
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring the effect of molecular size and framework functionalisation on transport in metal–organic frameworks using pulsed-field gradient nuclear magnetic resonance†
Shima
Zainal
 a,
Ahmed
Alsudani
b,
Ralph W.
Adams
b,
Mathias
Nilsson
b,
Xiaolei
Fan
*ac and
Carmine
D’Agostino
*ad
a,
Ahmed
Alsudani
b,
Ralph W.
Adams
b,
Mathias
Nilsson
b,
Xiaolei
Fan
*ac and
Carmine
D’Agostino
*ad
aDepartment of Chemical Engineering, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: carmine.dagostino@manchester.ac.uk; xiaolei.fan@manchester.ac.uk
bDepartment of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK
cNingbo China Beacons of Excellence Research and Innovation Institute, University of Nottingham Ningbo China, 211 Xingguang Road, Ningbo 315048, China
dDipartimento di Ingegneria Civile, Chimica, Ambientale e dei Materiali (DICAM), Alma Mater Studiorum – Università di Bologna, Via Terracini, 28, 40131 Bologna, Italy
First published on 31st May 2024
Abstract
Molecular transport is an important aspect in metal–organic frameworks (MOFs) as it affects many of their applications, such as adsorption/separation, drug delivery and catalysis. Yet probing the fundamental diffusion mechanisms in MOFs is challenging, and the interplay between the MOF's features (such as the pore structure and linker dynamics) and molecular transport remains mostly unexplored. Here, the pulsed-field gradient nuclear magnetic resonance (PFG NMR) technique is used to probe the diffusion of several probe molecules, i.e., water, xylenes and 1,3,5-triisopropylbenzene (TIPB), within the UiO-66 MOF and its derivatives (UiO-66NH2 and UiO-66Br). Exploiting differences in the size of probe molecules we were able to probe the diffusion rate selectively in the different pore environments of the MOFs. In particular, when relatively small molecules, such as water and small hydrocarbons, were used as probes, the PFG NMR log attenuation plots were non-linear with two distinctive diffusion regions, suggesting faster diffusion in the inter-crystalline space and slower diffusion within crystal aggregates, the latter occurring mostly inside the framework of the MOFs. Conversely, experiments with a larger probe molecule, i.e., TIPB, with a kinetic diameter of 0.95 nm, which makes it unable to access the framework windows of the MOF crystals, showed linear PFG NMR log attenuation plots, which indicates diffusion occurring in a single environment, most likely in the inter-crystalline space. Analysis of the apparent tortuosity values of the systems under investigation highlights the role of linker functionalisation in influencing the molecular diffusion of the probe molecules, which affects both intra-molecular interactions and pore accessibility within the MOF crystals. The findings of this work demonstrate that the diffusion behaviour of probe molecules within MOFs is influenced by the pore size, structure, functionalisation of the MOF linker and molecular interactions. Our study contributes to further advance the understanding of mass transport in MOFs by PFG NMR and provides insights that can inform the design and optimisation of MOF-based materials for various applications.
1. Introduction
Metal–organic frameworks (MOFs) are porous, crystalline solids made by combining organic linkers and metal-oxide clusters. In recent years, MOFs have gained prominence as versatile materials with applications ranging from gas storage to catalysis. Among these, UiO-66 (i.e., University of Oslo 66)1 and its derivatives have attracted significant attention due to their interesting properties and potential use.2,3 UiO-66 typically incorporates zirconium metal nodes into its structure forming strong bonds with organic linkers of terephthalic acid. These organic linkers create linkages between the metal nodes forming a crystalline three-dimensional framework. UiO-66 derivatives such as UiO-66NH2 and UiO-66Br are the linker modified versions of the original UiO-66, where specific functional groups are introduced to the structure (by functionalisation of the organic linker). These modifications can alter the properties of the UiO-66 MOFs, and thus their applications. In UiO-66NH2, the amino (–NH2) group is grafted to the organic linker in UiO-66, serving as additional binding sites for specific molecules, for example, adsorption of sulphur species and other contaminants in wastewater treatment applications.4 Incorporation of bromine (Br) atoms in UiO-66Br, altering its structure, can impact its structural properties significantly, thereby influencing chemical reactivity, adsorption capacity, and interactions with guest molecules. For instance, Br-modified MOFs were shown to be capable of removing elemental mercury (Hg0) from flue gas.5 Additionally, the introduction of –Br functional groups can induce steric hindrance, consequently modifying the MOF's electronic properties.6,7Although the applications of UiO-66 and its derivatives are well documented,8–10 insights into the microscopic mass transport of guest molecules within these materials remain mostly unexplored, and knowledge of mass transfer within the UiO-66 family is crucial for optimising their properties and applications as it enables the design of more efficient and tailored materials. Molecular diffusion in MOFs is influenced by a number of parameters, including the structure of the MOFs (such as pore size), functional groups on the linker, presence of defects and chemical interaction between guest molecules.11 Jacob et al. simulated acetone diffusion within pristine UiO-66 using molecular dynamics (MD) simulation, aiming to explore the effect of framework flexibility and hydrogen bonding (between acetone molecules and the hydroxyl groups present on the nodes of the UiO-66 framework) on the diffusion behaviour of acetone within UiO-66.12 The results demonstrate the critical significance of linker dynamics and provide insight into how hydrogen bonding affects acetone diffusivity under different loading conditions. Ramsahye et al. combined quasi-elastic neutron scattering (QENS) with MD simulation to investigate the diffusion of light hydrocarbons in UiO-66.13 These findings show a clear decrease in diffusion with an increase in the molecular size of the hydrocarbons, indicating the enhanced confinement effect within the UiO-66 framework due to the increase in the kinetic diameter of the guest molecules. Although simulation methods offer molecular insights, validation of the simulated results is challenging.
In addition to MD simulations, several experimental techniques can be used to measure diffusion in MOFs, such as gravimetric techniques and spectroscopy methods.11 Gravimetric techniques measure the mass changes, which are then correlated with the adsorption/desorption of guest molecules on/from MOFs, and hence diffusion rates.14 Although the bulk information obtained by gravimetric methods are useful for exploring diffusion behaviour in MOFs, they are slow and sensitive to variations in temperature and humidity. Spectroscopic methods, such as infrared spectroscopy (IR) and NMR, provide molecular-level information about the interaction between MOF components and diffusing species.15–17 These techniques offer insight into molecular interactions, diffusion pathways and guest–host interactions within MOFs.
Pulsed-field gradient nuclear magnetic resonance (PFG NMR) is a non-invasive and non-destructive technique which can probe the movement of molecules within porous materials. In addition, it allows quantitative measurement of diffusion coefficients,15,18 which is essential for understanding and comparing the transport phenomena within different porous materials. This method can also differentiate diffusion in different environments simultaneously, providing unique insights into the dynamics of molecular transport within porous materials.19 PFG NMR has been applied to various porous materials, such as diffusion of mixtures in microporous and mesoporous materials, including zeolites and heterogeneous catalysts. For example, PFG NMR diffusion studies were performed to investigate intra-crystalline diffusion in USY zeolites, and the findings challenge existing assumptions about the role of intra-crystalline mesopores in USY zeolites.20 Experimental evidence suggests that these mesopores do not significantly contribute to intra-crystalline diffusion and do not form a connected network within the crystals. PFG NMR was also applied to mesoporous SBA-15 and KIT-6 silicas to unravel mass transport and the effect of mesopore size on molecular diffusion and catalytic performance.21,22 The findings provide insights into how the morphology of SBA-15 silica (such as isolated fibres and bundles) influences molecular displacements and diffusion behaviour. In particular, the results show that the measured intra-particle NMR diffusivity agrees with the charge carrier diffusivity determined using the Nernst–Einstein equation.
In the context of MOFs, previous studies employing PFG NMR to study molecular self-diffusion within ZIFs and MOF-3 helped to establish a correlation between the molecular size and pore characteristics of the MOFs.23 These results demonstrate that light hydrocarbons can readily exchange between the intra-crystalline phase in a MOF and the surrounding environment. The findings above shed light on the diffusion dynamics of hydrocarbons within the MOF structure, which is essential for applications such as gas storage, separation, and catalysis. However, unlike zeolites and heterogeneous solid catalysts, the number of diffusion studies in MOFs using PFG NMR remains limited.
Herein, we present the results of a direct investigation into the molecular transport in UiO-66 and its linker functionalised derivatives by measuring the diffusion behaviour of relevant probe molecules using the PFG NMR technique. To characterise the type of diffusion, the experimental measurements were performed with probe molecules of different sizes focusing on investigating the effect of steric hindrance/intra-framework interaction over the length scale probed by PFG NMR.
2. Experimental
2.1 Synthesis of materials
All chemicals were of analytical grade and used without further purification. Anhydrous zirconium(IV) chloride, terephthalic acid (BDC), 2-amino terephthalic acid (BDC-NH2), bromoterephthalic acid (BDC-Br), dimethylformamide (DMF), hydrochloric acid (HCl), para (p)-, meta (m)-, ortho (o)-xylenes, methanol and 1,3,5-triisopropylbenzene (TIPB) were purchased from Sigma Aldrich and used without further purification. Deionised water was purified to a resistivity of 18.2 MΩ cm in a Millipore Milli-Direct Q water purification system.The UiO-66 MOFs were synthesized using the solvothermal method2 by reacting BDC (0.09 g, 0.5 mmol) with zirconium(IV) chloride (0.12 g, 0.5 mmol) in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in 25 ml of DMF. The solution was stirred for two hours, then transferred to a stainless-steel autoclave, sealed and placed in a preheated oven at 120 °C for 24 hours. After the synthesis, the reactor was cooled down, and the resulting white solid precipitate was washed with DMF and solvent exchanged with methanol before being dried under vacuum at 120 °C overnight. The synthesis procedure was then repeated for making UiO-66NH2 and UiO-66Br by changing the BDC linker to BDC-NH2 and BDC-Br, respectively.
:1 in 25 ml of DMF. The solution was stirred for two hours, then transferred to a stainless-steel autoclave, sealed and placed in a preheated oven at 120 °C for 24 hours. After the synthesis, the reactor was cooled down, and the resulting white solid precipitate was washed with DMF and solvent exchanged with methanol before being dried under vacuum at 120 °C overnight. The synthesis procedure was then repeated for making UiO-66NH2 and UiO-66Br by changing the BDC linker to BDC-NH2 and BDC-Br, respectively.
2.2 Material characterisation
Prior to characterisation all MOF materials were activated at 150 °C under vacuum. The powder X-ray diffraction (PXRD) patterns were acquired on a PANanalytical Empyrean XRD system with Cu-Kα1 radiation (λ = 1.54056 Å) in the 2θ range of 3° to 50° with a step size of 0.026° per second. The CryAlisPro software program was used for the integration of diffraction data.Nitrogen (N2) adsorption–desorption measurements were carried out using a Micromeritics ASAP 2020 analyser at 77 K. Before N2 physisorption, approximately 100 mg of the solid sample was degassed at 180 °C under vacuum for 12 h. The specific surface areas of the materials were determined using the Brunauer–Emmett–Teller (BET) method, and the pore size distribution was obtained by using the Barrett–Joyner–Halenda (BJH) method (based on the adsorption branch of the isotherms). Infrared spectroscopy was performed at 25 °C using an FTIR on a PerkinElmer Spectrum 2000 Spectrometer. Scanning electron microscopy (SEM) of the samples was conducted using a Tescan Mira small chamber instrument with a LaB6 emitter (operated at 10 kV).
2.3 PFG NMR measurements
PFG NMR experiments were performed using a Bruker Avance NEO 500 NMR spectrometer operating at a 1H frequency of 500 MHz with a diffusion probe (DiffBB). UiO-66, UiO-66NH2 and UiO-66Br particles were first dried under vacuum then soaked for 48 h in 2 mL of the probe molecules (i.e., water, p-xylene, m-xylene, o-xylene, n-octane and TIPB). The samples were then dried gently with filter paper to remove excess solvent and placed in a 5 mm NMR tube filled to a height of approximately 10 mm. Samples were placed in the spectrometer probe and left to equilibrate for 15 minutes before experiments. All experiments were performed at room temperature (approximately 298 K) using a 15 μs 1H 90° pulse, a 1.4 ms diffusion encoding time using half-sine gradient pulse shape with 16 to 32 amplitude increments of the gradient pulses between 2 and 15% of 17 T m−1, with a 100 ms diffusion time (Δ) and gradient recovery delays, τ of 0.2 ms. 64 scans per increment were acquired, with an inter-scan recycle delay of 5 s used for all experiments with each experiment taking between 45–90 minutes. The relaxation times T1 and T2 of the probe molecules in all three MOFs were measured to aid the choice of the acquisition parameters.NMR data was collected using a bipolar pulse pair STE pulse sequence and they were subsequently fitted to the Stejskal–Tanner model24 shown in eqn (1):
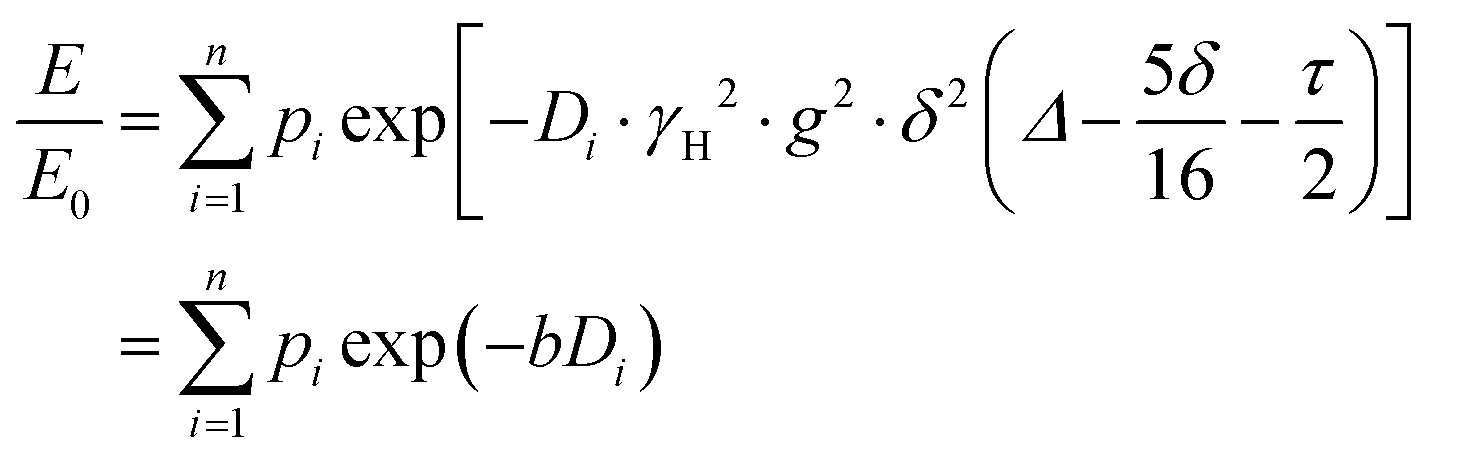 | (1) |
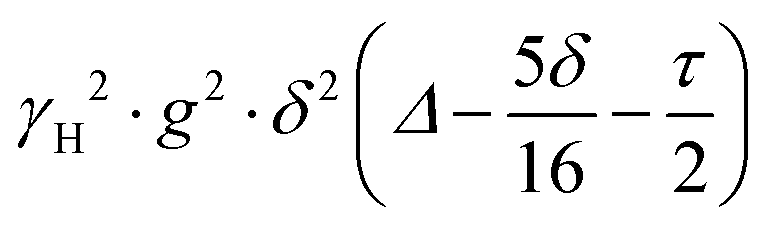 term is often referred to as the “b-factor”. In eqn (1), the number of different diffusion components in the system under study is represented by the value of n. The PFG NMR signal attenuation was analysed using the general NMR analysis toolbox (GNAT) software (Matlab, version R2021b).25
term is often referred to as the “b-factor”. In eqn (1), the number of different diffusion components in the system under study is represented by the value of n. The PFG NMR signal attenuation was analysed using the general NMR analysis toolbox (GNAT) software (Matlab, version R2021b).25
3. Results and discussion
3.1 Characterisation of UiO-66, UiO-66NH2 and UiO-66Br
The characterisation of UiO-66, UiO-66NH2 and UiO-66Br in this study builds upon previous investigations.2,3,7 The resulting MOFs exhibit different colours depending on the ligand used in the synthesis. UiO-66NH2 is yellow, UiO-66 and UiO-66Br are white, consistent with the results reported elsewhere.26 The bulk crystalline features of the MOFs were evaluated by PXRD to gain information on the crystallographic planes and structural similarity of these MOFs. As shown in Fig. 1, the diffraction peaks of UiO-66NH2 and UiO-66Br were consistent with that of the UiO-66 MOF, suggesting that all the MOFs under investigation are topologically identical. Sharp diffraction peaks at 2θ = 7.42, 8.6 and 25.7°, corresponding to the 111, 002 and 006 planes, are identified with comparable intensities in the XRD patterns of all three samples, suggesting that the linker functionalisation did not interfere with the crystallisation of the UiO-66 MOFs during the solvothermal synthesis.3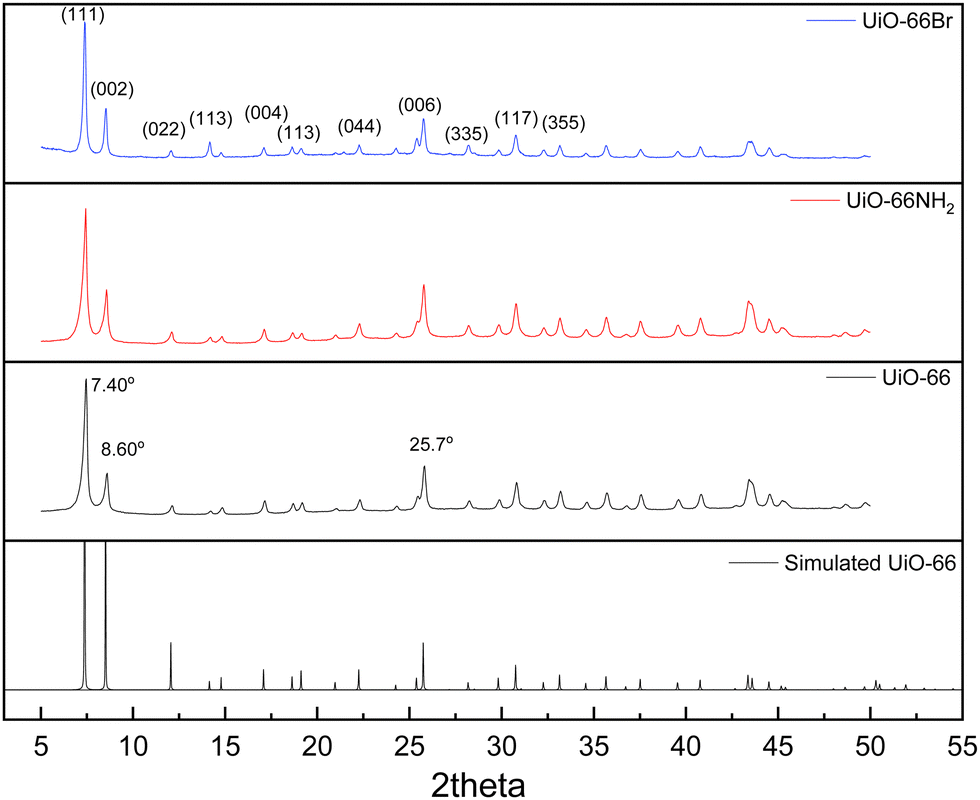 | ||
| Fig. 1 PXRD patterns of the synthesised UiO-66 and its derivatives. | ||
In addition, the PXRD patterns of the three MOFs were compared with the simulated patterns (based on the CIF file from the Cambridge Crystallography Open Database27). The comparison shows a very close resemblance between the XRD patterns of the synthesised MOFs and the simulated ones. Following these results, the UiO-66 structure is shown in Fig. 2. The structure consists of UiO-66 tetrahedral and octahedral cages, with sizes of 0.8 nm and 1.4 nm, respectively.28
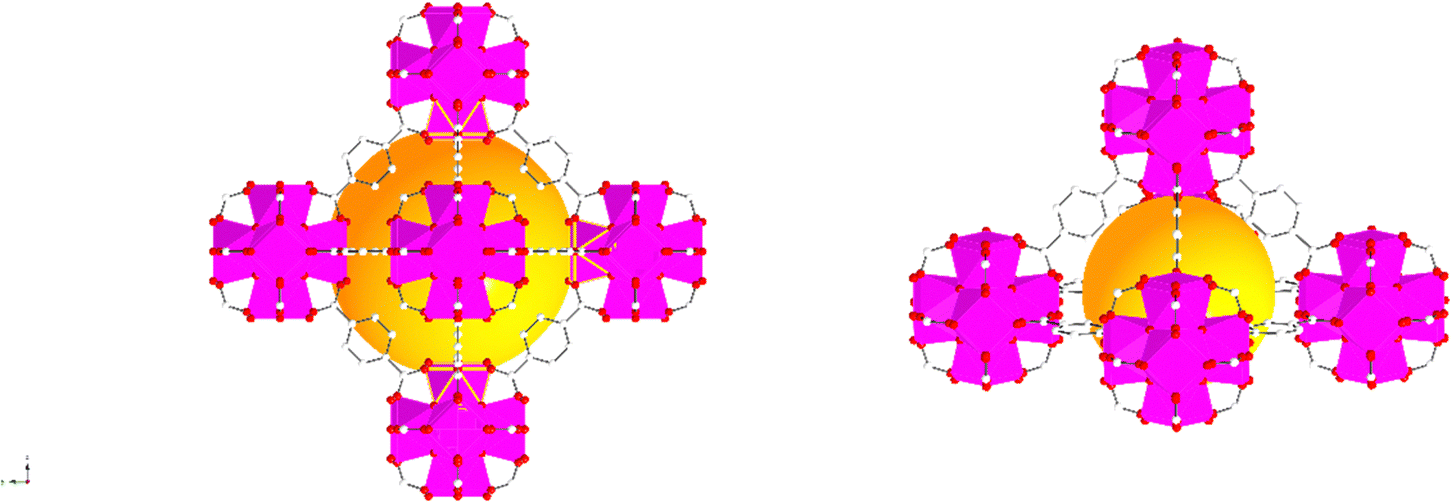 | ||
| Fig. 2 Simulated UiO-66 represented by balls and sticks showing an enlarged view of its octahedral (left) and tetrahedral (right) cages. Zr atoms are depicted as octahedra (Zr in pink, C in white, O in red). For clarity, guest molecules and hydrogen atoms have been removed from the structural diagrams. The figure was created using atomic coordinates from the literature.29 | ||
N2 adsorption–desorption isotherms of the MOFs are shown in Fig. S1 in the ESI,† showing that UiO-66 and UiO-66Br are microporous, whilst the isotherm of UiO-66NH2 shows the presence of a hysteresis loop. The presence of the hysteresis loop in the range of 0.9–1.0 p/p0 indicates that the sample may contain mesoscopic structural defects and/or inter-crystalline mesopores. Regarding the calculated specific surface area and pore volume, UiO-66 has the highest value of 953 m2 g−1 and 0.45 cm3 g−1, respectively (Table S1, ESI†). The functionalised UiO-66 maintains the same crystalline structure and topology of UiO-66 but with lower specific surface areas, as shown in Table S1 (ESI†), proving that the introduction of larger and heavier amine groups and bromine atoms to the linker decreased the surface area and the total pore volume for UiO-66NH2 and UiO-66Br. The calculated BET surface areas for all three samples in this study are consistent with those reported in the literature.2
The presence of –NH2 and –Br within the UiO-66 derivatives was confirmed by FTIR analysis (Fig. S2 and Table S2, ESI†). For UiO-66NH2 (red solid line in Fig. S2, ESI†), the two small peaks at 3507 cm−1 and 3384 cm−1 reflect the characteristic vibrations of the –NH2 group. Another two characteristic bands of the amino group can be identified at 1600 cm−1 and 1210 cm−1, respectively, for the N–H bending vibration and the C–N stretching of aromatic amines.30 Regarding UiO-66Br, its FTIR spectrum exhibits a specific peak at 680 cm−1, consistent with known vibrational modes associated with the presence of bromine functionalities. The appearance of these peaks support the effective introduction of bromine moieties onto the UiO-66 framework.30 The morphology of the as-synthesised MOFs was characterized by SEM (Fig. S3, ESI†), showing the typical polyhedral crystals for UiO-66 MOFs,31 and the particle sizes of the MOFs are comparable at <500 nm and <2 μm. The crystals clearly display irregularities and tend to form large aggregates of the order of several microns.
3.2 PFG NMR results
For the systems under investigation, diffusion may potentially occur inside the crystals (and within crystal aggregates), between the crystals (inter-crystalline) or as a combination of both.32 Diffusion within crystal aggregates, and in general intra-particle diffusion, may further be affected by the pore size distribution (i.e., presence of distinctive pore sizes within the same particle), which can lead to the observation of more than one diffusion environment. This has been previously reported for diffusion in activated carbon catalysts.33Fig. 3 shows the PFG NMR attenuation plots for different probe molecules diffusing through the porous structures of UiO-66 (Fig. 3(a)), UiO-66NH2 (Fig. 3(b)) and UiO-66Br (Fig. 3(c)). The data are plotted on a semi-log scale; hence a single exponential decay would appear on the graph as a straight line. In order to determine the effect of observation time, diffusion experiments were also carried out using observation times of 300 ms and 600 ms. The PFG attenuation plots and values of diffusivities (Fig. S5 and Table S6, ESI†) were in all cases very similar suggesting that the observation time does not significantly affect diffusion coefficients.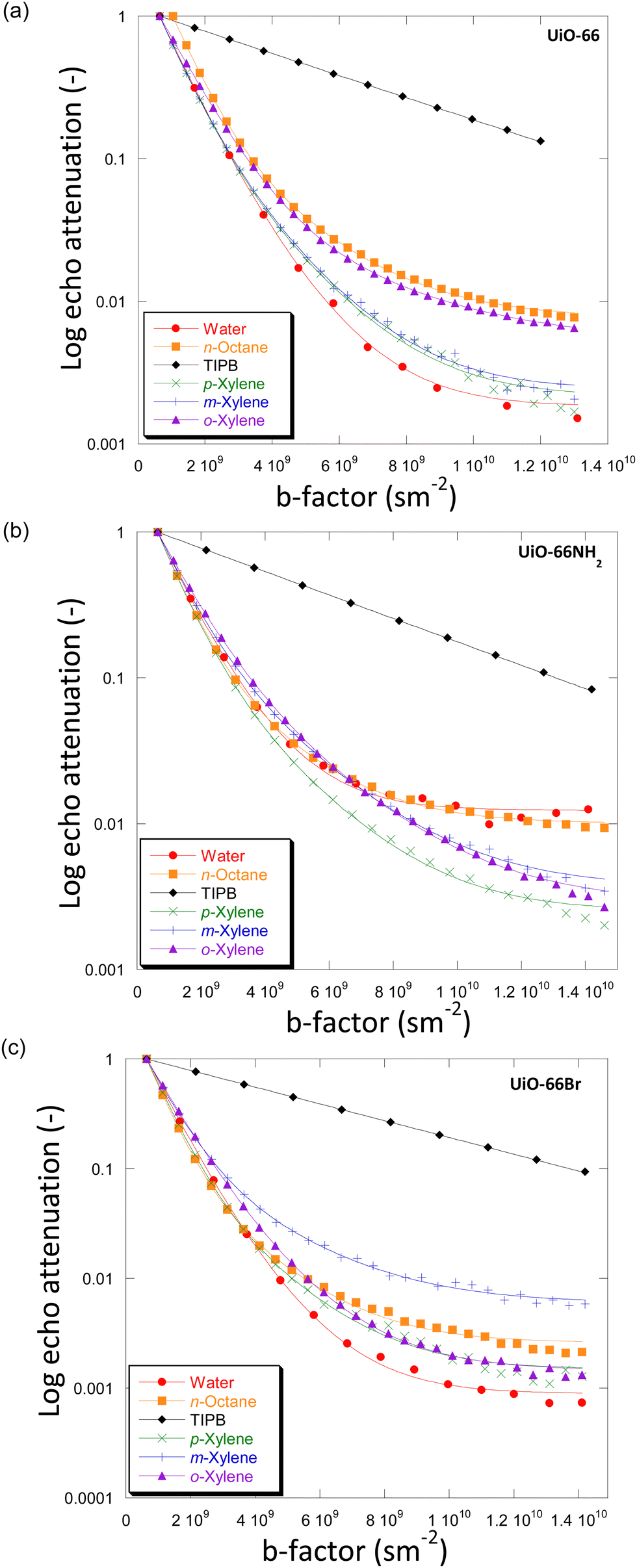 | ||
| Fig. 3 Diffusion attenuation plots for different probe molecules within the porous network of (a) UiO-66, (b) UiO-66NH2 and (c) UiO-66Br. The BBP-STE sequence was used in all experiments with a diffusion interval (Δ) of 100 ms. The solid lines are exponential fits using eqn (1). | ||
The plots for all MOFs when using small probe molecules, i.e., water (kinetic diameter, KD = 0.26 nm),34n-octane (KD = 0.48 nm)17 and xylenes (KD = 0.58, 0.68 and 0.68 nm for p-/m-/o-xylenes)35 all show non-linear behaviour. In more detail, the experimental data can be fitted well using a two-component diffusion model. Conversely, when the bulky molecule of TIPB (KD = 0.95 nm)36 was used, a linear and single-component diffusion behaviour was observed for the three MOFs under investigation. The results suggest that small molecules experience two distinct diffusion environments in slow-exchange with each other, whilst the larger TIPB molecule only experiences a single diffusion environment. Based on these observations, we attribute the slow-diffusion component of small probe molecules to diffusion within crystal aggregates, whilst the fast-diffusion component can be attributed to inter-crystalline diffusion (diffusion between crystals).
To understand the difference in the diffusion behaviour of different probe molecules, it is necessary to consider their molecular sizes, as well as the typical pore dimension of the MOFs under investigation. The small probe molecules of water, n-octane and xylenes with Kd values of 0.26–0.68 nm, possess lower Kd values than both the size of tetrahedral and octahedral cages (0.8 nm and 1.4 nm, respectively). This allows them to access the microporous networks within the UiO-66 MOF and its derivatives. Consequently, these molecules are expected to show a slow diffusivity component within the micropore network of the MOFs. Conversely, TIPB, with Kd = 0.95 nm, is too large to access the inner space of the MOFs through the tetrahedral windows, hence only inter-crystalline diffusion could be probed, which is what we observed in this study.
Using the bulk self-diffusion coefficients, Dself,bulk, of the probe molecules (Fig. S4 and Tables S3–S5, ESI†), which are consistent with those reported in the literature,20,37 we can make a relative comparison between those bulk values of diffusivity and the diffusivity values measured inside the MOFs. Compared to the Dself,bulk values of the probe molecules, the values of the fast-diffusion component within the MOFs, D1,self, are of the same order of magnitude but nearly halved, suggesting that the first diffusion regime of the probe molecules in the systems is that of bulk molecules hindered by the presence of the walls of the MOF crystals. Such drops in diffusivity are usually observed in mesoporous/macroporous materials38 and given the typical dimension of the MOF crystals studied in this work (approximately 100 nm), it is reasonable to assume that the inter-crystalline space will be formed of voids of similar dimension, hence explaining the values of D1,self observed. Conversely, the values of the slow-diffusion component, D2,self, of the small probe molecules are markedly smaller than that of the corresponding Dself,bulk, indicating molecular diffusion possibly occurring inside the microporous frameworks of the MOF. This assignment is supported by previous findings on PFG NMR diffusion coefficients in microporous materials.33,39,40 These findings further support our assignment, whereby we can assign D1,self to inter-crystalline diffusion and D2,self to diffusion within crystal aggregates, with the diffusion process being a slow-exchange between the two different diffusion environments.
It is important to observe that in Tables S3–S5 (ESI†), values of root mean square displacement (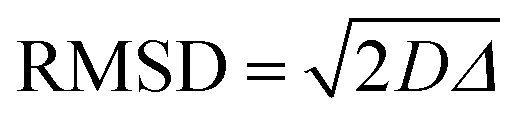 , where D is the self-diffusion coefficient and Δ is the observation/diffusion time) calculated for both D1,self and D2,self are in the order of a few to tens of microns, which is much larger than the typical size of the MOF crystals. We note however that the RMSD probed for the D2,self component is similar to the size of the crystal aggregates, as shown by the SEM images in Fig. S3 (ESI†). This implies that such a component is representative of diffusion inside such crystal aggregates rather than purely intra-crystalline diffusion. As molecules diffuse, they will traverse through crystal pores in a random walk pattern.18
, where D is the self-diffusion coefficient and Δ is the observation/diffusion time) calculated for both D1,self and D2,self are in the order of a few to tens of microns, which is much larger than the typical size of the MOF crystals. We note however that the RMSD probed for the D2,self component is similar to the size of the crystal aggregates, as shown by the SEM images in Fig. S3 (ESI†). This implies that such a component is representative of diffusion inside such crystal aggregates rather than purely intra-crystalline diffusion. As molecules diffuse, they will traverse through crystal pores in a random walk pattern.18
As shown in Tables S3–S5 (ESI†), the D1,self values of all the probe molecules within UiO-66Br are slightly higher than those measured in UiO-66 and UiO-66NH2, whilst the D2,self values for all probe molecules in all three MOFs differ significantly. The slightly higher D1,self observed in UiO-66Br may be caused by the differences in particle size of MOF crystals due to linker functionalisation. Although the SEM data (Fig. S3, ESI†) indicate that the average crystal size yielded by all three MOFs is approximately 100 nm, there may be some variation in crystallinity that affects its diffusivity.
As for the inter-crystalline diffusion process, the D2,self values can be affected by (i) the molecular size of a specific probe molecule, (ii) the pore size and topology of the MOF and (iii) the interaction between a probe molecule and the MOF framework, the latter affected by the presence of functional groups. As for aspect (ii), it is reasonable to assume that pore size and topology is similar across all MOFs studied, whereby we neglect the effect of additional linkers on pore size.
To further discuss (i) and (iii), we introduce the concept of apparent tortuosity, which is the ratio of the bulk diffusivity of a certain species to the diffusivity of the same species within the pore space, that is, a reduction in diffusivity inside the pore space compared to the bulk. In the MOF frameworks studied here we can identify two apparent tortuosities, ξ1 = Dself,bulk/D1,self and ξ2 = Dself,bulk/D2,self. Values of ξ1 and ξ2 are shown in Fig. 4 and Tables S3–S5 (ESI†). Fig. 4(a) shows that the ξ1 values for all probe molecules within the three MOFs is between 1.38–2.40. These values are consistent with diffusion occurring in a mesoporous network,17 hence this indeed suggests that this is diffusion occurring in the mesoscale space between the MOF crystals.
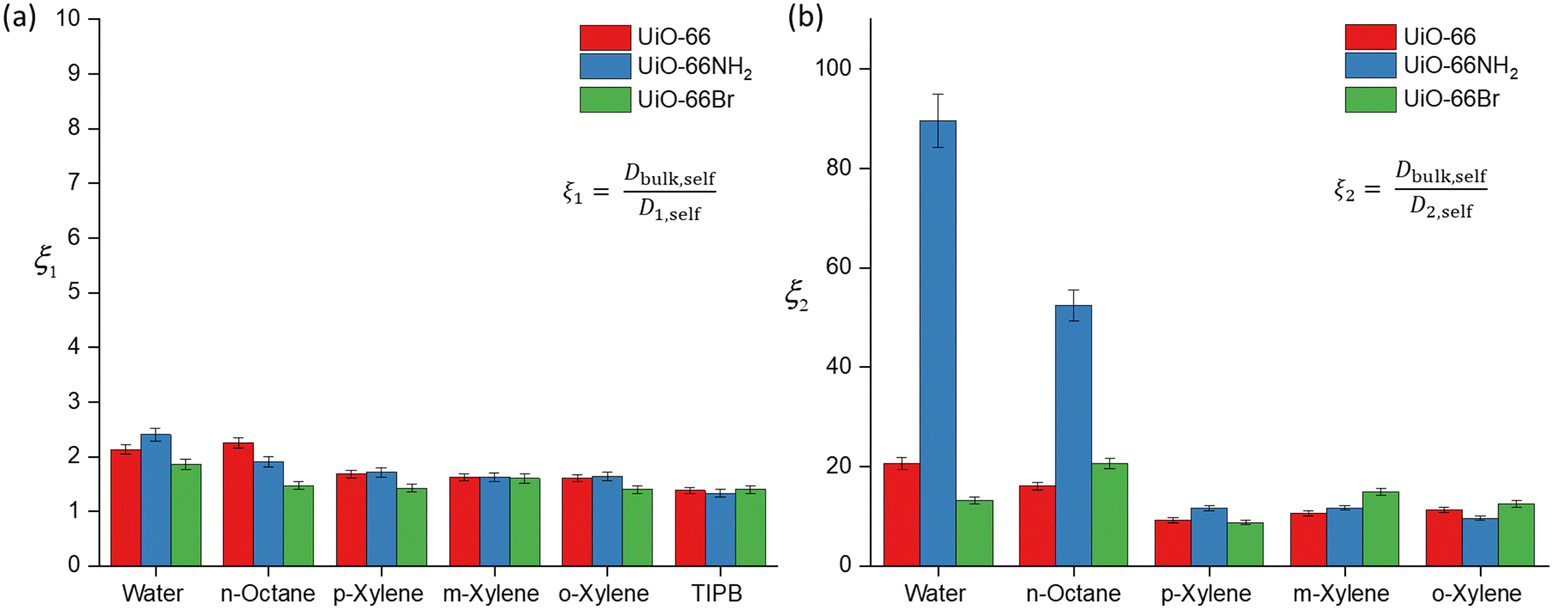 | ||
| Fig. 4 Apparent tortuosity values of the probe molecule–MOF systems under investigation obtained from PFG NMR. (a) Apparent tortuosity ξ1 values derived from the ratio of Dbulk,self/D1,self; (b) apparent tortuosity ξ2 obtained from ratio of Dbulk,self/D2,self. The relative error is approximately 5–6%. | ||
Values of the apparent tortuosity ξ2 are much higher, on average ten times higher than ξ1. Such values are consistent with molecules diffusing in much smaller micropores, which supports the assignment of D2,self to diffusion within crystal aggregates in MOF frameworks, whereby the typical measured pore sizes are less than 2 nm.41
We now compare ξ2 values across the different probe molecules used in the pristine UiO-66. TIPB is too big to access through the UiO-66 framework's triangular windows, hence no ξ2 can be defined in this case. Water and n-octane exhibit higher ξ2 (slower D2,self) compared to xylenes. The higher ξ2 values could be attributed to either entanglement effects during the diffusion process of these molecules within the internal pore environment of the MOFs or to the presence of interactions with the framework affecting the diffusion process. In MOFs, entanglement effects may arise as diffusion processes are affected by various factors of the framework materials such as pore geometry/topology and functional groups on linkers.42 Entanglement in this context arises from interactions between MOFs and diffusing probe molecules. MOFs can exhibit various types of interaction with probe molecules, including van der Waals, hydrophobic/hydrophilic and π–π interactions.11 These factors determine the guest–host interactions in MOFs, such as adsorbate-pore surface interaction and surface-to-pore diffusion, which play a significant role in determining the rate of diffusion.
For the case of water, the high values in ξ2 can be explained by considering the preferential interactions of water molecules within the frameworks. Given the geometry of UiO-66, the dimensions of its cages are suitable for confining water. Wang et al., conducted a study investigating water behaviour and confinement in UiO-66 using molecular modelling and simulations. Their findings indicate that water molecules tend to diffuse through tetrahedral cages due to stronger binding compared to octahedral cages, leading to confinement within.43 In this study, while we cannot definitively confirm through which cages water initially diffuses, we observe a similar behaviour characterised by low D2,self values (Table S3, ESI†). The slower diffusion suggests the presence of hydrogen bonding interactions between water and the UiO-66 framework influencing water diffusion within the MOFs. Similar phenomena were previously reported on water adsorption in UiO-66, where the presence of a metal oxyhydroxy cluster, which contains Zr–OH moieties, promotes coordination with small molecules, hence slowing down water diffusion.44
For straight-chain alkanes, like n-octane, the high ξ2 values can be attributed to the linear arrangement of the carbon chain, which may become entangled within the framework, leading to slower diffusion.45 As shown in Fig. 2, within UiO-66, each octahedral cage is surrounded by eight tetrahedral cages. This interconnected network of tetrahedral and octahedral cages enables the diffusion of n-octane from one cage to neighbouring cages. However, due to its linear arrangement, n-octane is prone to experiencing entanglement effects as it diffuses through the tetrahedral and octahedral cages.
Regarding the xylene isomers, the trend in ξ2 values indicates that o-xylene exhibits a slightly higher value (11.3), followed by m-xylene (10.6) and p-xylene (9.22). Although the differences in apparent tortuosity values are small, this indicates different preferential arrangements of these isomers within the MOFs. This discrepancy can be attributed to steric effects arising from the position of the two methyl groups on the benzene ring, leading to differences in the measured ξ2 values. UiO-66 also shows a reverse shape selectivity pattern,46 whereby the less bulky p-xylene is the least retained isomer, contrary to the typical shape selectivity based on molecular dimensions. Here, we observe a trend consistent with this pattern, with D2,self having the following trend, p-xylene > m-xylene > o-xylene. This interaction has been previously studied in the xylene isomers separation process.47
The calculated D2,self and ξ2 in UiO-66NH2 and UiO-66Br for all probe molecules differ from those obtained for the parent UiO-66. Observing the xylenes isomers, the ξ2 values for UiO-66NH2 show similar values to the pristine UiO-66. Differences are more marked for UiO-66Br when considering m-xylene and o-xylene, which show slightly higher values of ξ2 than those found for UiO-66, whilst for p-xylene there is no significant difference. These differences can be ascribed to changes in pore accessibility due to the introduction of the bromine group. Pristine UiO-66 showed ξ2 values of 11.3 for o-xylene and 10.6 for m-xylene. Following linker functionalisation with –Br, these values increased. This suggest that the presence of –Br in UiO-66 alters pore accessibility by modifying the materials structure and properties. When attached to the linker, –Br molecules undergo rotational movement within the pore system, hindering pore access. This rotational motion leads to steric hindrance, which obstructs the entry of probe molecules into the pores. This rotational linker has been previously investigated.48 As –Br linkers rotate, they can either enhance or restrict pore openings, consequently influencing the overall porosity of these materials. This effect is evidenced by the N2 physisorption results, showing that UiO-66Br exhibits a reduced specific surface area compared to pristine UiO-66.
The ξ2 value of water in functionalised UiO-66NH2 increases markedly to 83.9 from 20.6 (in UiO-66) but decreases to 13.2 in UiO-66Br. These changes are attributed to the changes in guest molecule–MOF interaction due to linker functionalisation. The grafting of hydrophilic –NH2 functional groups enhances the hydrogen bonding interaction with water, which can affect its diffusivity.49 While the geometry of UiO-66 is intrinsically conducive to water confinement, functionalising it with –NH2 groups intensifies the confinement effect not only due to geometry but also due to increased guest–host interactions.50 For UiO-66Br, the decrease in ξ2 is due to the steric hindrance caused by the –Br group. As discussed earlier, the introduction of –Br would hinder the pore access. While UiO-66 is inherently hydrophilic,51 functionalising it with –Br will alter its hydrophilicity, resulting in lower exchange of water within the framework.
The ξ2 value for n-octane increases from 16.1 in UiO-66 to 20.6 in UiO-66Br and 52.4 in UiO-66NH2. This trend is expected because the functionalisation of MOFs introduces steric hindrance, consequently slowing down the movement of molecules. Previous studies have demonstrated that grafting a polar functionality on UiO-66 reduces the linker's rotational ability, which hinders the accessibility of guest molecules.52 Our results highlight a significant impact of functionalising MOFs with –NH2 and –Br groups on the dynamics and framework flexibility within UiO-66.
4. Conclusions
In this work, we demonstrate the usefulness of the PFG NMR technique in unravelling the diffusion behaviour within UiO-66 and its derivatives. Furthermore, we investigate the impact of functionalisation on mass transport in these materials. The results show the presence of two diffusion environments, which are accessible to smaller probe molecules, D1,self associated with inter-crystalline diffusion and D2,self associated with diffusion within crystal aggregates. When molecules such as TIPB with a diameter larger than the diameter of the cage windows of UiO-66 and its derivatives are used as a probe for diffusion, only a single diffusion environment in the inter-crystalline space is observed, which is attributed to the inability of TIPB molecules to access the MOF framework. Functionalisation of the pristine MOF with amino and bromine groups leads to a significant change in diffusion within crystal aggregates, D2,self, due to changes in intra-framework interactions, particularly for water and n-octane, whereas does not significantly affect transport of molecules in the inter-crystalline space. This indicates that the diffusion properties within MOF structures can be influenced by factors such as rotational barriers of functionalised linker molecules and interactions between probe molecules and the MOF structure.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation program under grant agreement no. 872102 and the EPSRC (EP/N033949/1 and EP/R018790/1). S. Z. would like to thank Majlis Amanah Rakyat (MARA) for funding her PhD research.References
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Chem. Mater., 2010, 22, 6632–6640 CrossRef CAS.
- L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud and C. Lamberti, Chem. Mater., 2011, 23, 1700–1718 CrossRef CAS.
- J. Guo, X. Fan, J. Wang, S. Yu, M. Laipan, X. Ren, C. Zhang, L. Zhang and Y. Li, Chem. Eng. J., 2021, 425, 130588 CrossRef CAS.
- S. Zhao, W. Huang, J. Xie, Z. Qu and N. Yan, Fuel, 2021, 289, 119807 CrossRef CAS.
- W. Xiang, Y. Zhang, Y. Chen, C.-J. Liu and X. Tu, J. Mater. Chem. A, 2020, 8, 21526–21546 RSC.
- A. Dhakshinamoorthy, A. Santiago-Portillo, A. M. Asiri and H. Garcia, ChemCatChem, 2019, 11, 899–923 CrossRef CAS.
- R. Vakili, R. Gholami, C. E. Stere, S. Chansai, H. Chen, S. M. Holmes, Y. Jiao, C. Hardacre and X. Fan, Appl. Catal., B, 2020, 260, 118195 CrossRef CAS.
- R. Vakili, E. K. Gibson, S. Chansai, S. Xu, N. Al-Janabi, P. P. Wells, C. Hardacre, A. Walton and X. Fan, ChemCatChem, 2018, 10, 4238–4242 CrossRef CAS PubMed.
- R. Vakili, S. Xu, N. Al-Janabi, P. Gorgojo, S. M. Holmes and X. Fan, Microporous Mesoporous Mater., 2018, 260, 45–53 CrossRef CAS.
- C. H. Sharp, B. C. Bukowski, H. Li, E. M. Johnson, S. Ilic, A. J. Morris, D. Gersappe, R. Q. Snurr and J. R. Morris, Chem. Soc. Rev., 2021, 50, 11530–11558 RSC.
- J. J. Wardzala, J. P. Ruffley, I. Goodenough, A. M. Schmidt, P. B. Shukla, X. Wei, A. Bagusetty, M. De Souza, P. Das, D. J. Thompson, C. J. Karwacki, C. E. Wilmer, E. Borguet, N. L. Rosi and J. K. Johnson, J. Phys. Chem. C, 2020, 124, 28469–28478 CrossRef CAS.
- N. A. Ramsahye, J. Gao, H. Jobic, P. L. Llewellyn, Q. Yang, A. D. Wiersum, M. M. Koza, V. Guillerm, C. Serre, C. L. Zhong and G. Maurin, J. Phys. Chem. C, 2014, 118, 27470–27482 CrossRef CAS.
- Z. Zhao, Z. Li and Y. Lin, Ind. Eng. Chem. Res., 2009, 48, 10015–10020 CrossRef CAS.
- F. Stallmach and P. Galvosas, Annu. Rep. NMR Spectrosc., 2007, 51–131, DOI:10.1016/s0066-4103(07)61102-8.
- C. D'Agostino, R. C. Harris, A. P. Abbott, L. F. Gladden and M. D. Mantle, Phys. Chem. Chem. Phys., 2011, 13, 21383–21391 RSC.
- C. D’Agostino, J. Mitchell, L. F. Gladden and M. D. Mantle, J. Phys. Chem. C, 2012, 116, 8975–8982 CrossRef.
- J. Kärger, D. Freude and J. Haase, Processes, 2018, 6, 147 CrossRef.
- F. Stallmach and J. Kärger, Adsorption, 1999, 5, 117–133 CrossRef CAS.
- S. V. P. Kortunov, J. Kärger, R. Valiullin, P. Gottschalk, M. FéElía, M. S. C. M. Perez, B. Drescher, G. McElhiney, C. Berger, R. Gläser and J. Weitkamp, J. Am. Chem. Soc., 2005, 10, 13055–13059 CrossRef PubMed.
- S. Naumov, R. Valiullin, J. Kärger, R. Pitchumani and M.-O. Coppens, Microporous Mesoporous Mater., 2008, 110, 37–40 CrossRef CAS.
- N. Dvoyashkina, C. F. Seidler, M. Wark, D. Freude and J. Haase, Microporous Mesoporous Mater., 2018, 255, 140–147 CrossRef CAS.
- F. Stallmach, S. Gröger, V. Künzel, J. Kärger, O. Yaghi, M. Hesse and U. Müller, Angew. Chem., Int. Ed., 2006, 45, 2123–2126 CrossRef CAS PubMed.
- D. Sinnaeve, Concepts Magn. Reson., Part A, 2012, 40A, 39–65 CrossRef CAS.
- L. Castañar, G. D. Poggetto, A. A. Colbourne, G. A. Morris and M. Nilsson, Magn. Reson. Chem., 2018, 56, 546–558 CrossRef PubMed.
- D. Zou and D. Liu, Mater. Today Chem., 2019, 12, 139–165 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- M. Kim and S. M. Cohen, CrystEngComm, 2012, 14, 4096–4104 RSC.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- S. Zhao, W. Huang, J. Xie, W. Liu, Z. Qu and N. Yan, Fuel, 2021, 289, 119807 CrossRef CAS.
- R. Vakili, S. Xu, N. Al-Janabi, P. Gorgojo, S. M. Holmes and X. Fan, Microporous Mesoporous Mater., 2018, 260, 45–53 CrossRef CAS.
- J. Kärger, D. M. Ruthven and D. N. Theodorou, Diffusion in nanoporous materials, Wiley Online Library, 2012 Search PubMed.
- C. D'Agostino, Y. Ryabenkova, P. J. Miedziak, S. H. Taylor, G. J. Hutchings, L. F. Gladden and M. D. Mantle, Catal. Sci. Technol., 2014, 4, 1313–1322 RSC.
- T. C. Bowen, R. D. Noble and J. L. Falconer, J. Membr. Sci., 2004, 245, 1–33 CrossRef CAS.
- A. Kurganov, S. Marmé and K. Unger, in Zeolites and Related Microporous Materials: State of the Art 1994 – Proceedings of the 10th International Zeolite Conference, Garmisch-Partenkirchen, Germany, 17–22 July 1994, 1994, pp. 1299–1306 DOI:10.1016/s0167-2991(08)63671-x.
- A. Bazyari, A. A. Khodadadi, N. Hosseinpour and Y. Mortazavi, Fuel Process. Technol., 2009, 90, 1226–1233 CrossRef CAS.
- T. M. O. Popp, A. Z. Plantz, O. M. Yaghi and J. A. Reimer, ChemPhysChem, 2020, 21, 32 CrossRef PubMed.
- Y. Jiao, L. Forster, S. Xu, H. Chen, J. Han, X. Liu, Y. Zhou, J. Liu, J. Zhang and J. Yu, Angew. Chem., Int. Ed., 2020, 59, 19478–19486 CrossRef CAS PubMed.
- J. Kärger and S. Vasenkov, Microporous Mesoporous Mater., 2005, 85, 195–206 CrossRef.
- P. Kortunov, S. Vasenkov, J. Kärger, R. Valiullin, P. Gottschalk, M. Fé Elía, M. Perez, M. Stöcker, B. Drescher and G. McElhiney, J. Am. Chem. Soc., 2005, 127, 13055–13059 CrossRef CAS PubMed.
- Q. Zhu, G. D. Moggridge, M. Ainte, M. D. Mantle, L. F. Gladden and C. D’Agostino, Chem. Eng. J., 2016, 306, 67–76 CrossRef CAS.
- O. M. Yaghi, Nat. Mater., 2007, 6, 92–93 CrossRef CAS PubMed.
- S. Wang, G. Zhou, Y. Sun and L. Huang, AIChE J., 2021, 67, e17035 CrossRef CAS.
- T. G. Grissom, C. H. Sharp, P. M. Usov, D. Troya, A. J. Morris and J. R. Morris, J. Phys. Chem. C, 2018, 122, 16060–16069 CrossRef CAS.
- D. Schuring, A. Jansen and R. Van Santen, J. Phys. Chem. B, 2000, 104, 941–948 CrossRef CAS.
- P. S. Bárcia, D. Guimarães, P. A. Mendes, J. A. Silva, V. Guillerm, H. Chevreau, C. Serre and A. E. Rodrigues, Microporous Mesoporous Mater., 2011, 139, 67–73 CrossRef.
- P. Silva, S. M. Vilela, J. P. Tome and F. A. Almeida Paz, Chem. Soc. Rev., 2015, 44, 6774–6803 RSC.
- J. T. Damron, J. Ma, R. Kurz, K. Saalwächter, A. J. Matzger and A. Ramamoorthy, Angew. Chem., Int. Ed., 2018, 130, 8814–8817 CrossRef.
- C. Caratelli, J. Hajek, F. G. Cirujano, M. Waroquier, F. X. L. i Xamena and V. Van Speybroeck, J. Catal., 2017, 352, 401–414 CrossRef CAS.
- J. Zhang, F. Paesani and M. Lessio, J. Mater. Chem. C, 2023, 11, 10247–10258 RSC.
- X. Liu, X. Wang and F. Kapteijn, Chem. Rev., 2020, 120, 8303–8377 CrossRef CAS PubMed.
- S. Devautour-Vinot, G. Maurin, C. Serre, P. Horcajada, D. Paula da Cunha, V. Guillerm, E. N. de Souza Costa, F. Taulelle and C. Martineau, Chem. Mater., 2012, 24, 2168–2177 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp00447g |
| This journal is © the Owner Societies 2024 |