
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Branched polyesters from radical ring-opening polymerization of cyclic ketene acetals: synthesis, chemical hydrolysis and biodegradation†
Maryam
Mousa
a,
Magnus
Jonsson
b,
Olivia
Wilson
a,
Roy
Geerts
c,
Helena
Bergenudd
b,
Christoffer
Bengtsson
 d,
Anna
Larsson Kron
b and
Eva
Malmström
*ae
d,
Anna
Larsson Kron
b and
Eva
Malmström
*ae
aKTH Royal Institute of Technology, School of Engineering Science in Chemistry, Biotechnology and Health, Department of Fibre and Polymer Technology, Division of Coating Technology, Teknikringen 56-58, SE-100 44 Stockholm, Sweden. E-mail: mavem@kth.se; Tel: +46 70 4287225
bNouryon Pulp and Performance Chemicals AB, Box 13000, 850 13 Sundsvall, Sweden
cNouryon Specialty Chemicals, P.O. Box 10, 7400 AA Deventer, The Netherlands
dDrug Discovery and Development Platform, Science for Life Laboratory, Department of Organic Chemistry, Stockholm University, Solna, Sweden
eKTH Royal Institute of Technology, School of Engineering Science in Chemistry, Biotechnology and Health, Department of Fibre and Polymer Technology, Wallenberg Wood Science Center, Teknikringen 56-58, SE-100 44 Stockholm, Sweden
First published on 18th October 2023
Abstract
Herein, we report a new synthetic route to the cyclic ketene acetal, 2-methylene-4-methyl-1,3-dioxepane (Me-MDO) as a way to expand the tool box of synthesis procedures for cyclic ketene acetals and actualize them as realistic alternatives for synthesizing biodegradable polymers. In this work, 2-methylene-1,3-dioxepane (MDO) and Me-MDO were polymerized by radical ring-opening polymerization to synthesize degradable polyesters. NMR and SEC were used to monitor the polymerization while DSC was used to study the thermal properties. Poly(2-methylene-1,3-dioxepane) (PMDO) showed increased degree of branching with higher conversion, subsequently decreasing crystallinity. The effect of branching and the introduction of side-groups on the chemical hydrolysis rate and biodegradability of the polyesters was assessed using a chemical hydrolysis test and the OECD 301D ready biodegradability screening test, respectively. A significant reduction in the chemical hydrolysis rate and biodegradability was observed upon the introduction of a side group in the poly(2-methylene-4-methyl-1,3-dioxepane) (PMe-MDO) polyester. Less obvious effects on the hydrolysis rate and biodegradability were observed as a result of the polyester branching.
Introduction
The accumulation of plastics in the environment has emerged as a rapidly accelerating challenge over the last decade, as approximately 14 million tons of plastics end up in the ocean every year.1 Recycling is one solution to tackle this problem, however, it is economically and practically challenging. According to recent reports, more than half of the plastics produced since the 1950s have been disposed in landfill sites where most of the plastics are non-degradable and will remain for thousands of years.2–4 To mitigate this, the design of degradable polymers has been a focus in polymer research in recent years to achieve the goal of a circular economy. However, the development of plastics that are both durable and can be recycled by degradation is challenging.5 Therefore, it is important to find robust synthetic routes to degradable polymers by first acquiring readily accessible building blocks/monomers on a large scale, followed by developing polymerization mechanisms that result in polymers with various applications in regard to durability and biodegradability. It is also important to assess the end-life of such biodegradable polymers in regards to their ultimate biodegradation (where the compound, under aerobic conditions, is utilized by micro-organisms resulting in the production of carbon dioxide, water, mineral salts and biomass)6 in nature or under controlled conditions. Finally, knowledge on the controlled chemical hydrolysis of polymers can help to improve polymer recycling on a large scale where degradation products can be recovered and reconstructed into new materials with alternative uses to support the transition to a circular material flow.Aliphatic polyesters, such as polycaprolactone (PCL) and poly(lactic acid) (PLA), are easily synthesized by ring-opening polymerization (ROP) of their corresponding cyclic esters and are readily degradable which can limit their potential application areas.7–9 The more durable vinyl polymers constitute 45–50% of all polymers and are usually synthesized by free radical polymerization (FRP) which is one of the most versatile polymerization methods due to its robustness and mild conditions, resulting in hydrolytically stable polymers with persistent carbon–carbon bonds in the polymer backbones.10,11 Radical ring-opening polymerization (rROP) combines the advantages of ROP and FRP to introduce a heteroatom or a cleavable functional group in the main chain and thereby, constitute a pathway to biodegradable vinyl polymers.12
The monomers used in rROP are cyclic compounds containing heteroatoms and an exocyclic double bond, acting as a radical acceptor. Bailey and co-workers were pioneers in polymerizing cyclic ketene acetals (CKAs) by rROP, enabling the introduction of ester functionalities into the polymer backbone.13–16 The rROP of CKAs may occur by two mechanisms with different outcomes; either through a ring-opening reaction where the addition of the radical to the exocyclic double bond leads to the formation of an ester and a propagating radical (Scheme 1a), or by a ring-retaining path where a carbon–carbon bond is formed instead (Scheme 1b). The former path is more desirable as it opens the possibility to (co)polymerize CKAs with traditional vinyl monomers, such as styrene or methyl methacrylate (MMA), to introduce degradable sites in the polymer. 7-Membered cyclic monomers are more reactive towards rROP compared to 5- and 6-membered rings and will favour the ring-opening mechanism. However, due to the higher ring strain the 7-membered cyclic monomers are more challenging to synthesize than the more stable 5-, and 6-membered rings for which the occurrence of the two competing mechanisms is more pronounced during rROP (Scheme 1).12,13,17
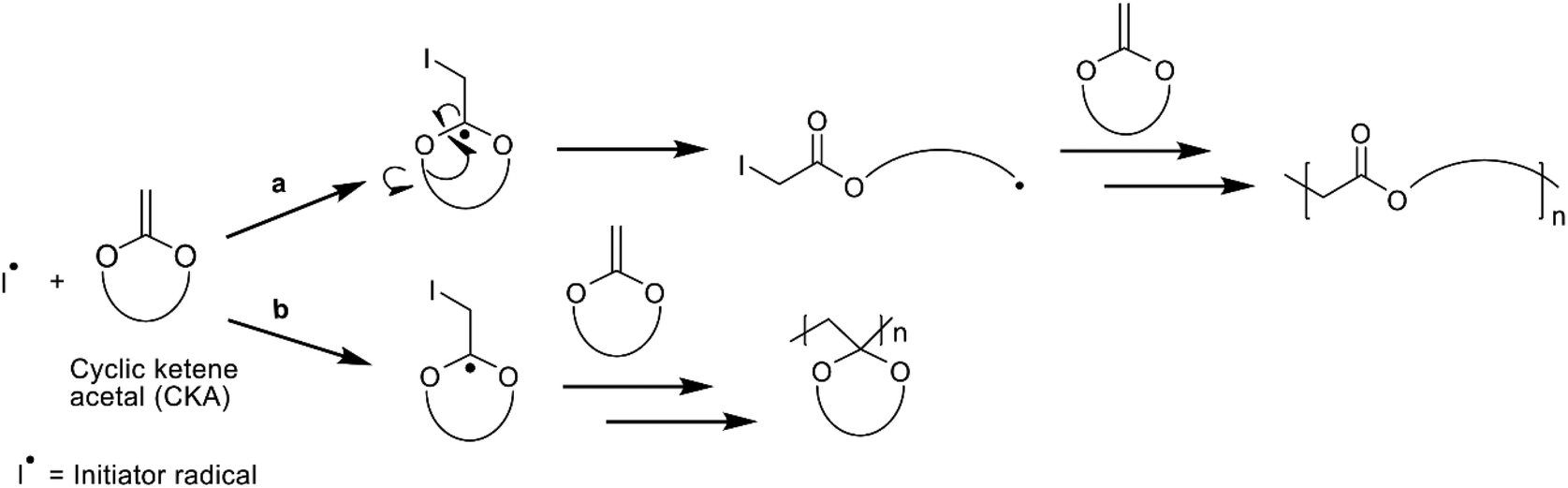 | ||
| Scheme 1 The reaction mechanisms of radical ring-opening polymerization of the CKA monomer, MDO, showing the two alternative outcomes; (a) ring-opening and (b) ring-retaining of the monomer unit. | ||
The synthesis and rROP of different CKAs, especially the 7-membered 2-methylene-1,3-dioxepane (MDO) have been investigated previously.13,18–32 The structural analysis of poly(2-methylene-1,3-dioxepane) (PMDO) reveals a polymer that has a similar structure to conventional polycaprolactone (PCL), but with branches due to intramolecular hydrogen atom transfer (back-biting) reactions.25,33 A branched polyester can be beneficial, especially when used in biomedical applications where linear polyesters can have some limitations such as low surface functionality and low encapsulation efficiency.34 Besides, amorphous biodegradable polymers can be valuable in packaging industry.35 Branched PCL can also result in polymers having different physical and mechanical properties compared to linear PCL as branching will decrease crystallinity as well as melt viscosity.36 In addition, the copolymerization of MDO and other comonomers, such as styrene or MMA, has shown large differences in reactivity ratios between MDO and comonomers which influence the final composition of the copolymers.23,24,37–41
2-Methylene-4-methyl-1,3-dioxepane (Me-MDO) is a structurally related 7-membered CKA-monomer synthesized from 1,4-pentanediol.42,43 The difference between Me-MDO and MDO is the presence of the methyl substituent in Me-MDO, which reduces the back-biting reaction due to the formed, more stabilized, secondary radical resulting in a less branched structure. The methyl group also results in a polyester (PMe-MDO) that is less crystalline than PMDO. Another effect of the methyl substituent is the reduced reactivity compared to MDO, as the methyl group can contribute to steric hindrance around the propagating radical. The structural differences can be compared to the reactivity differences between methyl acrylate (MA) and MMA where the propagating radical of MA is more reactive than MMA.44 The polymerization mechanism and structural analysis of PMe-MDO have been studied by Hiraguri et al.42
CKAs are scarcely available due to demanding synthetic routes restricting the variety of reagents and low yields, as well as their susceptibly towards hydrolysis.17 Therefore, it is of significant relevance to further develop new synthetic pathways toward CKA monomers as realistic alternatives to form degradable vinyl polymers.
In this paper, a new synthetic route to Me-MDO and the rROP polymerization of MDO and Me-MDO have been investigated. The polymerizations were monitored by monomer conversion, and the final polymers were characterized with respect to structure, molecular weight and thermal properties. Furthermore, PMDO and PMe-MDO were tested for chemical hydrolysis and ready biodegradability to investigate whether the degree of branching in PMDO and the methyl substituent in PMe-MDO have effects on the degree of hydrolysis and/or biodegradability.
Experimental
Material
2-Methylene-1,3-dioxepane (MDO, >95%) and 1,4-pentanediol (>99%) were purchased from Ambeed/Chemtronica. Trioctylmethylammonium chloride (Aliquat 336) was purchased from TCI/Chemtronica. Potassium hydroxide (KOH, 90%), 2,2-′azobisisobutyronitrile (AIBN, >98%), polycaprolactone (PCL, average Mn = 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and 140000 when assessed by SEC), potassium tert-butoxide (t-BuOK, ≥98%), N-bromosuccinimide (NBS) and ethyl vinyl ether (>98%) were purchased from Sigma Aldrich. Chloroform (CHCl3, >99%), methanol (MeOH, ≥99.8%, VWR), toluene and acetonitrile were purchased from VWR, anhydrous tetrahydrofuran (THF, >99%) was purchased from thermo scientific. All chemicals were used as received, unless otherwise stated.
000 and 140000 when assessed by SEC), potassium tert-butoxide (t-BuOK, ≥98%), N-bromosuccinimide (NBS) and ethyl vinyl ether (>98%) were purchased from Sigma Aldrich. Chloroform (CHCl3, >99%), methanol (MeOH, ≥99.8%, VWR), toluene and acetonitrile were purchased from VWR, anhydrous tetrahydrofuran (THF, >99%) was purchased from thermo scientific. All chemicals were used as received, unless otherwise stated.
Characterization
000 g mol−1.
Monomer synthesis
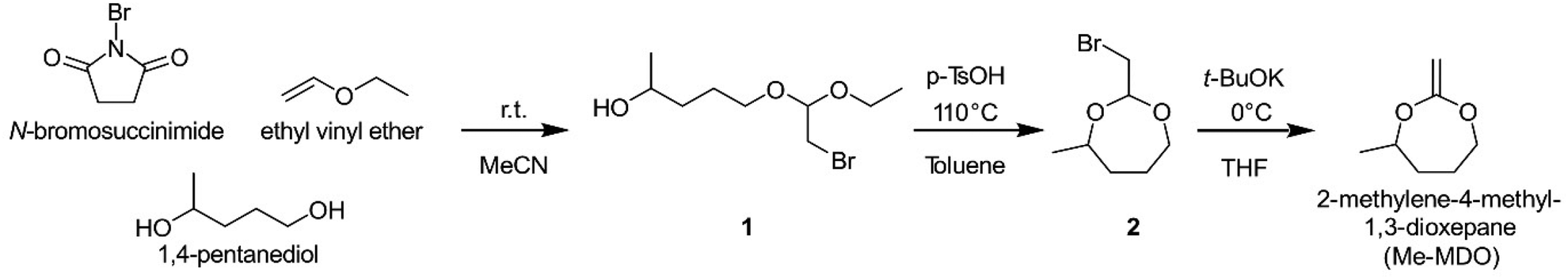 | ||
| Scheme 2 Synthetic route to 2-methylene-4-methyl-1,3-dioxepane (Me-MDO). | ||
Toluene was removed under reduced pressure and the crude product was purified with vacuum distillation to yield in a diastereomeric mixture of a transparent oil. Yield: 73%. 1H NMR (CDCl3, 400 MHz): δ 1.23, 1.25 (two doublets, 3H, –CH3–), 1.43–1.81 (m, 4H, –OCH3CHC![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2C2–), 3.32, 3.34 (two doublets, 2H, BrCH2–), 3.59–4.09 (m, 3H, CH3CO–, –OC2–), 4.91, 4.93 (two triplets, 1H, –OC(CH2Br)O–). 13C NMR (CDCl3, 100 MHz): δ 22.3 and 22.5 (major/minor, CH3–), 28.4 and 29.0 (major/minor, –CH2
2C2–), 3.32, 3.34 (two doublets, 2H, BrCH2–), 3.59–4.09 (m, 3H, CH3CO–, –OC2–), 4.91, 4.93 (two triplets, 1H, –OC(CH2Br)O–). 13C NMR (CDCl3, 100 MHz): δ 22.3 and 22.5 (major/minor, CH3–), 28.4 and 29.0 (major/minor, –CH2![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2CH2–), 32.3 and 32.8 (minor/major, –H2CH2CH2O–), 36.2 and 36.3 (major/minor, BrCH2–), 64.7 and 76.4 (major/minor, –OCH2–), 69.0 and 69.5 (major/minor, –OH(CH3)–), 99.1, 100.10 (minor/major, –OH (CH2Br)O–).
H2CH2–), 32.3 and 32.8 (minor/major, –H2CH2CH2O–), 36.2 and 36.3 (major/minor, BrCH2–), 64.7 and 76.4 (major/minor, –OCH2–), 69.0 and 69.5 (major/minor, –OH(CH3)–), 99.1, 100.10 (minor/major, –OH (CH2Br)O–).
1H NMR (CDCl3, 400 MHz): δ 1.30 (d, J = 6.4 Hz, 3H, CH3–), 1.49–1.81 (m, 4H, –OCH2CH2–), 3.47 (s, 2H, CH2 ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ), 3.81–4.07 (m, 3H, –OC(CH3)C2–). 13C NMR (CDCl3, 100 MHz): δ 22.2 (CH3–), 28.6 (–CH2H2CH2–), 35.8 (–OCH2CH2H2), 67.6 (CH2), 69.5 (–OCH2–), 78.1 (–OH(CH3)–), 163.4 (CH2).
), 3.81–4.07 (m, 3H, –OC(CH3)C2–). 13C NMR (CDCl3, 100 MHz): δ 22.2 (CH3–), 28.6 (–CH2H2CH2–), 35.8 (–OCH2CH2H2), 67.6 (CH2), 69.5 (–OCH2–), 78.1 (–OH(CH3)–), 163.4 (CH2).
Polymerization
MDO (8.7 mmol, 1.0 g) or Me-MDO (8.7 mmol, 1.1 g) was added to a vial followed by the addition of 1 mol% of initiator (AIBN) (0.08 mmol, 14.3 mg). The vial, capped with a septum, was placed in liquid nitrogen and degassed with argon for 15 min. Afterwards, the vial was immersed in a preheated oil bath (65 °C). Aliquots were withdrawn during the reaction to follow the monomer conversion by 1H NMR. The polymerization was quenched by diluting the reaction mixture with THF and subsequently precipitated in MeOH. The polymer was filtered off and was washed twice with MeOH to remove unreacted monomer.PMDO – 1H NMR (CDCl3, 400 MHz): δ 0.91–0.95 (m, 3H, –CH3, branch), 1.33–1.43 (m, 2H, –OCH2CH2C2CH2–), 1.59–1.69 (m, 4H, –OCH2C2CH2C2–), 2.30 (t, J = 7.5 Hz, 2H, OCCH2–), 2.77–2.80 (m, 1H, –CH–, branch site), 4.02–4.10 (m, 2H, –OCH2–). 13C NMR (CDCl3, 100 MHz): δ 13.9 (–CH3, branch), 19.3 (–CH2H2CH3, branch), 24.7 (–OCH2CH2CH2H2–), 25.7 (–OCH2CH2H2CH2–), 28.5 (–OCH2H2CH2CH2–), 30.8 (–H2CH2CH3), branch, 34.3 (OCH2–), 64.3 (–OCH2–), 173.6 (OC).
PMe-MDO – 1H NMR (CDCl3, 400 MHz): δ 0.95 (d, J = 6.6 Hz, 3H, CH3–), 1.22–1.42 (m, 2H, –CH2C2CH–), 1.55–1.73 (m, 2H, –CH2C2CH2–), 1.92–2.01 (m, 1H, –CCH3–), 2.09–2.33 (m, 2H, OCCH2–), 4.01–4.08 (m, 2H, –OCH2–), 4.86–4.93 (m, 1H, –OCCH3a′ when polymer 2 forms). 13C NMR (CDCl3, 100 MHz): δ 19.7 (CH3–), 26.3 (–CH2H2CH2–), 30.2 (–HCH3–), 33.1 (CH2H2CH–), 41.8 (OCH2–), 64.5 (–OCH2–), 173.1 (OC).
Chemical hydrolysis
100 mg of polymer was dissolved in THF (8 mL), followed by the addition of 1.25 mL of 0.5wt% KOH in MeOH. The reaction was left to stir at room temperature for up to 5 h with aliquots taken after 10, 30 min and 5 h to monitor the degree of hydrolysis. The pH of the aqueous phases was adjusted to <7 using HCl (10%) followed by extraction with chloroform (3 × 5 mL). The organic phase was washed with brine (1 × 5 mL), dried with anhydrous MgSO4 (s) whereafter the solvent was evaporated under reduced pressure. The hydrolysis products were characterized by NMR and SEC.Biodegradation
The biodegradation potential of the PMDO, PMe-MDO and PCL polyesters was assessed in ready biodegradation screening tests performed according to the OECD 301D test guideline with minor modifications.45,46 The polyesters were exposed to aerated undiluted river water supplemented with the mineral salts of the OECD nutrient medium or to homogenized activated sludge diluted (2 mg dry weight per L) in aerated OECD nutrient medium. Ammonium chloride was omitted from the nutrient medium to prevent additional oxygen consumption due to nitrification that is not related to the biodegradation of the test substance. The origin of both inocula and the preconditioning of the inocula prior use in the OECD 301D test is described in the ESI.†The tests were performed in 0.3 L biological oxygen demand (BOD) bottles with glass stoppers. The polyesters are poorly water soluble and were therefore dosed with the help of dichloromethane (DCM) which was removed before the start of the test.47 The polyesters were dosed to the bottles from a 1.5 g L−1 DCM stock. The bottles were subsequently placed for 48 h on a roller plate in the fume hood to completely volatilize the DCM and obtain an even distribution of the polyester substances on the inside of the bottles. The final concentration of the polyesters in the test bottles was 2.5 mg L−1. Control bottles containing only the inocula, and prepared in the same way as the test bottles using DCM without the polyesters, were included. Two biological replicates, triplicate tests of each and control BOD bottles were incubated in the dark at 22–24 °C for 28–196 days. The biodegradation of the polyesters was followed by oxygen measurements in the bottles using an oxygen electrode, oxygen meter and a special funnel. The funnel enabled repeated oxygen measurements in the bottles without loss of the medium.46 Biodegradation of the polyesters was calculated as the ratio of biological oxygen demand to theoretical oxygen demand ratio.45 The theoretical oxygen demand (ThOD) of the polyesters was calculated from their molecular formulas. The used molecular formulas and corresponding theoretical oxygen demand values for the polyester substances can be found in Table S1.†
Results and discussion
Monomer synthesis
When synthesizing Me-MDO according to a previously published procedure utilizing bromoacetaldehyde dimethyl acetal in excess of 1,4-pentanediol,13,42 the yield was only ∼30% for the intermediate 2 (Scheme 2). This led us to design a modified synthetic procedure to give the cyclic bromoacetal 2 in an overall yield of 65%. In the first step, NBS is reacted with ethyl vinyl ether to form a bromonium ion which is in equilibrium with its corresponding oxonium ion. The oxonoium ion is attacked by 1,4-pentanediol to form the bromoacetal intermediate, 1, which undergoes an acid-catalyzed ring-closure to form 2-(bromomethyl)-4-methyl-1,3-dioxepane, 2. This reaction was possible to perform on a multigram scale (10–20 g).The cyclic bromoacetal 2 was converted to Me-MDO in an elimination reaction for which 2 h at 0 °C were found to be the optimum conditions. Higher temperatures and longer times resulted in undesired side-reactions reducing the yield, as observed by NMR analysis. In the final step of the work-up, the tert-butanol formed in the process was removed under reduced pressure. This was found to be a critical step in preventing the hydrolysis of Me-MDO.
Polymerization of MDO and Me-MDO
The rROP of MDO and Me-MDO was performed in bulk using AIBN as an initiator at 65 °C and NMR was used to investigate the structures of the polymers. The quaternary carbons in the cyclic acetals have a discrete shift in 13C NMR at ∼100 ppm which can be used to monitor the progress of the desired ring-opening polymerization. The polymerizations of both MDO and Me-MDO seemed to result in nearly complete ring-opening, as concluded by the absence of the quaternary carbon signal in 13C NMR (∼100 ppm) (Fig. S5 and Fig. S6†).48,49ROP of ε-caprolactone and rROP of MDO should result in polymers with identical structures (Fig. 1), and for this reason the structure–property characteristics of PMDO were evaluated and compared to commercial PCL as a reference. 1H and 13C NMR spectra of PMDO show few additional peaks compared to PCL (Fig. 1) due to the presence of branching in PMDO. This is a consequence of the highly reactive propagating primary radical which may undergo back-biting by 1,4-hydrogen atom transfer (H-transfer) and 1,7-H-transfer as proposed by Gonsalves et al., creating branch points in the polymer chain (Scheme S1† and Fig. 1) distinguishable by NMR.22 In our work, the 1,7-H-transfer is suggested to be the dominant back-biting reaction as shown by the appearance of a peak at 2.77–2.80 ppm in 1H NMR, the branching point, (marked with f in Fig. 1) which was also concluded by Agarwal et al.25 The peak emanating from the terminal methyl end-group of the branch after back-biting can be observed in both 1H NMR (0.91–0.95 ppm) and 13C NMR (13.9 ppm) and confirmed by 2D-NMR (Fig. S9†). The branching point from 1,4-H-transfer gives rise to a peak at around at 70 ppm in 13C NMR which could not be observed in our spectra.22
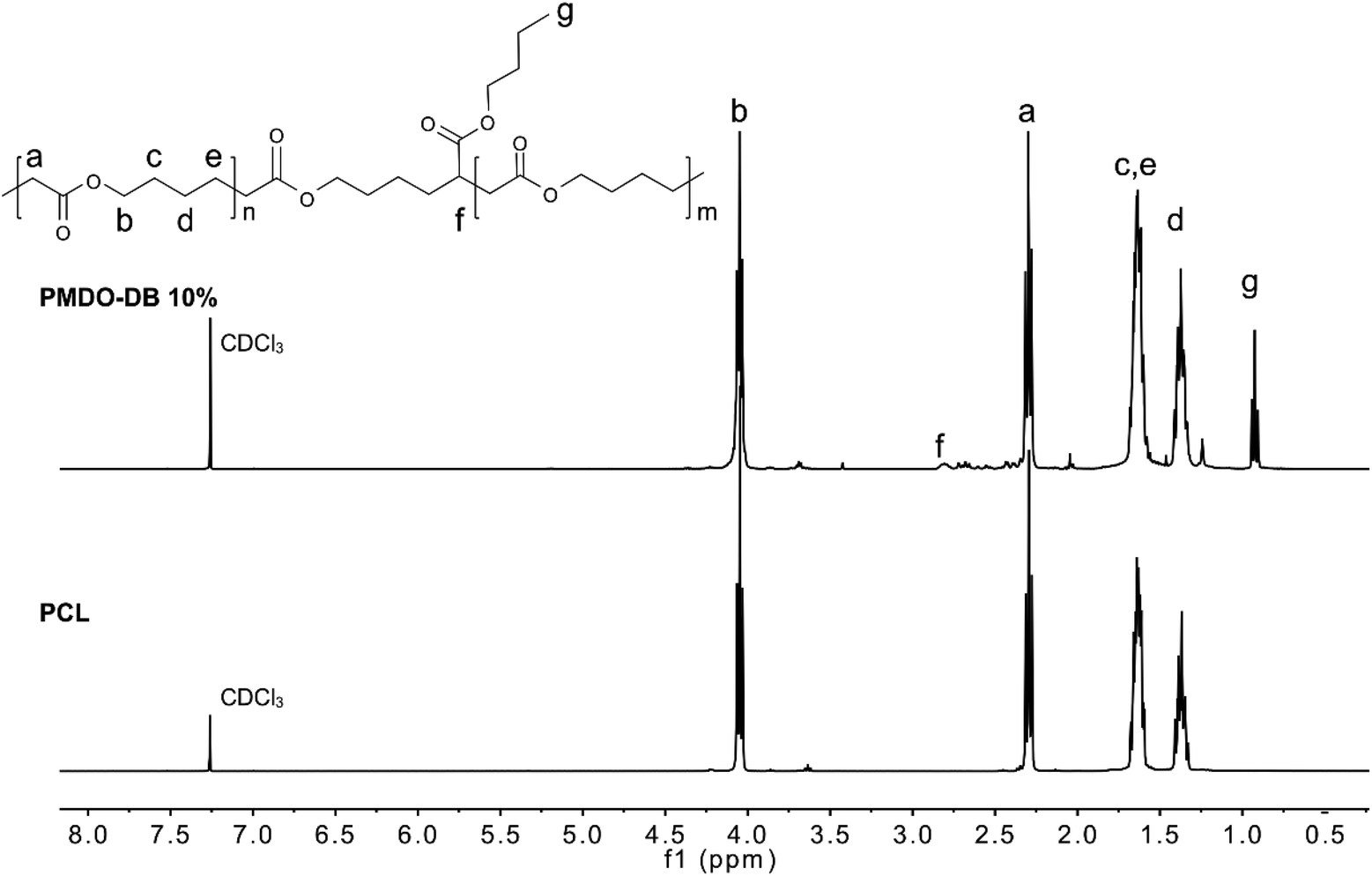 | ||
| Fig. 1 1H NMR spectra of PMDO-DB 10% and PCL in CDCl3. | ||
The degree of branching is defined as the ratio of the number of branched repeating units to the total number of repeating units. For PMDO, the branching density was calculated by comparing the integral of the methyl group g on the branch (0.91–0.95 ppm) (Fig. 1) to the rest of the peaks. The degree of branching was found to increase with increasing conversion of MDO (Table 1). A plausible explanation for this could be that as less monomer is available, more back-biting occurs which increases the number of branches along the polymer chain.
| Polymer-branching % | Time (h) | Conv.a (%) | M n (kg mol−1) | Đ | T g (°C) | T m (°C) | Degree of branchingd (%) | Degree of crystallinityc(%) |
|---|---|---|---|---|---|---|---|---|
a Determined by 1H NMR on crude samples.
b Determined by SEC on worked-up samples using polystyrene standards for calibration.
c Determined by DSC on worked-up samples, 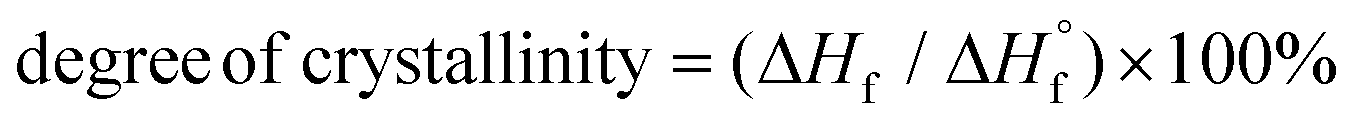 , where ΔHf and ΔHf are the apparent heat of fusion per gram of the sample and the thermodynamic heat of fusion per gram of 100% crystalline PCL, respectively. , where ΔHf and ΔHf are the apparent heat of fusion per gram of the sample and the thermodynamic heat of fusion per gram of 100% crystalline PCL, respectively. 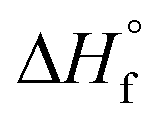 is 135.31 J g−1.50
d Determined by 1H NMR on worked-up samples.
e Conversion calculations are based on the comparison of the methyl group integration in the monomer and in the methyl group in most dominant polymer (formed from secondary radical) as the other methyl group formed from the primary radical overlaps with other peak. Therefore, the conversion is most likely underestimating the actual conversion. is 135.31 J g−1.50
d Determined by 1H NMR on worked-up samples.
e Conversion calculations are based on the comparison of the methyl group integration in the monomer and in the methyl group in most dominant polymer (formed from secondary radical) as the other methyl group formed from the primary radical overlaps with other peak. Therefore, the conversion is most likely underestimating the actual conversion.
|
||||||||
| PCL | — | — | 22 | 1.5 | −64 | 42–62 | — | 52 |
| PMDO-DB 8% | 5 | 57 | 21 | 2.8 | −63 | 14–43 | 8 | 34 |
| PMDO-DB 10% | 7 | 68 | 26 | 3.5 | −63 | 1–35 | 10 | 26 |
| PMDO-DB 15% | 12 | 99 | 31 | 4.7 | −63 | −5–29 | 15 | 17 |
| PMDO-DB 18% | 24 | 99 | 41 | 11.5 | −62 | — | 18 | — |
| PMe-MDOe | 48 | 53e | 10 | 1.7 | −56 | — | — | — |
The molecular weights were assessed by CHCl3-SEC and the results showed a general increase in molecular weight of PMDO as well as molecular weight distribution with longer reaction time (Table 1 and Fig. 2). A multimodal distribution can be observed in the chromatograms where the peaks shift towards higher molecular weights with increasing reaction time, which can especially be observed for PMDO-DB 18% (Fig. 2). It is indeed unexpected as the monomer conversion after 12 h is nearly 100%. However, there is still AIBN present in the reaction mixture generating radicals and although AIBN-radicals are not known as potent hydrogen abstractors, they may give rise to radicals that can result in chain–chain couplings which can eventually lead to crosslinking in a step-wise manner, explaining the steep increase in molecular weight.
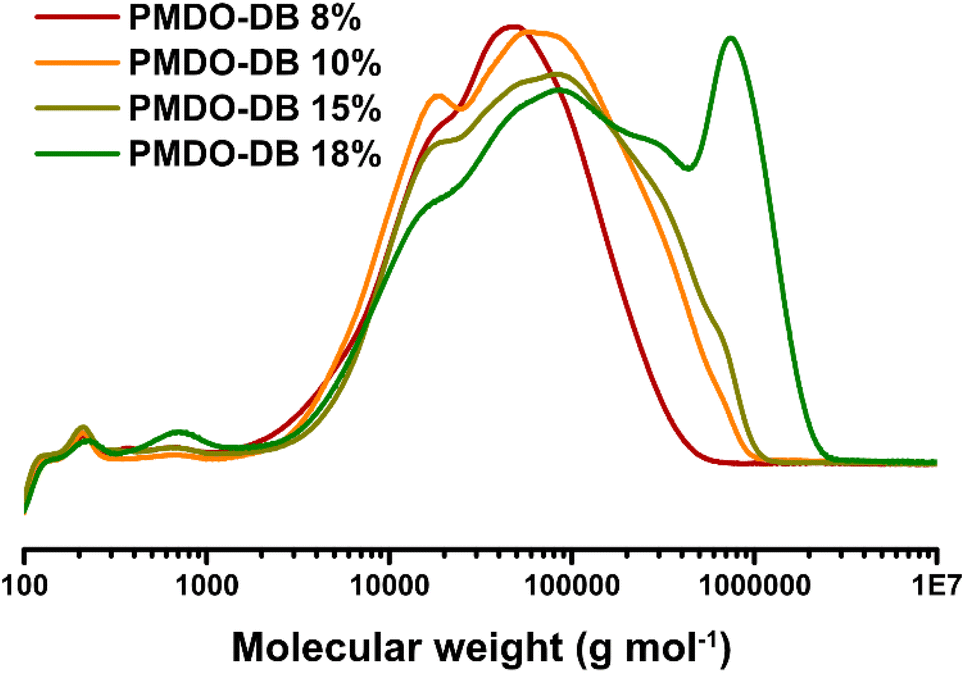 | ||
| Fig. 2 SEC chromatograms of PMDO-DB 8%, PMDO-DB 10%, PMDO-DB 15% and PMDO DB 18%. | ||
The polymerization mechanism of Me-MDO differs from that of MDO due to the presence of the methyl-substituent. The ring-opening and radical formation can occur at either side of the double bond (Scheme 3). However, the polymerization route involving a secondary, more stable, propagating radical (Scheme 3a) is more favoured.42 When the 1H NMR of PMe-MDO (Fig. 3) is scrutinized, the peak a′ is observed, emanating from the methine proton in the repeating unit formed from polymerization through the primary propagating radical (Scheme 3b). The ratio between the two different repeating units, originating from the two polymerization routes, was assessed by comparing integral ratios between peaks a and a′, where it was calculated that 85% of the repeating units were formed by the secondary propagating radical. As a result, 1,7-transfer appear be less pronounced in PMe-MDO compared to PMDO (Fig. 3).42
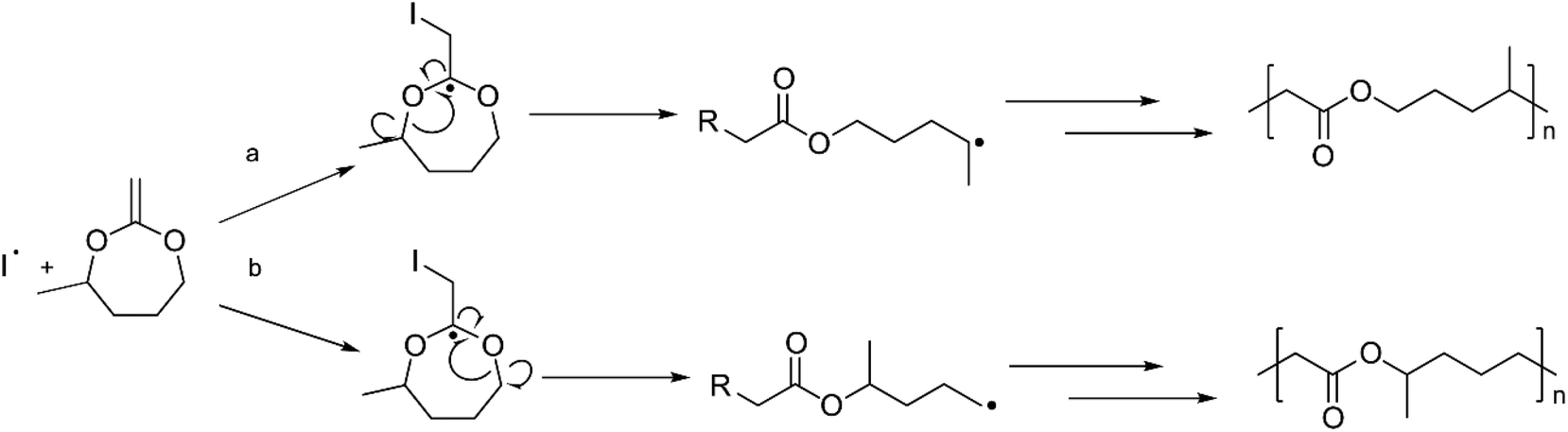 | ||
| Scheme 3 The two polymer structures that can form when polymerizing Me-MDO. | ||
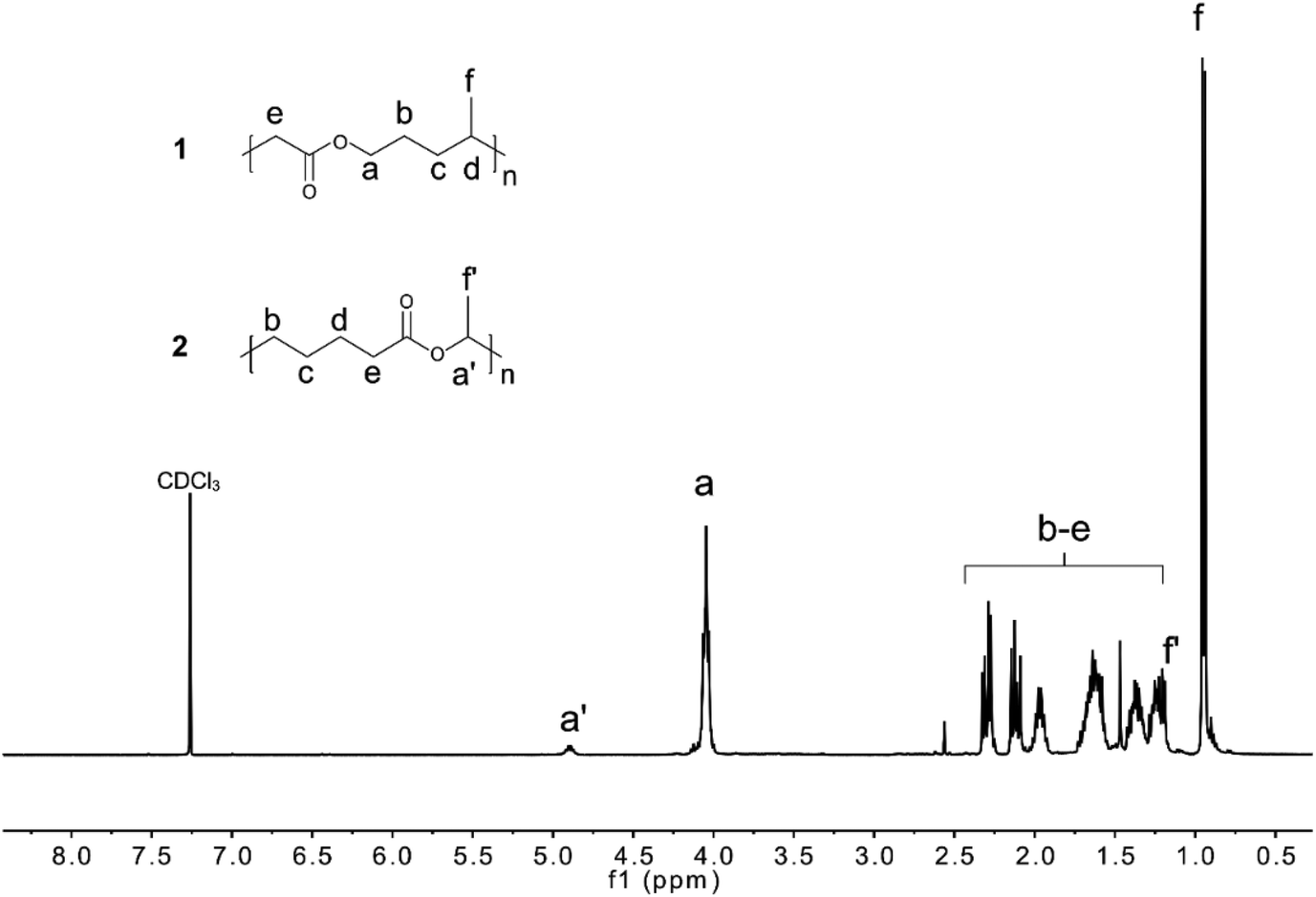 | ||
| Fig. 3 1H NMR spectrum of PMe-MDO in CDCl3. | ||
The structural analysis of PMDO and PMe-MDO is, however, more complex than previously discussed. When enlarging the 1H NMR and 13C NMR spectra, a plethora of small peaks in the aliphatic regions can be observed, suggesting the formation of additional polymerization products that cannot be interpreted by NMR due their low abundance (Fig. S6 and Fig. S8†). To a small extent the presence of unidentified peaks may interfere with assessment of monomer conversion and the degree of branching.
Thermal properties
Differential scanning calorimetry (DSC) was used to assess the thermal properties of the synthesized polymers from the second heating. As expected, all PMDOs exhibited a glass transition (Tg) at −60 °C (Table 1) in accordance with PCL. PCL is a semicrystalline polymer with a crystalline melting transition at 60 °C (Fig. 4) whose degree of crystallinity can be calculated from the melting enthalpy,50,51 which was found to be 52% in the commercial PCL used as a reference in this study. Both PMDO-DB 8% and PMDO-DB 10% also exhibited semicrystalline morphologies as indicated by multimodal melting transitions, however at slightly lower temperatures. Their degrees of crystallinity were calculated to 34- and 26% respectively, which can be explained by the increased degree of branching calculated as, 8- and 10% respectively. The formation of a branched backbone increases the chain irregularity and thereby reduces the chain's capability to crystallize. As reported in previous literature, lower molecular weight PCLs may give rise to multimodal melting peaks, reflecting the coexistence of various crystal types and sizes.52 PMDO-DB 15%, also exhibited a crystallization peak at around −30 °C, reflecting the influence of the slightly different chain configuration as a consequence of an even higher degree of branching (17%). Interestingly, the sample with the highest molecular weight and the highest degree of branching, PMDO-DB 18%, only gave rise to a glass transition and no melting point (Table 1). Based on these results, it can be concluded that rROP of MDO can be used as an alternative to ROP of ε-CL to alter the chain configuration, and thereby the morphology of PCL which can potentially expand the applications window for this material.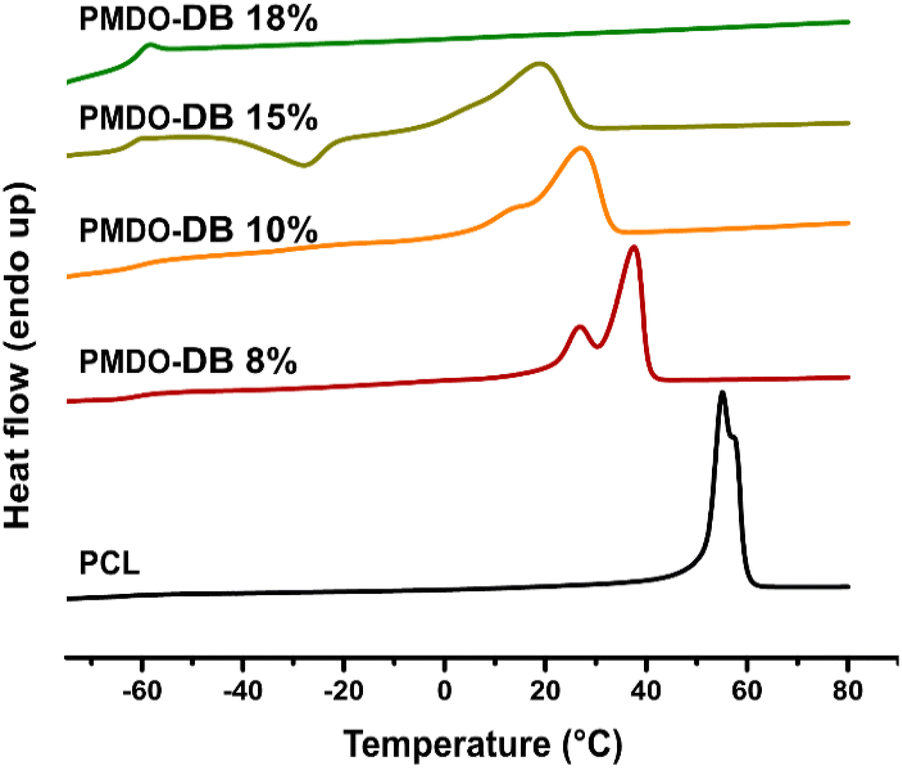 | ||
| Fig. 4 DSC thermograms of comm. PCL, PMDO-DB 8%, PMDO-DB 10%, PMDO-DB 15% and PMDO-DB 18%. | ||
The thermal analysis of PMe-MDO only revealed a glass transition at slightly higher temperature than for PCL, −56 °C (Fig. S10†). The absence of crystallinity can be explained by the methyl-group which prevents the chain from organizing into regular domains.
Chemical hydrolysis
We were interested in investigating the hydrolysis of the synthesized polymers under controlled conditions, to elucidate the effect of the degree of branching. Aliphatic polyesters are susceptible to hydrolysis due to the ester bond in the main chain, and their hydrolysis can be monitored either by immersing solid specimens in aqueous media or by dissolving the polymer in an appropriate solvent/solvent mixture. Previously reported hydrolysis protocols27,33,37,53 were initially explored, utilizing KOH in MeOH (5 wt%). However, as the hydrolysis was found to occur too fast to allow for structural elucidation, the procedures were slightly modified. For example, the OH− concentration was significantly reduced enabling monitor of the degree of hydrolysis of the polymer. Moreover, in an effort to erase surface and bulk effects, the hydrolysis of polymers dissolved in THF was investigated instead of using solid samples. The hydrolysis was monitored by NMR and SEC to ultimately assess the effect of the degree of branching. The alkaline hydrolysis of the polyester in MeOH results, first, in the formation of the corresponding methyl ester. This can already be observed in the 1H NMR after ten minutes of PMDO hydrolysis, by the appearance of a singlet at 3.66 ppm (peak 6, Fig. 5(B)) and a triplet at 3.64 ppm (peak 1, Fig. 5(B)), originating from the methylene protons next to the hydroxyl group of the hydrolysed product A (Fig. 5(A)). After 5 h, most of the polymer is hydrolysed, and the hydrolysis products are proposed in (Fig. 5(A)). The hydrolysis product of the branched unit of PMDO may result in the formation of B (Fig. 5(A)) where the protons at 3.69 ppm (Fig. 5(B)) can be assigned to the methyl esters protons marked with Me. 1H NMR of the PCL hydrolysis did not show any additional peaks around 3.69 ppm (Fig. S13†) as PCL is not branched which is further evidence that B is a hydrolysis product of PMDO. Moreover, the SEC chromatograms (Fig. S12†) reveal the occurrence of the hydrolysis as the original peak has completely disappeared and shifted to significantly lower molecular weights.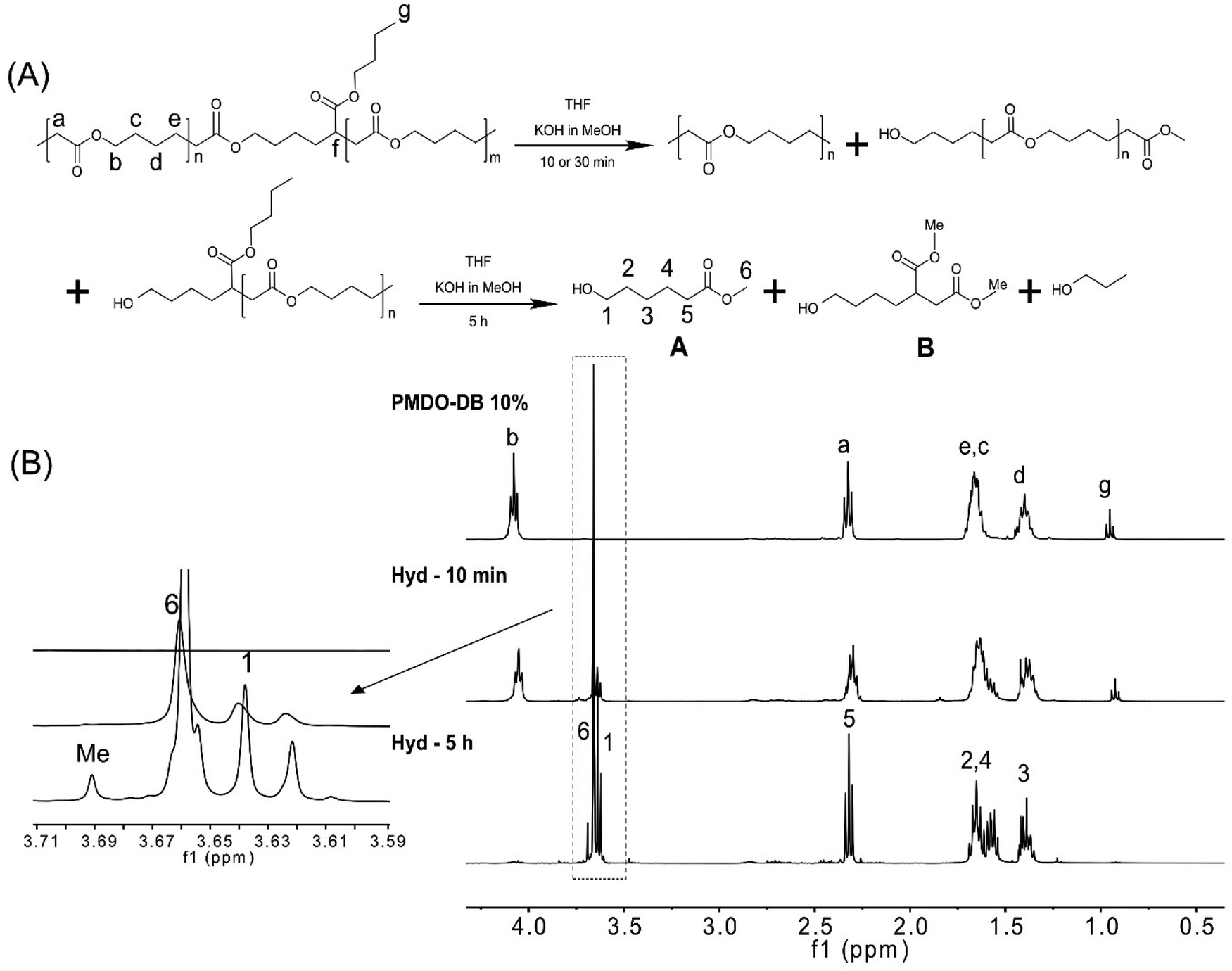 | ||
| Fig. 5 (A) The hydrolysis mechanism and the proposed hydrolysis products. (B) 1H NMR spectra of PMDO-DB 10% and the hydrolysis products of PMDO-DB 10% after 10 min and after 5 h, respectively in CDCl3. | ||
Furthermore, to understand how the rate of hydrolysis is affected by the degree of branching of the polymer, the hydrolysis and the molecular weight were monitored by NMR and SEC with time. In the case of PMDO and PCL where the difference lies in the degree of branching, only slight differences were observed (Fig. 6). The degree of hydrolysis was slightly slower with a higher degree of branching, compared to PCL, which can be attributed to steric hindrance around the ester bonds in the main chain because of the branching. The hydrolysis of PMe-MDO is expected to follow a similar mechanism (Fig. 5(A)). Interestingly, the hydrolysis of PMe-MDO was found to be significantly slower compared to the hydrolysis of PMDO and PCL (Fig. 6), which is attributed to the methyl substituent in PMe-MDO causing steric hinderance.
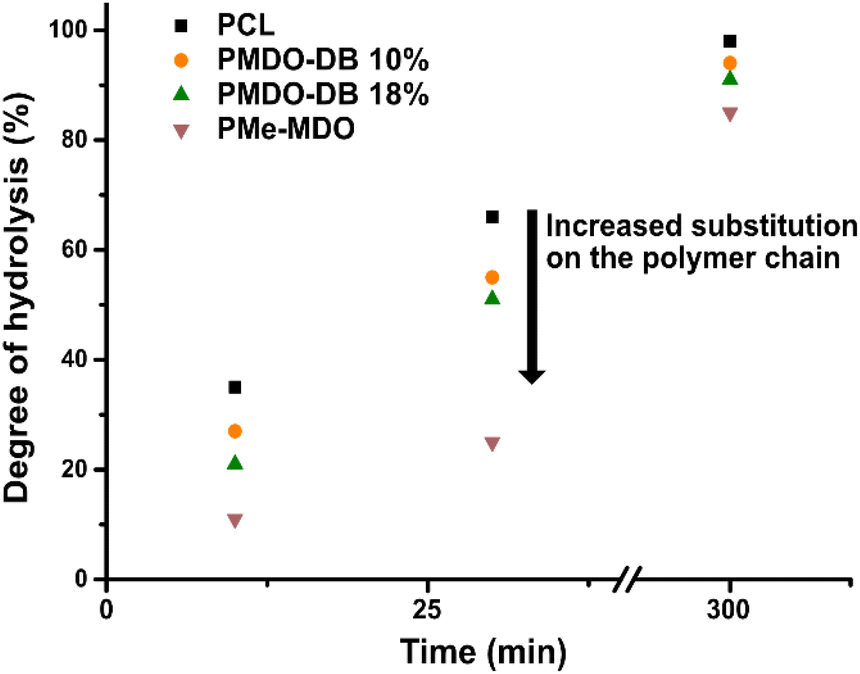 | ||
| Fig. 6 Degree of hydrolysis of PCL, PMDOs and PMe-MDO as a function of time. | ||
Biodegradability
Ready biodegradability tests are regarded as stringent and substances passing these tests give unequivocal proof for a rapid and ultimate biodegradation (mineralization) in most environments including wastewater treatment plants.6 Readily biodegradability is a classification that demonstrates the ultimate biodegradation of a chemical. A sample is considered readily biodegradable when it reaches 60% biodegradation or more within a time period of 28 days.45 The biodegradability of PMDO, PMe-MDO, and PCL polyesters was assessed using the (prolonged) OECD 301D ready biodegradation screening test.The PMDO-DB 10% polyester achieved a ready biodegradation result in the Closed Bottle Test (CBT) with river water inoculum in one of the biological replicates, resulting in 62% biodegradation at day 28 (Table 2). In the same test using the river water inoculum, PMDO-DB 18% achieved 58% biodegradation at day 28. The standard deviations of the replicates for both PMDO samples show that the approximate time to reach 60% degradation is around 28 days and the degradation rates for both PMDO samples are therefore regarded comparable. The results from the second river water biological replicate for both PMDO-DB 10% and PMDO-DB 18% showed a slower biodegradation rate in general where ultimate biodegradation was reached after 56 and 112 days, respectively. The CBT results from the activated sludge inoculum indicate that biodegradation of both PMDO samples was slower compared to biodegradation results in river water inoculum. Ultimate biodegradation achieved for PMDO-DB 10% and PMDO-DB 18% in the activated sludge was achieved at day 56 and day 84, respectively (Table 2).
| Inoculum | Polyester | Biodegradation percentage (Mean ± SD)a | |||
|---|---|---|---|---|---|
| Day 28 | Day 56 | Day 84 | %/[Day]c | ||
| a Mean and standard deviation (SD) are calculated based on the biodegradation achieved in three replicate bottles for two biological replicate per inoculum. b n.a. = not analyzed. c The number in brackets is the day when the biodegradation test was stopped. | |||||
| River water | PCL | 28 ± 14 | 61 ± 9 | n.a.b | n.a. |
| 29 ± 19 | 60 ± 12 | n.a. | n.a. | ||
| PMDO-DB 10% | 62 ± 16 | 66 ± 5 | n.a. | n.a. | |
| 47 ± 6 | 62 ± 2 | 66 ± 3 | n.a. | ||
| PMDO-DB 18% | 58 ± 18 | 66 ± 3 | n.a. | n.a. | |
| 23 ± 12 | 34 ± 17 | 51 ± 13 | 63 ± 7 [112] | ||
| PMe-MDO | 2 ± 1 | 10 ± 7 | 24 ± 12 | 50 ± 11 [196] | |
| 4 ± 1 | 9 ± 4 | 24 ± 23 | 60 ± 8 [140] | ||
| Activated sludge | PCL | 34 ± 28 | 64 ± 1 | n.a. | n.a. |
| 17 ± 13 | 54 ± 7 | 61 ± 5 | n.a. | ||
| PMDO-DB 10% | 38 ± 4 | 62 ± 9 | n.a. | n.a. | |
| 18 ± 5 | 55 ± 16 | 62 ± 9 | n.a. | ||
| PMDO-DB 18% | 13 ± 5 | 43 ± 21 | 61 ± 2 | n.a. | |
| 34 ± 19 | 52 ± 2 | 57 ± 4 | 60 ± 2 [112] | ||
| PMe-MDO | 6 ± 4 | 20 ± 14 | 36 ± 14 | 53 ± 3 [196] | |
| 3 ± 2 | 23 ± 8 | 32 ± 9 | n.a. | ||
PCL was biodegraded by >60% at day 56–84 in the prolonged CBT inoculated with river water and activated sludge (Table 2). These results demonstrate that PCL was mineralized, however, not within 28 days and thus does not account for a ready biodegradability classification. Ready biodegradability of PCL has nevertheless been demonstrated with both river water in an OECD 301C test and activated sludge in the OECD 301C and OECD 301F test.54–56 The observed differences in the biodegradation rates of PCL reported in literature and found in this study cannot be explained, however a relatively fast biodegradation of PCL is expected since PCL-degrading micro-organisms are omnipresent in the environment57 and various polyesterases are known to be capable of performing the initial hydrolysis step of the polyester bonds.58
The biodegradation rate of the PMe-MDO was much slower compared to the PMDO and PCL (Table 2). In the prolonged CBT only 10% and 20% biodegradation were achieved at day 56 with the river water and activated sludge inoculum, respectively. Ultimate biodegradation was reached for one of the biological replicates of PMe-MDO in river water inoculum after 140 days. In the other test, PMe-MDO may eventually biodegrade completely over time as a plateau in the biodegradation curve has clearly not been reached and biodegradation appears to slowly increase after day 196 (Fig. S15†). Ultimate biodegradation was demonstrated in this study for both the branched PMDO, linear PCL polyester samples in both inoculum (river water and activated sludge), and for PMe-MDO only in river water. The approximate time to reach ultimate biodegradation for the branched PMDO and linear PCL in the OECD 301D test probably varies between 28 and 60 days. Although the PMe-MDO is a less branched structure compared to the PMDO polyesters, the introduction of a methyl group in the MDO building blocks clearly slows down the biodegradation rate. Mineralization of the PMe-MDO was demonstrated in only of the OECD 301D tests after 140 days and was almost demonstrated in the other tests. The slow but continuous increase of the biodegradation in the other tests also suggest that the PMe-MDO polyester is capable of ultimate biodegradation.
Conclusions
The current work explored polymers obtained by radical ring-opening polymerization (rROP) of 2-methylene-1,3-dioxepane (MDO) and 2-methylene-4-methyl-1,3-dioxepane (Me-MDO). A new robust synthesis method for Me-MDO, utilizing ethyl vinyl ether and N-bromosuccinimide as reagents, was developed as an important step towards synthesizing CKA-monomers more efficiently, thereby increasing the accessibility to CKAs as versatile building blocks for degradable polymers. Increasing monomer conversion led to increased molecular weights as well as higher degrees of branching reducing polymer crystallinity. As a consequence of the methyl substituent, PMe-MDO was found to be fully amorphous. The degradation of PMDO and PMe-MDO was compared to that of linear PCL where PMDO showed slight changes in the rate of hydrolysis and biodegradation. The presence of side-groups in PMe-MDO decreased the rate of hydrolysis and biodegradation significantly. The more detailed understanding of the rROP of CKAs, their synthesis and the corresponding polymers’ behaviours under hydrolytic conditions may accelerate the use of CKAs as potential (co)monomers in vinyl polymerizations to mitigate plastic accumulation after end-of-life. Furthermore, the ability to synthesize branched polyesters with reduced, or no, crystallinity may be interesting for various applications. Understanding the effects of the polymer structure on the hydrolysis and biodegradation is an important aspect when designing new polymers with regards to their end-use.Author contributions
Maryam Mousa: methodology, validation, investigation, writing – original draft, writing – review & editing, visualization; Magnus Jonsson: methodology, validation, investigation, writing – review & editing, visualization; Olivia Wilson: methodology, validation, investigation; Roy Geerts: methodology, validation, investigation, writing – original draft, visualization; Helena Bergenudd: methodology, supervision; Christoffer Bengtsson: methodology, supervision, writing – review & editing; Anna Larsson Kron: conceptualization, methodology, validation, investigation, resources, writing – original draft, writing – review & editing, supervision, project administration, funding acquisition; Eva Malmström: conceptualization, methodology, validation, investigation, resources, writing – original draft, writing – review & editing, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the financial support from the Swedish Foundation for Strategic Research (ID18-0075).References
- Marine Plastic Pollution: International Union for Conservation of Nature; 2021 [Available from: https://www.iucn.org/resources/issues-briefs/marine-plastic-pollution#:~:text=At%20least%2014%20million%20tons,waters%<?pdb_no 20to?>20to<?pdb END?>%20deep%2Dsea%20sediments.
-
European Commission, Directorate-General for Research and Innovation, R. Koopmans, K. Doorsselaer, C. Velis, et al., A Circular Economy for Plastics: Insights from Research and Innovation to Inform Policy and Funding Decisions, Publications Office, 2019 Search PubMed
.
- R. C. Thompson, C. J. Moore, F. S. vom Saal and S. H. Swan, Plastics, The Environment and Human Health: Current Consensus and Future Trends, Philos. Trans. R. Soc., B, 2009, 364(1526), 2153–2166 CrossRef CAS PubMed
- P. G. Ryan, C. J. Moore, J. A. van Franeker and C. L. Moloney, Monitoring The Abundance of Plastic Debris in The Marine Rnvironment, Philos. Trans. R. Soc., B, 2009, 364(1526), 1999–2012 CrossRef CAS PubMed
- A.-C. Albertsson and M. Hakkarainen, Designed to Degrade, Science, 2017, 358(6365), 872–873 CrossRef CAS PubMed
- U. Pagga, Testing biodegradability with standardized methods, Chemosphere, 1997, 35(12), 2953–2972 CrossRef CAS PubMed
-
P. Lecomte and C. Jérôme, Recent Developments in Ring-Opening Polymerization of Lactones, in Synthetic Biodegradable Polymers, ed. B. Rieger, A. Künkel, G. W. Coates, R. Reichardt, E. Dinjus and T. A. Zevaco, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 173–217 Search PubMed
- A.-C. Albertsson and I. K. Varma, Recent Developments in Ring Opening Polymerization of Lactones for Biomedical Applications, Biomacromolecules, 2003, 4(6), 1466–1486 CrossRef CAS PubMed
-
S. Dutta, W.-C. Hung, B.-H. Huang and C.-C. Lin, Recent Developments in Metal-Catalyzed Ring-Opening Polymerization of Lactides and Glycolides: Preparation of Polylactides, Polyglycolide, and Poly(lactide-co-glycolide), in Synthetic Biodegradable Polymers, ed. B. Rieger, A. Künkel, G. W. Coates, R. Reichardt, E. Dinjus and T. A. Zevaco, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 219–283 Search PubMed
-
P. Nesvadba, Radical Polymerization in Industry, Encyclopedia of Radicals in Chemistry, Biology and Materials, 2012 Search PubMed
- A. W. Jackson, S. R. Mothe, L. R. Chennamaneni, A. van Herk and P. Thoniyot, Unraveling the History and Revisiting the Synthesis of Degradable Polystyrene Analogues via Radical Ring-Opening Copolymerization with Cyclic Ketene Acetals, Materials, 2020, 13(10), 2325 CrossRef CAS PubMed
- A. Tardy, J. Nicolas, D. Gigmes, C. Lefay and Y. Guillaneuf, Radical Ring-Opening Polymerization: Scope, Limitations, and Application to (Bio)Degradable Materials, Chem. Rev., 2017, 117(3), 1319–1406 CrossRef CAS PubMed
- W. J. Bailey, Z. Ni and S. R. Wu, Synthesis of Poly-ε-Caprolactone Via a Free Radical Mechanism. Free Radical Ring-Opening Polymerization of 2-Methylene-1,3-dioxepane, J. Polym. Sci., Polym. Chem. Ed., 1982, 20(11), 3021–3030 CrossRef CAS
- W. J. Bailey, Z. Ni and S. R. Wu, Free Radical Ring-Opening Polymerization of 4,7-Dimethyl-2-methylene-1,3-dioxepane and 5,6-Benzo-2-methylene-1,3-dioxepane, Macromolecules, 1982, 15(3), 711–714 CrossRef CAS
- T. Endo, M. Okawara, W. J. Bailey, K. Azuma, K. Nate and H. Yokono, Photoinitiated Ring-Opening Polymerization of 2-Methylene-1,3-dioxepane, J. Polym. Sci., Polym. Lett. Ed., 1983, 21(5), 373–380 CrossRef CAS
- W. J. Bailey, J. L. Chou, P. Z. Feng, B. Issari, V. Kuruganti and L. L. Zhou, Recent Advances in Free-Radical Ring-Opening Polymerization, J. Macromol. Sci., Part A: Pure Appl.Chem., 1988, 25(5–7), 781–798 CrossRef CAS
- T. Pesenti and J. Nicolas, 100th Anniversary of Macromolecular Science Viewpoint: Degradable Polymers from Radical Ring-Opening Polymerization: Latest Advances, New Directions, and Ongoing Challenges, ACS Macro Lett., 2020, 9(12), 1812–1835 CrossRef CAS PubMed
- A. Díaz-Ortiz, P. Prieto, A. Loupy and D. Abenhaïm, A Short and Efficient Synthesis of Ketene O,O- and S,S-Acetals Under Focused Microwave Irradiation and Solvent-Free Conditions, Tetrahedron Lett., 1996, 37(10), 1695–1698 CrossRef
- M. R. Hill, T. Kubo, S. L. Goodrich, C. A. Figg and B. S. Sumerlin, Alternating Radical Ring-Opening Polymerization of Cyclic Ketene Acetals: Access to Tunable and Functional Polyester Copolymers, Macromolecules, 2018, 51(14), 5079–5084 CrossRef CAS
- A. B. Argade and B. R. Joglekar, A Short and Simple Synthesis of Ketene Acetals, Synth. Commun., 1993, 23(14), 1979–1984 CrossRef CAS
- A. Díaz-Ortiz, E. Díez-Barra, A. de la Hoz and P. Prieto, Preparation of Racemic and Enantiomerically Pure Cyclic Ketene Acetals, Synth. Commun., 1993, 23(14), 1935–1942 CrossRef
- S. Jin and K. E. Gonsalves, A Study of the Mechanism of the Free-Radical Ring-Opening Polymerization of 2-Methylene-1,3-dioxepane, Macromolecules, 1997, 30(10), 3104–3106 CrossRef CAS
- G. E. Roberts, M. L. Coote, J. P. A. Heuts, L. M. Morris and T. P. Davis, Radical Ring-Opening Copolymerization of 2-Methylene 1,3-Dioxepane and Methyl Methacrylate: Experiments Originally Designed To Probe the Origin of the Penultimate Unit Effect, Macromolecules, 1999, 32(5), 1332–1340 CrossRef CAS
- L. F. Sun, R. X. Zhuo and Z. L. Liu, Synthesis and Enzymatic Degradation of 2-Methylene-1,3-dioxepane and methyl acrylate copolymers, J. Polym. Sci., Part A: Polym. Chem., 2003, 41(18), 2898–2904 CrossRef CAS
- S. Agarwal, Microstructural Characterisation and Properties Evaluation of Poly (methyl methacrylate-co-ester)s, Polym. J., 2007, 39(2), 163–174 CrossRef CAS
- S. Agarwal, R. Kumar, T. Kissel and R. Reul, Synthesis of Degradable Materials Based on Caprolactone and Vinyl Acetate Units Using Radical Chemistry, Polym. J., 2009, 41(8), 650–660 CrossRef CAS
- J. Undin, T. Illanes, A. Finne-Wistrand and A.-C. Albertsson, Random Introduction of Degradable Linkages into Functional Vinyl Polymers by Radical Ring-Opening Polymerization, Tailored for Soft Tissue Engineering, Polym. Chem., 2012, 3(5), 1260–1266 RSC
- G. G. Hedir, C. A. Bell, R. K. O'Reilly and A. P. Dove, Functional Degradable Polymers by Radical Ring-Opening Copolymerization of MDO and Vinyl Bromobutanoate: Synthesis, Degradability and Post-Polymerization Modification, Biomacromolecules, 2015, 16(7), 2049–2058 CrossRef CAS PubMed
- D. Ding, X. Pan, Z. Zhang, N. Li, J. Zhu and X. Zhu, A Degradable Copolymer of 2-Methylene-1,3-dioxepane and Vinyl Acetate by Photo-Induced Cobalt-Mediated Radical Polymerization, Polym. Chem., 2016, 7(33), 5258–5264 RSC
- P. Ang, S. R. Mothe, L. R. Chennamaneni, F. Aidil, H. H. Khoo and P. Thoniyot, Laboratory-Scale Life-Cycle Assessment: A Comparison of Existing and Emerging Methods of Poly(ε-caprolactone) Synthesis, ACS Sustainable Chem. Eng., 2021, 9(2), 669–683 CrossRef CAS
- J.-B. Lena and A. M. Van Herk, Toward Biodegradable Chain-Growth Polymers and Polymer Particles: Re-Evaluation of Reactivity Ratios in Copolymerization of Vinyl Monomers with Cyclic Ketene Acetal Using Nonlinear Regression with Proper Error Analysis, Ind. Eng. Chem. Res., 2019, 58(46), 20923–20931 CrossRef CAS
- J. Folini, C.-H. Huang, J. C. Anderson, W. P. Meier and J. Gaitzsch, Novel Monomers in Radical Ring-Opening Polymerisation for Biodegradable and pH Responsive Nanoparticles, Polym. Chem., 2019, 10(39), 5285–5288 RSC
- P. Xu, X. Huang, X. Pan, N. Li, J. Zhu and X. Zhu, Hyperbranched Polycaprolactone through RAFT Polymerization of 2-Methylene-1,3-dioxepane, Polymers, 2019, 11(2), 318 CrossRef PubMed
- E. Christodoulou, M. Notopoulou, E. Nakiou, M. Kostoglou, P. Barmpalexis and D. N. Bikiaris, Branched Poly(ε-caprolactone)-Based Copolyesters of Different Architectures and Their Use in the Preparation of Anticancer Drug-Loaded Nanoparticles, Int. J. Mol. Sci., 2022, 23(23), 15393 CrossRef CAS PubMed
- S. Agarwal and C. Speyerer, Degradable Blends of Semi-Crystalline and Amorphous Branched Poly(caprolactone): Effect of Microstructure on Blend Properties, Polymer, 2010, 51(5), 1024–1032 CrossRef CAS
- M. Bednarek, Branched aliphatic polyesters by ring-opening (co)polymerization, Prog. Polym. Sci., 2016, 58, 27–58 CrossRef CAS
- H. Wickel and S. Agarwal, Synthesis and Characterization of Copolymers of 5,6-Benzo-2-methylene-1,3-dioxepane and Styrene, Macromolecules, 2003, 36(16), 6152–6159 CrossRef CAS
- W. J. Bailey, P. Y. Chen, S.-C. Chen, W.-B. Chiao, T. Endo and B. Gapud,
et al. Free Radical Ring-Opening Polymerization and Its Use to Make Biodegradable Polymers and Functionally Terminated Oligomers, Makromol. Chem., Macromol. Symp., 1986, 6(1), 81–100 CrossRef CAS
- T. Zeng, W. You, G. Chen, X. Nie, Z. Zhang and L. Xia,
et al. Degradable PE-Based Copolymer with Controlled Ester Structure Incorporation by Cobalt-Mediated Radical Copolymerization under Mild Condition, iScience, 2020, 23(3), 100904 CrossRef CAS PubMed
- S. Agarwal and R. Kumar, Synthesis of High-Molecular-Weight Tulipalin-A-Based Polymers by Simple Mixing and Heating of Comonomers, Macromol. Chem. Phys., 2011, 212(6), 603–612 CrossRef CAS
- F. Wenzel, S. Hamzehlou, L. Pardo, M. Aguirre and J. R. Leiza, Kinetics of Radical Ring Opening Polymerization of the Cyclic Ketene Acetal 2-Methylene-1,3-dioxepane with Vinyl Monomers, Ind. Eng. Chem. Res., 2021, 60(29), 10479–10488 CrossRef CAS
- Y. Hiraguri and S. Aiba, Synthesis and Radical Polymerization of Bio-based New Cyclic Ketene Acetal, 2-Methylene-4-methyl-1,3-dioxepane, J. Macromol. Sci., Part A: Pure Appl.Chem., 2014, 51(7), 582–588 CrossRef CAS
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Gamma-Valerolactone, a Sustainable Platform Molecule Derived from Lignocellulosic Biomass, Green Chem., 2013, 15(3), 584–595 RSC
- C. Arias, M. M. C. López-González, M. Fernández-García, J. M. Barrales-Rienda and E. L. Madruga, Free-radical copolymerization of methyl acrylate with methyl methacrylate in benzene solution, Polymer, 1993, 34(8), 1786–1789 CrossRef CAS
- OECD, Test No. 301: Ready Biodegradability1992.
- C. G. van Ginkel and C. A. Stroo, Simple method to prolong the closed bottle test for the determination of the inherent biodegradability, Ecotoxicol. Environ. Saf., 1992, 24(3), 319–327 CrossRef CAS PubMed
-
Water quality—Preparation and treatment of poorly water-soluble organic compounds for the subsequent evaluation of their biodegradability in an aqueous medium, International Organization for Standardization, Geneva, ISO, 2018 Search PubMed
- S. Agarwal, Q. Jin and S. Maji, Biobased Polymers from Plant-Derived Tulipalin A, ACS Symp. Ser., 2012, 1105, 197–212 CrossRef CAS
- S. Maji, F. Mitschang, L. Chen, Q. Jin, Y. Wang and S. Agarwal, Functional Poly(Dimethyl Aminoethyl Methacrylate) by Combination of Radical Ring-Opening Polymerization and Click Chemistry for Biomedical Applications, Macromol. Chem. Phys., 2012, 213(16), 1643–1654 CrossRef CAS
- C. G. Pitt, F. I. Chasalow, Y. M. Hibionada, D. M. Klimas and A. Schindler, Aliphatic polyesters. I. The degradation of poly(ε-caprolactone) in vivo, J. Appl. Polym. Sci., 1981, 26(11), 3779–3787 CrossRef CAS
- H. Hatakeyama, T. Yoshida and T. Hatakeyama, Effect of Side Chain Association on Thermal and Viscoelastic Properties of Cellulose Acetate Based Polycaprolactones, J. Therm. Anal. Calorim., 2000, 59(1), 157–168 CrossRef CAS
- L. Peponi, I. Navarro-Baena, J. E. Báez and J. M. Kenny, Marcos-Fernández A, Effect of the Molecular Weight on the Crystallinity of PCL-b-PLLA di-Block Copolymers, Polymer, 2012, 53(21), 4561–4568 CrossRef CAS
- E. Guégain, C. Zhu, E. Giovanardi and J. Nicolas, Radical Ring-Opening Copolymerization-Induced Self-Assembly (rROPISA), Macromolecules, 2019, 52(10), 3612–3624 CrossRef
- K.-i. Kasuya, K.-i. Takagi, S.-i. Ishiwatari, Y. Yoshida and Y. Doi, Biodegradabilities of various aliphatic polyesters in natural waters, Polym. Degrad. Stab., 1998, 59(1), 327–332 CrossRef CAS
-
Y. Yakabe and M. Kitano, Evaluation of biodegradability of biodegradable plastics in activated sludge, in Studies in Polymer Science, ed. Y. Doi and K. Fukuda, Elsevier, 1994, vol. 12, pp. 331–336 Search PubMed
- R. Nabeoka, H. Suzuki, Y. Akasaka, N. Ando and T. Yoshida, Evaluating the Ready Biodegradability of Biodegradable Plastics, Environ. Toxicol. Chem., 2021, 40(9), 2443–2449 CrossRef CAS PubMed
- H. Nishida and Y. Tokiwa, Distribution of poly(β-hydroxybutyrate) and poly(ε-caprolactone)aerobic degrading microorganisms in different environments, J. Environ. Polym. Degrad., 1993, 1(3), 227–233 CrossRef CAS
- A. Gricajeva, A. K. Nadda and R. Gudiukaite, Insights into polyester plastic biodegradation by carboxyl ester hydrolases, J. Chem. Technol. Biotechnol., 2022, 97(2), 359–380 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Additional NMR spectra, DSC thermograms, SEC traces and biodegradation data. See DOI: https://doi.org/10.1039/d3py00630a |
| This journal is © The Royal Society of Chemistry 2023 |