
Oxygenase mimicking immobilised iron complex catalysts for alkane hydroxylation with H2O2†
Seiya
Sakakura
a,
Ryunosuke
Kitamoto
a,
Kazuki
Goto
a,
Seito
Miura
a,
Takamasa
Takeda
a,
Masaya
Okamura‡
 a,
Arisa
Fukatsu
b,
Shinobu
Itoh
b and
Shiro
Hikichi‡
*a
a,
Arisa
Fukatsu
b,
Shinobu
Itoh
b and
Shiro
Hikichi‡
*a
aDepartment of Material and Life Chemistry, Faculty of Engineering, Kanagawa University, Yokohama, 221-8686, Japan. E-mail: hikichi@kanagawa-u.ac.jp
bDepartment of Molecular Chemistry, Division of Applied Chemistry, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, 565-0871, Japan
First published on 11th July 2023
Abstract
Immobilised iron complex catalysts with hydrophobic reaction fields that mimic the active sites of alkane hydroxylating enzymes were constructed into the mesopores of an SBA-15 type silicate support. The reaction of a chelating ligand (= L) anchored SBA-15 type support with (EtO)3SiC2H4CnF2n+1 and (Me3Si)2NH yielded the corresponding fluoroalkyl (= FC(n)) and trimethylsilyl group (= TMS)-modified supports L-SBA-FC(n)TMS with n = 4, 6, and 8. The ligand-anchored supports reacted with Fe(OTf)2 or FeCl3 to yield the corresponding iron complex-immobilised catalysts. The structure, stability, and catalytic activity of the formed iron complexes depended on the anions of the used iron sources and the lengths of the fluoroalkyl chains. Examination of the cyclohexane oxidation with H2O2 revealed that the support decorated by longer fluoroalkyl chains and TMS was effective in improving the activity and alcohol selectivity of the iron complex immobilised catalysts. In a series of catalysts derived from Fe(OTf)2, the longest fluoroalkyl chain (= FC(8)) modified catalyst was the most reactive and stable. In the FeCl3-derived double-hydrophobised catalysts, the FC(6) modified one exhibited higher activity compared to the FC(8) derivative. Propane oxidation catalysis of mononuclear iron complex-immobilised catalysts Fe(OTf)2/L-SBA-FC(n)TMS (where n = 6 or 8) demonstrated the substrate condensation effect of the hydrophobic pocket formed by the longer fluoroalkyl pillars. Formation of not only 2-propanol and acetone but also 1-propanol and propionaldehyde suggested the synergy of the strong radical characteristics of the generated active oxidant and the substrate concentration effect. The most active catalyst for the cyclohexane oxidation, FeCl3/L-SBA-FC(6)TMS, catalysed methane oxidation with H2O2: the products were methanol, formic acid, and methyl hydroperoxide, whereas no alkyl hydroperoxides formed in the oxidation of propane. Higher bond dissociation energy (= BDEC–H) of methane compared to propane resulted in decelerating the H atom abstraction (HAT) from methane by the oxidant formed on the iron complex while relatively accelerating the decomposition of H2O2.
Introduction
In living organisms, many oxygenase enzymes catalyse the hydroxylation of aliphatic C–H substrates. The active sites of such oxygenase enzymes are composed of an iron or copper complex with organic ligands such as porphyrin and amino acid residues involving N, O, or S donor atoms.1–4 The resulting metal complexes activate O2 to yield an active oxidant such as high-valent metal-oxido species. The metal complex active sites are located in the hydrophobic pocket.5–7 The hydrophobicity of the spheres surrounding the active sites increases their affinity for hydrophobic substrates such as alkanes, rapidly removing hydrophilic oxygenation products, i.e. alcohols, from the active sites and inhibiting over-oxidation. To date, several synthetic metal complex catalysts with alkane oxidation activity designed to reproduce the structure and electronic properties of the catalytically active sites of oxygenase have been reported. For example, Kojima et al. reported an iron complex with an NHC ligand having both the electronic property necessary to generate high-valent iron-oxido species and a hydrophobic pocket that can trap alkane molecules, showing high activity and selectivity for the hydroxylation reaction of gaseous alkanes.8 To achieve high catalytic activity similar to that of enzymes in artificial complex catalysts, therefore, it is necessary to consider the structure and properties of the environment surrounding the complex catalyst molecules.Immobilisation of metal complex catalyst molecules on a solid surface leads to preventing deactivation due to structural changes caused by chemical equilibrium in solution or disproportionation reactions between complex catalyst molecules.9–15 In addition to such an improvement effect on stability, immobilised metal complex catalysts have the advantage in which different functional groups can be easily added to the solid support. This additional functionalisation will control the structural properties surrounding the catalytically active metal centre as well as the affinity of the catalysts toward substrates. In line with these concepts, our group collaborated with Kojima's group to develop immobilised metal complex catalysts for alkene epoxidation and alkane hydroxylation.15 The catalytically active sites are the cationic iron complexes with NHC-ligands, which do not carry any hydrophobic substituents for the surrounding vacant site on the iron centres. Noteworthily, the hydrophobic environment surrounding the active sites is achieved by the introduction of fluoroalkyl chains onto the surface of the cation-exchangeable mesoporous aluminosilicate. The resulting hydrophobic catalysts exhibit the high activity of alkene epoxidation and alkane hydroxylation with H2O2.
We have been developing immobilised metal complex catalysts with an SBA-15 type mesoporous silica support.16–20 Through the reaction of a ligand having a linker connecting group that shows specific reactivity toward the organic functional group implanted on the wall of the mesopore of the SBA-15 type silica support, the metal-supporting ligand can be covalently bonded to the support. The immobilised metal complex catalyst prepared by the reaction of metal ions with the ligands anchored onto the support has excellent stability without the leaching of the complex molecules themselves. On the immobilised metal-complex catalysts with the silicate support, the existence of silanol (Si–OH) groups on the silica surface is an important factor in controlling the properties of the support because they interact with various chemicals, including hydrophilic molecules and metal ions. Modification of the silica surface is then achieved by the reaction of the silanol groups with appropriate silane coupling reagents. In this study, we planned to use silane coupling reagents with long-chain fluoroalkyl groups to create hydrophobic columns surrounding the active sites of the complexes anchored to the supports.15 Controlling the coordination space by introducing substituents that cover the metal centre onto the ligand is often attempted in the molecular design of complex catalysts. From the synthetic chemistry viewpoint, however, introducing the substituent groups on the ligand is sometimes not so easy. In this study, therefore, the design of the metal-supporting ligand is maintained, and the space-controlling groups are anchored onto the support surface. The coordination space of the metal centre of the complex is considered to be sterically constrained by covering the periphery of the complex bound to the support with columns of long-chain alkyl fluoride. Due to the nature of the long chain fluoroalkyl groups, the formed coordination space is hydrophobic. This is expected to create an environment of catalytically active sites similar to that of the oxygenase enzymes.
Results and discussion
Design and preparation of the hydrophobic-functionalised support and iron catalysts
We have developed a ligand-anchored SBA-15 type support by the click reaction of bis(pyridylmethyl)propargylamine with organic azide groups immobilised on the silica wall through one-pot condensation of Si(OEt)4 and (EtO)3SiC3H6N3.21 To eliminate the coordination ability of the remaining silanol groups of the support surface, end-capping with a trimethylsilyl (= TMS) group was applied, and the resulting TMS-capped surface exhibits a hydrophobic nature. This support, namely L-SBA-TMS, was applied to various d-block metal complex catalysts M/L-SBA-TMS. Their catalytic activity for cyclohexane oxidation with meta-chloroperoxybenzoic acid (mCPBA) depended on the metal, and its order was Ni ∼ Co > Fe > Mn > Cu.18 When L is fixed at high density on the supports, two ligand molecules coordinate with the metal ion to form a coordinatively saturated, catalytically inactive complex.16 Therefore, the lower the ligand fixation density on the support, the more efficiently the catalytically active sites can be constructed. During the reaction with mCPBA as the oxidant, unfortunately, the leaching of metals could not be avoided. Therefore, we decided that the oxidant applied to the alkane hydroxylation was switched from mCPBA to H2O2. The ligand L, which is a tertiary amine with one triazolyl and two pyridyl groups working as nitrogen donors, can be regarded as an analogue of tris(2-methylpyridyl)amine, namely TPA. Que et al. have reported that iron complexes with TPA and its derivatives react with hydrogen peroxide to form mononuclear iron(III)–OOH complexes and are active in the catalytic oxidation of alkanes with H2O2.22,23 In addition, our preliminary examination revealed that Fe and Cu/L-SBA-TMS were active for the alkane oxidation with H2O2, but a large amount of Cu ions leached out during the reaction. Thus, we have focused on the Fe catalyst. As noted above, the support with a lower ligand-immobilised density is more efficient in constructing catalytically active sites. In that case, there is a space for additional chemical modification of the surrounding silica wall to which the ligands are anchored.The ligand-anchored support L-SBA was prepared by the method reported previously.16 The reaction of L-SBA with (EtO)3SiC2H4CnF2n+1, which are silane coupling reagents composed of fluoroalkyl groups with different carbon chain lengths, yielded the corresponding fluoroalkyl-modified supports L-SBA-FC(n) with n = 4, 6, and 8. Loading amounts of the fluoroalkyl groups were estimated by the analysis of TG curves. On L-SBA-FC(n), many Si–OH groups remained. Therefore, additional modification with the TMS capping through the reaction with (Me3Si)2NH. The resulting double hydrophobized supports are named L-SBA-FC(n)TMS. TG analyses of these supports indicated the increasing organic groups on the supports (Fig. S1†). Increasing the organic functional groups led to the reduction of the surface area (Table 1 and Fig. S2†), although the original structure of the silicate framework was retained on the functionalization as indicated by the XRD patterns (Fig. S3†). The incorporation of the hydrophobic functional groups on the support was also supported by the comparison of the IR spectra of the dried L-SBA, L-SBA-FC(6) and L-SBA-FC(6)TMS (Fig. S4†). Increasing the hydrophobic characteristics reflected in the decreasing intensity of νO–H attributed to the surface Si–OH groups and absorbed H2O. The symmetric stretching bands of CF3 and CF2 groups of the fluoroalkyl chain might be overlapped on the Si–O–Si bands, while the bands of HOOCC5H10C6F13 are known to appear at around 1319 cm−1 and 1147 cm−1, respectively.24 In contrast, νC–H and νC–Si bands attributed to the trimethylsilyl groups were observed at 2964 cm−1 and 849 cm−1, respectively, on the double hydrophobised support L-SBA-FC(6)TMS.
| Modification | Loading amount/mmol g−1 | Surface area/m2 g−1 | Pore volume/cm3 g−1 | Pore diameter/nm | ||
|---|---|---|---|---|---|---|
| L | FC(n) | TMS | ||||
| FC(8) | 0.058 | 0.42 | — | 354 | 0.41 | 4.1 |
| FC(6) | 0.058 | 0.35 | — | 403 | 0.65 | 6.3 |
| FC(4) | 0.052 | 0.96 | — | 518 | 0.83 | 7.3 |
| FC(8)TMS | 0.058 | 0.42 | 2.48 | 284 | 0.34 | 4.2 |
| FC(6)TMS | 0.058 | 0.35 | 2.57 | 349 | 0.55 | 6.4 |
| FC(4)TMS | 0.052 | 0.96 | 2.55 | 296 | 0.49 | 7.3 |
The reactions of the ligand-anchored supports with Fe(OTf)2 or FeCl3 yielded the corresponding iron complex-immobilised catalysts (Fig. 1). The loading amounts of iron depended on the anions of the used iron compounds, the lengths of the fluoroalkyl chains, and the TMS-capping of the silica surface (with or without) (Table 2). The loading amounts of iron on the support with only TMS capping were larger than those on the anchored ligand, whether Fe(OTf)2 or FeCl3 was applied as the iron source. On the fluoroalkyl-functionalised supports, the iron loadings derived from Fe(OTf)2 depended on the lengths of the carbon chain of the fluoroalkyl groups; the shortest one (n = 4) resulted in an exceeding amount of the ligand, whereas the ratio of iron to the ligand was close to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 on the longer fluoroalkyl chain-modified supports (n = 6 and 8). The short chain of FC(4) could not cover L and that resulted in the formation of multinuclear iron complexes with bridging ligands such as OTf− and OH−. Notably, FeCl3 yielded the higher iron-loaded catalysts with an Fe/L ratio of ca. 1.6 on the supports of both n = 6 and 8. In the case of the double-hydrophobised supports, the coordination of iron onto the surface of the supports would be negligible. Therefore, the iron loadings exceeding the anchored ligand could be attributed to the formation of a dinuclear complex with a single ligand. We reported two molecular structures of the iron(III)–chlorido complexes with the non-anchored L′, which was synthesised by the reaction of bis(pyridylmethyl)propargylamine with tert-butyl azide.18 On a trichloride complex, [FeCl3L′], the triazolyl group of L′ was not coordinated to the iron centre. In contrast, a cationic dichloride complex, [FeCl2L′](BF4), was L′ coordinated to the iron(III) centre in a tetradentate manner.18 Therefore, the structural flexibility of L would make it possible to work as a bridging ligand to form a dinuclear complex as shown in Fig. 2(c).25
:1 on the longer fluoroalkyl chain-modified supports (n = 6 and 8). The short chain of FC(4) could not cover L and that resulted in the formation of multinuclear iron complexes with bridging ligands such as OTf− and OH−. Notably, FeCl3 yielded the higher iron-loaded catalysts with an Fe/L ratio of ca. 1.6 on the supports of both n = 6 and 8. In the case of the double-hydrophobised supports, the coordination of iron onto the surface of the supports would be negligible. Therefore, the iron loadings exceeding the anchored ligand could be attributed to the formation of a dinuclear complex with a single ligand. We reported two molecular structures of the iron(III)–chlorido complexes with the non-anchored L′, which was synthesised by the reaction of bis(pyridylmethyl)propargylamine with tert-butyl azide.18 On a trichloride complex, [FeCl3L′], the triazolyl group of L′ was not coordinated to the iron centre. In contrast, a cationic dichloride complex, [FeCl2L′](BF4), was L′ coordinated to the iron(III) centre in a tetradentate manner.18 Therefore, the structural flexibility of L would make it possible to work as a bridging ligand to form a dinuclear complex as shown in Fig. 2(c).25
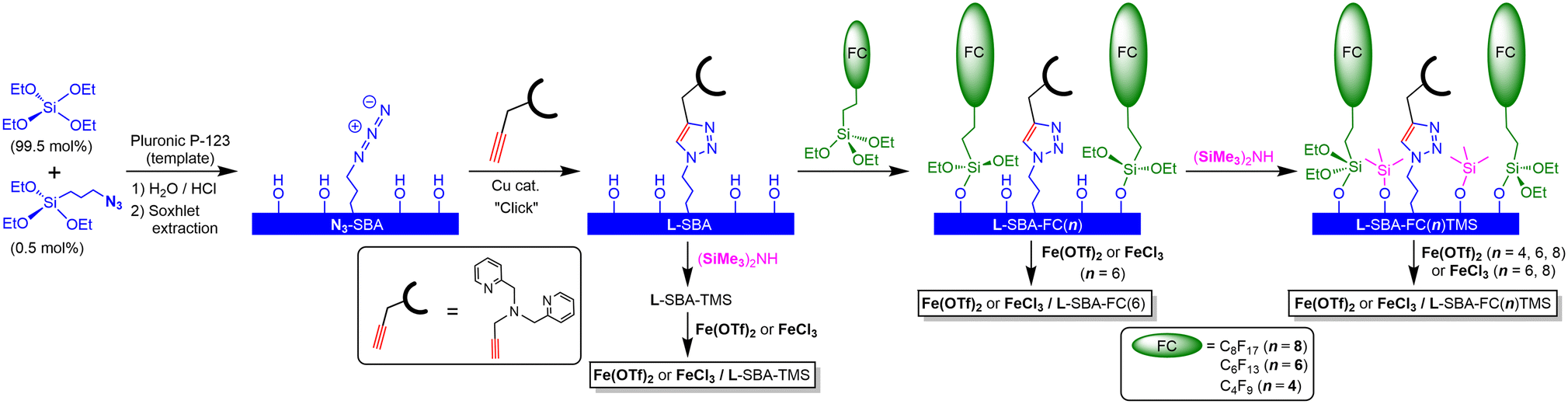 | ||
| Fig. 1 Preparation of the supports and the catalysts. | ||
| Catalyst | Loading amount/mmol g−1 | Fe/L | |
|---|---|---|---|
| L | Fe | ||
| (Fe source: Fe(OTf)2) | |||
| Fe(OTf)2/L-SBA-FC(8)TMS | 0.0584 | 0.0537 | 0.92 |
| Fe(OTf)2/L-SBA-FC(6)TMS | 0.0584 | 0.0544 | 0.93 |
| Fe(OTf)2/L-SBA-FC(4)TMS | 0.0521 | 0.1302 | 2.50 |
| Fe(OTf)2/L-SBA-TMS | 0.0422 | 0.0740 | 1.75 |
| Fe(OTf)2/L-SBA-FC(6) | 0.0265 | 0.0265 | 1.00 |
| (Fe source: FeCl3) | |||
| FeCl3/L-SBA-FC(8)TMS | 0.0376 | 0.0580 | 1.54 |
| FeCl3/L-SBA-FC(6)TMS | 0.0290 | 0.0472 | 1.63 |
| FeCl3/L-SBA-TMS | 0.0599 | 0.0986 | 1.65 |
| FeCl3/L-SBA-FC(6) | 0.0265 | 0.0648 | 2.44 |
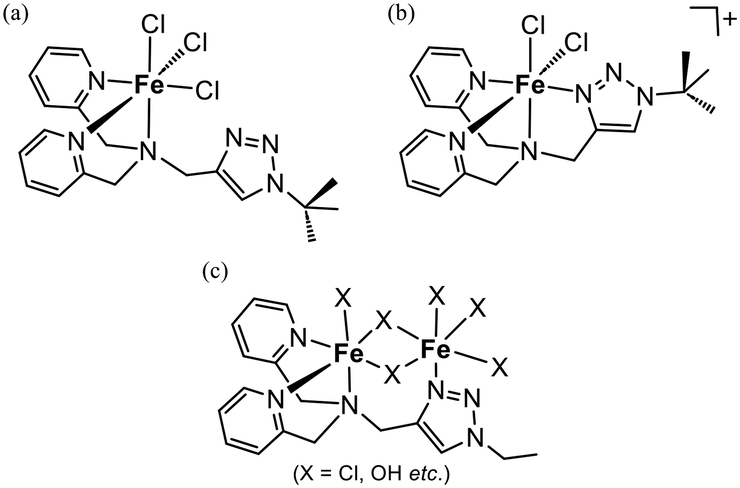 | ||
| Fig. 2 Schematic drawings of the coordination structures of the iron(III) complexes. (a) [FeCl3L′] (ref. 18), (b) [FeCl2L′]+ (ref. 18), and (c) possible structures of the iron complex on the FeCl3-derived catalysts. | ||
Catalytic activity for the cyclohexane oxidation with H2O2
To explore the effect of the hydrophobised supports, the activities of the iron catalysts toward cyclohexane oxygenation with H2O2 were examined. The yields of cyclohexanol and cyclohexanone obtained after 6 hours are summarised in Table 3. The comparison with the activity of a series of iron catalysts derived from Fe(OTf)2 demonstrated that the double-hydrophobised supports were effective in improving the activity and alcohol selectivity. The catalysts with the single-hydrophobised supports with TMS (entry 4) or C2H4C6F13 (i.e. n = 6; entry 5) yielded cyclohexanone as the major product. The TONs and A/K values of the single-hydrophobised catalysts were similar to those of [Fe(TPA)(MeCN)2](OTf)2, which is a prototype of our immobilized complex, in the homogeneous reaction (entry 10). According to the previous report on the catalysis of [Fe(TPA)(MeCN)2](OTf)2, the slow addition of H2O2 (using a syringe pump) leads to high alcohol selectivity.23 The higher concentration of H2O2 under our reaction conditions resulted in the fast progress of the cyclohexane oxidation with low A/K (Fig. S5†) concomitant with the change in the solution colour from pale yellow to purple to yellow again and the generation of bubbles attributed to H2O2 degradation. In contrast, the double-hydrophobised support catalysts showed extremely high alcohol selectivity with the A/K values exceeding 20 (entries 1–3). The alcohol yields derived from Fe(OTf)2/L-SBA-FC(n)TMS were 8 to 16 times higher than that on the catalyst with only TMS modification. The activity of the shortest fluoroalkyl chain-modified catalyst (n = 4; entry 3) was slightly lower than that of the longer chain-modified catalysts (n = 6 and 8; entries 2 and 1, respectively). The reason for such a difference might be attributed to the difference in the initial structure of the iron centres and their stability. In fact, the leaching of 47% of iron occurred in the catalyst with n = 4, whereas the leaching of iron could be negligible in the catalyst with n = 8. On Fe(OTf)2/L-SBA-FC(4)TMS, the immobilised iron complexes might have multinuclear iron cores as indicated by the exceeding loading amount of iron over that of L. Although the observed performance, as well as the loading ratio of Fe/L of the double-hydrophobised catalysts with n = 6 and 8, is close, 13% of iron leached out from Fe(OTf)2/L-SBA-FC(6)TMS. Such a difference of the stability of the immobilised iron complexes might lead to a wrapping effect of the longer fluoroalkyl chain of FC(8). Although partial iron leaching occurred during the first use of Fe(OTf)2/L-SBA-FC(6)TMS, the stability and reusability of this catalyst was confirmed; during the second use, the product yield was 88% of the first reaction and the selectivity to cyclohexanol decreased. However, in the third run, both TON and A/K were similar to the second run (Table S1†). The fact that the reaction was terminated by the filtration of Fe(OTf)2/L-SBA-FC(6)TMS (first application; Fig. S6†) supported that the reaction was catalysed by an immobilised complex. Only trace amounts of iron were detected in the obtained filtrate. These results might indicate that the iron complexes in Fe(OTf)2/L-SBA-FC(6)TMS have several types of different structures and stability, and that the unstable complexes leach iron during the first use but the stable iron complex species remain immobilised on the support. Noteworthily, the existence of O2 in the gas phase of the reaction vessel did not affect the catalytic activity. The catalytic reactions by Fe(OTf)2/L-SBA-FC(6)TMS under different atmospheres (argon, air, O2) exhibited similar profiles (Table S2 and Fig. S7†). It is known that the formation of long-lived alkyl radicals in the presence of oxygen significantly reduces the A/K ratio because of the one-to-one formation of alcohols and ketones by a Russell-type termination process due to the coupling of alkyl peroxyl radicals derived from the reaction of the corresponding alkyl radicals with oxygen.26 There are many reports on reduced alcohol selectivity in alkane oxidation using metal complexes as catalysts and peroxides as oxidants in a homogeneous liquid phase reaction in the presence of O2.27 In our system, therefore, the retention of the high alcohol selectivity even in the presence of O2 indicates that the major source of the oxygen atom in the products is H2O2 and autoxidation through the cyclohexyl radical diffused from the hydrophobic active site to the liquid phase would be a minor reaction pathway. The efficiency of H2O2 utilization, however, is quite low due to the non-productive decomposition of H2O2 proceeding. H2O2 (2 mmol) being in contact with Fe(OTf)2/L-SBA-FC(8)TMS (2 μmol of Fe) in 4 mL of MeCN without cyclohexane resulted in the slow generation of O2 bubbles at ambient temperature, and ca. 10% of H2O2 was decomposed in 3 h estimated from the volume (2.4 ± 0.1 mL) of the collected gas.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Fe/L | Products/TONa | A/K | Leaching of Fe/% | |
| A | K | |||||
| a TON = [Product]/[Fe]. b Homogeneous reaction. | ||||||
| 1 | Fe(OTf)2/L-SBA-FC(8)TMS | 0.92 | 29.5 | 1.2 | 24.6 | <0.1 |
| 2 | Fe(OTf)2/L-SBA-FC(6)TMS | 0.93 | 27.2 | 0.8 | 36.5 | 13.9 |
| 3 | Fe(OTf)2/L-SBA-FC(4)TMS | 2.50 | 15.1 | 0.6 | 28.6 | 46.8 |
| 4 | Fe(OTf)2/L-SBA-TMS | 1.75 | 1.8 | 4.5 | 0.4 | Not measured |
| 5 | Fe(OTf)2/L-SBA-FC(6) | 1.00 | 3.5 | 4.2 | 0.9 | 31.3 |
| 6 | FeCl3/L-SBA-FC(8)TMS | 1.54 | 40.6 | 1.8 | 23.3 | 0.6 |
| 7 | FeCl3/L-SBA-FC(6)TMS | 1.63 | 62.7 | 2.2 | 28.5 | <0.1 |
| 8 | FeCl3/L-SBA-TMS | 1.65 | 8.2 | 7.1 | 1.2 | 3.6 |
| 9 | FeCl3/L-SBA-FC(6) | 2.44 | 8.4 | 11.9 | 0.7 | 50.5 |
| 10b | [Fe(TPA)(MeCN)2](OTf)2 | 1.00 | 5.0 | 6.1 | 0.8 | — |

A similar double-hydrophobised effect on the improvement of the activity and the product selectivity was observed on the catalysts derived from FeCl3. Although the loading amounts of iron were higher than those of L, the activity of the double-hydrophobised catalyst derived from FeCl3 was higher than that of the corresponding catalyst derived from Fe(OTf)2 (as found in the comparison of entries 6 versus 1 and 7 versus 2 in Table 3). The higher activity of the FeCl3-derived catalysts might arise from the intrinsic high activity of the partially-formed dinuclear iron species compared to the mononuclear iron species. In fact, the active site of soluble methane monooxygenase (sMMO) is a dinuclear iron complex, and no mononuclear iron site which catalyzes methane hydroxylation has not been confirmed so far.2,5,6 In the dinuclear iron site, two-electron transfer from the two iron centres to the bound peroxide generates the active high-valent iron-oxide species efficiently. In the FeCl3-derived double-hydrophobised catalysts, the TONs of the products derived from the catalyst with a longer fluoroalkyl chain (n = 8; entry 6) were lower than those on the catalyst with FC(6) (i.e. entry 7), although the leaching of iron from both the catalysts were negligible. As described above, some of the immobilised iron complexes might be dinuclear species with single L bridging two iron centres. In the case of dinuclear iron complexes formed from FeCl3, the effect of steric hindrance due to the carbon chain of the fluoroalkyl group is reflected in differences in the accessibility of the substrate and oxidant. In contrast, in the mononuclear iron complexes obtained from Fe(OTf)2, the difference in carbon chain length led to differences in the stability of the complexes themselves. Such differences in the effects depending on the iron sources are thought to be based on the differences in the local structure, such as the orientation of the iron complexes in the hydrophobic pocket.
Applicability for the gaseous alkane oxygenation
The catalytic activity of the developed catalysts for the oxidation of gaseous alkanes was explored. At first, the activity of the mononuclear iron complex-immobilised catalysts derived from the double-hydrophobised supports having longer fluoroalkyl chains, Fe(OTf)2/L-SBA-FC(n)TMS where n = 6 or 8, was examined. To clarify the effect of the hydrophobic reaction field, sterically less bulky n-propane was used as a substrate and water was applied as the reaction solvent. The catalyst modified with FC(8) was more active than the shorter ones (n = 6), possibly due to the effect of substrate enrichment on the hydrophobic field surrounding the iron centre (Table 4). Both catalysts exhibited similar product selectivity. The major product was acetone, the oxidized form of the secondary C–H with smaller BDEC–H (98.1 kcal mol−1).28 Noteworthily, the terminal methyl group with larger BDEC–H (99.9 kcal mol−1)28 was also oxidized to yield 1-propanol and propionaldehyde. Such a low regioselectivity might arise from the strong radical characteristics of the generated active oxidant. In addition, the concentration of n-propane in the hydrophobic pocket formed by the fluoroalkyl modifier led to the random access of C–H to the oxidant formed on the iron centre. If alkyl hydroperoxides are involved in the oxidized products, it is known that the apparent yield of alcohols is increased by the action of a reducing agent (Na2SO3 or triphenylphosphine-3,3′,3′′-trisulfonic acid trisodium salt; TPPTS) before the analysis of the reaction solution by GC. In this work, therefore, we compared the yields of products with and without the reducing agent treatment. No increase in the yields of alcohols was observed even when the reducing agent treatment was carried out. It is considered that no alkyl hydroperoxides were formed in the presented propane oxidation. The total yields of the oxidized products were higher when not treated with the reducing agent. That might be attributed to the action of the remaining hydrogen peroxide during GC analysis.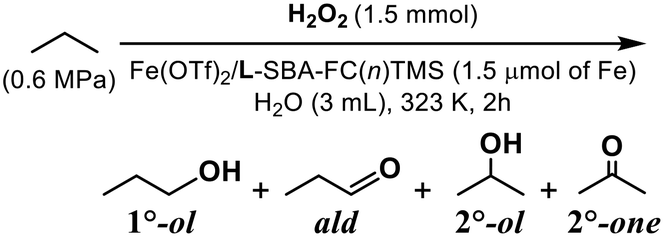
Finally, the methane oxidation activity of FeCl3/L-SBA-FC(6)TMS, which showed the highest catalytic activity for the cyclohexane oxidation, was examined. CD3CN was applied as the reaction solvent because (1) the oxidation of the solvent would be avoided as much as possible, (2) aqueous hydrogen peroxide was used for the admixture, and (3) the solubility of methane in acetonitrile was higher than that in water. The 1H NMR spectrum of the reaction solution revealed that the products were CH3OOH, CH3OH and HCOOH, with the yields (TON) of 4.7, 8.3 and 27.6, respectively (Fig. S8†). As described above, no alkyl hydroperoxides were formed in the oxidation of propane. The formation of methyl hydroperoxide might be due to the difference in BDEC–H between methane and propane. Hydrogen atom abstraction (HAT) from methane by the oxidant formed on the iron complex was decelerated due to higher BDEC–H (105.0 kcal mol−1)28 of methane, and that resulted in the relative acceleration of the decomposition of H2O2. As a result, the concentration of hydroperoxyl radicals (˙OOH) increased,29 and coupling between ˙OOH and ˙CH3 occurred. The production of methyl hydroperoxide through a Fenton-like mechanism is reported on the methane oxidation by an Fe–zeolite catalyst with H2O2.30,31 Also, the silica-immobilized Fe–TPA complexes yielded methyl hydroperoxide.32 On the other hand, HAT from propane, which has a lower BDEC–H than methane, would proceed preferentially, and ˙OOH would not reach the concentration for the required generation of alkyl hydroperoxides.
Experimental
Materials and methods
Nitrogen sorption/desorption studies were performed at liquid nitrogen temperature (77 K) using a Microtrac BELSORP MINI X. Before the adsorption experiments, the samples were outgassed under reduced pressure for 3 h at 333 K. Inductively coupled plasma mass spectrometry (ICP-MS) was performed on an Agilent Technologies 7700 Series ICP-MS. Thermogravimetric analysis was performed on a Rigaku Thermo plus EVO. Atomic absorption analysis was performed on a Shimadzu AA-6200. NMR spectra were recorded on a JEOL ECA-500 spectrometer (1H, 500.0 MHz). Gas chromatography (GC) analyses were conducted on Shimadzu GC-2010 and GC-2025 instruments with a flame ionization detector equipped with RESTEK Rtx-5 (on GC-2010) and Rtx-WAX (on GC-2025) capillary columns (Restek, length = 30 m, i.d. = 0.25 mm, thickness = 0.25 μm). GC-MS analyses were conducted on a Shimadzu GS-MS QP 2010 Plus instrument equipped with a GL Science InertCapWAX capillary column (30 m × 0.25 mm). Oxidation reactions of propane were conducted in a high pressure reactor equipped with a high-performance liquid chromatograph pump to introduce liquid samples into a pressurized reaction vessel at the designed time (Taiyo-System, Co., Ltd.). Substrate gas was supplied to the reactor with a continuous flow, and the pressure of the reactor was controlled with a vent valve. The reaction temperature was monitored with a thermometer set in the reactor and controlled with a feedback circuit.The commercially-available chemicals were used without further purification.
The double-hydrophobised support L-SBA-FC(n)TMS was prepared by the treatment of L-SBA-FC(n) with 1,1,1,3,3,3-hexamethyldisilazane, (Me3Si)2NH, with a similar procedure for the preparation of L-SBA-FC(n). The applied amount of (Me3Si)2NH was 0.38 mmol per 100 mg of L-SBA-FC(n).
Immobilised iron was quantified by atomic absorption spectrometry. 5 mg of catalyst was dissolved in 1 mL of 10% aq. potassium hydroxide by heating for 5 min. The resulting solution was then acidified with 2 mL of conc. aq. nitric acid and diluted with deionized H2O to a total volume of 10 mL. The solution was passed through a syringe filter prior to its analysis.
Conclusions
The reaction of a TPA-analog ligand anchored SBA-15 type support with (EtO)3SiC2H4CnF2n+1 and (Me3Si)2NH yielded the corresponding fluoroalkyl (= FC(n)) and TMS-modified supports L-SBA-FC(n)TMS with n = 4, 6, and 8. The ligand-anchored supports reacted with Fe(OTf)2 or FeCl3 to yield the corresponding iron complex-immobilised catalysts; however, the structure, stability, and catalytic activity of the formed iron complex depended on the anions of the used iron compounds and the local structure of the surface of the supports. Examination of the cyclohexane oxidation with H2O2 revealed that the supports hydrophobised by longer fluoroalkyl chains and TMS were effective in improving the activity and alcohol selectivity of the iron complex immobilised catalysts. In a series of catalysts derived from Fe(OTf)2, the longest fluoroalkyl chain (= FC(8)) modified catalyst was the most reactive and stable. In the FeCl3-derived double-hydrophobised catalysts, the FC(6) modified one exhibited higher activity compared to the FC(8) derivative. Such differences in the effects of the length of the fluoroalkyl chain depending on the iron sources are thought to be the differences in the local structure, such as the orientation of the iron complexes in the hydrophobic pocket. The comparison of the propane oxidation catalysis of the mononuclear iron complex-immobilised catalysts Fe(OTf)2/L-SBA-FC(n)TMS (where n = 6 or 8) demonstrated the substrate condensation effect of the hydrophobic pocket formed by the longer fluoroalkyl pillars. Formation of not only 2-propanol and acetone but also 1-propanol and propionaldehyde suggested the synergy of the strong radical characteristics of the generated active oxidant and the substrate concentration effect. The most active catalyst for the cyclohexane oxidation, FeCl3/L-SBA-FC(6)TMS, catalysed methane oxidation with H2O2 to give methanol, formic acid, and methyl hydroperoxide. No alkyl hydroperoxides were formed in the oxidation of propane. Therefore, the formation of methyl hydroperoxide might be due to the difference in BDEC–H between methane and propane.The presented results demonstrate that the hydrophobic reaction field is efficient in improving oxidation catalysis. To construct a high-performance methane oxidation catalyst, however, an intrinsic activity of the oxidant generated on the metal centre must be higher and the sophisticated design of the hydrophobic pocket surrounding the active site is required.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by CREST (grant no. JPMJCR16P1) from Japan Science and Technology Agency (JST) and Kanagawa University (ordinary budget: 411).Notes and references
- V. C.-C. Wang, S. Maji, P. P.-Y. Chen, H. K. Lee, S. S.-F. Yu and S. I. Chan, Chem. Rev., 2017, 117, 8574–8621 CrossRef CAS PubMed.
- C. E. Tinberg and S. J. Lippard, Acc. Chem. Res., 2011, 44, 280–288 CrossRef CAS PubMed.
- M. O. Ross and A. C. Rosenzweig, J. Biol. Inorg. Chem., 2017, 22, 307–319 CrossRef CAS.
- J. K. Stanfield and O. Shoji, Chem. Lett., 2021, 50, 2025–2031 CrossRef CAS.
- D. A. Whittington, A. C. Rosenzweig, C. A. Frederick and S. J. Lippard, Biochemistry, 2001, 40, 3476–3482 CrossRef CAS PubMed.
- R. Banerjee and J. D. Lipscomb, Acc. Chem. Res., 2021, 54, 2185–2195 CrossRef CAS.
- K.-Y. Ng, L.-C. Tu, Y.-S. Wang, S. I. Chan and S. S.-F. Yu, ChemBioChem, 2008, 9, 1116–1123 CrossRef CAS.
- H. Fujisaki, T. Ishizuka, H. Kotani, Y. Shiota, K. Yoshizawa and T. Kojima, Nature, 2023, 616, 476–481 CrossRef CAS PubMed.
- Y. Yamada, K. Morita, T. Sugiura, Y. Toyoda, N. Mihara, M. Nagasaka, H. Takaya, K. Tanaka, T. Koitaya, N. Nakatani, H. Ariga-Miwa, S. Takakusagi, Y. Hitomi, T. Kudo, Y. Tsuji, K. Yoshizawa and K. Tanaka, JACS Au, 2023, 3, 823–833 CrossRef CAS PubMed.
- Y. Yamada, J. Kura, Y. Toyoda and K. Tanaka, Dalton Trans., 2021, 50, 6718–6724 RSC.
- M. Mukherjee and A. Dey, Chem. Commun., 2020, 56, 11593–11596 RSC.
- Y. Aratani, Y. Yamada and S. Fukuzumi, Chem. Commun., 2015, 51, 4662–4665 RSC.
- M. Yamada, K. D. Karlin and S. Fukuzumi, Chem. Sci., 2016, 7, 2856–2863 RSC.
- I. A. S. Matias, A. P. C. Ribeiro and L. M. D. R. S. Martins, Molecules, 2020, 25, 5642 CrossRef CAS PubMed.
- H. Fujisaki, M. Okamura, S. Hikichi and T. Kojima, Chem. Commun., 2023, 59, 3265–3268 RSC.
- J. Nakazawa, T. Hori, T. D. P. Stack and S. Hikichi, Chem. – Asian J., 2013, 8, 1191–1199 CrossRef CAS.
- J. Nakazawa, A. Yata, T. Hori, T. D. P. Stack, Y. Naruta and S. Hikichi, Chem. Lett., 2013, 42, 1197–1199 CrossRef CAS.
- J. Nakazawa, Y. Doi and S. Hikichi, Mol. Catal., 2017, 443, 14–24 CrossRef CAS.
- T. Tsuruta, T. Yamazaki, K. Watanabe, Y. Chiba, A. Yoshida, S. Naito, J. Nakazawa and S. Hikichi, Chem. Lett., 2015, 44, 144–146 CrossRef CAS.
- A. Nakamizu, T. Kasai, J. Nakazawa and S. Hikichi, ACS Omega, 2017, 2, 1025–1030 CrossRef CAS.
- J. Nakazawa, B. J. Smith and T. D. P. Stack, J. Am. Chem. Soc., 2012, 134, 2750–2759 CrossRef CAS.
- R. Y. N. Ho, G. Roelfes, B. L. Feringa and L. Que, Jr., J. Am. Chem. Soc., 1999, 121, 264–265 CrossRef CAS.
- K. Chen and L. Que, Jr., J. Am. Chem. Soc., 2001, 123, 6327–6337 CrossRef CAS PubMed.
- T. Hasegawa, Chem. Rec., 2017, 17, 903–917 CrossRef CAS PubMed.
- T. N. Sorrell, F. C. Pigge and P. S. White, Inorg. Chim. Acta, 1993, 210, 87–90 CrossRef CAS.
- G. A. Russell, J. Am. Chem. Soc., 1957, 79, 3871–3877 CrossRef CAS.
- (a) S. Rana, J. P. Biswas, A. Sen, M. Clémancey, G. Blondin, J. M. Latour, G. Rajaraman and D. Maiti, Chem. Sci., 2018, 9, 7843–7858 RSC; (b) Y. Morimoto, S. Hanada, R. Kamada, A. Fukatsu, H. Sugimoto and S. Itoh, Inorg. Chem., 2021, 60, 7641–7649 CrossRef CAS PubMed; (c) J. Cheng, Y. Shiota, M. Yamasaki, K. Izukawa, Y. Tachi, K. Yoshizawa and H. Shimakoshi, Inorg. Chem., 2022, 61, 9710–9724 CrossRef CAS PubMed; (d) Y. Sato, M. Okamura and S. Hikichi, Eur. J. Inorg. Chem., 2023, 26, e202200728 CrossRef CAS.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energy, CRC Press, Boca Raton, London, New York, 2007 Search PubMed.
- B. Ensing, F. Buda and E. J. Baerends, J. Phys. Chem. A, 2003, 107, 5722–5731 CrossRef CAS.
- C. Hammond, N. Dimitratos, J. A. Lopez-Sanchez, R. L. Jenkins, G. Whiting, S. A. Kondrat, M. H. Ab Rahim, M. M. Forde, A. Thetford, H. Hagen, E. E. Stangland, J. M. Moulijn, S. H. Taylor, D. J. Willock and G. J. Hutchings, ACS Catal., 2013, 3, 1835–1844 CrossRef CAS.
- C. Hammond, I. Hermans and N. Dimitratos, ChemCatChem, 2015, 7, 434–440 CrossRef CAS.
- S. A. Ikbal, C. Colomban, D. Zhang, M. Delecluse, T. Brotin, V. Dufaud, J.-P. Dutasta, A. B. Sorokin and A. Martinez, Inorg. Chem., 2019, 58, 7220–7228 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy00698k |
| ‡ Present affiliation: Department of Applied Chemistry, Faculty of Chemistry and Biochemistry, Kanagawa University, Japan. |
| This journal is © The Royal Society of Chemistry 2023 |