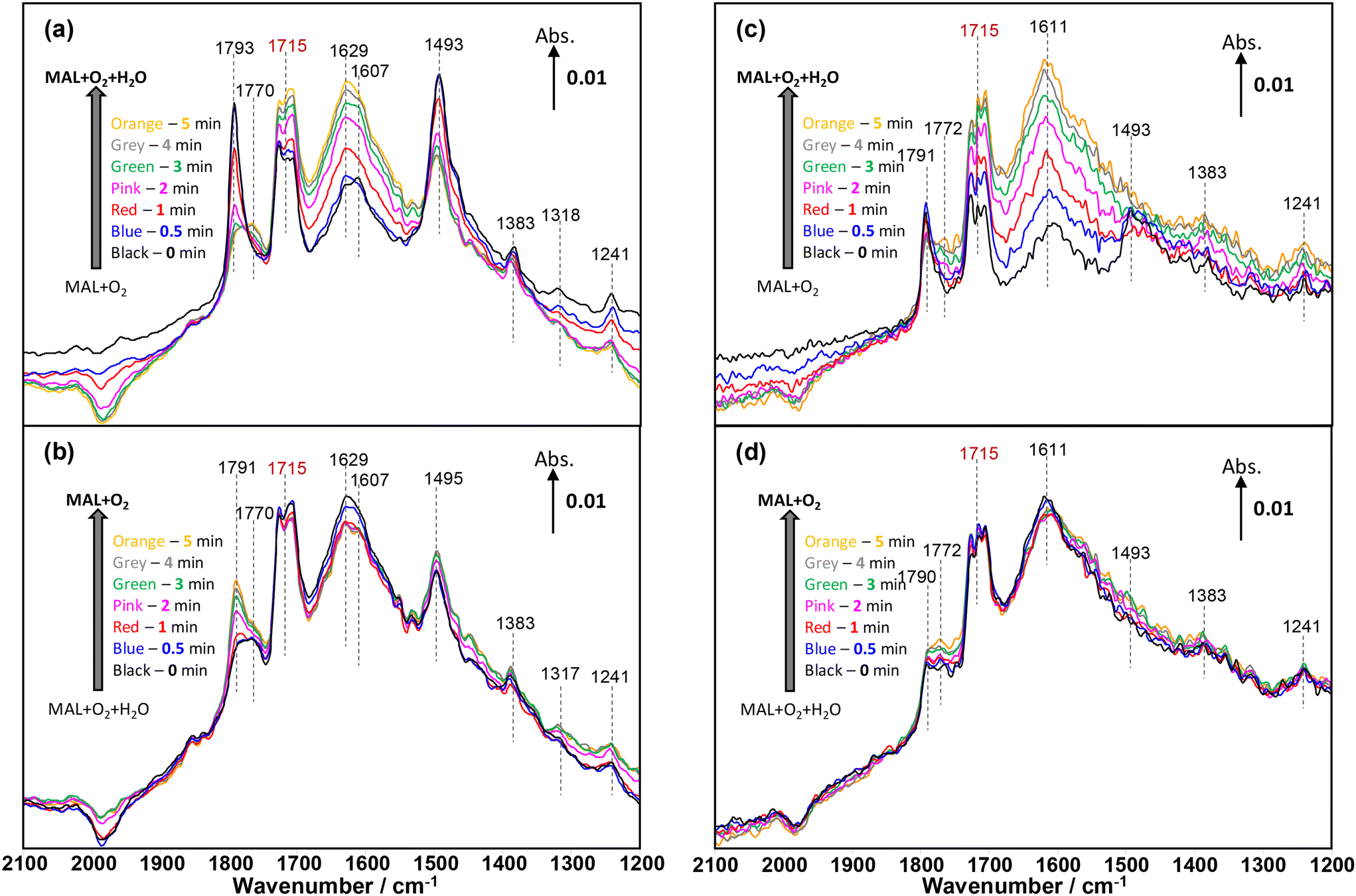 | ||
| Fig. 7 Changes in in situ DRIFT spectra (2100–1200 cm−1) corresponding to the formation of MAA formation shown in Fig. 3 as a function of time on stream over HPMo (a and b) and HPMoV (c and d) catalysts at 320 °C during the 2nd cycle of switches of H2O in and out of the MAL + O2 gas feed. The gas feed is composed of 3500 ppm MAL, 7000 ppm O2, 7000 ppm H2O (when added), and Ar balance and the total flow rate is 100 cm3 min−1. | ||
From Fig. 6, regardless of the HPA used, the production of MAA was promoted by adding H2O into the MAL + O2 gas feed and this is consistent with previous studies.36,37 On exposure to MAL + O2 reaction conditions for 1 min, MAA was formed at the start of the reaction before adding H2O. However, an increase in MAA was noticeable over the course of 5 min of reaction when H2O was added. On removing H2O from the MAL + O2 feed, a gradual decrease in the concentration of MAA was observed. Further switches of water resulted in a similar behaviour with a slow decrease in overall concentration of MAA over the course of 30 min. As noted previously, the formation of MAA over the HPMoV catalyst was significantly higher than that over HPMo.
Fig. 7 displays the changes in in situ DRIFT spectra corresponding to the changes shown in Fig. 6 during the 1st cycle of H2O switching in and out of the MAL + O2 feed over both HPMo (Fig. 7a and b) and HPMoV (Fig. 7c and d) catalysts at 320 °C. Differences in in situ DRIFT spectra calculated between the H2O switches are also provided in Fig. S7† for the comparison with Fig. 7. In Fig. 7a, over the HPMo catalyst, a significant formation of lactone-type adsorbed species (Ads1 and Ads2) in the IR range of 1800–1780 cm−1 was initially observed; however, on addition of H2O to the MAL + O2 feed, a rapid decrease in these surface species together with the bidentate methacrylate surface species (Ads3) at 1493 cm−1 was observed. It was reported2,49,51 that the addition of H2O (–OH group) re-oxidised the reduced MoO3 surfaces and this may have facilitated the decrease in those adsorbed species as a result of being transformed into more active intermediates, i.e. monodentate methacrylate adsorbed species (Ads4). It should also be noted that although the bidentate methacrylate surface species (Ads3) at 1493 cm−1 decreased in the presence of H2O, these species remained over the HPMo catalyst.
Chansai et al.37 reported in situ DRIFTS-MS analysis in the region of the Keggin structure between 1200 and 700 cm−1 during the transient H2O/D2O switching experiments on the HPMo catalyst at 320 °C and reported the occurrence of H–D exchange, possibly on the bridging oxygen of the Mo–O–Mo unit, where the H/D atom of water is abstracted. Therein, the H/D recombined with the monodentate carboxylate-type intermediates (proposed as monodentate methacrylate (Ads4) in the current work) to produce gas phase MAA. The current DRIFTS analysis shows that the formation of monodentate methacrylate species (Ads4) is possible and it is an important intermediate in the final step to form gaseous MAA.
When H2O was removed from the MAL + O2 feed in Fig. 7b, there is little change in the monodentate methacrylate species (Ads4) at ∼1770 cm−1 and bidentate methacrylate adsorbed species (Ads3) and/or π-complex (Ads5) at 1495–1493 cm−1. Only the surface lactone-type species (Ads1 and Ads2) at 1791 cm−1 substantially increased due to the adsorption of MAL under MAL + O2 reaction conditions. Once H2O was added again, the oxidation of MAL was enhanced, resulting in an increase of carbonyl of monodentate methacrylate adsorbed species at 1770 cm−1, hence facilitating the formation of gas phase MAA. It is important to point out that the accumulation of surface species under the reaction conditions is predominant on the catalyst surfaces and suppresses an observation of evident change in surface species after a couple of experimental cycles.
Similar changes to those surface species over the HPMo catalyst were observed for the HPMoV catalyst (Fig. 7c and d). In the case of the HPMoV catalyst, both adsorbed lactone-type species (Ads1 and Ads2) in the IR range of 1800–1780 cm−1 and bidentate methacrylate species (Ads3) at 1493 cm−1 are smaller than those found over HPMo. After H2O was removed, both adsorbed lactone-type species (Ads1 and Ads2) and monodentate methacrylate species (Ads4) increased slightly but the bidentate methacrylate species (Ads3) were undetected at 1493 cm−1, probably converted to monodentate methacrylate species (Ads4) and/or swamped by the IR signals of H2O bending vibrations between 1670 and 1400 cm−1.
Fig. 6 shows that the formation of gas phase MAA over HPMo is less than that over HPMoV; it is interesting to note from Fig. 7 that both lactone-type adsorbed species (Ads1 and Ads2) at 1793 cm−1 and bidentate methacrylate species (Ads3) at 1493 cm−1 are evidently more pronounced over HPMo than over the HPMoV catalyst. This suggests that the formation of surface species (Ads1, Ads2, and Ads3) may partially block the active sites for the oxidation reaction of MAL and possibly suppress the formation of MAA to some extent. Conversely, Fig. 7 also reveals that the monodentate methacrylate adsorbed species (Ads4) at 1770 cm−1, which is proposed to be important and associated with MAA product, is more predominant over HPMoV than over HPMo.
To gain an insight into the difference in surface species between the two HPA catalysts, the changes of the two IR ranges of 1800–1780 cm−1, assigned to possibly unreactive lactone-type surface spectators (Ads1 and Ads2), and of 1780–1760 cm−1, assigned to possibly active monodentate methacrylate intermediates (Ads4), were compared. Fig. 8 shows the comparison of the evolution of the ratio of monodentate methacrylate adsorbed species (Ads4) to lactone-type adsorbed species (Ads1 and Ads2) during H2O switches in and out of the MAL + O2 feed at 320 °C. It has been found that the difference in the profile of those surface species between HPMo and HPMoV catalysts is somewhat interesting. It is clearly seen that the HPMoV catalyst demonstrates a continuous rise, while the HPMo catalyst shows both an increase and a decrease in the ratio of those species with respect to the switches of H2O in and out of the MAL + O2 feed over the course of 30 min.
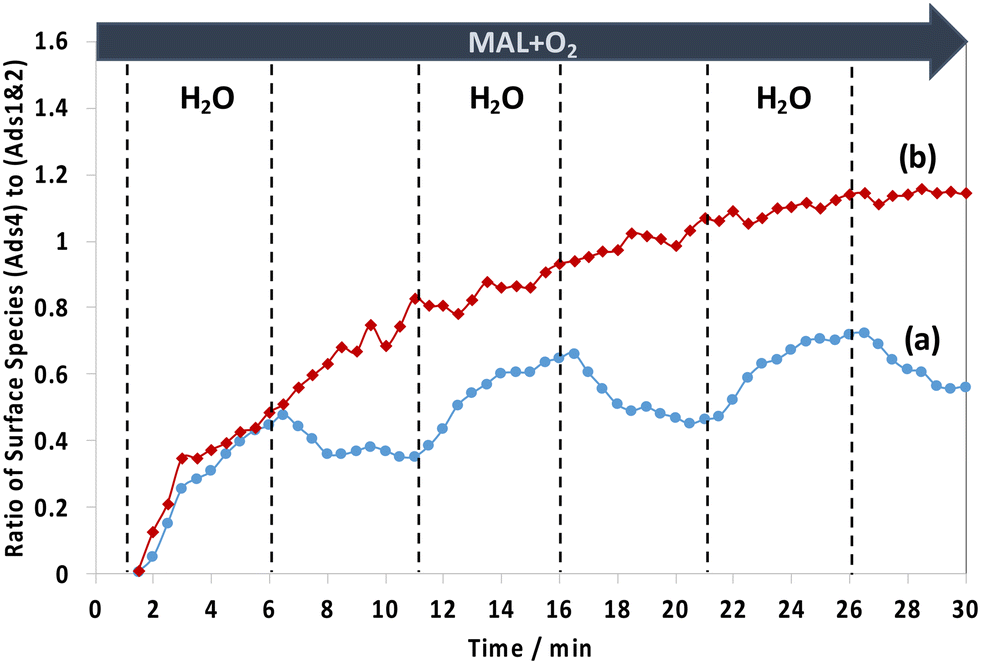 | ||
| Fig. 8 Changes in the ratio of monodentate methacrylate adsorbed species (Ads4) in the IR range of 1780–1760 cm−1 to lactone-type adsorbed species (Ads1 and Ads2) in the IR range of 1800–1780 cm−1 as a function of time on stream at 320 °C over HPMo (a) and HPMoV (b) catalysts during the switches of H2O in and out of the MAL + O2 feed. The gas feed is composed of 3500 ppm MAL, 7000 ppm O2, 7000 ppm H2O (when added), and Ar balance and the total flow rate is 100 cm3 min−1. | ||
Examining Fig. 8 between 1 and 6 min when H2O is first introduced to the MAL + O2 gas feed, it is found that the ratio of monodentate methacrylate adsorbed species (Ads4) to lactone-type adsorbed species (Ads1 and Ads2) for HPMoV shows an initial rise which is slightly faster than that found for HPMo. This means that the formation of monodentate methacrylate adsorbed species (Ads4) is more pronounced than that of lactone-type adsorbed species (Ads1 and Ads2). This suggests that the faster the formation of monodentate methacrylate surface species (Ads4), the more rapid the formation of gaseous MAA product, as seen in Fig. 5, where gaseous MAA is produced more significantly over HPMoV than over HPMo. Interestingly, our DRIFTS-MS analysis strongly suggests that the formation of monodentate methacrylate adsorbed species (Ads4) as potentially important intermediates has a significant role in facilitating the formation of gas phase MAA product.
In contrast, in the absence of H2O between 6 and 11 min, the ratio of monodentate methacrylate adsorbed species (Ads4) to lactone-type adsorbed species (Ads1 and Ads2) on the HPMo catalyst was decreased, indicative of a more significant increase of lactone-type adsorbed species (Ads1 and Ads2) than monodentate methacrylate adsorbed species (Ads4). It is worth noting that a different trend was observed over the HPMoV catalyst, in which the ratio of monodentate methacrylate adsorbed species (Ads4) to lactone-type adsorbed species (Ads1 and Ads2) continues to rise, indicating that monodentate methacrylate adsorbed species (Ads4) are more pronounced than lactone-type adsorbed species (Ads1 and Ads2). The difference in these ratios between HPMo and HPMoV catalysts suggests that the presence of V in the Keggin structure of HPA has a direct influence on the formation of monodentate methacrylate surface species (Ads4) rather than that of lactone-type adsorbed species (Ads1 and Ads2) under reaction conditions at 320 °C.
Interestingly, there is a difference in how the surface monodentate methacrylate species (Ads4) change over HPMo and HPMoV catalysts, as shown in Fig. 8. The continuous rise of the ratio of monodentate methacrylate adsorbed species (Ads4) to lactone-type adsorbed species (Ads1 and Ads2) for the HPMoV catalyst during H2O switches in and out of the MAL + O2 reaction over HPMoV is proposed to be due to differences in the amount of V4+ species present. The V 2p3/2 XPS spectra (Fig. S8†) show two vanadyl species present in the Keggin structure, i.e. V5+ at 517.7 eV and V4+ at 516.5 eV,29,30,35 and there is a difference in the ratio of V4+ to V5+ species between both fresh and spent HPMoV catalyst, which is 0.29 and 0.82, respectively. This increase in the ratio of V4+ to V5+ species between the fresh and spent HPMoV samples after 60 min under MAL + O2 + H2O reaction conditions at 320 °C is consistent with the proposal that the amount of V4+ in HPMoV could be responsible for the formation of methacrylic acid (MAA) and the formation of monodentate methacrylate species (Ads4) under reaction conditions with and without H2O. Zhou et al.30 also reported that the amount of V4+ is correlated with methacrylic acid formation. This change in oxidation state of vanadyl species from V5+ to V4+ species may also explain the continuous rise of the ratio of monodentate methacrylate adsorbed species (Ads4) to lactone-type adsorbed species (Ads1 and Ads2) for HPMoV due to (1) an increase in V4+/VO2+ species as proposed by Zhou et al.30 and (2) the high reaction rate constant and low activation energy to form the monodentate methacrylate species as recently reported by Tian and co-workers.48
Additionally, we have investigated the acid properties of both HPMo and HPMoV catalysts using in situ DRIFTS technique and pyridine adsorption at 320 °C. The comparison of in situ DRIFT spectra of pyridine adsorption at 320 °C (recorded at 10 min under Ar purge after the pyridine adsorption at 30 °C for 60 min) for both HPMo and HPMoV catalysts is shown in Fig. S9.† Interestingly, the ratio of Brønsted to Lewis acid sites on HPMoV (57.7) is much greater than that on HPMo (14.9). Consistent with this study, previous studies52–56 were carried out to investigate the acid properties of HPA compounds and showed that strong Brønsted acid sites facilitated the activation of MAL due to an increase in the electrophilicity of the carbonyl carbon of MAL.55 This led to an enhancement of catalytic oxidation activity and the formation of desirable MAA product via the repulsive interaction between Brønsted sites and acidic MAA product. These Brønsted acid sites perhaps explain the observation of the present work that the HPMoV catalyst exhibits faster and more formation of active monodentate methacrylate adsorbed species (Ads4) to produce gas phase MAA than the HPMo catalyst as presented in Fig. 6–8.
It is important to note that the continuous rise of the ratio of monodentate methacrylate adsorbed species (Ads4) to lactone-type adsorbed species (Ads1 and Ads2) for the HPMoV catalyst indicates that the surfaces of HPMoV can also act as a reservoir of monodentate methacrylate adsorbed species (Ads4). This is likely due to (1) a greater number of V4+/VO2+ active sites as reported by Zhou and co-workers30 that the substitution of Mo with V in the Keggin structure can increase the abundance of V4+/VO2+ active sites, which influenced the redox property for better redox active sites through faster reduction and re-oxidation steps and somewhat had a direct relation to the catalytic activity and the selectivity of MAA, and (2) Brønsted acid sites present in the HPMoV catalyst, where both inactive and active surface species with the same chemical composition and structure detected by in situ DRIFTS are more rapidly formed than in the HPMo catalyst. Only a small portion of monodentate methacrylate adsorbed species (Ads4) is converted or reacted to produce MAA on the HPMoV catalyst.
4. Conclusions
The current work reports, for the first time, an in situ DRIFTS-MS analysis to gain an insight into the role and effect of V on the formation of MAA during the selective oxidation of MAL to MAA over HPA catalysts. Under different reaction conditions at 320 °C, the MS results show that the presence of V in the HPMoV catalyst yields higher formation of gas phase MAA product than that in the HPMo catalyst. Simultaneously, the in situ DRIFTS analysis reveals that several possible HC-adsorbed species have been formed and tentatively identified: a 5-membered ring lactone-type species (Ads1), a 6-membered ring lactone-type species (Ads2), bidentate methacrylate species (Ads3), monodentate methacrylate species (Ads4), and π-adsorbed MAL species (Ads5). Interestingly, the transient switching of H2O in and out of the MAL + O2 gas feed reveals that the substitution of Mo with V in the Keggin primary structure selectively and substantially facilitates the formation of monodentate methacrylate adsorbed species (Ads4) in the final step before forming gas phase MAA. It is important to point out that the current work reports the influence of V in the HPMoV catalyst on the surface adsorbed species under various reaction conditions in comparison with the HPMo catalyst. However, the reaction mechanism of MAL to MAA between the two catalysts is not differentiated. It is likely that the same mechanism of selective oxidation of MAL occurs but in the presence of V in the Keggin primary structure, the HPMoV catalyst facilitates an increased formation of reactive monodentate methacrylate intermediates (Ads4), resulting in further enhancement of gaseous MAA formation when compared with the HPMo catalyst.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support of this work from Mitsubishi Chemical Corporation.References
- Y. Konishi, K. Sakata, M. Misono and Y. Yoneda, J. Catal., 1982, 77, 169–179 CrossRef CAS.
- V. Ernst, Y. Barbaux and P. Courtine, Catal. Today, 1987, 1, 167–180 CrossRef CAS.
- K. Eguchi, T. Seiyama, N. Yamazoe, S. Katsuki and H. Taketa, J. Catal., 1988, 111, 336–344 CrossRef CAS.
- L. M. Deuβer, J. W. Gaube, F.-G. Martin and H. Hibst, in 11th International Congress On Catalysis - 40th Anniversary, ed. J. W. Hightower, W. Nicholas Delgass, E. Iglesia, A. T. B. T. Bell in S. S. and C., Elsevier, 1996, vol. 101, pp. 981–990 Search PubMed.
- K. Inumaru, A. Ono, H. Kubo and M. Misono, J. Chem. Soc., Faraday Trans., 1998, 94, 1765–1770 RSC.
- A. M. Herring and R. L. McCormick, J. Phys. Chem. B, 1998, 102, 3175–3184 CrossRef CAS.
- C. Marchal-Roch, N. Laronze, N. Guillou, A. Tézé and G. Hervé, Appl. Catal., A, 2000, 199, 33–44 CAS.
- C. Marchal-Roch, N. Laronze, N. Guillou, A. Tézé and G. Hervé, Appl. Catal., A, 2000, 203, 143–150 CAS.
- K. Krauß, A. Drochner, M. Fehlings, J. Kunert and H. Vogel, J. Mol. Catal. A: Chem., 2000, 162, 413–422 CrossRef.
- S. Ganapathy, M. Fournier, J. F. Paul, L. Delevoye, M. Guelton and J. P. Amoureux, J. Am. Chem. Soc., 2002, 124, 7821–7828 CrossRef CAS PubMed.
- C. Marchal-Roch, N. Laronze, R. Villanneau, N. Guillou, A. Tézé and G. Hervé, J. Catal., 2000, 190, 173–181 CrossRef CAS.
- M. Misono, Chem. Commun., 2001, 1141–1152 RSC.
- I. K. Song and M. A. Barteau, J. Mol. Catal. A: Chem., 2004, 212, 229–236 CAS.
- N. Ballarini, F. Candiracci, F. Cavani, H. Degrand, J.-L. Dubois, G. Lucarelli, M. Margotti, A. Patinet, A. Pigamo and F. Trifirò, Appl. Catal., A, 2007, 325, 263–269 CrossRef CAS.
- H. Kim, J. C. Jung, S. H. Yeom, K.-Y. Lee and I. K. Song, J. Mol. Catal. A: Chem., 2006, 248, 21–25 CrossRef CAS.
- H. Kim, J. C. Jung, D. R. Park, S.-H. Baeck and I. K. Song, Appl. Catal., A, 2007, 320, 159–165 CAS.
- R. Bayer, C. Marchal-Roch, F. X. Liu, A. Tézé and G. Hervé, J. Mol. Catal. A: Chem., 1996, 114, 277–286 CrossRef CAS.
- V. F. Odyakov, E. G. Zhizhina and K. I. Matveev, J. Mol. Catal. A: Chem., 2000, 158, 453–456 CrossRef CAS.
- P. Villabrille, G. Romanelli, P. Vázquez and C. Cáceres, Appl. Catal., A, 2004, 270, 101–111 CrossRef CAS.
- G. Mestl, T. Ilkenhans, D. Spielbauer, M. Dieterle, O. Timpe, J. Kröhnert, F. Jentoft, H. Knözinger and R. Schlögl, Appl. Catal., A, 2001, 210, 13–34 CAS.
- C. Marchal-Roch, C. Julien, J. F. Moisan, N. Leclerc-Laronze, F. X. Liu and G. Hervé, Appl. Catal., A, 2004, 278, 123–131 CrossRef CAS.
- T. Ressler, O. Timpe, F. Girgsdies, J. Wienold and T. Neisius, J. Catal., 2005, 231, 279–291 CAS.
- X.-K. Li, J. Zhao, W.-J. Ji, Z.-B. Zhang, Y. Chen, C.-T. Au, S. Han and H. Hibst, J. Catal., 2006, 237, 58–66 CrossRef CAS.
- A. Brückner, G. Scholz, D. Heidemann, M. Schneider, D. Herein, U. Bentrup and M. Kant, J. Catal., 2007, 245, 369–380 CrossRef.
- J. E. Molinari, L. Nakka, T. Kim and I. E. Wachs, ACS Catal., 2011, 1, 1536–1548 CrossRef CAS.
- Y.-L. Cao, L. Wang, L.-L. Zhou, G.-J. Zhang, B.-H. Xu and S.-J. Zhang, Ind. Eng. Chem. Res., 2017, 56, 653–664 CrossRef CAS.
- M. Kanno, T. Yasukawa, W. Ninomiya, K. Ooyachi and Y. Kamiya, J. Catal., 2010, 273, 1–8 CrossRef CAS.
- M. Kanno, Y. Miura, T. Yasukawa, T. Hasegawa, W. Ninomiya, K. Ooyachi, H. Imai, T. Tatsumi and Y. Kamiya, Catal. Commun., 2011, 13, 59–62 CrossRef CAS.
- F. Jing, B. Katryniok, F. Dumeignil, E. Bordes-Richard and S. Paul, J. Catal., 2014, 309, 121–135 CAS.
- L. Zhou, L. Wang, S. Zhang, R. Yan and Y. Diao, J. Catal., 2015, 329, 431–440 CrossRef CAS.
- L. Zhou, L. Wang, Y. Cao, Y. Diao, R. Yan and S. Zhang, Mol. Catal., 2017, 438, 47–54 CAS.
- Y.-L. L. Cao, L. Wang, B.-H. H. Xu and S.-J. J. Zhang, Chem. Eng. J., 2018, 334, 1657–1667 CrossRef CAS.
- Y. Geng, S. Xiong, B. Li, Y. Liao, X. Xiao and S. Yang, Ind. Eng. Chem. Res., 2018, 57, 856–866 CrossRef CAS.
- X. Li, J. Zhang, F. Zhou, Y. Wang, X. Yuan and H. Wang, Mol. Catal., 2018, 452, 93–99 CrossRef CAS.
- L. Zhou, S. Zhang, Z. Li, J. Scott, Z. Zhang, R. Liu and J. Yun, RSC Adv., 2019, 9, 34065–34075 CAS.
- S. Yasuda, J. Hirata, M. Kanno, W. Ninomiya, R. Otomo and Y. Kamiya, Appl. Catal., A, 2019, 570, 164–172 CrossRef CAS.
- S. Chansai, Y. Kato, W. Ninomiya and C. Hardacre, Faraday Discuss., 2021, 229, 443–457 RSC.
- L. Zhang, S. Paul, F. Dumeignil and B. Katryniok, Catalysts, 2021, 11, 769 CrossRef CAS.
- M. J. da Silva, A. A. Rodrigues and N. P. G. Lopes, Inorganics, 2023, 11, 162 CrossRef CAS.
- M. Misono, K. Sakata, Y. Yoneda and W. Y. Lee, in Studies in Surface Science and Catalysis, ed. T. Seiyama and K. Tanabe, Elsevier, 1981, vol. 7, pp. 1047–1059 Search PubMed.
- N. Essayem, G. Coudurier, M. Fournier and J. C. Védrine, Catal. Lett., 1995, 34, 223–235 CrossRef CAS.
- W. Rachmady and M. A. Vannice, J. Catal., 2002, 208, 170–179 CrossRef CAS.
- S. Kohl, A. Drochner and H. Vogel, Catal. Today, 2010, 150, 67–70 CrossRef CAS.
- G. Y. Popova, Y. A. Chesalov, E. M. Sadovskaya and T. V. Andrushkevich, J. Mol. Catal. A: Chem., 2012, 357, 148–153 CAS.
- J. A. Schaidle, J. Blackburn, C. A. Farberow, C. Nash, K. X. Steirer, J. Clark, D. J. Robichaud and D. A. Ruddy, ACS Catal., 2016, 6, 1181–1197 CrossRef CAS.
- M. Zhou, H. Zhang, H. Ma and W. Ying, Fuel Process. Technol., 2016, 144, 115–123 CrossRef CAS.
- H. Alalwan and A. Alminshid, Spectrochim. Acta, Part A, 2020, 229, 117990 CAS.
- Y. Tian, H. Yan, J. Li, Y. Yang, T. Guo, X. Zhang, S. Xiang and C. Li, Ind. Eng. Chem. Res., 2024, 63, 4807–4816 CrossRef CAS.
- H. Böhnke, J. Gaube and J. Petzoldt, Ind. Eng. Chem. Res., 2006, 45, 8794–8800 CrossRef.
- H. Böhnke, J. Gaube and J. Petzoldt, Ind. Eng. Chem. Res., 2006, 45, 8801–8806 CrossRef.
- S. Kwon, P. Deshlahra and E. Iglesia, J. Catal., 2018, 364, 228–247 CAS.
- I. K. Song, M. S. Kaba and M. A. Barteau, J. Phys. Chem., 1996, 100, 17528–17534 CrossRef CAS.
- M. J. Janik, R. J. Davis and M. Neurock, Catal. Today, 2005, 105, 134–143 CrossRef CAS.
- A. M. Alsalme, P. V. Wiper, Y. Z. Khimyak, E. F. Kozhevnikova and I. V. Kozhevnikov, J. Catal., 2010, 276, 181–189 CrossRef CAS.
- S. Yasuda, A. Iwakura, J. Hirata, M. Kanno, W. Ninomiya, R. Otomo and Y. Kamiya, Catal. Commun., 2019, 125, 43–47 CrossRef CAS.
- C. Pezzotta, V. S. Marakatti and E. M. Gaigneaux, Catal. Sci. Technol., 2020, 10, 7984–7997 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: http://doi.org/10.1039/d4cy00552j |
| This journal is © The Royal Society of Chemistry 2024 |