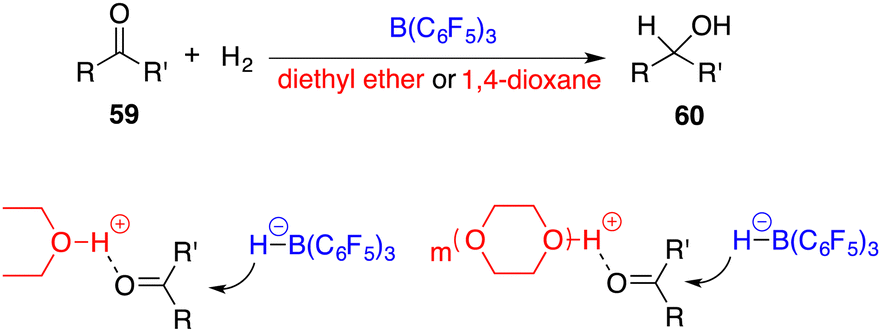DOI:
10.1039/D3CS00701D
(Review Article)
Chem. Soc. Rev., 2023,
52, 8580-8598
Asymmetric catalysis with FLPs
Received
28th August 2023
First published on 14th November 2023
Abstract
Chiral catalysts play a crucial role in the realm of asymmetric catalysis. Since their breakthrough discovery in 2006, chiral frustrated Lewis pairs (FLPs) have risen as a novel catalyst category for a broad range of metal-free asymmetric reactions. This review provides an overview of the remarkable progress made in this field over the past 15 years. The design and synthesis of chiral FLPs and their applications in hydrogenation, hydrosilylation, transfer hydrogenation, and various other reactions are summarized and highlighted.

Xiangqing Feng
| Xiangqing Feng received her BSc from Xinyang Normal University in 2006, her MSc in 2009 from Nankai University, and her PhD from the Institute of Chemistry, Chinese Academy of Sciences, under the supervision of Professor Haifeng Du in 2012. She joined the Institute of Chemistry, Chinese Academy of Sciences, as an Assistant Professor in 2012. |

Wei Meng
| Wei Meng received his BSc from Shanxi University in 2006 and PhD from Tianjin University in 2012 under the supervision of Prof. Jun-An Ma. He joined Peking University as a postdoctoral fellow in 2012 under the supervision of Prof. Zhi-Xiang Yu. He joined the Institute of Chemistry, Chinese Academy of Sciences, as an Assistant Professor in 2014 and was promoted to Associate Professor in 2017. |

Haifeng Du
| Haifeng Du received his BSc in 1998 from Nankai University, MSc from Nankai University and the Shanghai Institute of Organic Chemistry under the supervision of Professors Jiben Meng and Kuiling Ding in 2001, and PhD from the Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, under the supervision of Professor Kuiling Ding in 2004. He joined Colorado State University as a postdoctoral fellow with Professor Yian Shi in 2004. In October of 2008, he joined the Institute of Chemistry, Chinese Academy of Sciences, as a Professor. His interests include the development of novel catalysts for asymmetric catalysis and selective synthesis. |
Introduction
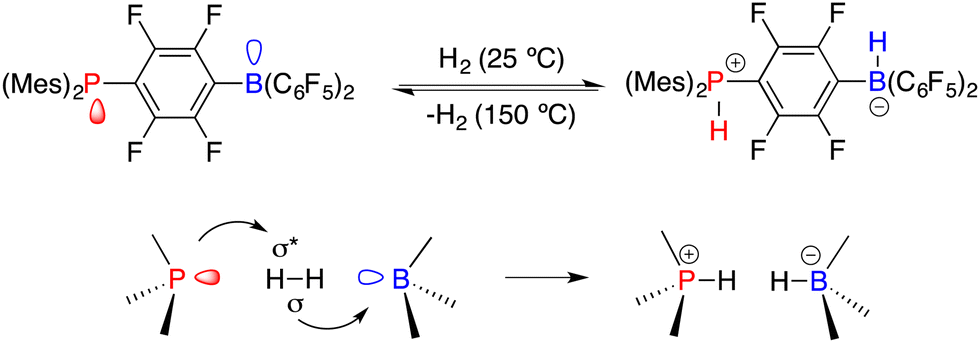
Asymmetric catalysis represents the most efficient and powerful pathway for the synthesis of optically active compounds, which are in growing demand for chemical, pharmaceutical, agricultural, and material science applications. Generally, chiral catalysts play a key role in asymmetric catalysis, and the pursuit of innovative catalysts stands as one of the most important subjects of this field.1 In 2006, Stephan and co-workers found that sterically encumbered Lewis acids and bases could form unquenched Lewis pairs deviating from the conventional Lewis acid–base adducts, which were later vividly named frustrated Lewis pairs (FLPs) (Scheme 1).2 An unusual property of FLPs is that they could activate dihydrogen reversibly, which belongs to the inherent advantage of transition metals for a very long period. Shortly thereafter, a pioneering example of the FLP-catalyzed hydrogenation of imines was disclosed, which paved a metal-free way for catalytic hydrogenation.3 A significant advancement in the rapid expansion of FLP catalysts and substrate scope for the catalytic hydrogenation has been achieved over the past 16 years.4
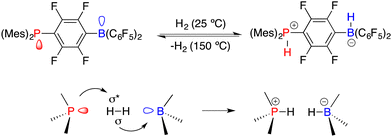 |
| | Scheme 1 Reversible activation of H2 with FLPs. | |
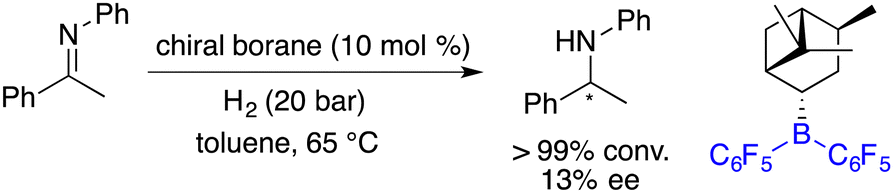
Another pioneering work was reported by Klankermayer and co-workers in 2008. An asymmetric hydrogenation of an imine was realized by using a chiral borane generated from (+)-α-pinene and HB(C6F5)2, yielding the desired amine product with 13% ee (Scheme 2).5 Despite its modest enantioselectivity, it marks the commencement of chiral FLPs stepping into asymmetric catalysis as a novel class of catalysts.
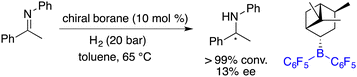 |
| | Scheme 2 Initial asymmetric hydrogenation catalyzed by a chiral borane. | |
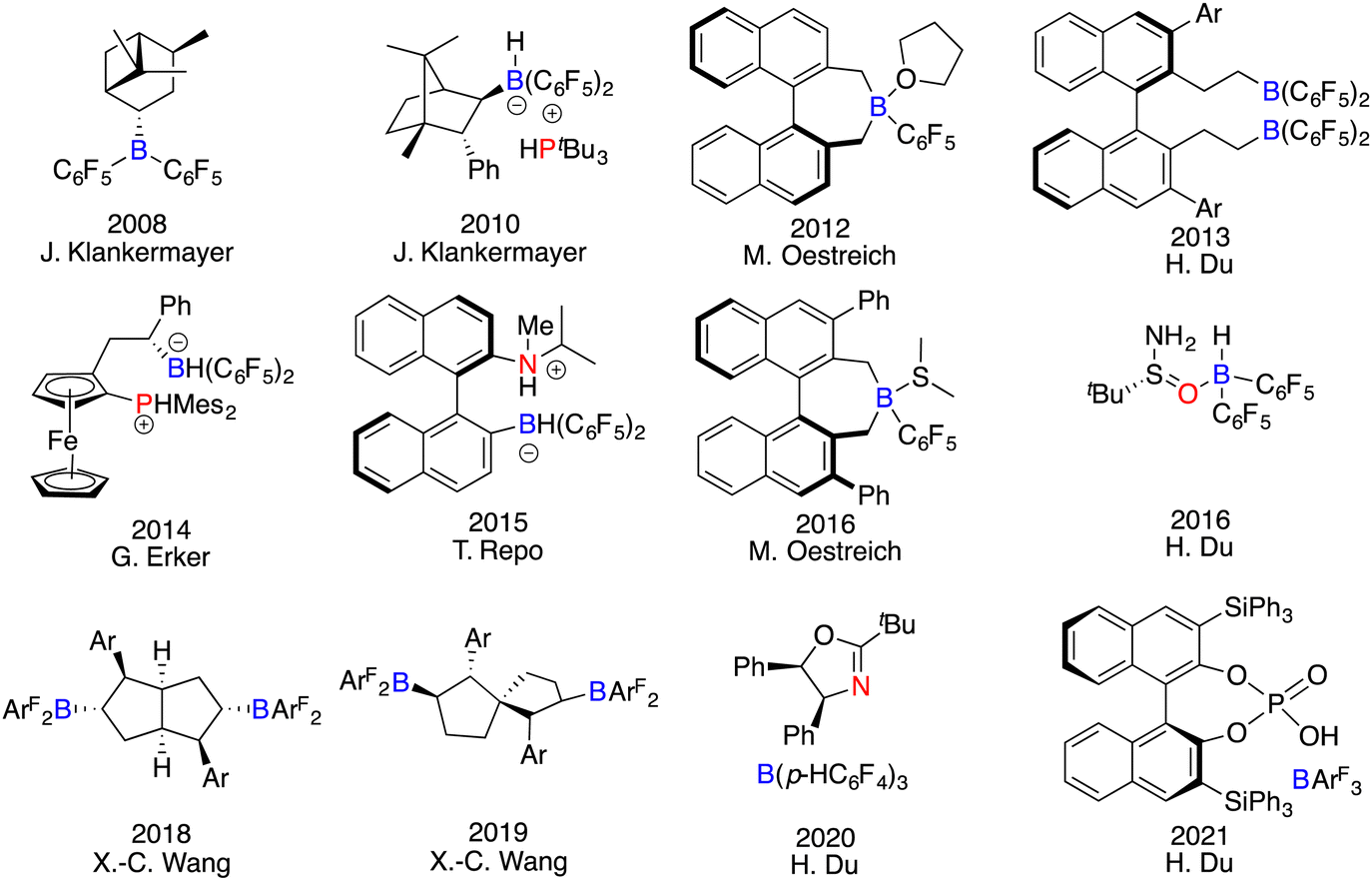
As a dual-component class of catalysts, the apparent simplicity and versatility in the design of FLPs may suggest that either the Lewis acid, the Lewis base, or both could serve as the chiral element. However, the actual process of developing chiral FLPs is a formidable undertaking, dictated by their intrinsic characteristics. In the realm of Lewis acid components, chiral boranes deviate obviously from metallic Lewis acids, which could rely on numerous chiral ligands to form structurally diverse complexes. In contrast, chiral frameworks must be integrated into the boron centre through covalent bonds. The absence of secondary interactions between the substrate and catalyst makes it difficult to find an effective chiral framework to govern the enantioselectivity. Moreover, their strong acidity, bulky structures, and sensitivity to moisture render the synthesis and purification of chiral boranes a formidable task. Conversely, the landscape of Lewis base components unveils a different facet. Chiral Lewis bases manifest robust stability and convenient accessibility indeed. Nevertheless, asymmetric induction with chiral Lewis bases remains a conundrum, primary due to the inherent mechanism of hydrogenation and the unavoidable competition of the substrates as achiral Lewis bases. Despite these obstacles, an assorted array of chiral FLPs have been successfully developed and applied in asymmetric reactions (Fig. 1).6
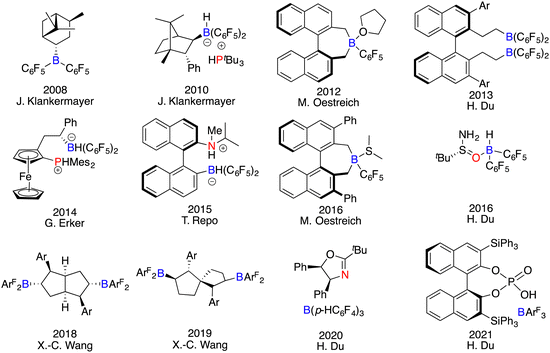 |
| | Fig. 1 Representative chiral catalysts for asymmetric catalysis. | |
The development of chiral FLP catalysts can be roughly delineated into three stages: chiral Lewis acid-driven FLPs (commencing from 2008), chiral Lewis base-driven FLPs (initiated from 2016), and achiral FLPs in concert with other types of chiral catalysts (commencing from 2021). The synthesis method of chiral boranes can be categorized into three protocols. The first one is the hydroboration of chiral alkenes with Piers’ borane HB(C6F5)2. The second one is the substitution reaction of (C6F5)nBCl3−n with chiral organometallic reagents. The last one is the in situ hydroboration of chiral alkenes or alkynes with Piers’ borane obviating the need for purification, which stands as a more efficient and convenient method. The realm of asymmetric reactions catalyzed by FLPs has also been transcended from hydrogenation to hydrosilylation, transfer hydrogenation, Mannich-type reactions, and cyclization reactions. Moreover, the advent of heterogeneous chiral FLP catalyst systems further augments the scope of this catalytic modality.
In this review, we provide an overview of the significant progress accomplished over the past 15 years. The architecture of this review is according to the reaction categories, including asymmetric hydrogenation, asymmetric hydrosilylation, asymmetric transfer hydrogenation, and other asymmetric reactions. Examples in each reaction category were summarized according to the substrate type. We highlight both the merits and limitations of FLP-catalysis and wish to provide some useful information for future development.
1. Asymmetric hydrogenation
The asymmetric hydrogenation of unsaturated compounds has achieved great success and become one of the most reliable tools for the synthesis of optically active compounds. This field has been traditionally predominated by chiral transition-metal catalysts. Since the seminal discovery by Stephan and co-workers, chiral FLPs have risen to prominence as a distinct category of metal-free catalysts for the asymmetric hydrogenation. Notably, they exhibit some distinctive merits. The substrate scope was also expanded from the initial imines to encompass alkenes, heteroarenes, and ketones.
1.1 Asymmetric hydrogenation of imines
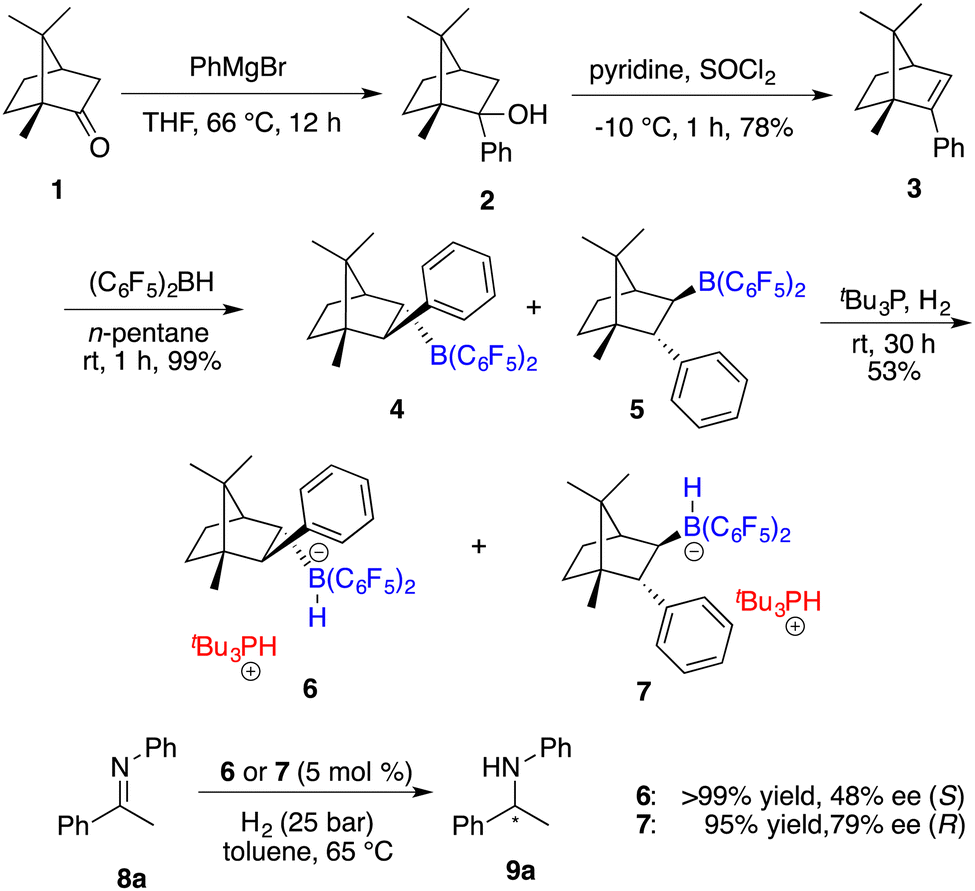
Having reported the initial example of asymmetric hydrogenation with a chiral borane, Klankermayer and co-workers devoted their efforts toward the design and synthesis of more effective chiral FLPs.7 As illustrated in Scheme 3, the reaction of (1R)-(+)-camphor (1) with phenylmagnesium bromide followed by dehydration yielded chiral alkene 3.
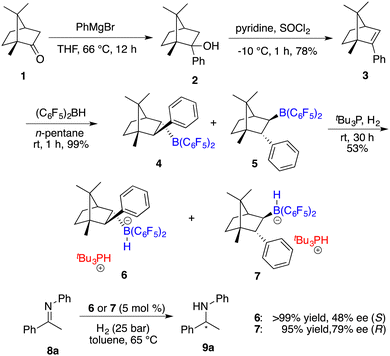 |
| | Scheme 3 Synthesis of chiral boranes and their application in asymmetric hydrogenation. | |
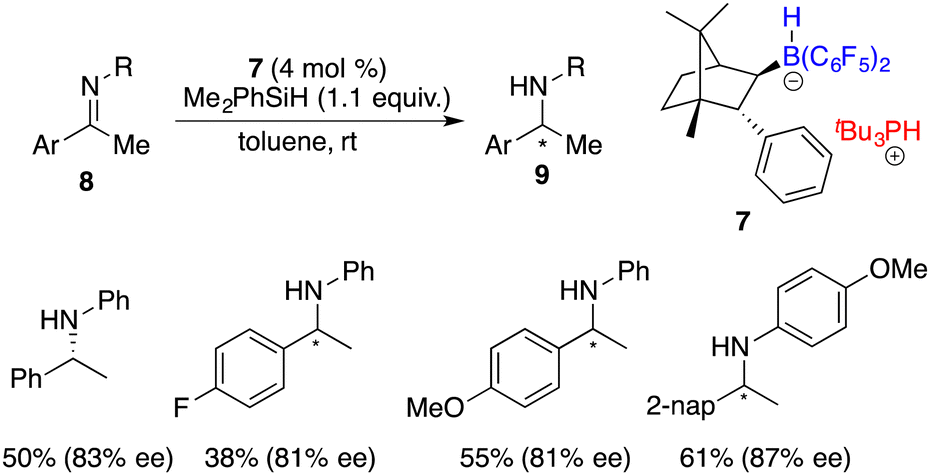
Subsequent hydroboration of 3 with HB(C6F5)2 produced a pair of chiral borane diastereomers 4 and 5 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 ratio, which brought about difficulty in terms of their separation at this point. Fortunately, a resolution was eventually attained through a partitioning of salts 6 and 7, generated by the splitting of H2 with tri-tert-butylphosphine and boranes. The X-ray analysis of salts 6 and 7 unveiled multiple C–H⋯F interactions between the phosphonium and hydridoborate moieties. Salt 7 proved to be a more effective catalyst, delivering the desired amine products 9 in high yields with 74–83% ee, thus representing the first highly enantioselective example achieved in the field of FLP catalysis. In succession, the same research group developed a stable intramolecular phosphine–borane FLP salt 10, which could be recycled under ambient conditions and reused in the asymmetric hydrogenation of imines 4 times without loss of any activity and enantioselectivity (Scheme 4).8
:4 ratio, which brought about difficulty in terms of their separation at this point. Fortunately, a resolution was eventually attained through a partitioning of salts 6 and 7, generated by the splitting of H2 with tri-tert-butylphosphine and boranes. The X-ray analysis of salts 6 and 7 unveiled multiple C–H⋯F interactions between the phosphonium and hydridoborate moieties. Salt 7 proved to be a more effective catalyst, delivering the desired amine products 9 in high yields with 74–83% ee, thus representing the first highly enantioselective example achieved in the field of FLP catalysis. In succession, the same research group developed a stable intramolecular phosphine–borane FLP salt 10, which could be recycled under ambient conditions and reused in the asymmetric hydrogenation of imines 4 times without loss of any activity and enantioselectivity (Scheme 4).8
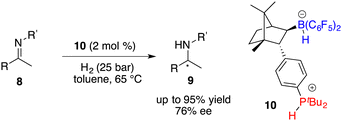 |
| | Scheme 4 Asymmetric hydrogenation with a recyclable chiral FLP catalyst. | |
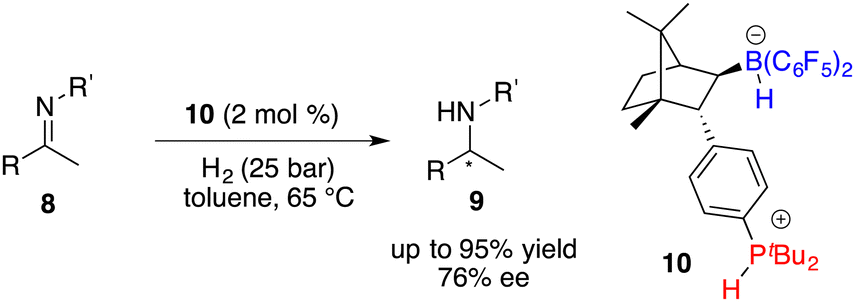
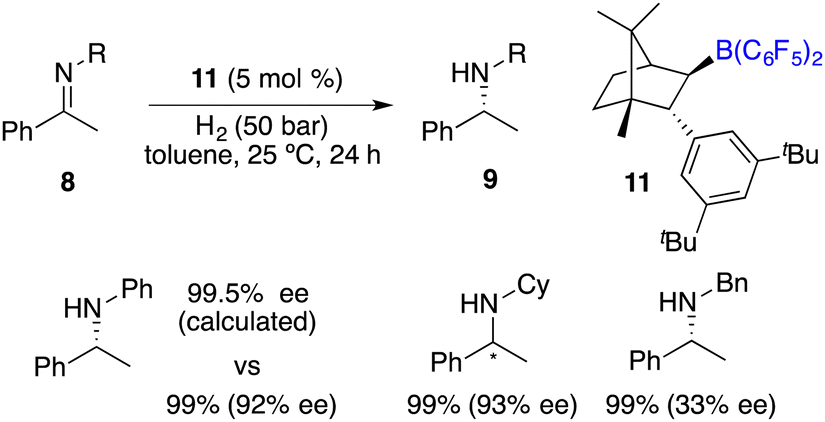
In the year 2020, Repo, Pápai and co-workers made a modification of this category of chiral boranes using density functional theory (DFT) calculations. Employing chiral borane 11, the asymmetric hydrogenation of imines 8 could produce amine products 9 in high yields with up to 95% ee (Scheme 5).9 Impressively, the predicted DFT analysis was in close agreement with the experimental results. Specific aryl–aryl and aryl–alkyl catalyst–substrate interactions are found to be essential for stereoinduction.
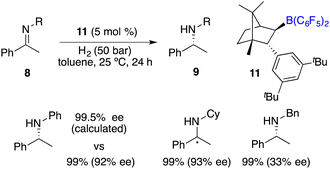 |
| | Scheme 5 Asymmetric hydrogenation of imines with chiral borane 11. | |
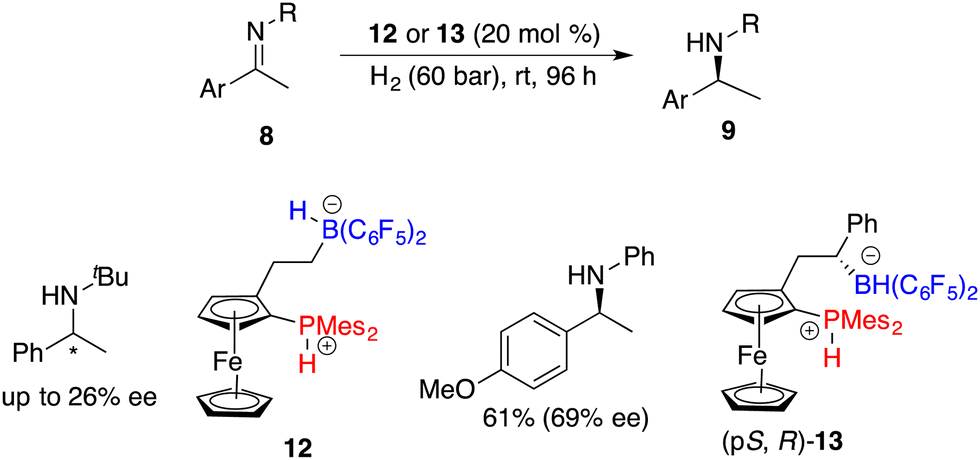
The Erker research group developed a planar-chiral ferrocene-based phosphonium/hydridoborate 12 through the hydroboration of terminal alkenes with HB(C6F5)2 for the asymmetric hydrogenation of imines, but only 26% ee was achieved (Scheme 6).10 The introduction of a phenyl group into the alkene engendered the formation of chiral borane diastereomers during the hydroboration process.11 After being treated with H2, phosphonium/hydridoborate 13 could be easily obtained as the major isomer. Notably, the presence of an adjacent chiral stereogenic center to the boron of 13 is helpful to improve the enantioselectivity.
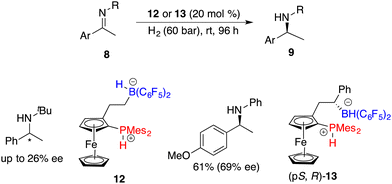 |
| | Scheme 6 Planar-chiral FLP-catalyzed asymmetric hydrogenation of imines. | |
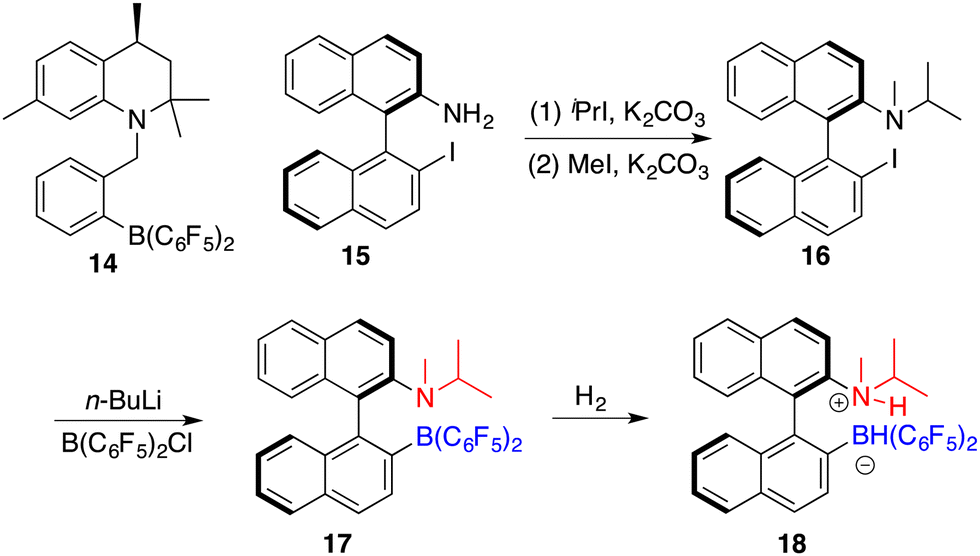
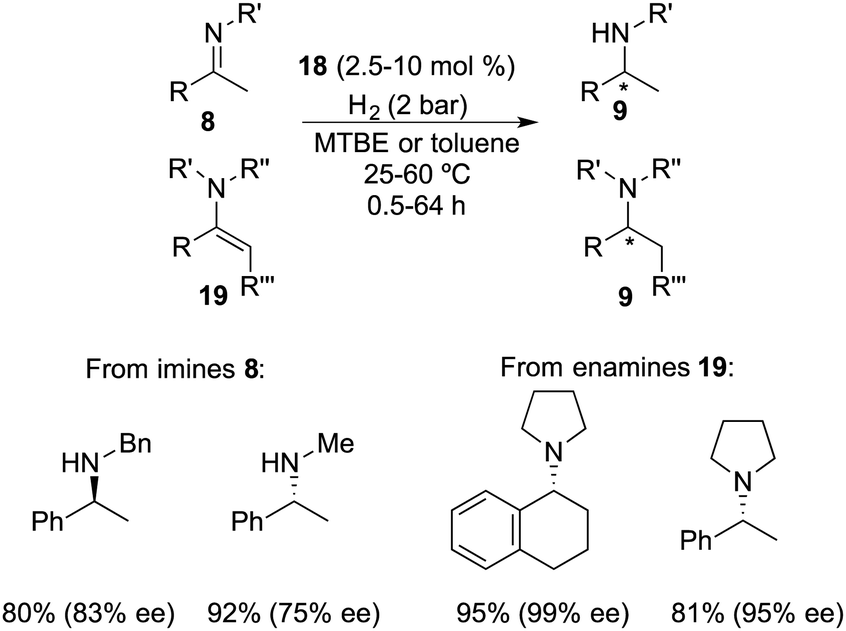
Repo and co-workers developed a chiral ansa-ammonium borate catalyst 14 for the metal-free hydrogenation of imines 8, yielding the corresponding amines 9 with up to 35% ee (Scheme 7).12 In 2015, the same research group disclosed the synthesis of a chiral binaphthyl-linked aminoborane and its application in the asymmetric hydrogenation of imines and enamines.13 The stepwise introduction of methyl and isopropyl groups into the nitrogen atom of 15 followed by the substitution of the iodine atom with ClB(C6F5)2 delivered intramolecular aminoborane 17. Subsequent treatment of 17 with H2 afforded ammonium borohydride salt 18, which is a highly effective catalyst for the enantioselective hydrogenation of unhindered imines 8 and enamines 19, delivering the corresponding amines 9 in moderate to good yields with up to 99% ee (Scheme 8). DFT calculations indicate that both repulsive steric and stabilizing intramolecular non-covalent forces are very important in the hydride transfer step of the catalytic cycle.
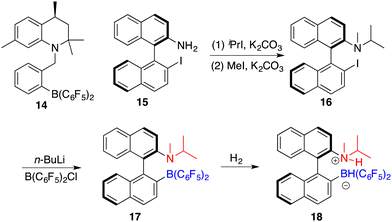 |
| | Scheme 7 Synthesis of chiral ansa-ammonium borate catalysts. | |
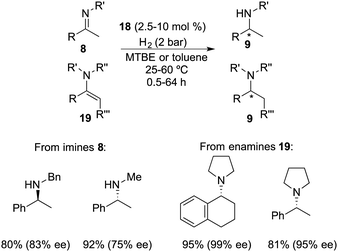 |
| | Scheme 8 Asymmetric hydrogenation of imines and enamines with ansa-ammonium borate catalysts. | |
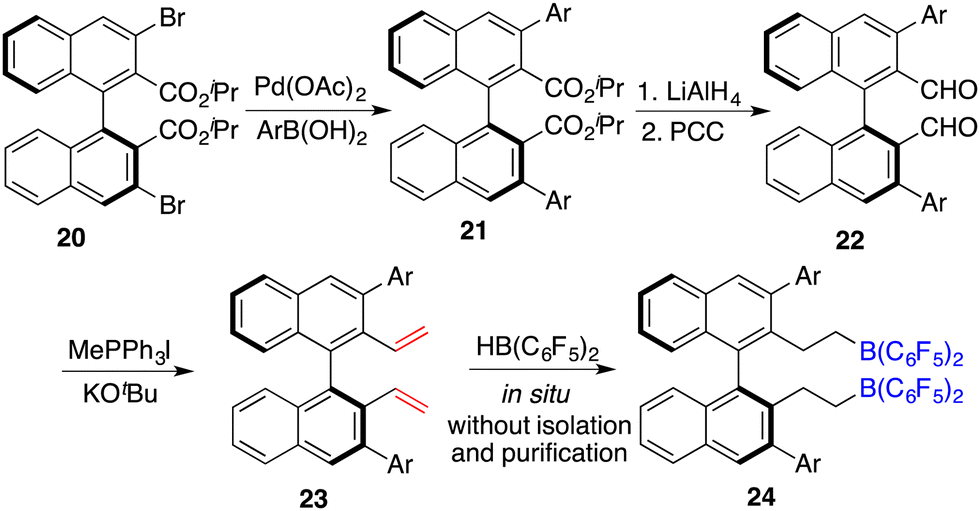
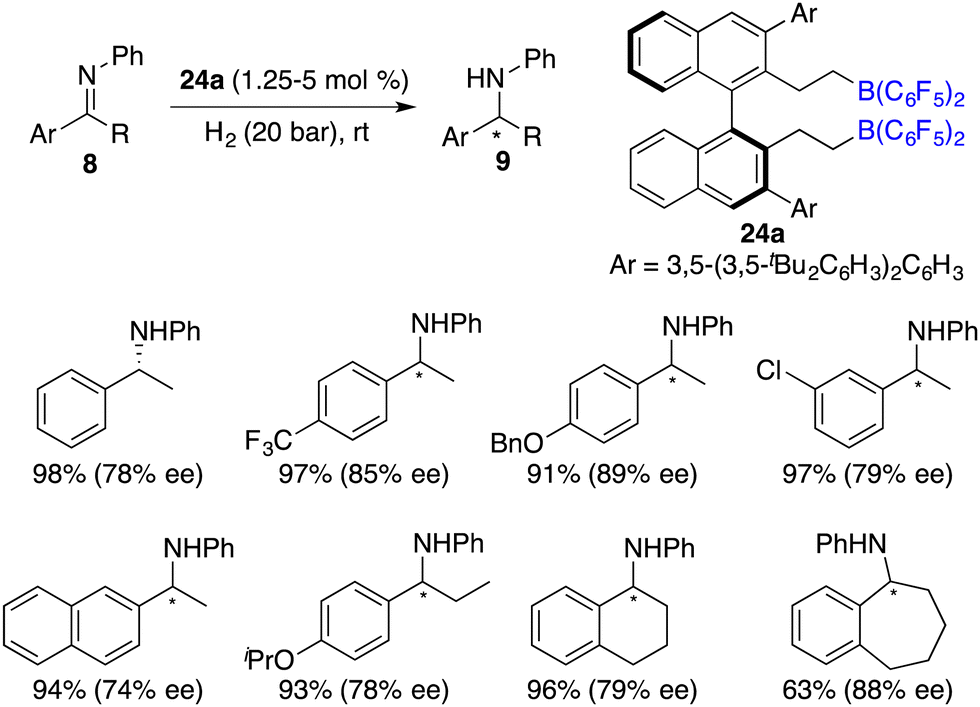
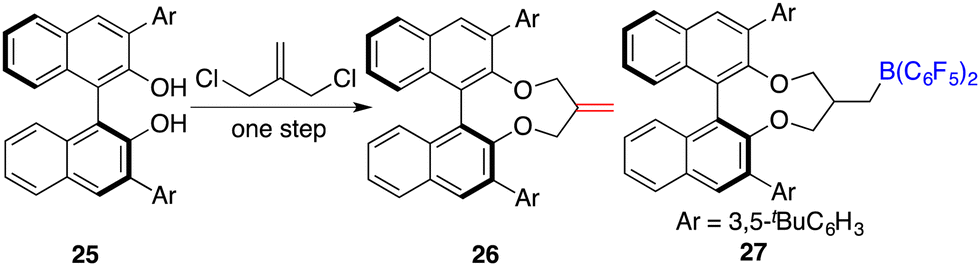
To circumvent the tedious isolation and purification of sensitive chiral boranes, Du and co-workers introduced a pioneering strategy. This novel approach pivoted on the in situ hydroboration of chiral dienes with HB(C6F5)2.14 The incorporation of various substituents into binaphthyl-based dicarboxylate 20via a Suzuki coupling reaction followed by reduction with LiAlH4 and oxidation with PCC could afford chiral dialdehydes 22. Via a Wittig reaction, chiral dienes 23 can be easily obtained (Scheme 9).15 Employing terminal alkenes in lieu of internal alkenes obviates the generation of superfluous diastereomers. The hydroboration of chiral dienes 23in situ engendered the formation of chiral boranes 24. Due to the flexible structure of chiral boranes 24, bulky substituents on the binaphthyl-framework were required to attain commendable enantioselectivities for the asymmetric hydrogenation of imines 8. Chiral borane 24a is a highly effective catalyst, giving a variety of optically active secondary amines 9 in high yields with up to 89% ee (Scheme 10).16 To shorten the synthetic routes towards chiral alkenes, a spectrum of binaphthyl-based chiral mono-alkenes 26 were prepared from readily available diols 25. Chiral borane 27 derived from the corresponding chiral alkene is also an effective catalyst for the asymmetric hydrogenation of imines 8, and the value of ee could reach up to 89% (Scheme 11).17
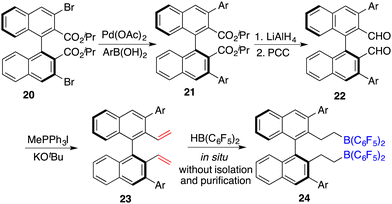 |
| | Scheme 9 The synthesis of chiral dienes and the development of chiral boranes. | |
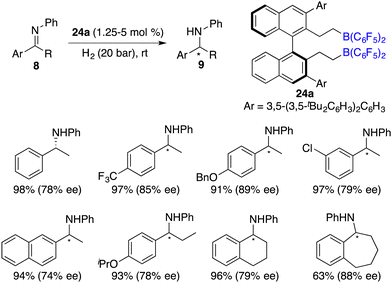 |
| | Scheme 10 Asymmetric hydrogenation of imines with chiral bisboranes. | |
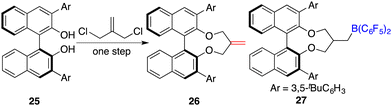 |
| | Scheme 11 Synthesis of chiral mono-alkene-derived boranes. | |
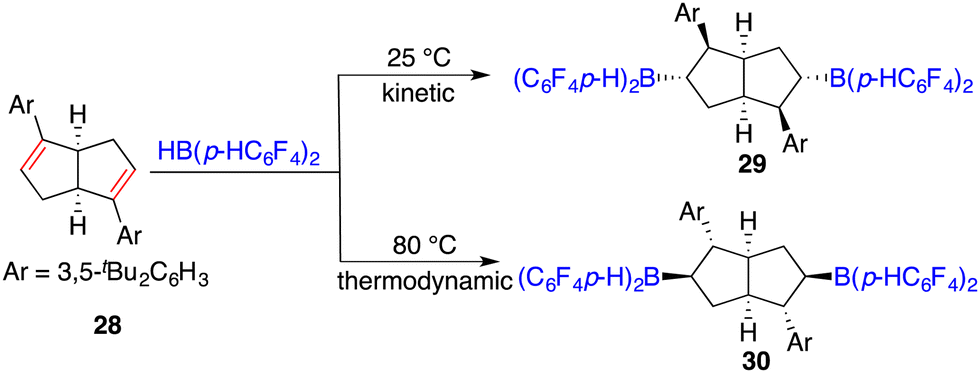
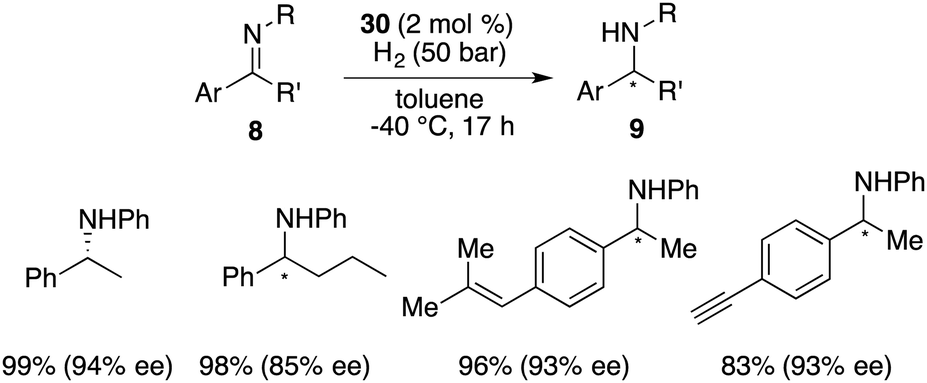
The hydroboration of internal alkenes with HB(C6F5)2 conventionally results in a mixture of diastereomers, posing challenges for subsequent purification endeavours. However, the presence of an adjacent stereogenic center to the boron atom holds promise for facilitating asymmetric induction. In 2018, Wang and co-workers developed a new class of chiral C2-symmetric bicyclic bisborane catalysts, originating from the hydroboration of internal dienes 28 with HB(C6F5)2 or HB(p-C6F4H)2 (Scheme 12).18 By judiciously varying reaction temperatures, the kinetic and thermodynamic products, 29 and 30, respectively, could be selectively accessed. Bisborane catalyst 30 exhibited remarkable activity and selectivity for the asymmetric hydrogenation of imines 8, delivering a wide range of optically active amines 9 in high yields with up to 95% ee (Scheme 13). Significantly, both C–C double bonds and triple bonds could be maintained under hydrogenation conditions, which is an advantage of the FLP catalysis.
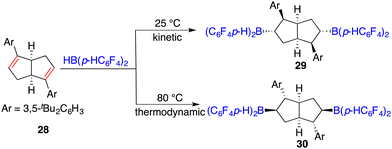 |
| | Scheme 12 Synthesis of chiral C2-symmetric bicyclic bisboranes derived from internal dienes. | |
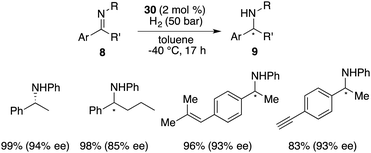 |
| | Scheme 13 Asymmetric hydrogenation of imines with chiral C2-symmetric bicyclic bisboranes. | |
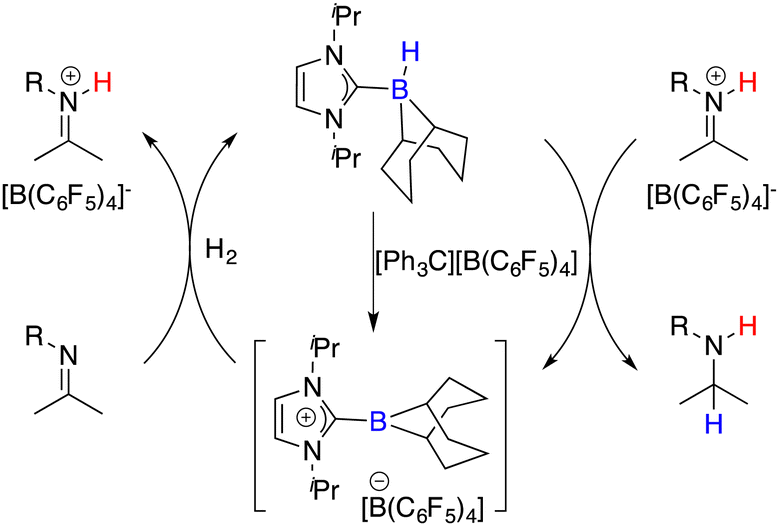
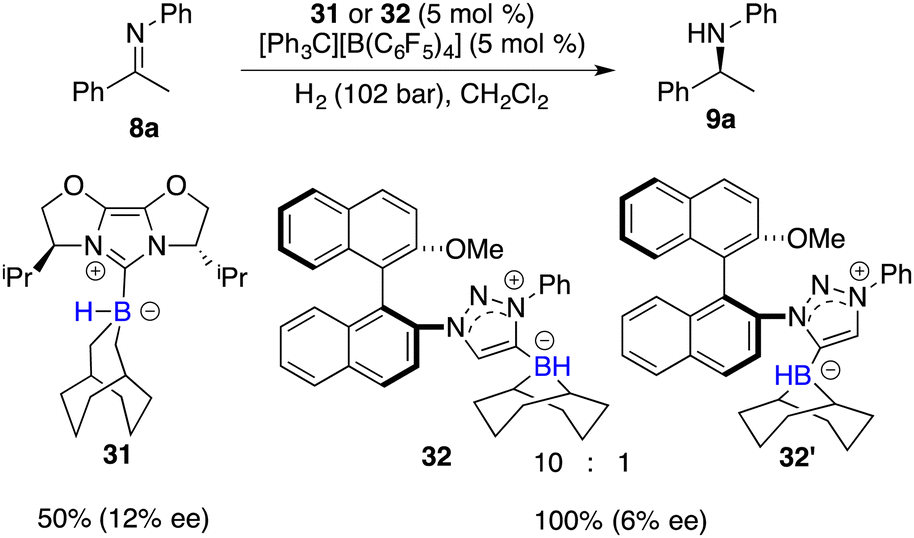
Beyond the extensively studied borane Lewis acids, borenium species derived from carbene–borane adducts have also been harnessed as Lewis acid components within FLPs. As shown in Fig. 2, a hydride abstraction of carbene adduct precursors with [Ph3C][B(C6F5)4] engenders the formation of borenium cations, which can split dihydrogen coupled with imine Lewis bases to form iminium cations. These cations, in turn, engage in dihydrogen splitting, concomitant with the participation of imine Lewis bases to furnish iminium cations. A subsequent hydride transfer from carbene adducts to the generated iminium cations yields amines and simultaneously regenerates borenium cations (Fig. 2). Chiral carbene–borane adducts, characterized by stability and facile accessibility, are promising catalyst candidates for the FLP catalysis. Stephan and co-workers have prepared an array of chiral carbene–borane adduct precursors, exemplified by 31 and 32, and examined their utility in the asymmetric hydrogenation of imines 8 (Scheme 14).19 Unfortunately, the outcomes were not satisfactory, yielding enantioselectivities ranging from 1 to 20% ee.
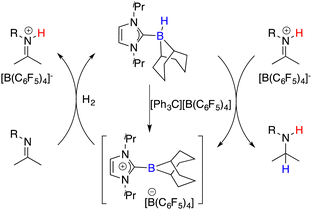 |
| | Fig. 2 Catalytic cycle for hydrogenation with borenium cations. | |
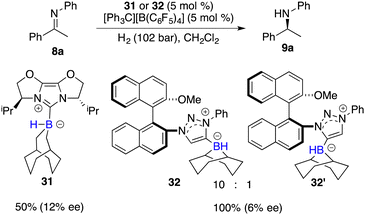 |
| | Scheme 14 Chiral borenium-based hydrogenation of imines. | |
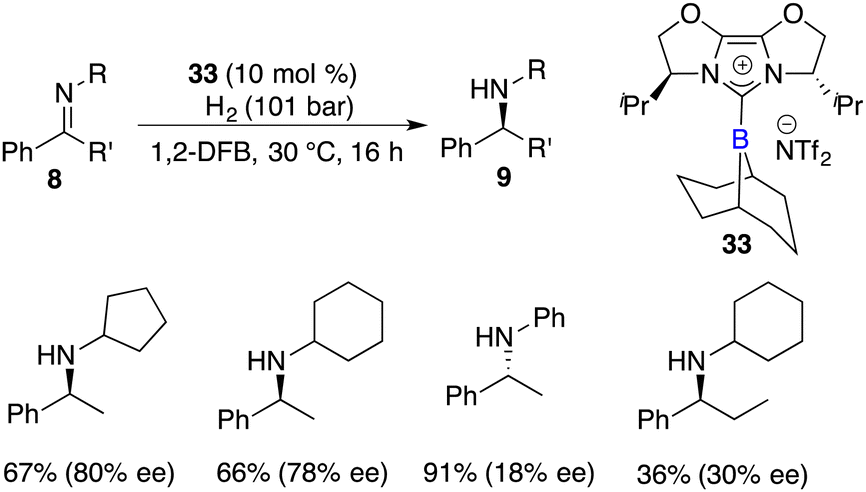
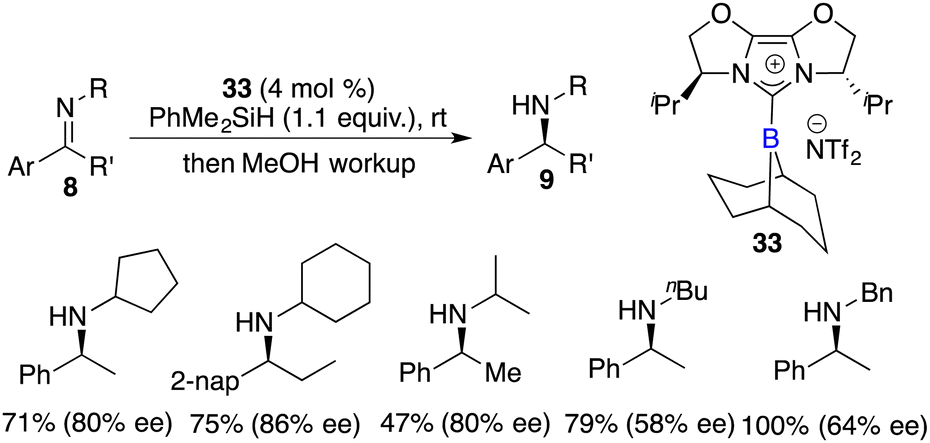
Fuchter and co-workers reported an enantioselective hydrogenation of ketimines 8 using chiral N-heterocyclic carbene-stabilized borenium cation 33 as a catalyst. Optically active secondary amines 9 were obtained in moderate to high yields with up to 80% ee (Scheme 15).20 The deviation from Stephan's work lies in the choice of borane and anion, which leads to a substantial improvement in enantioselectivity. The use of elevated hydrogen pressure remains a requisite element for this reaction to proceed.
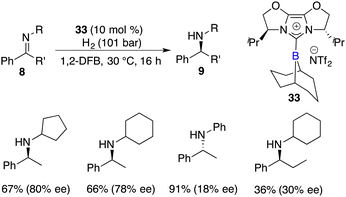 |
| | Scheme 15 Enantioselective reduction of N-alkyl ketimines with chiral borenium. | |
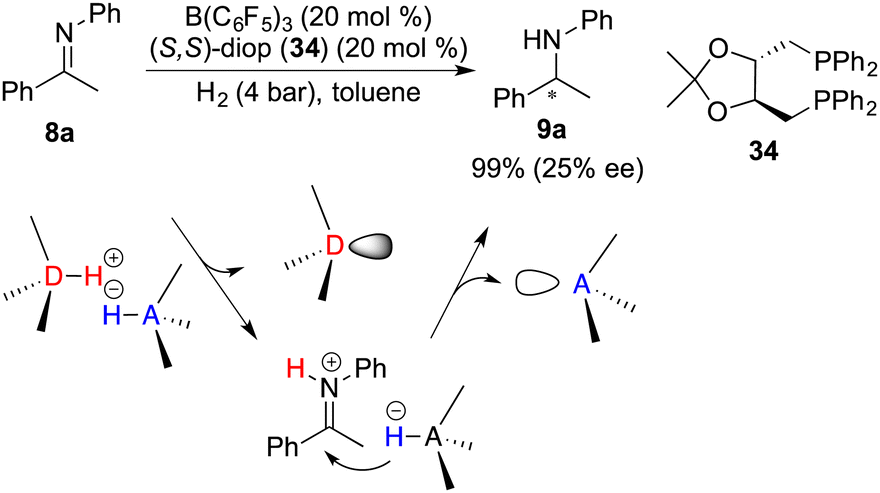
In spite of the favourable attributes such as stability and accessibility associated with chiral Lewis bases, the development of chiral Lewis base-driven FLP catalysts remains relatively scant. In 2011, Stephan and co-workers unveiled an asymmetric hydrogenation of imine 8a facilitated by a chiral FLP comprising B(C6F5)3 and diop 34. The desired amine 9a was obtained in 99% yield with 25% ee (Scheme 16).21 In this context, an inevitable rivalry emerges involving imine 8a as a prospective Lewis base component, ultimately leading to racemic hydrogenation. Notably, in view of the hydrogenation mechanism, the proton transfer occurs prior to the hydride transfer and forms an iminium cation. Concomitantly, the Lewis base is prone to dissociate from the reaction center, thereby resulting in a challenge to effect the following asymmetric induction process with a chiral Lewis base.
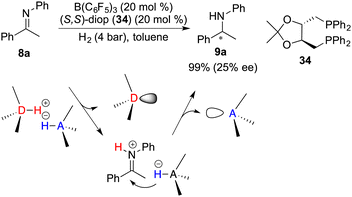 |
| | Scheme 16 Asymmetric hydrogenation with a chiral Lewis base-driven FLP. | |
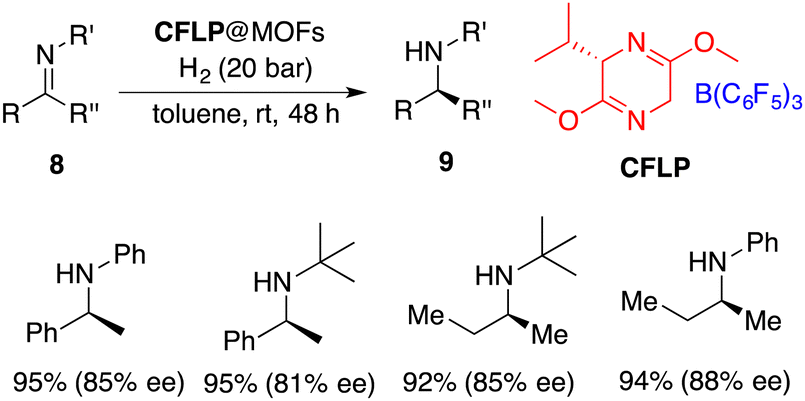
Heterogeneous catalysis emerges as a potential remedy to address this intrinsic challenge. Recently, Tang and Ma incorporated a chiral FLP (CFLP) comprising a chiral Lewis base and B(C6F5)3 into an achiral porous metal–organic framework (MOF), thereby achieving a heterogeneous catalyst CFLP@MOF. It is a highly effective catalyst for the asymmetric hydrogenation of imines 8, delivering the desired amines 9 in excellent yields with 77–88% ee (Scheme 17).22 Significantly, alkyl–alkyl imines were also suitable substrates for this reaction. The CFLP@MOF composite inherits the merits intrinsic to both homogeneous and heterogeneous catalysts, exhibiting not only high activity and selectivity, but also good recyclability.
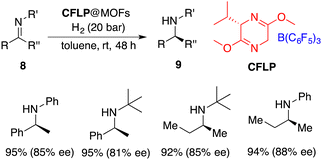 |
| | Scheme 17 Asymmetric hydrogenation of imines with a heterogeneous FLP catalyst. | |
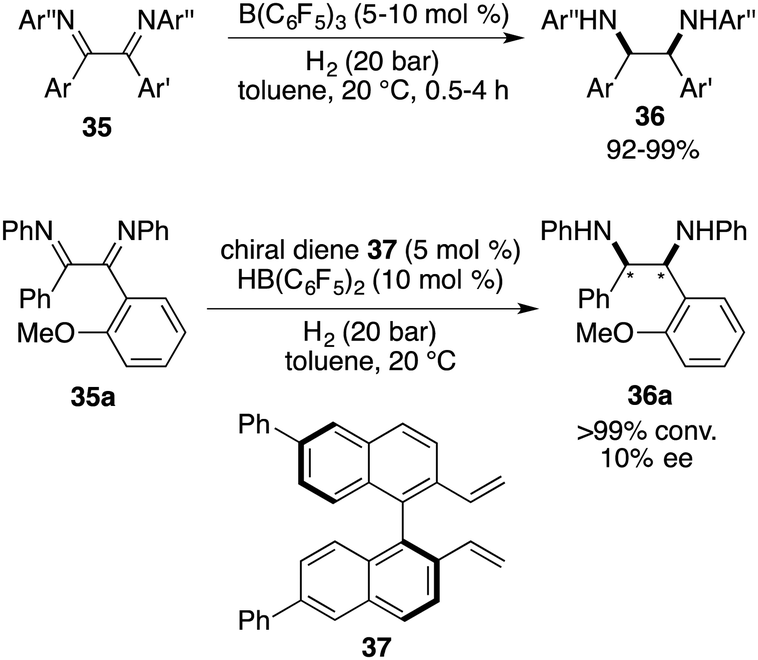
An extension of the imine scope for FLP-catalyzed asymmetric hydrogenation has achieved important progress. Vicinal diamines are important building blocks, and the direct asymmetric hydrogenation of vicinal diimines provides a straightforward way for their synthesis. However, such an asymmetric transformation has not been reported, which might be attributed to the steric hindrance and/or deactivation of the metal catalysts. In 2015, Du and co-workers reported a B(C6F5)3-catalyzed hydrogenation of 1,2-diaryl-1,2-diimines 35, furnishing a wide range of cis-1,2-diaryl-1,2-diamines 36 in 92–99% yields as single isomers (Scheme 18).23 Unfortunately, after evaluation of a wide range of chiral diene-derived boranes, only 10% ee was obtained using a chiral diene 37 derived borane catalyst. The difficulty may be attributed to the cis-selectivity of this reaction, rendering a subtle distinction between two sides of imines.
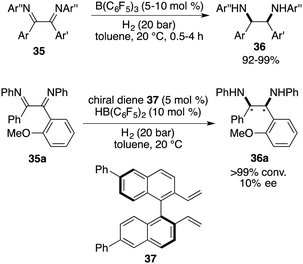 |
| | Scheme 18 Catalytic hydrogenation of diimines. | |
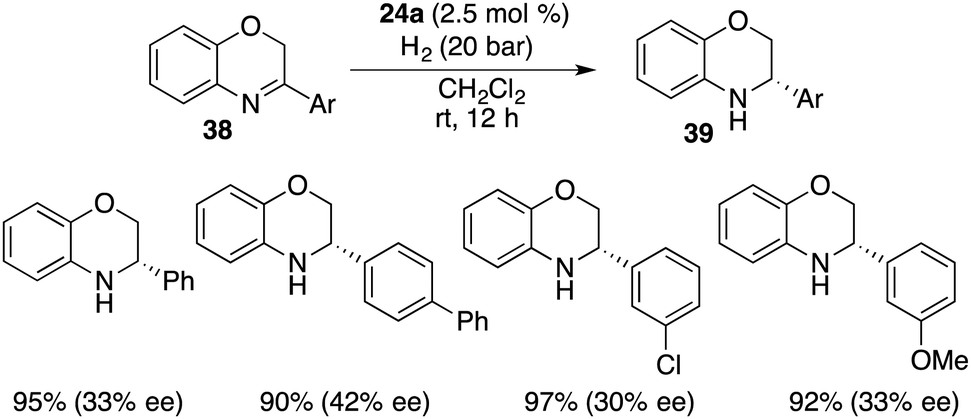
Cyclic imines were seldom employed as substrates for FLP-catalyzed hydrogenation. Du and co-workers extended the application of bulky borane 24a to the asymmetric hydrogenation of 3-substituted 2H-1,4-benzoxazines 38, delivering optically active 3,4-dihydro-2H-1,4-benzoxazines 39 in 90–97% yields with up to 42% ee (Scheme 19).24
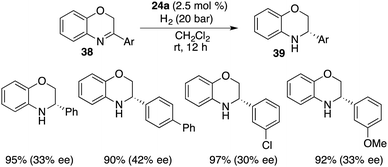 |
| | Scheme 19 Asymmetric hydrogenation of 1,4-benzoxazines. | |
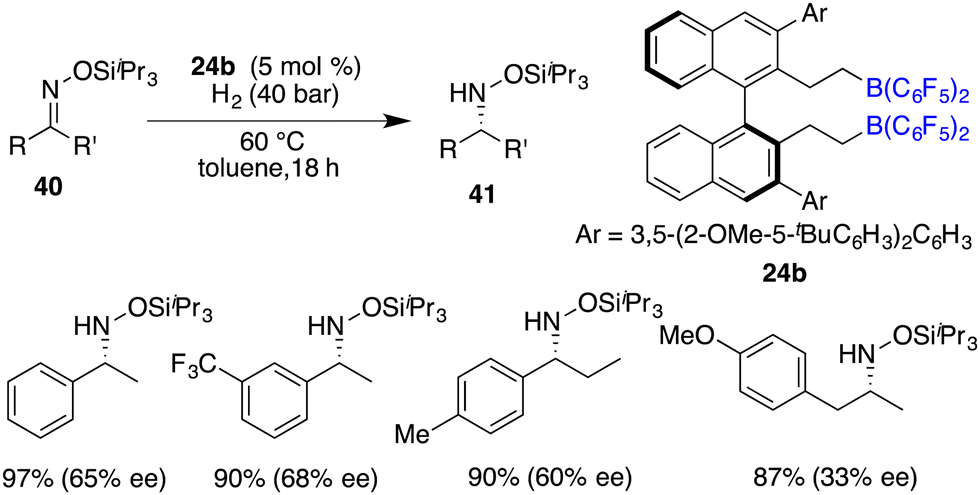
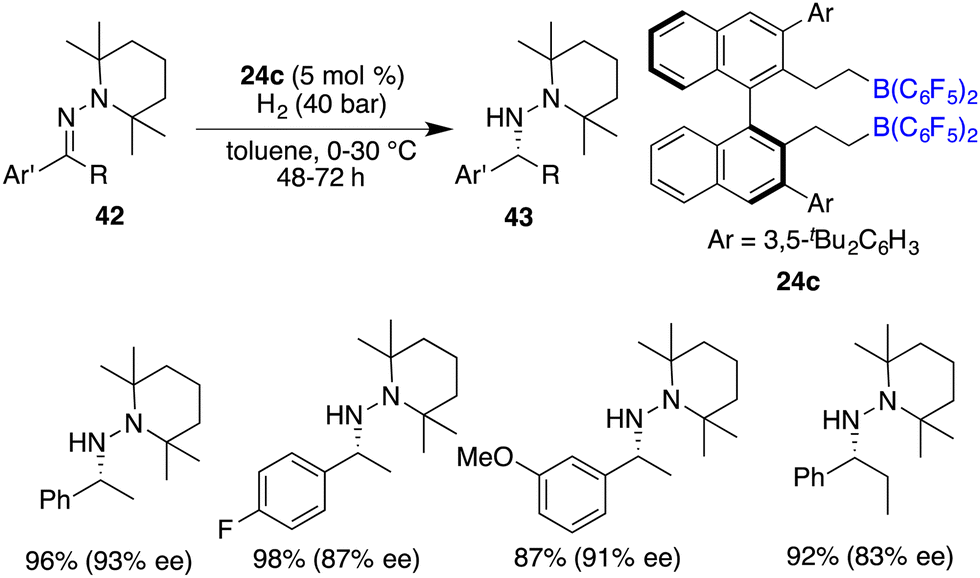
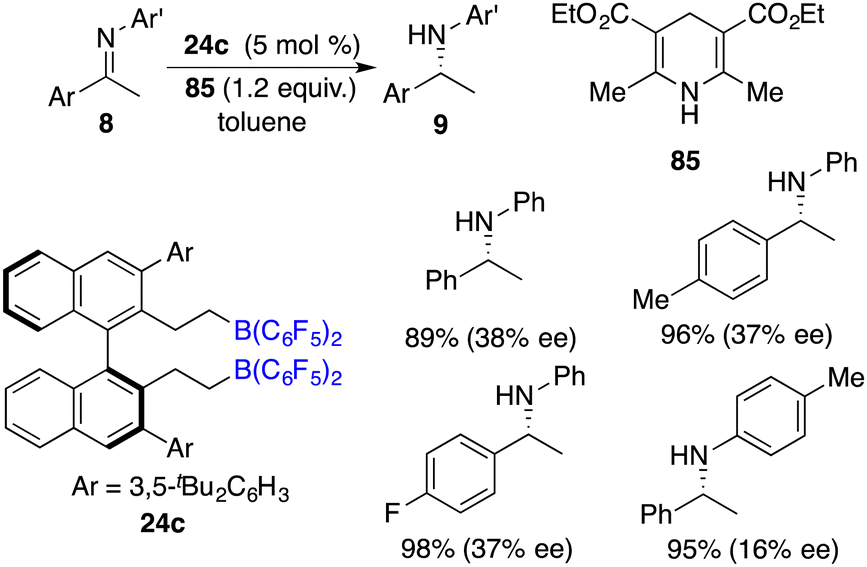
The asymmetric hydrogenation of oxime derivatives provides an attractive and straightforward route for accessing optically active hydroxylamine derivatives, but it usually leads to primary amines instead of hydroxylamines owing to the sensitive N–O bond. In 2014, Oestreich and co-workers reported a B(C6F5)3-catalyzed hydrogenation of oxime ethers to obtain hydroxylamine derivatives in the presence of H2 (100 bar).25 More recently, Du and co-workers achieved an enantioselective hydrogenation of TIPS-protected oximes 40 by using a very bulky chiral borane catalyst 24b (Scheme 20).26 A wide range of hydroxylamine derivatives 41 could be afforded in 84–99% yields with 33–68% ee. As a subsequent advancement, the same research group employed chiral borane 24c for the asymmetric hydrogenation of hydrazones 42, affording a broad range of optically active hydrazines 43 in 87–99% yields with 75–93% ee (Scheme 21).27 The 2,2,6,6-tetramethylpiperidinyl moiety was found to be essential for the attainment of the high enantioselectivities observed.
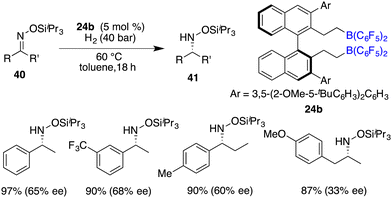 |
| | Scheme 20 Asymmetric hydrogenation of TIPS-protected oximes. | |
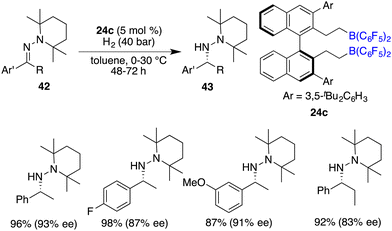 |
| | Scheme 21 Asymmetric hydrogenation of hydrozones. | |
1.2. Asymmetric hydrogenation of N-heteroarenes
The pioneering FLP-catalyzed asymmetric hydrogenation of N-heteroarenes was reported by Repo and co-workers. Through the unitization of chiral ansa-ammonium borate catalysts 14, the asymmetric hydrogenation of 2-substituted quinolines yielded the desired products with up to 37% ee.12 In recent years, considerable progress has been accomplished for the asymmetric hydrogenation of N-heteroarenes. In particular, chiral FLPs have exhibited some distinctive catalytic activities and selectivities during the hydrogenation of these types of substrates.
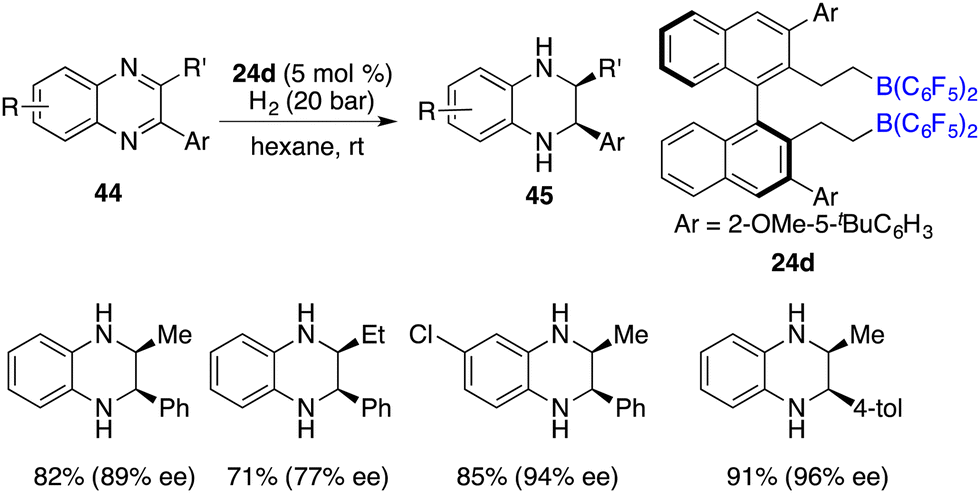
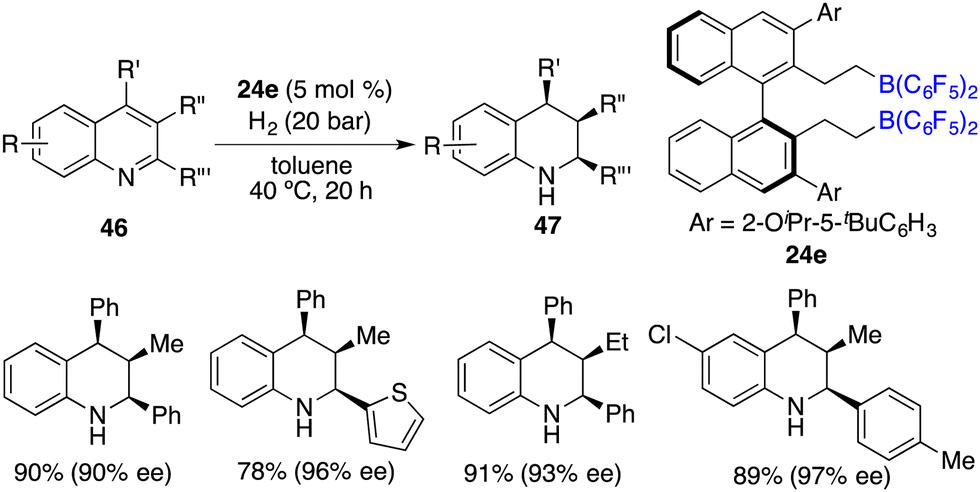
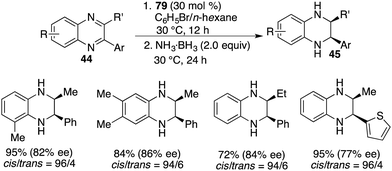
The asymmetric hydrogenation of 2-substituted quinoxalines has been well developed with chiral transition metal catalysts. Conversely, scarce examples have been documented concerning the asymmetric hydrogenation of 2,3-disubstituted quinoxalines to access optically active tetrahydroquinoxalines containing two contiguous stereogenic centers. In 2015, Du and co-workers reported an enantioselective hydrogenation of 2,3-disubstituted quinoxalines 44 with chiral borane 24d as a catalyst (Scheme 22).28 A variety of optically active cis-2,3-disubstituted 1,2,3,4-tetrahydroquinoxalines 45 were obtained in high yields with up to >99/1 dr and 96% ee. However, diaryl or dialkyl substituted quinoxalines were inert substrates for this asymmetric hydrogenation, despite that they were effective substrates for the B(C6F5)3-catalyzed reaction.
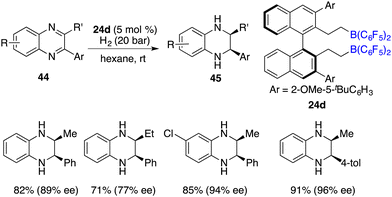 |
| | Scheme 22
cis-Selective and enantioselective hydrogenation of 2,3-disubstituted quinoxalines. | |
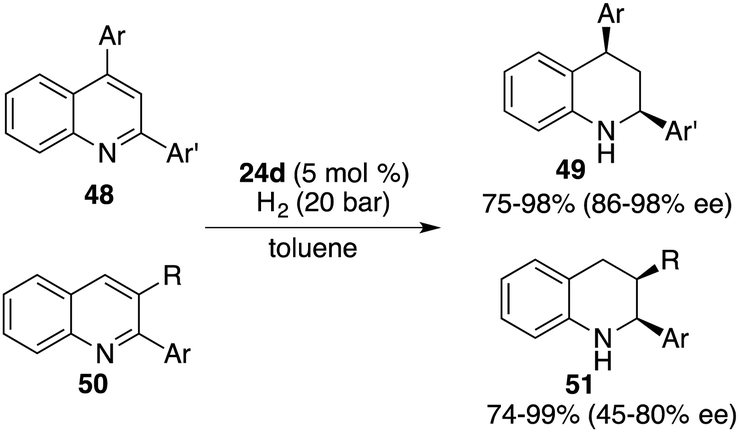
Through a subtle modification of the chiral diene, an asymmetric hydrogenation of 2,3,4-trisubstituted quinolines 46 was also successfully realized. A wide range of cis,cis-2,3,4-trisubstituted tetrahydroquinolines 47 bearing three contiguous stereogenic centers were furnished in 76–99% yields with 82–99% ee (Scheme 23).29 Remarkably, 2,4-disubstituted quinolines 48 were also effective substrates for chiral borane 26d catalyzed asymmetric hydrogenation, delivering a variety of 2,4-disubstituted tetrahydroquinolines 49 in 75–98% yields with 95/5–99/1 dr and 86–98% ee (Scheme 24).30 As for the domain of 2,3-disubstituted quinolines 50, despite their high reactivities, the outcomes remain somewhat modest. The asymmetric hydrogenation of 2-substituted quinolines was also examined, revealing enantioselectivities of markedly lower magnitude.31 This points to the pivotal role played by substituents at the 4-position of quinolines in engendering the observed high enantioselectivities. It is noteworthy, however, that 4-substituted quinolines continue to present a challenge within the realm of transition metal catalysis.
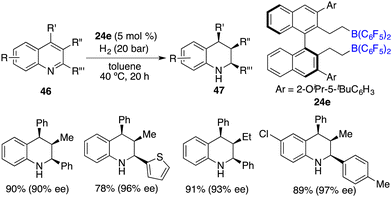 |
| | Scheme 23
cis-Selective and enantioselective hydrogenation of 2,3,4-trisubstituted quinolines. | |
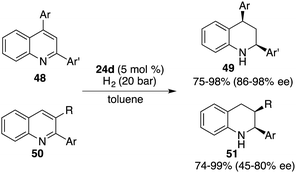 |
| | Scheme 24 Enantioselective hydrogenation of disubstituted quinolines. | |
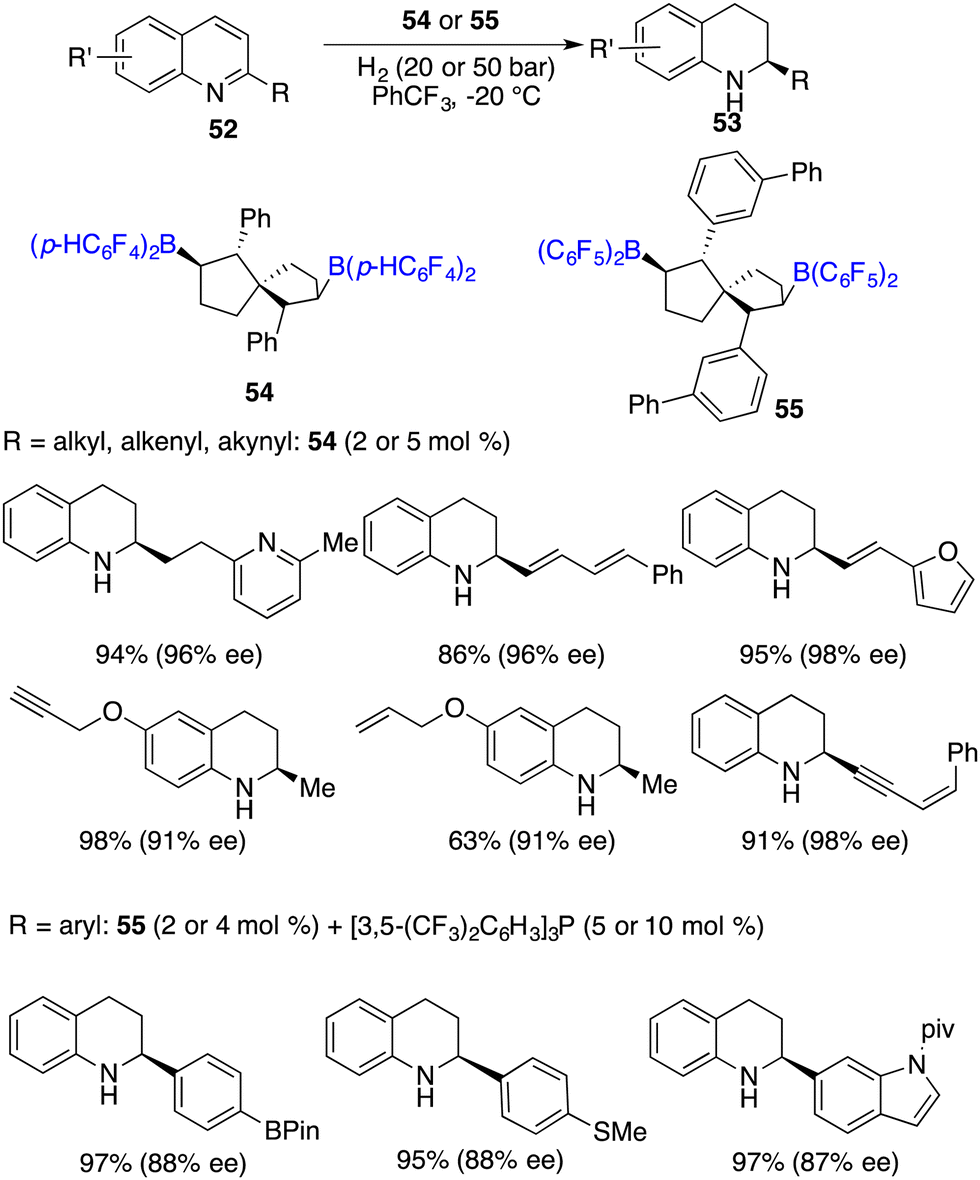
Wang and co-workers developed chiral spiro-bicyclic bisboranes 54 and 55 by means of the hydroboration of C2-symmetric spiro-bicyclic dienes with HB(C6F5)2 and HB(p-C6F4H)2. These chiral boranes were remarkably efficient for the hydrogenation of 2-substituted quinolines 52, affording a variety of tetrahydroquinolines 53 in 80–99% yields with 86–99% ee (Scheme 25).32 In the case of 2-aryl substituted quinolines, the introduction of an additional phosphine Lewis base was needed. Notably, a remarkable turnover number (TON) of up to 460 could be reached for this reaction, which stands as the highest one within the realm of FLP-catalyzed asymmetric reactions. It is noteworthy to point out that a wide range of functional groups, including carbon–carbon double bonds or triple bonds, pyridinyl, boryl, and methylthio moieties, were all well tolerated for the metal-free asymmetric hydrogenation.
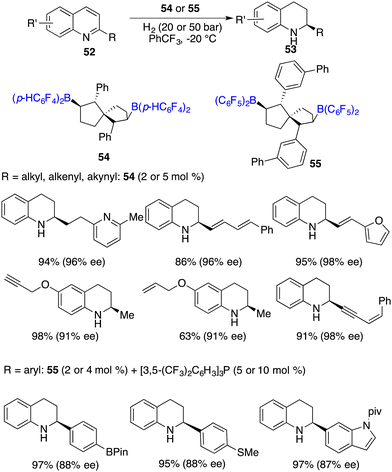 |
| | Scheme 25 Enantioselective hydrogenation of quinolines with chiral spiro-bicyclic bisboranes. | |
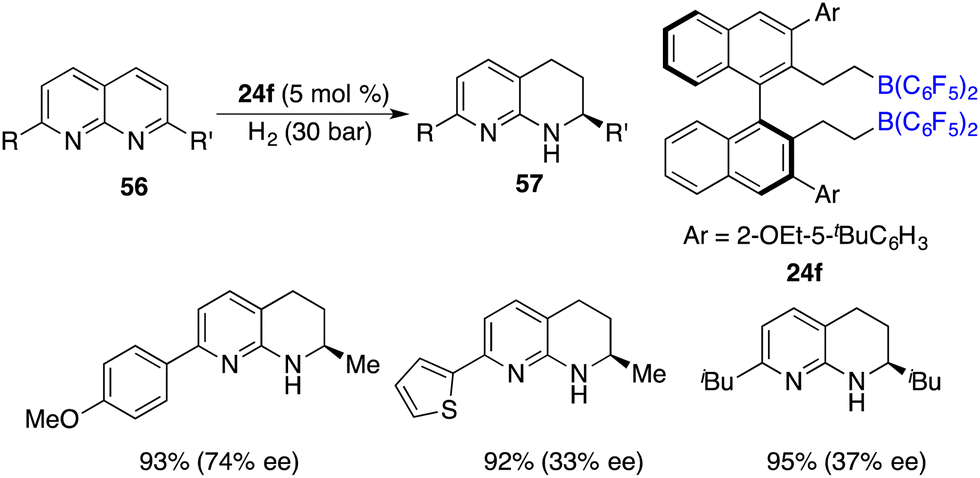
In the year 2016, a metal-free hydrogenation of 2,7-disubstituted 1,8-naphthyridines 56 was accomplished through the utilization of an in situ generated borane catalyst by the hydroboration of pentafluorostyrene with HB(C6F5)2, furnishing 1,2,3,4-tetrahydro-1,8-naphthyridines 58 in high yields. The employment of chiral borane 24f as a catalyst engendered the production of 1,2,3,4-tetrahydro-1,8-naphthyridines 58 in high yields with up to 74% ee (Scheme 26).33 It is intriguing to observe that alkyl-substituted rings within 1,8-naphthyridines were more reactive to be reduced exclusively. The asymmetric hydrogenation of simple pyridines still remains an unsolved problem. Du and co-workers utilized a pentafluorostyrene-derived borane as a catalyst for the hydrogenation of pyridines, furnishing the desired piperidines in high yields with excellent cis selectivity.34 Regrettably, the chiral borane proved to be ineffective for the asymmetric hydrogenation, leading to only less than 10% ee.
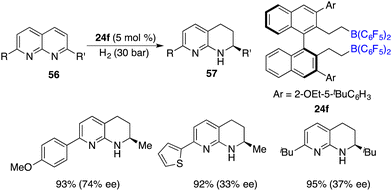 |
| | Scheme 26 Enantioselective hydrogenation of 1,8-naphthyridines. | |
1.3 Asymmetric hydrogenation of silyl enol ethers, simple ketones, and enones
Great success has been achieved for the asymmetric hydrogenation of ketones by using a diverse range of chiral transition metal catalysts for the synthesis of optically active secondary alcohols. However, simple ketones were less reactive substrates for the FLP catalysis. Silyl enol ethers can be conveniently prepared from ketones. Erker and Paradies reported the catalytic hydrogenation of silyl enol ethers by using a combination of phosphine and borane as the FLP catalyst, respectively,35,36 which provides a detour for the synthesis of secondary alcohols.
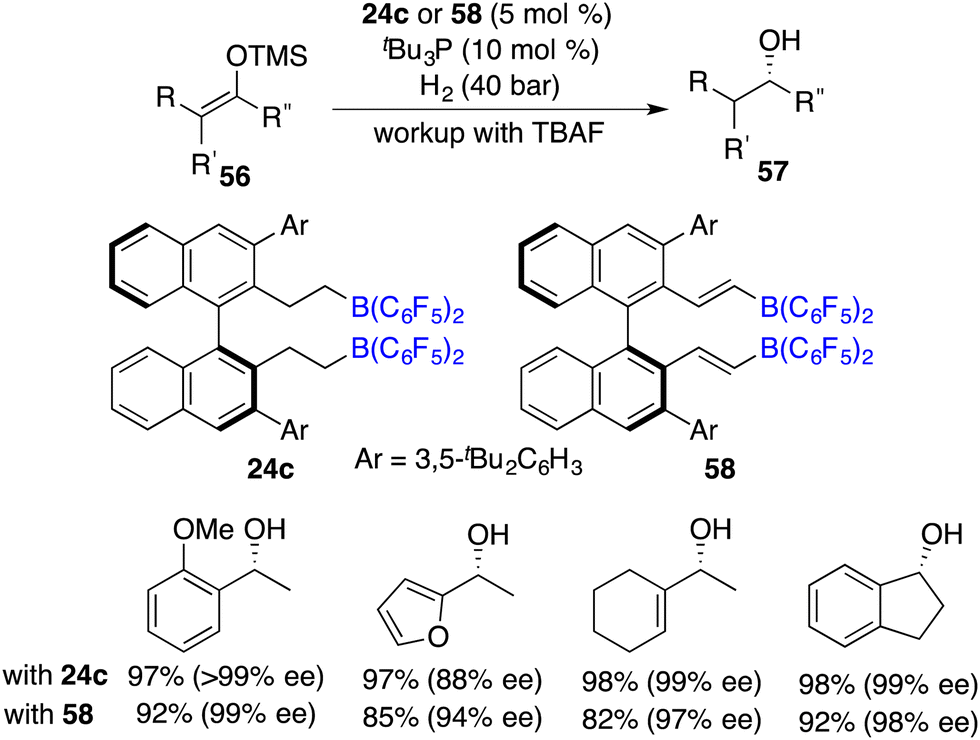
Subsequently, Du and co-workers realized the first highly enantioselective hydrogenation of silyl enol ethers 56 by using a combination of tBu3P and chiral borane 24c as an ingenious FLP catalyst. A wide range of optically active alcohols were furnished in excellent yields with remarkably high ees (Scheme 27).37 Lewis bases were required and found to have a substantial impact on both the reactivity and the enantioselectivity. Notably, carbon–carbon double bonds can be well maintained under hydrogenation conditions. In 2015, the same group developed a novel chiral alkenylborane 58 stemming from the in situ-hydroboration of chiral diyne with HB(C6F5)2. It was also an effective catalyst for the asymmetric hydrogenation of silyl enol ethers 56 (Scheme 27).38
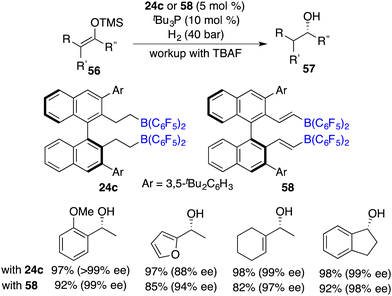 |
| | Scheme 27 Asymmetric hydrogenation of silyl enol ethers. | |
In 2014, the FLP-catalyzed hydrogenations of ketones 59 were successfully unlocked by Stephan and Ashley, respectively (Scheme 28).39,40 The choice of Lewis base is crucial for this success. As weak Lewis bases, diethyl ether and 1,4-dioxane can be transformed as strong Brønsted acids after the splitting of H2 with B(C6F5)3. The possible activation models are outlined in Scheme 28, in which the proton tethered to the oxygen atom activates the ketone and facilitates the nucleophilic hydride attack to yield alcohols 60. This provides a good possibility to use a chiral FLP as a catalyst for achieving the asymmetric reaction.
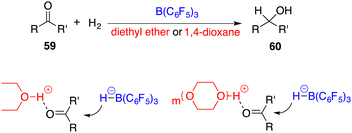 |
| | Scheme 28 Catalytic hydrogenation of simple ketones. | |
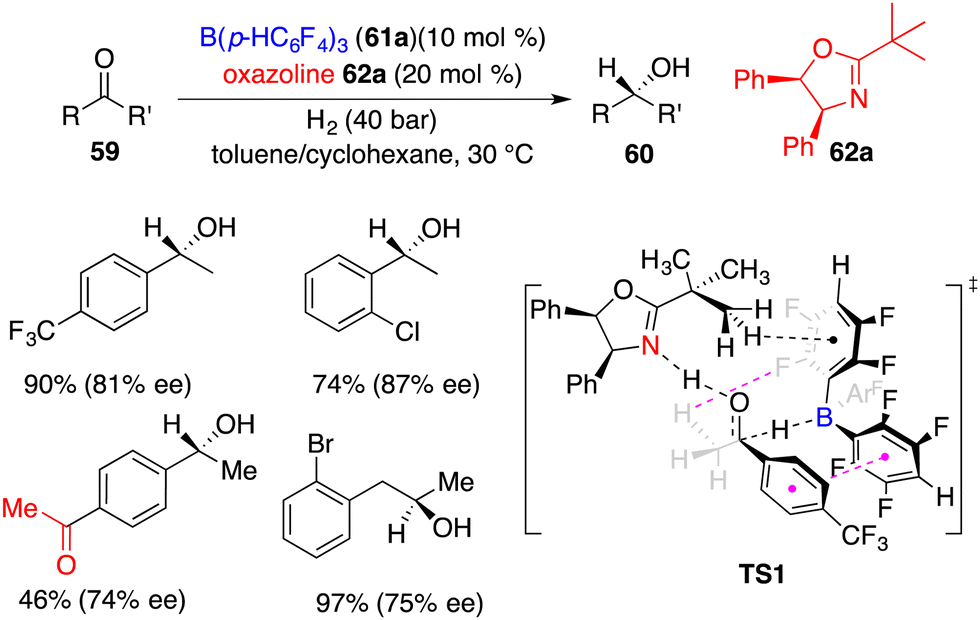
Du and co-workers developed a novel FLP of chiral oxazoline Lewis base 62a and achiral boron Lewis acid 61a for asymmetric hydrogenation of simple ketones 59 (Scheme 29).41 A variety of optically active secondary alcohols 60 were obtained in moderate to high yields with up to 87% ee. Electron-withdrawing substituents on the phenyl group were preferred to increase the reactivity. Interestingly, the asymmetric hydrogenation of 1,4-diacetylbenzene furnished the product in 46% yield with 74% ee without any over-reduced diol by-products. Significantly, dialkyl ketones were also suitable substrates to obtain reasonable results. A DFT-calculation reveals that there are hydrogen bonding and π–π stacking interactions in the transition state and a sequential concerted H-transfer process of proton and hydride (TS1). An unavoidable competition of ketone as a Lewis base is attributed to the obvious dwindling ee between calculation and experiment.
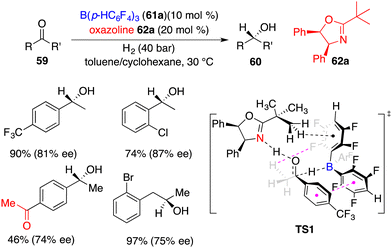 |
| | Scheme 29 Asymmetric hydrogenation of simple ketones. | |
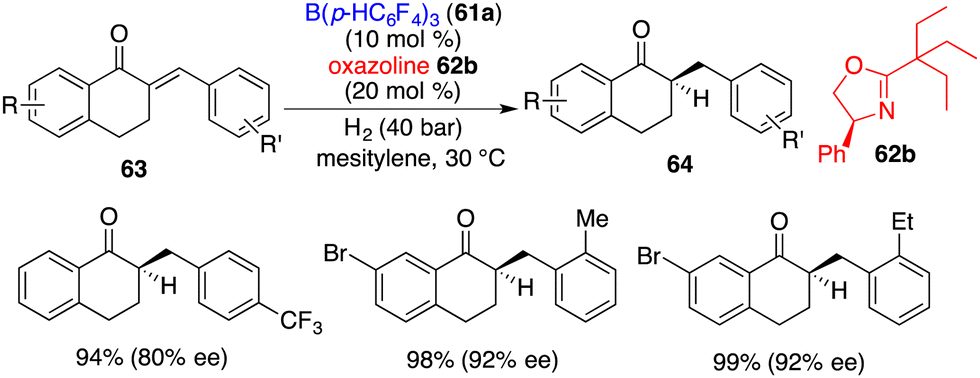
The application of this chiral Lewis base-driven FLP was further extended to the asymmetric hydrogenation of 1-tetralone-derived enones 63 (Scheme 30).41 With the utilization of oxazoline 62b, all hydrogenation reactions proceeded smoothly to produce optically active ketones 64 in high yields with 63–92% ee. The over-reduction of the resulting ketones was not observed.
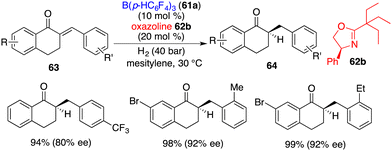 |
| | Scheme 30 Asymmetric hydrogenation of enones. | |
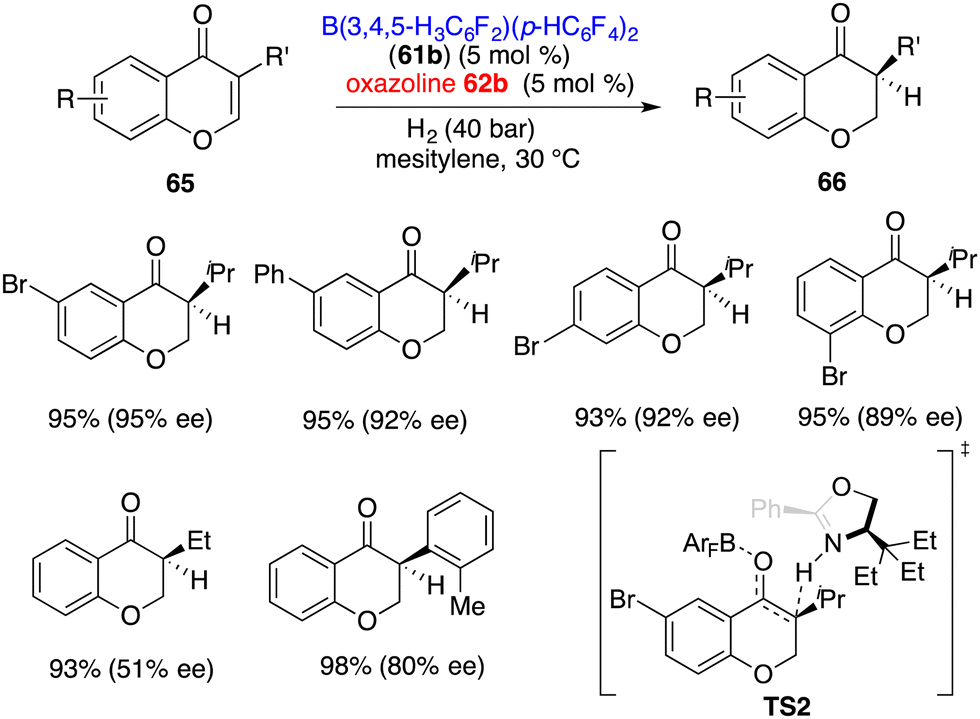
3-Substituted chromones 65 were more effective substrates for the asymmetric hydrogenation by using an FLP comprising 61b and 62b, giving the corresponding products 66 in 91–98% yields with up to 95% ee (Scheme 31).41 The ee was highly depended on the steric hindrance of chromones. A sharp decrease of ee to 51% was observed when using a less bulky ethyl group instead of the isopropyl group. Distinct from ketones, the hydrogen-transfer for the hydrogenation of enones is stepwise. The protonation of the enolate is the configuration step (TS2). A 2.2 kcal mol−1 activation free energy difference between two transition states indicates the formation of an S-isomer favourably, which is well consistent with experimental results.
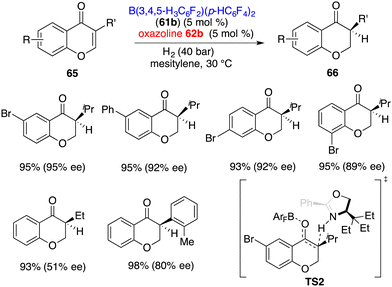 |
| | Scheme 31 Asymmetric hydrogenation of chromones. | |
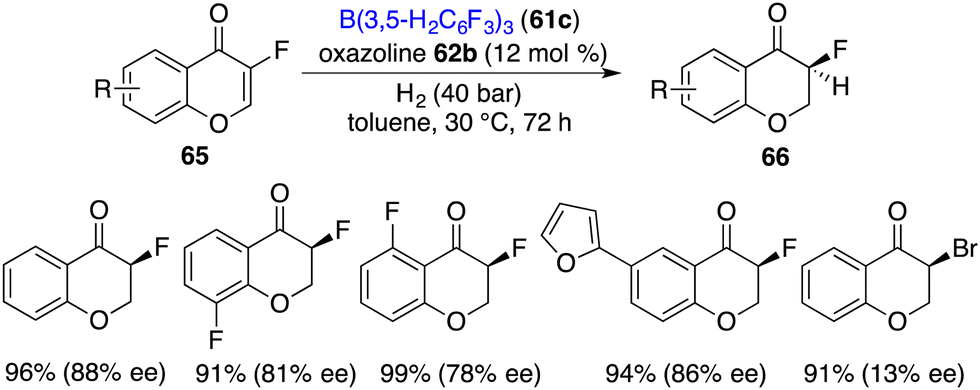
Unexpectedly, with fluorine as a small size substituent, 3-fluorinated chromones 65 were also suitable substrates for the asymmetric hydrogenation. (Scheme 32).42 By using borane 61c and chiral oxazoline 62b as an FLP catalyst, a variety of 3-fluorochroman-4-ones 66 were obtained in 89–99% yields with 75–88% ee. However, when the fluorine substituent was changed to a bromine atom, the ee dropped sharply to 13%, which indicates that there might be interactions between the fluorine atom and the FLP catalyst.
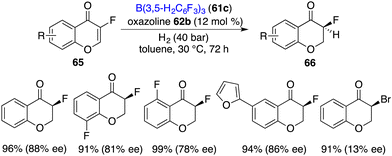 |
| | Scheme 32 Asymmetric hydrogenation of 3-fluorinated chromones. | |
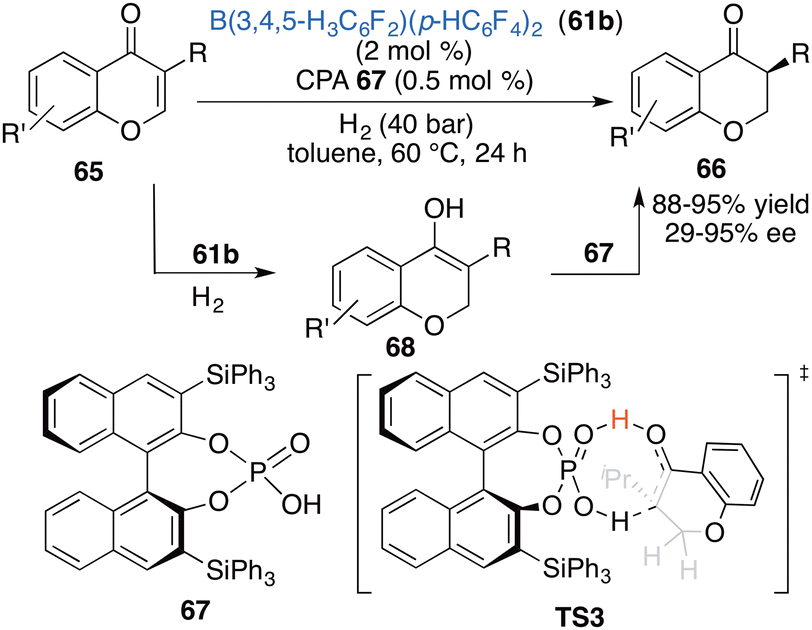
Inspired by the aforementioned work, Du and co-workers put forward a novel strategy of relay catalysis with a combination of an achiral FLP and a chiral proton shuttle. An asymmetric hydrogenation of chromones 65 was successfully accomplished by using achiral borane 61b and chiral phosphoric acid 67 to obtain the desired products 66 in 88–98% yields with up to 95% ee (Scheme 33).43 Achiral borane 61b and chiral phosphoric acid 67 are well compatible in this reaction. The reactivity is mainly determined by borane 61a, while the enantioselectivity is controlled by CPA 67 as a chiral proton shuttle. A DFT calculation indicates that there exists a 6.1 kcal mol−1 energy difference between two transition states during the 1,3-proton transfer process leading to (S)-stereoselectivity.
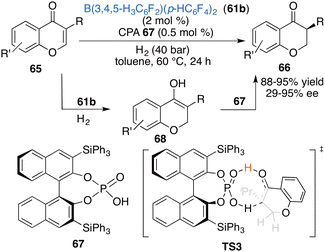 |
| | Scheme 33 Asymmetric relay hydrogenation of chromones. | |
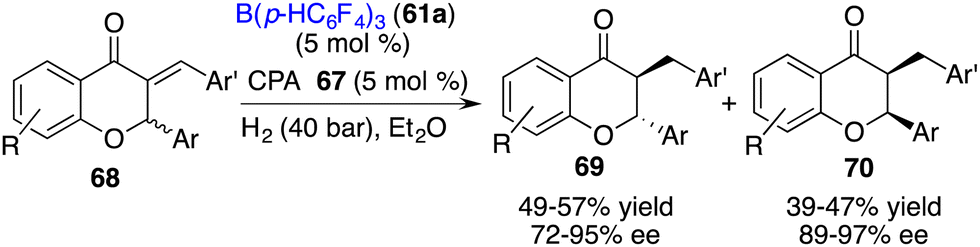
The direct asymmetric hydrogenation of 2,3-disubsituted chromones to access flavanones or chromanones containing two contiguous stereogenic centers remains a challenge. As a detour, an asymmetric hydrogenation of racemic 3-alkylidene flavanones 68 for the stereodivergent synthesis of 3-substituted flavanones has been accomplished by Du and co-workers via a relay catalysis by borane 61a and chiral phosphoric acid 67. A wide range of trans- and cis-flavanones 69 and 70 were obtained with high levels of yields and ee values (Scheme 34).44
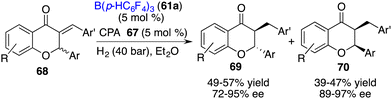 |
| | Scheme 34 Asymmetric hydrogenation of 3-alkylidene flavanones. | |
2. Asymmetric hydrosilylation
In 1996, Piers and co-workers unveiled a catalytic hydrosilylation of aromatic aldehydes, ketones, and esters by using B(C6F5)3 as a catalyst, wherein the boron Lewis acid activates the Si–H bond instead of the substrates to form zwitterions of silylium and hydridoborate.45 This type of hydrosilylation was therefore called Piers-type hydrosilylation and has attracted both synthetic and mechanistic interest from chemists.
2.1 Asymmetric hydrosilylation of imines
In 2009, Oestreich and co-workers attempted the hydrosilylation of imines with a silicon-stereogenic silane. However, racemic amines were produced.46 Klankermayer and co-workers reported a highly enantioselective hydrosilylation of imines 8 by using chiral FLP salt 7 as a catalyst (Scheme 35).47 A variety of chiral secondary amines 9 were furnished in moderate to high yields with 81–87% ee. The addition of phosphine was revealed to be crucial for achieving high enantioselectivity. In contrast, employing the enantiopure borane itself as a catalyst, despite its high reactivity, only resulted in a racemic product.
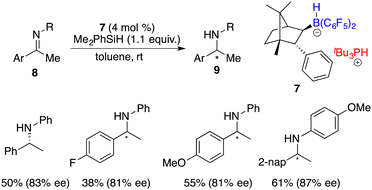 |
| | Scheme 35 Hydrosilylation of imines with a chiral FLP catalyst. | |
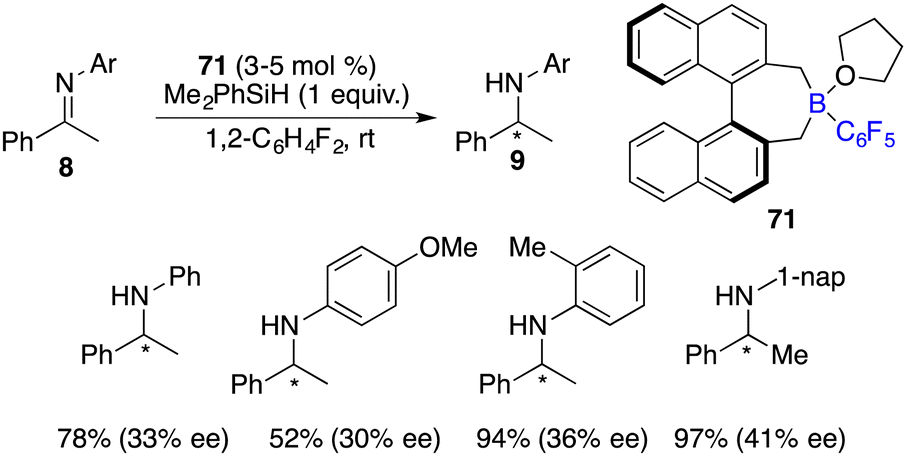
Oestreich and co-workers developed an axially chiral C6F5-substituted borane 71 for asymmetric hydrosilylation of imines 8 (Scheme 36).48 For the benzyl-substituted imine, a promising 62% ee was achieved. With a combination of borane 71 and axially chiral silanes, obvious matched/mismatched phenomena were observed, but the enantioselectivity was not improved.
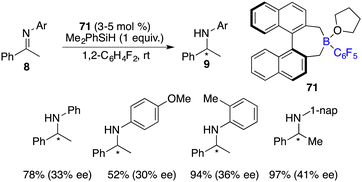 |
| | Scheme 36 Enantioselective hydrosilylation of imines with a chiral borane. | |
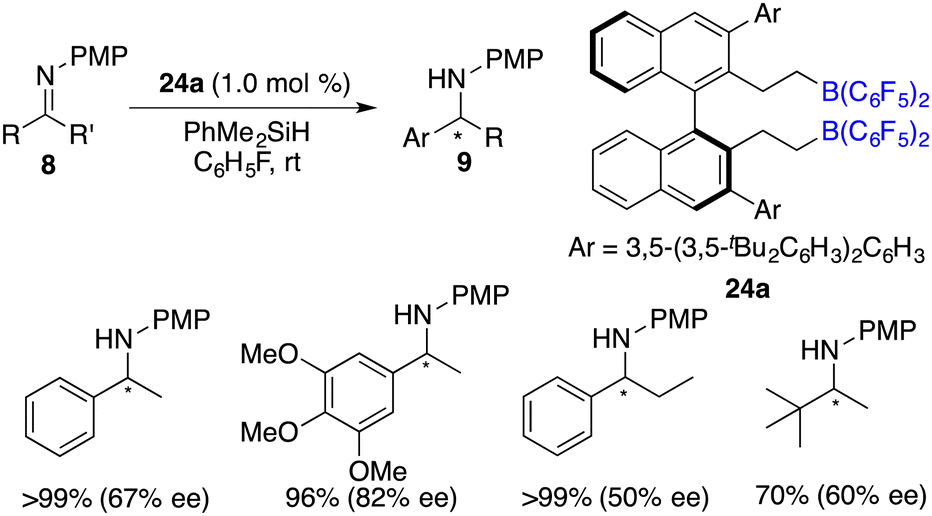
Du and co-workers described an enantioselective hydrosilylation of imines 8 using chiral borane catalyst 24a, giving secondary amines 9 in 70–>99% yields and 44–82% ee (Scheme 37).49 An additional Lewis base was not required for this reaction. The racemization of products with chiral borane 24a did not occur, which might be attributed to its bulky steric hindrance. Very recently, the same research group expanded the imine scope to N-sulfonyl ketimines and vicinal diimines. In particular, optically active cis-vicinal diamines could be afforded with up to 70% ee.50 Moreover, chiral borenium cation 33 was also an effective catalyst for asymmetric hydrosilylation of N-alkyl imines 8 (Scheme 38).20 The desired amine products 9 were obtained in moderate to high yields with up to 86% ee.
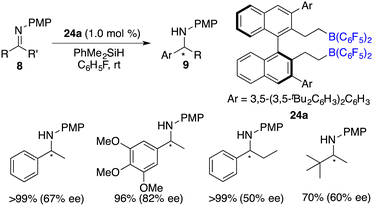 |
| | Scheme 37 Asymmetric hydrosilylation of imines with a chiral bisborane. | |
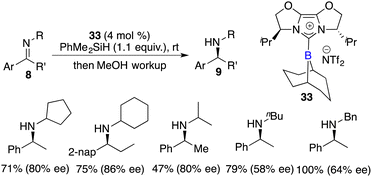 |
| | Scheme 38 Enantioselective hydrosilylation of N-alkyl ketimines with chiral borenium. | |
2.2 Asymmetric hydrosilylation of ketones
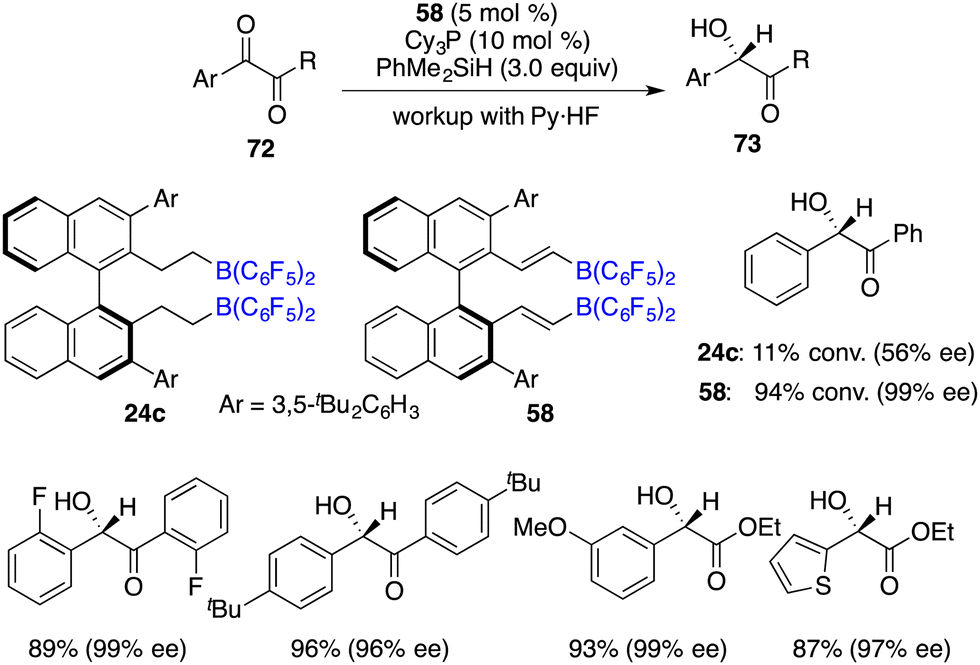
In comparison to imines, the progress of the asymmetric hydrosilylation of ketones was relatively slow. Oestreich and co-workers reported a B(C6F5)3-catalyzed hydrosilylation of acetophenone with a chiral silane. The chiral silane was recovered in 89% yield with 84% ee and chiral alcohol was simultaneously produced in 67% yield with 38% ee.51 In 2016, Du and co-workers disclosed an asymmetric hydrosilylation of 1,2-dicarbonyl compounds using chiral alkenylboranes 58 and Cy3P as an FLP catalyst. A wide range of optically active α-hydroxy carbonyl compounds were furnished in 52–98% yields with 86–99% ee (Scheme 39).52 Notably, a Lewis base was required for this reaction, and no diol by-products were observed even with excess amounts of PhMe2SiH. Chiral alkenylborane 58 exhibited noticeable advantages over chiral alkylborane 24c in terms of both reactivity and enantioselectivity. However, the hydrosilylation of simple ketones under the current conditions gave only moderate ee.
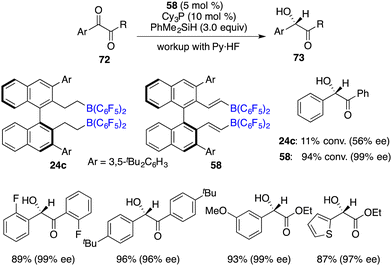 |
| | Scheme 39 Enantioselective hydrosilylation of 1,2-dicarbonyl compounds. | |
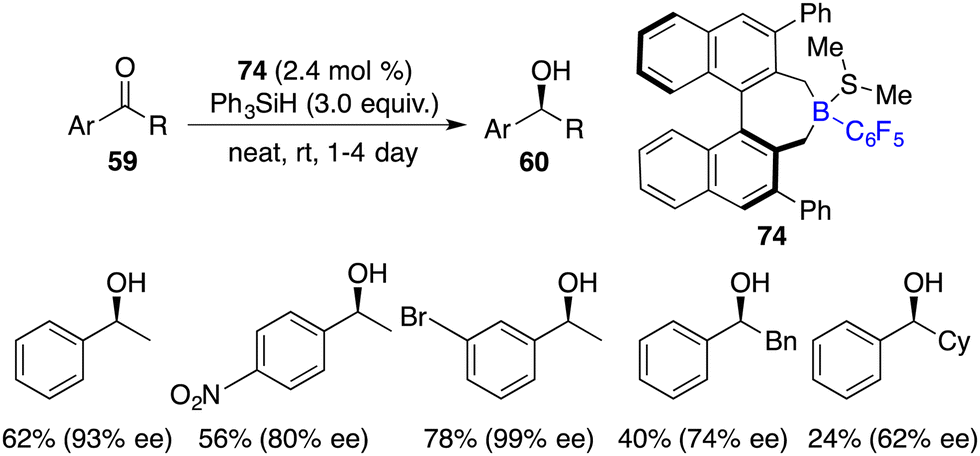
In the same year, Oestreich and co-workers developed an axially chiral borane 74, which was a highly effective catalyst for the enantioselective hydrosilylation of simple ketones 59 with PhSiH3 without the addition of a Lewis base. The desired chiral secondary alcohols 60 were accessed in 12–87% yields with up to 99% ee (Scheme 40).53 The success is likely attributed to the bulky steric hindrance of chiral borane 74 and the use of reactive trihydrosilane as a reductant.
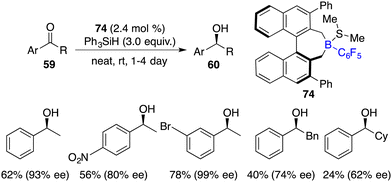 |
| | Scheme 40 Enantioselective hydrosilylation of simple ketones. | |
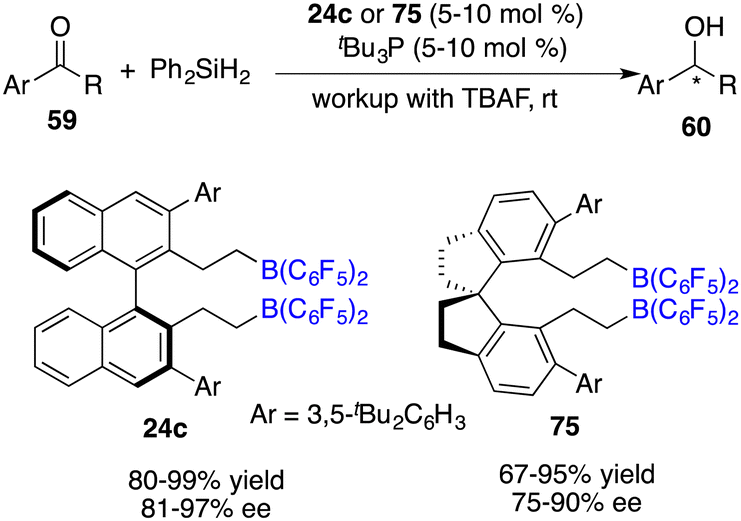
Utilizing the silane Ph2SiH2 as a reductant, Du and co-workers also realized the asymmetric hydrosilylation of simple ketones 59 under the catalysis of tBu3P and chiral borane 24c (Scheme 41).54 An array of optically active secondary alcohols 60 were furnished in 80–99% yields with 81–97% ee. The introduction of a C2-symmetric 1,1’-spirobiindane framework into a chiral borane offered an effective catalyst 75 for the asymmetric hydrosilylation, affording up to 90% ee.55
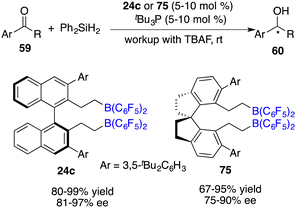 |
| | Scheme 41 Asymmetric hydrosilylation of ketones with chiral bisboranes. | |
2.3 Asymmetric hydrosilylation of enones
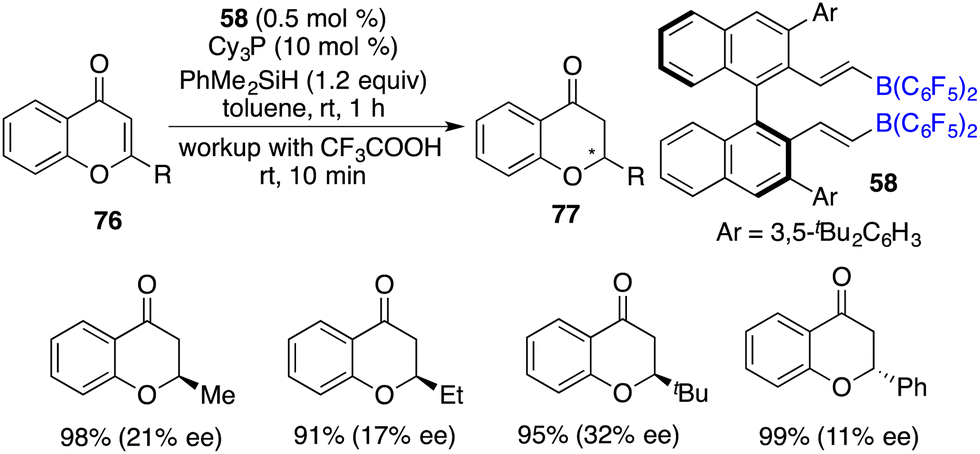
Du and co-workers reported a highly efficient hydrosilylation of chromones and flavones 76 with as low as 0.1 mol% borane catalyst derived from pentafluorostyrene and HB(C6F5)2, giving a variety of chromanones and flavanones 77 in 60–99% yields. Subsequently, the asymmetric reaction was realized with chiral alkenylborane 58 and Cy3P as an FLP catalyst, delivering optically active chromanones and flavanones 77 in high yields with up to 32% ee (Scheme 42).56
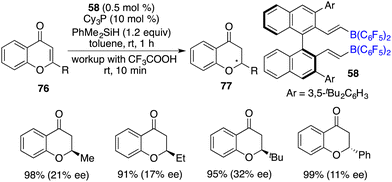 |
| | Scheme 42 Asymmetric hydrosilylation of chromones and flavones. | |
2.4 Asymmetric tandem cyclization/hydrosilylation
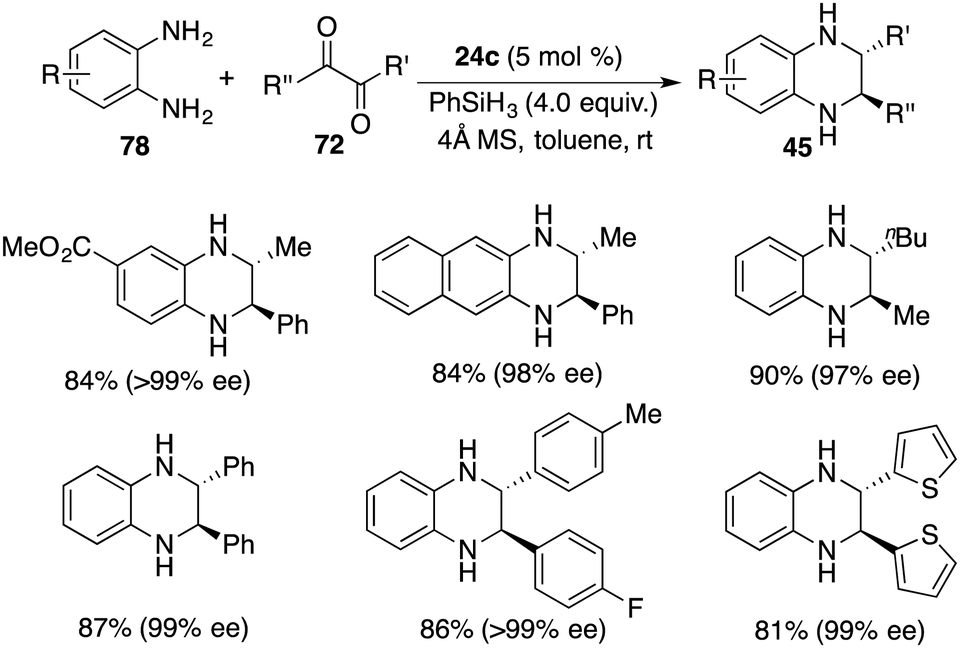
In a recent revelation, Xu and co-workers unveiled the discovery of a one-pot tandem cyclization/hydrosilylation of 1,2-diaminobenzenes 78 and 1,2-diketones 72 by using chiral borane 24c. A wide range of trans-2,3-disubstituted 1,2,3,4-tetrahydroquinoxalines 45 were furnished in high yields with up to >20:1 dr and up to >99% ee (Scheme 43).57 Alkyl–alkyl, alkyl–aryl, and aryl–aryl substituents were all tolerated in this reaction. Mechanistic studies reveal that the hydride transfer might be involved in the rate-limiting step. Moreover, the transformation of the cis-isomer to trans-isomer was also ruled out according to the control experiments.
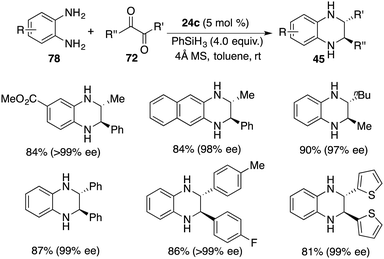 |
| | Scheme 43 Enantio- and diastereoselective synthesis of trans-2,3-disubstituted 1,2,3,4-tetrahydroquinoxalines. | |
3. Asymmetric transfer hydrogenation
Asymmetric transfer hydrogenation stands as a mild reduction method for the synthesis of optically active compounds. Several hydrogen sources have been employed for both catalytic and asymmetric transfer hydrogenation of unsaturated compounds with FLPs as catalysts.
3.1 Asymmetric transfer hydrogenation with ammonia borane
Ammonia borane (NH3·BH3) possesses several favourable attributes, rendering it an excellent candidate for hydrogen storage due to its substantial storage capacity (19.6% weight H), low molecular weight (30.87 g mol−1), and robust stability. Moreover, it holds the potential to serve as a hydrogen source, offering both proton and hydride transfer capabilities in synthetic processes. Despite these advantages, the application of ammonia borane as a hydrogen donor in the context of asymmetric transfer hydrogenation has garnered relatively limited attention in the literature.
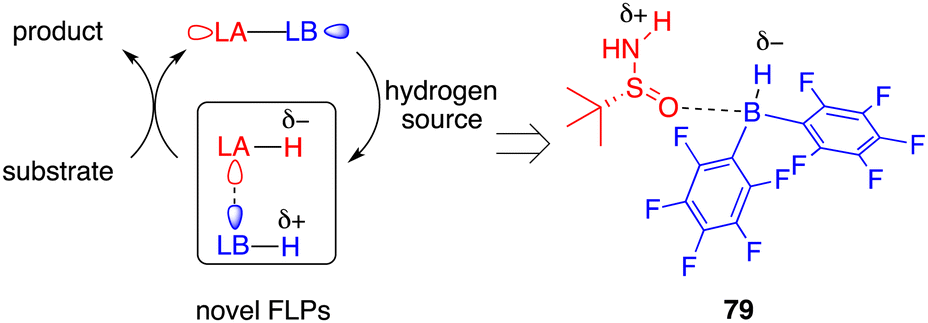
Taking inspiration from the well-established zwitterion species commonly associated with FLPs, Du and co-workers devised an innovative variation of FLP, one that incorporates an active acidic hydrogen atom within the Lewis acid and a hydridic hydrogen atom within the Lewis base. This strategic design was formulated to curtail the displacement of the Lewis base from the reaction center after proton transfer (Fig. 3).58 Accordingly, they introduced a novel chiral Lewis base-driven FLP 79 through the in situ pairing of HB(C6F5)2 and chiral tert-butylsulfinamide. This development has opened up new avenues for controlling enantioselectivity by harnessing the potential of chiral Lewis bases.
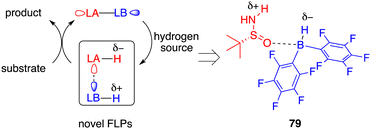 |
| | Fig. 3 Design of a novel type of FLP catalysis. | |
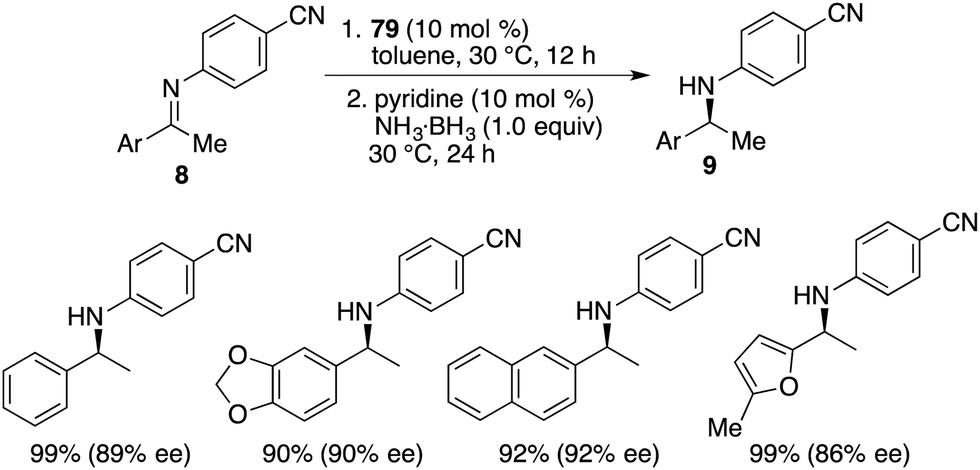
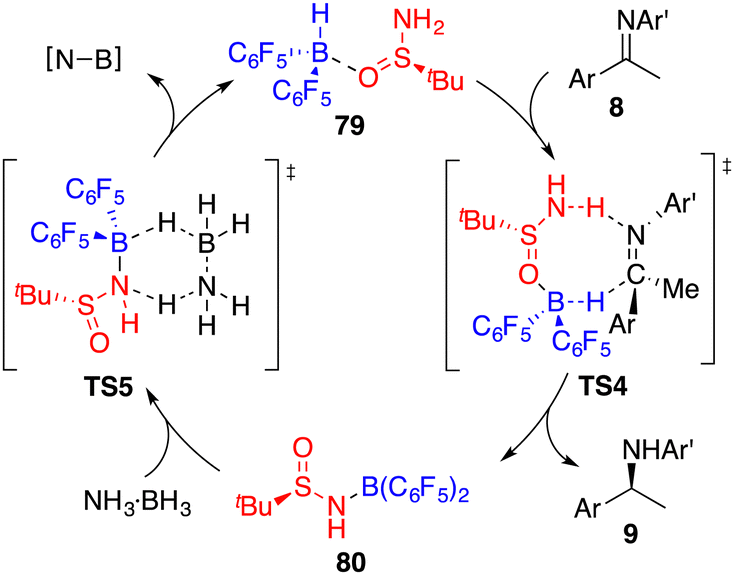
By utilizing chiral FLP 79 as a catalyst, a highly enantioselective transfer hydrogenation of imines 8 was successfully achieved, leading to the formation of optically active amines 9 in 78–99% yields with 84–95% ees (Scheme 44).58 The incorporation of a pyridine additive in this reaction was observed to play a crucial role in preventing the racemic reaction of imines 8 led by HB(C6F5)2, effectively capturing free HB(C6F5)2 in the reaction mixture. A plausible mechanism for this transformation is postulated based on both experimental observations and DFT calculations (Fig. 4). The transfer of hydrogen takes place through an 8-membered ring transition state TS4, and the regeneration of catalyst 79 from compound 80, utilizing ammonia borane, occurs via a 6-membered ring transition state TS5.
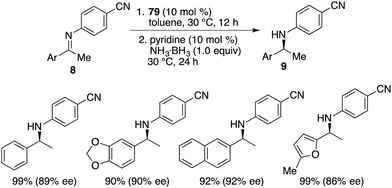 |
| | Scheme 44 Asymmetric transfer hydrogenation of imines. | |
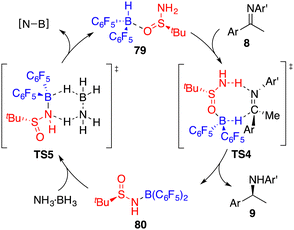 |
| | Fig. 4 Catalytic cycle for transfer hydrogenation of imines. | |
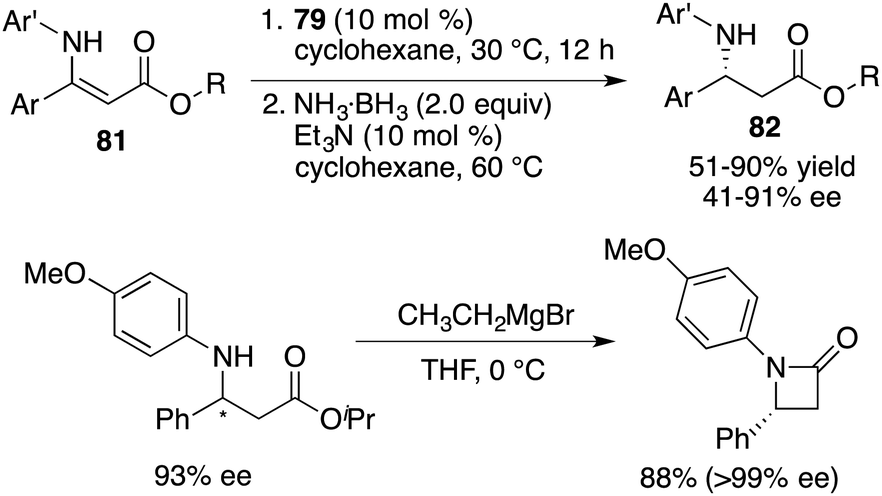
Building upon these findings, Du and co-workers extended the application of this catalyst to the asymmetric transfer hydrogenation of β-N-substituted enamino esters 81. This effort afforded the desired β-amino esters 82 in moderate to good yields with up to 91% ee (Scheme 45).59 An enantiopure β-lactam could be easily prepared from a β-amino ester via a single-step Grignard reaction. Furthermore, these studies served as inspiration for a novel approach in chiral ammonia borane regeneration. This strategy hinges on the liberation of H2 through interactions between CPA and ammonia borane, and the subsequent regeneration is facilitated through the hydrolysis of the resulting boro-phosphate. This regenerable ammonia borane system was also successfully applied to asymmetric transfer hydrogenation.60
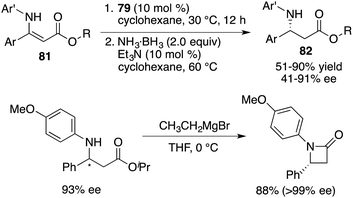 |
| | Scheme 45 Asymmetric transfer hydrogenations of β-N-substituted enamino esters. | |
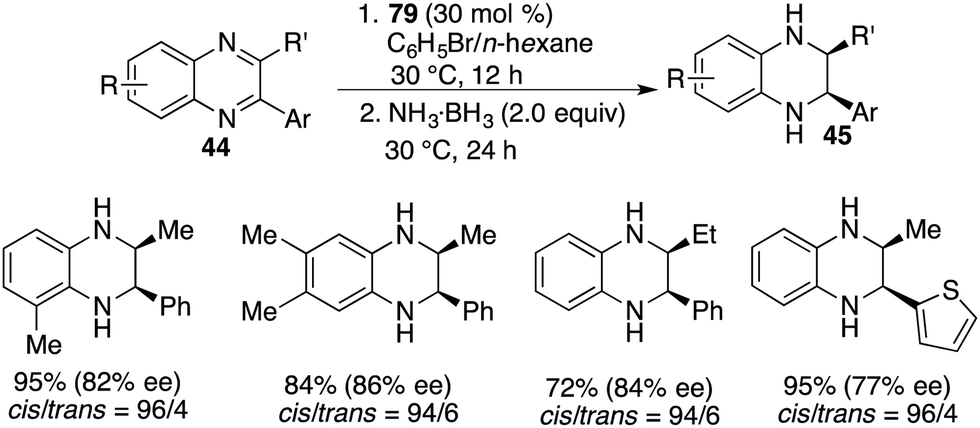
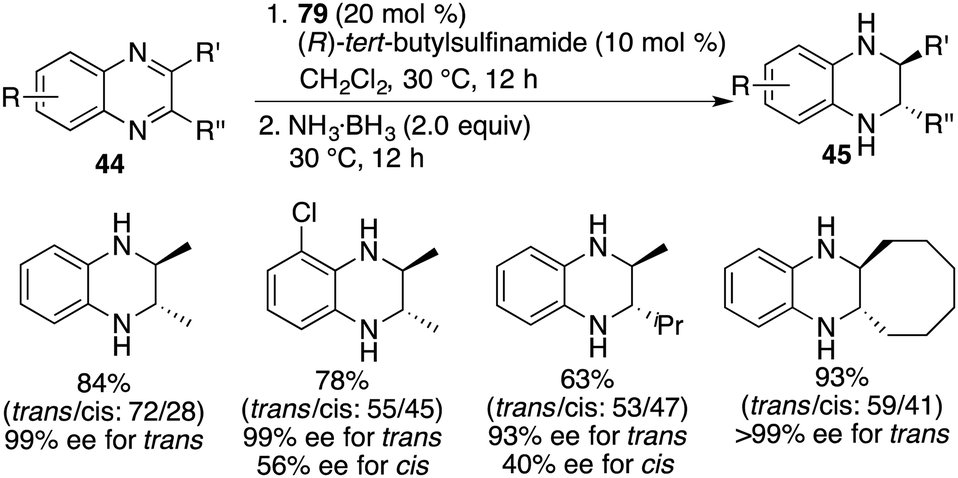
Chiral FLP 79 has also demonstrated its effectiveness as a catalyst for the asymmetric transfer hydrogenation of 2,3-disubstituted quinoxalines 44. In the cases of 2-alkyl-3-arylquinoxalines, the reaction predominantly led to cis-tetrahydroquinoxalines 45 as major products, exhibiting high reactivity and notable cis-stereoselectivity (Scheme 46).61 It's worth noting that the enantioselectivities achieved in the transfer hydrogenation were relatively low compared to the direct hydrogenation process. On the other hand, 2,3-dialkylquinoxalines, which were unreactive in the context of asymmetric hydrogenation, became suitable substrates for asymmetric transfer hydrogenation with ammonia borane as the reductant. This transformation furnished the trans isomers as the major products in moderate to good yields and with up to >99% ee (Scheme 47).61
 |
| | Scheme 46 Asymmetric transfer hydrogenation of 2-alkyl-3-arylquinoxalines. | |
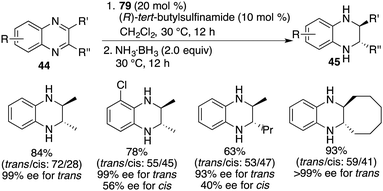 |
| | Scheme 47 Asymmetric transfer hydrogenation of 2,3-dialkylquinoxalines. | |
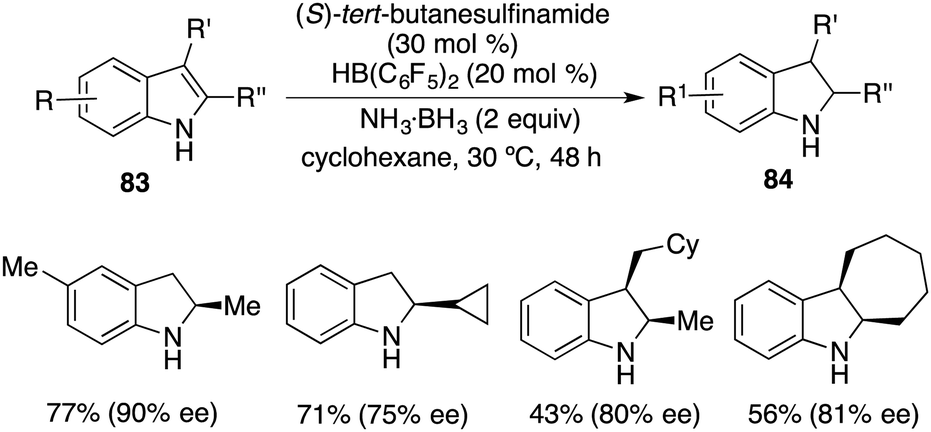
More recently, a notable advancement was reported involving the asymmetric transfer hydrogenation of N-unprotected indoles 83. This was facilitated by employing an FLP catalyst consisting of (S)-tert-butylsulfinamide and HB(C6F5)2. A variety of optically active indolines 84 were obtained in 40–78% yields with up to 90% ee (Scheme 48).62
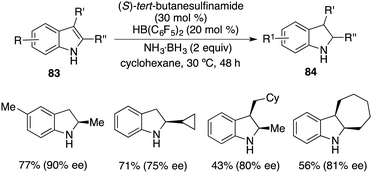 |
| | Scheme 48 Asymmetric transfer hydrogenation of N-unprotected indoles. | |
3.2 Asymmetric transfer hydrogenation with Hantzsch esters
Hantzsch esters have been widely employed as hydrogen donors in the context of asymmetric transfer hydrogenation reactions. Du and co-workers described an efficient transfer hydrogenation of imines using Hantzsch esters as a hydrogen source. With as low as 0.1 mol % B(C6F5)3, a variety of amines were furnished in 80–99% yields. Furthermore, by employing chiral borane 24c as the catalyst, the asymmetric transfer of imines 8 with Hantzsch ester 85 went smoothly to afford the corresponding amine products 9 in high yields. However, the ee values achieved in these reactions were in the range of low to moderate values (Scheme 49).63
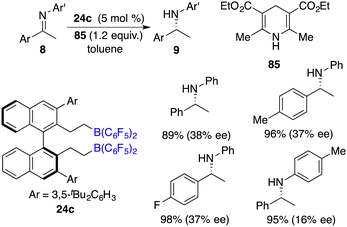 |
| | Scheme 49 Asymmetric transfer hydrogenation of imines with Hantzsch esters. | |
3.3 Asymmetric transfer hydrogenation with H2
Dinucleotide (NAD(P)H) mimics such as Hantzsch esters, dihydrophenanthridine, and benzothiazoline have been widely employed as stoichiometric hydrogen donors for asymmetric transfer hydrogenations. The transfer hydrogenation with catalytic amounts of hydrogen donors, which could be regenerated by the hydrogenation with H2 stands as a very attractive way. Zhou and Beller have already developed a practical strategy for the regenerable hydrogen donors facilitated by transition-metal catalyzed hydrogenation, respectively.64,65
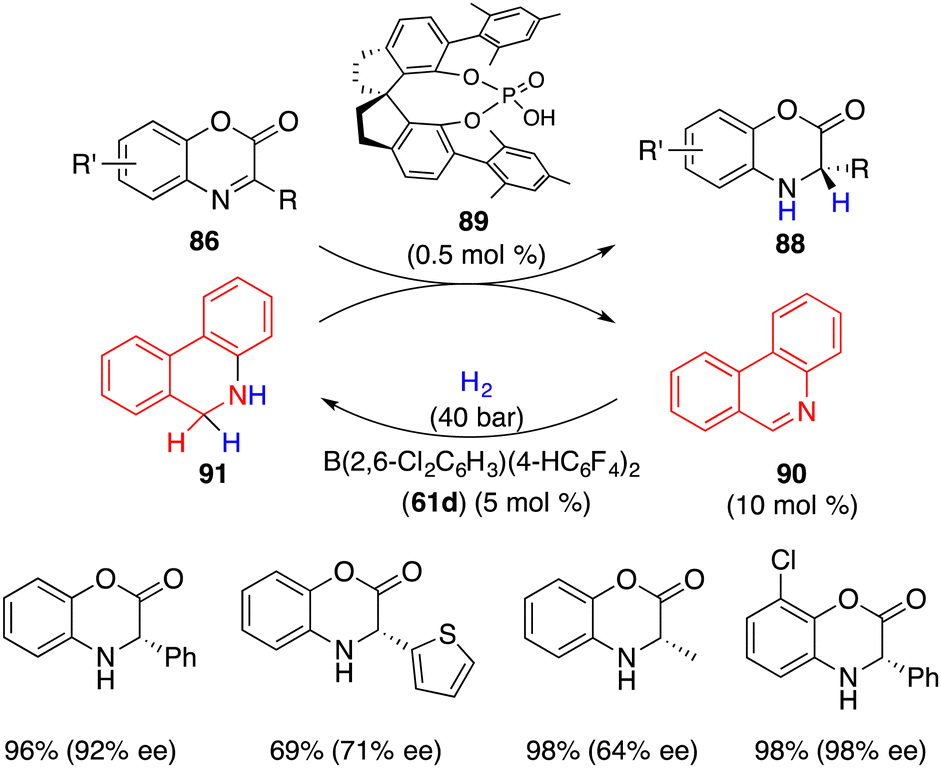
Recently, Du and co-workers reported a highly enantioselective transfer hydrogenation of electron-deficient benzoxazinones 86 with chiral phosphoric acid 89 under H2, wherein borane 61 promoted the hydrogenation of phenanthridine 90 to regenerate dihydrophenanthridine 91 as the hydrogen donor (Scheme 50).66 A wide range of desired products 88 were accessed in 69–99% yields with 64–99% ees. Notably, benzoxazinones 86 were inert substrates for the borane-catalyzed hydrogenation, and only 0.5 mol % chiral phosphoric acid 89 was enough for achieving excellent enantioselectivity. This work provides a possible resolution of unreactive substrates in the asymmetric FLP catalysis.
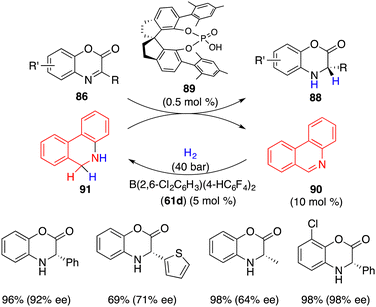 |
| | Scheme 50 Asymmetric transfer hydrogenation of benzoxazinones. | |
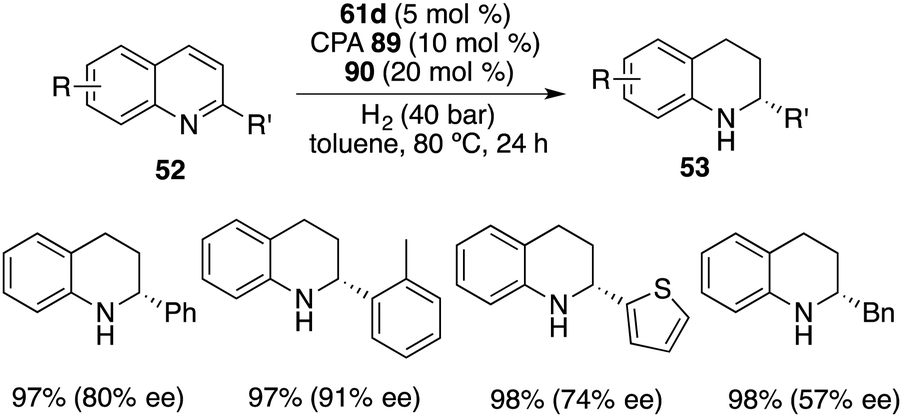
In contrast to benzoxazinones 86, 2-substituted quinolines 52 exhibited notable reactivity as substrates in the context of borane-catalyzed hydrogenation. Interestingly, the realm of asymmetric transfer hydrogenation was successfully extended to 2-substituted quinolines 52 through the utilization of borane 61d with chiral phosphoric acid 89 and 20 mol % phenanthridine 90. The reaction proceeded well to afford a variety of tetrahydroquinolines 53 in 95–99% yields with 39–91% ee (Scheme 51). Despite involving a borane-driven racemic pathway, satisfactory ee values could still be obtained.67
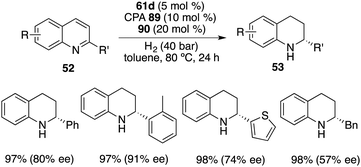 |
| | Scheme 51 Asymmetric transfer hydrogenation of quinolines. | |
4. Other asymmetric reactions
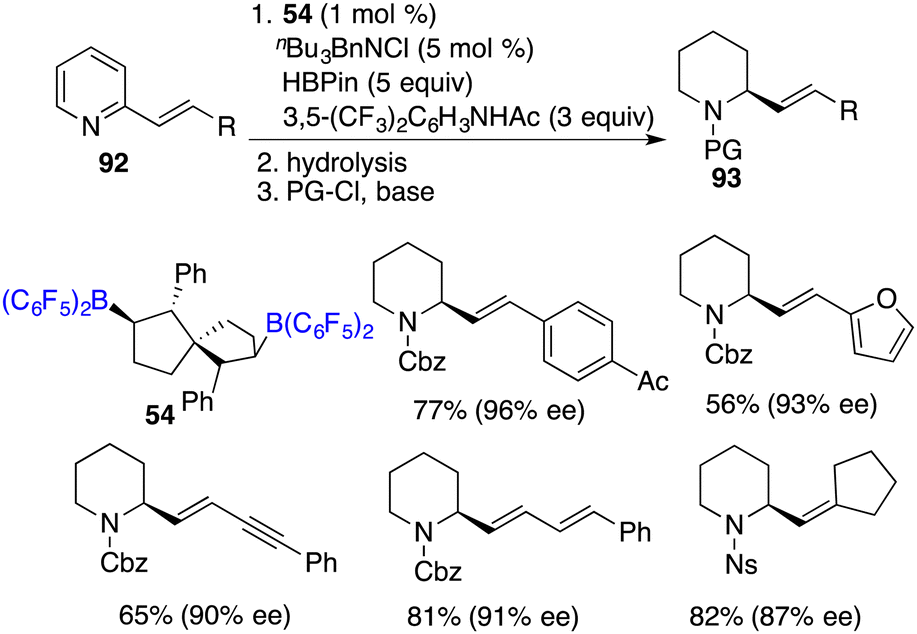
Recently, Wang and co-workers unveiled a highly chemoselective and enantioselective reduction of 2-vinyl-substituted pyridines 92 by the utilization of chiral spiro-bicyclic bisborane 54 as a catalyst and HBpin and an acidic amide as reducing reagents (Scheme 52).68 A diverse array of 2-vinyl-substituted piperidines 93 containing various functional groups were furnished in moderate to high yields with up to 96% ee. The reactions proceed through a cascade process involving 1,4-hydroboration followed by the transfer hydrogenation of a dihydropyridine intermediate. In this reduction, the double bond was retained and could be conveniently converted into natural products and other useful heterocyclic compounds.
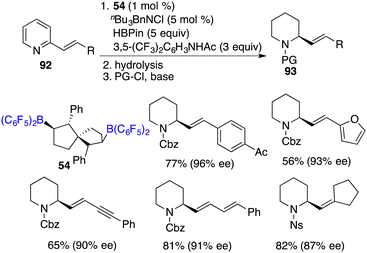 |
| | Scheme 52 Metal-free enantioselective reduction of 2-vinylpyridines. | |
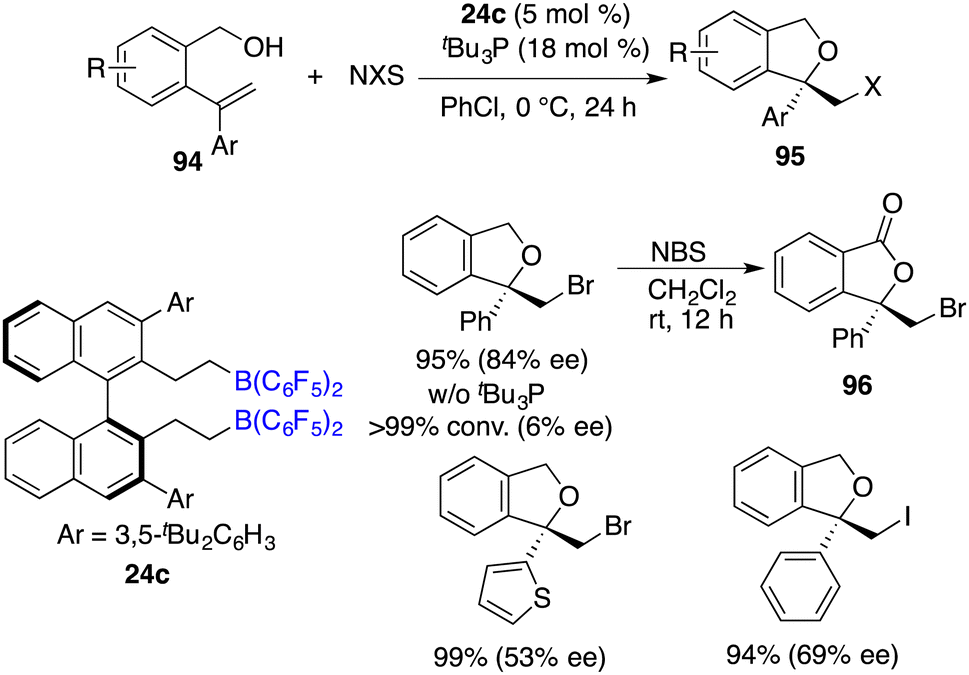
Asymmetric halocyclization of alkenes stands as a potent avenue for accessing chiral heterocyclic compounds. In this realm, Du and co-workers described an asymmetric halocyclization of 2-vinylbenzyl alcohols 94 with NBS or NIS by using a combination of chiral borane 24c and tBu3P as a catalyst (Scheme 53).69 A wide range of chiral 1,3-dihydroisobenzofuran derivatives 95 were obtained in good to excellent yields with 53–87% ee. A notable facet is the requisite involvement of a Lewis base, which emerges as an essential factor in achieving the high enantioselectivities. Chiral borane 24c exhibited remarkable activity, while the enantioselectivity was observed to be profoundly low. Moreover, the utilization of NBS as the halogen source facilitates an efficient and clean oxidation of compound 94, yielding the corresponding ester 96 without any loss of ee.
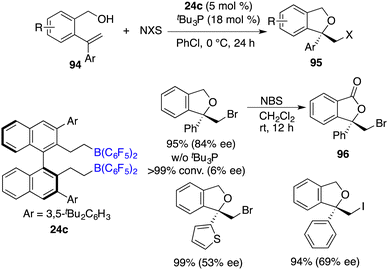 |
| | Scheme 53 Asymmetric halocyclization of 2-vinylbenzyl alcohols with chiral FLPs. | |
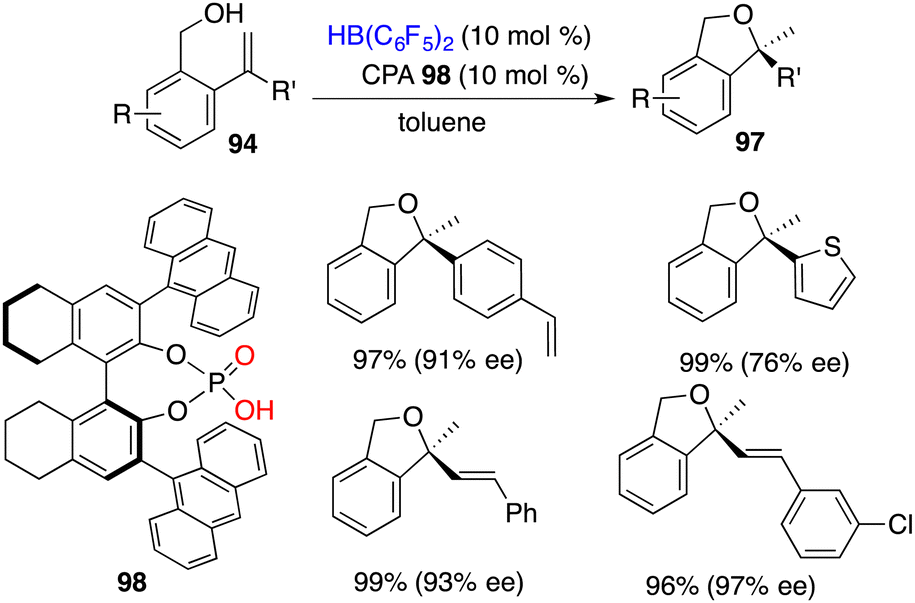
The asymmetric intramolecular hydroalkoxylation of alkenes represents an efficient and straightforward avenue for synthesizing optically active cyclic ethers. Very recently, Du and co-workers realized a highly efficient cyclization of 2-vinylbenzyl alcohols 94 employing 0.05 mol % B(C6F5)3 as a catalyst, delivering the desired products 97 in 82–99% yields.70 Chiral boranes derived from chiral dienes were not effective catalysts for the asymmetric hydroalkoxylation, leading to only less than 30% ee. A novel intramolecular FLP 98 derived in situ from chiral phosphoric acid and HB(C6F5)2 was designed and prepared. It proved to be an effective chiral catalyst for the hydroalkoxylation of 94, delivering optically active 1,3-dihydroisobenzofurans 97 in 78–99% yields with 60–97% ee (Scheme 54). For chiral boro-phosphates, the oxygen atom of P = O acts as a Lewis base and the boron atom acts as a Lewis acid, which serve to activate the O–H bond cooperatively and further promote the subsequent hydroalkoxylation process.
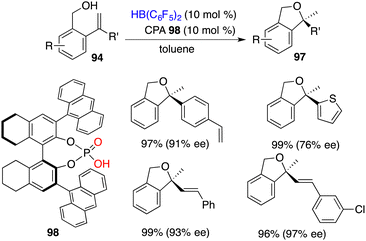 |
| | Scheme 54 Asymmetric hydroalkoxylation with CPA-derived FLPs. | |
5. Asymmetric reactions with chiral FLPs as bifunctional catalysts
FLPs have attractive dual roles in catalysis, one lies in their ability to activate the covalent bonds, thus facilitating the following transformation, and the other rests in their capacity to serve as Lewis bases and acid bifunctional catalysts.
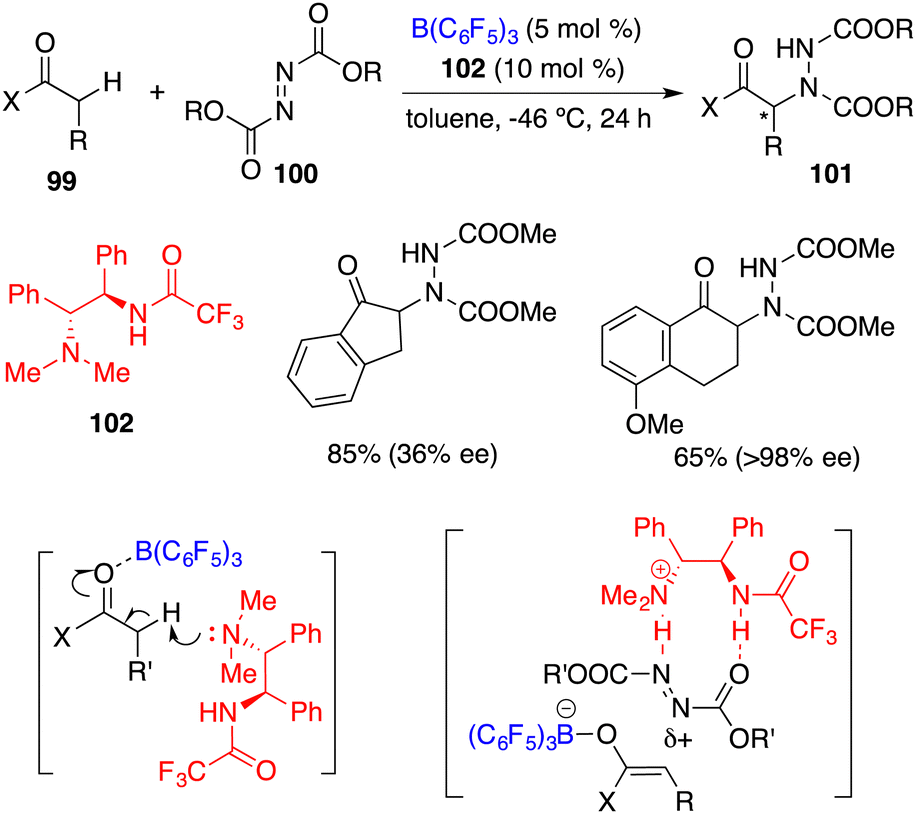
Wasa and co-workers reported an FLP comprising chiral base 102 and B(C6F5)3 for the enantioselective direct α-amination of carbonyl compounds 99 with azodicarboxylates 100, delivering the desired products 101 in 47–95% yields with up to 98% ee (Scheme 55).71 The acidity of the α-C–H bond of 99 was increased through the coordination of the carbonyl group with B(C6F5)3, which was subsequently deprotonated by the hindered amine 102 to form a tight ion pair consisting of a boron enolate and an ammonium cation. The ammonium cation served as the hydrogen-bond donor to activate 100 and facilitated the following nucleophilic attack by the boron enolate.
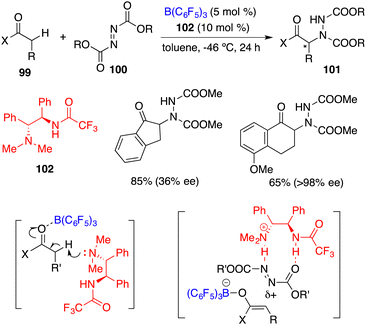 |
| | Scheme 55 Direct enantioselective α-amination of carbonyl compounds. | |
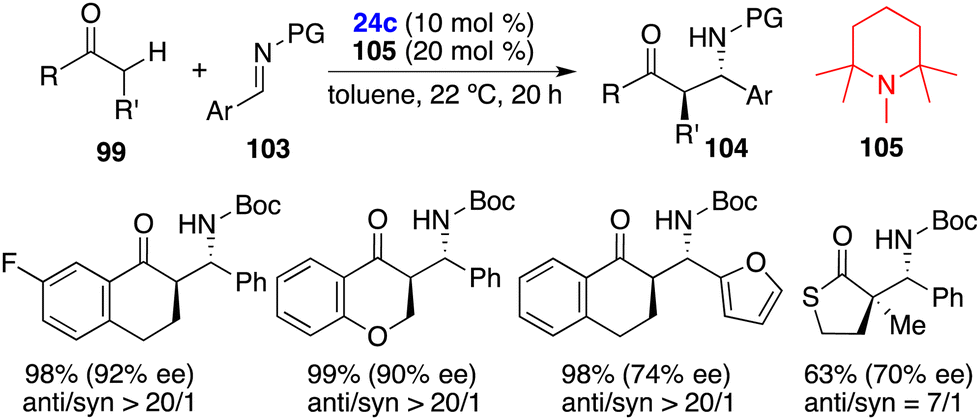
Furthermore, the same group advanced their exploration of an enantioselective direct Mannich-type reaction of carbonyl compounds 99 and aldimines 103 by using an FLP of chiral Lewis borane 24c and amine 105 (Scheme 56).72 A variety of optically active β-aminocarbonyl compounds 104 were obtained in high yields with up to >20/1 dr and 96% ee. The cooperative interactions of carbonyl compounds 99 with an unquenched Lewis pair of chiral borane 24c and amine 105 accelerate the generation of enolate and promote the subsequent addition to hydrogen-bond-activated aldimines 103.
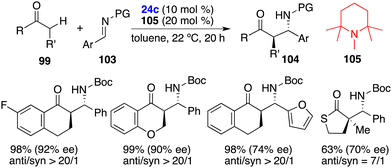 |
| | Scheme 56 Enantioselective direct Mannich-type reactions. | |
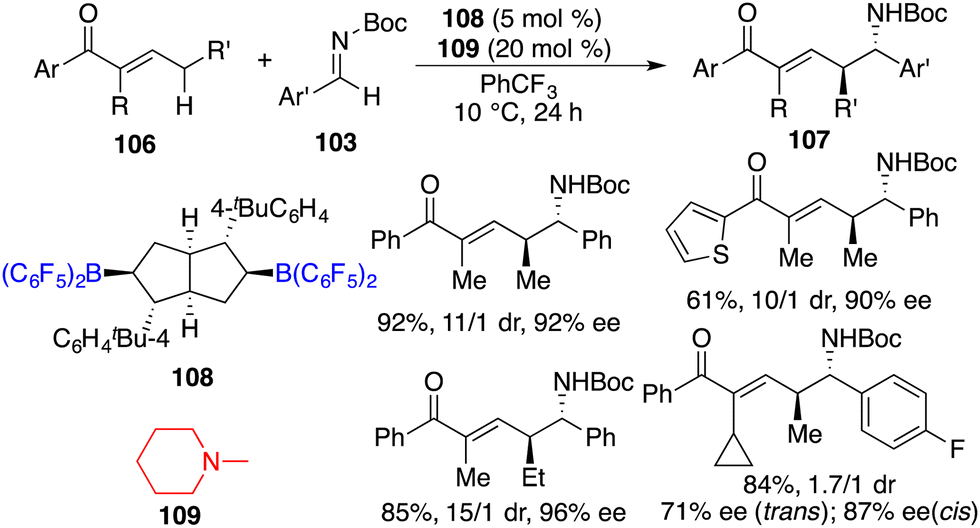
In a recent work, Wang and co-workers unveiled highly regio-, diastereo-, and enantioselective direct asymmetric vinylogous Mannich reactions of acyclic α,β-unsaturated ketones 106 and aldimines 103 under the cooperative catalysis of chiral bicyclic bisborane 108 and amine 109 (Scheme 57).73 A wide range of optically active δ-amino-α,β-unsaturated carbonyl derivatives 107 were delivered in good yields with up to 96% ee. Strong Lewis acidity, bulky steric hindrance, and structural rigidity of borane 108 catalysts were pivotal for the initiation of the reaction and for achieving high selectivities.
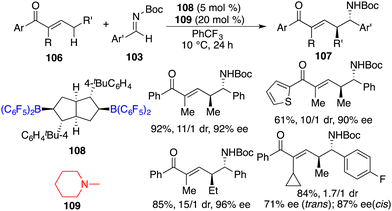 |
| | Scheme 57 Direct asymmetric vinylogous Mannich reactions of acyclic α,β-unsaturated ketones. | |
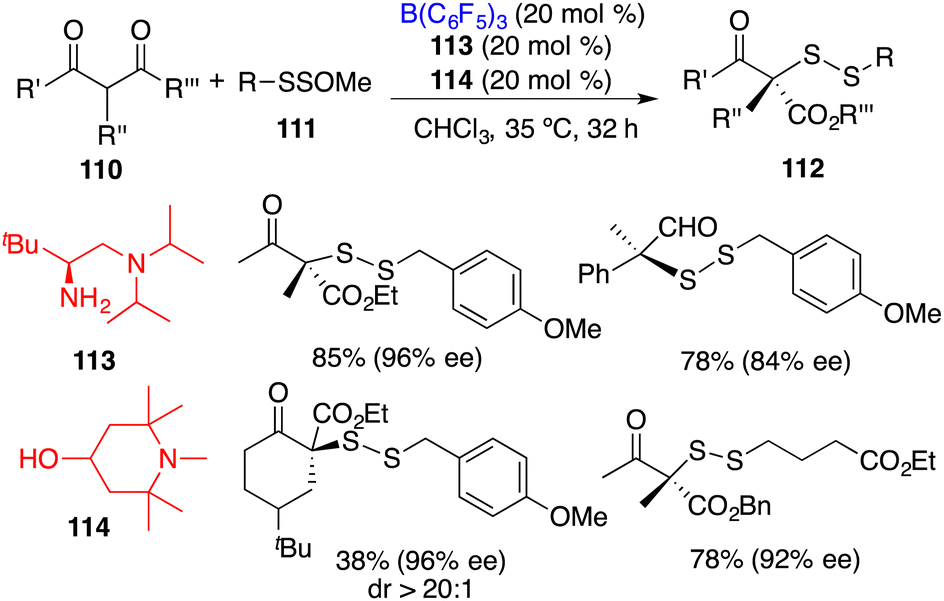
Luo and co-workers recently reported an asymmetric disulfuration of β-ketocarbonyls 110 with 111 by utilizing the combination of B(C6F5)3, chiral amine 113, and achiral amine 114 as a three-component Lewis acid–base catalyst (Scheme 58).74 A wide range of optically active disulfides 112 bearing S-containing tetrasubstituted carbon stereocenters were obtained in moderate to good yields with high levels of enantioselectivities. The bulky tertiary amine 114 served to activate the bulky primary–tertiary diamine–borane Lewis pair for enamine catalysis via frustrated interactions.
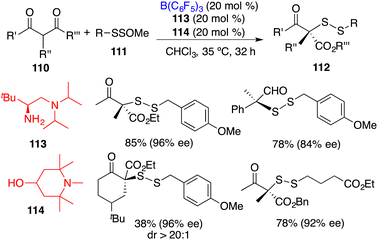 |
| | Scheme 58 Asymmetric disulfuration with β-ketocarbonyls. | |
Summary and outlook
The advent of FLP chemistry provides a novel category of catalysts in the realm of asymmetric catalysis, leading to remarkable advancements in recent years. As dual-component catalysts, both chiral Lewis acid-driven FLPs and chiral Lewis base-driven FLPs have been designed and successfully accessed. Furthermore, an innovative strategy has emerged wherein achiral FLPs synergize with other types of chiral catalysts, effectively broadening the scope of FLP applications. The domain of metal-free asymmetric hydrogenation has undergone a remarkable expansion encompassing a diverse array of unsaturated compounds, such as imines, enamines, N-heteroarenes, silyl enol ethers, simple ketones, and enones. FLP catalysis has been extended from hydrogenation to a diverse set of enantioselective reactions, such as hydrosilylation, transfer hydrogenation, and cyclization. The versatility of chiral FLPs extends to their utility as unquenched Lewis acid–base bifunctional catalysts for an array of asymmetric transformations. It is noteworthy that chiral FLPs have exhibited some interesting and distinctive catalytic activities and stereoselectivities, presenting a promising avenue for their increasing application in asymmetric catalysis.
Despite the significant progress made in the field, several formidable challenges remain in the realm of FLP catalysis. These challenges encompass catalytic efficiency, substrate scope, and the diversity of reaction types, all of which require further attention and innovative solutions in the future. A notable disparity exists between the catalytic efficiency of chiral FLPs and that of transition-metal catalysts. While transition-metal catalysts have demonstrated remarkable efficiency, chiral FLPs still lag behind in this regard. Addressing the underlying factors responsible for the deactivation of FLP catalysts and designing more efficient catalysts accordingly are important research subjects in this field. Additionally, expanding the repertoire of reaction types and extending the substrate scope are still in demand. A particularly promising avenue is the integration of chiral FLPs into heterogeneous catalysis, which also represents an important direction in the future.
Author contributions
All authors participated in the discussion and the writing of the review.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (21825108, 22271290, and 22331011).
Notes and references
-
(a)
Comprehensive Asymmetric Catalysis, ed. E. N. Jacobson, A. Pfaltz and H. Yamamoto, Springer-Verlag, Heidelberg, 1999 Search PubMed;
(b)
Privileged Ligands and Catalysts, ed. Q.-L. Zhou, Wiley-VCH, Weinheim, Germany, 2011 Search PubMed.
- G. C. Welch, R. R. San Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed.
- P. A. Chase, G. C. Welch, T. Jurca and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 46, 8050–8053 CrossRef CAS PubMed.
- For leading reviews, see:
(a) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS PubMed;
(b) T. Soos, Pure Appl. Chem., 2011, 83, 667–675 CAS;
(c) G. Erker, Pure Appl. Chem., 2012, 84, 2203–2217 CrossRef CAS;
(d) J. Paradies, Angew. Chem., Int. Ed., 2014, 53, 3552–3557 CrossRef CAS PubMed;
(e) D. W. Stephan, Acc. Chem. Res., 2015, 48, 306–316 CrossRef CAS PubMed;
(f) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed;
(g) D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018–10032 CrossRef CAS PubMed;
(h) D. W. Stephan, Science, 2016, 354, aaf7229 CrossRef PubMed;
(i) S. Arndt, M. Rudolph and A. S. K. Hashmi, Gold Bull., 2017, 50, 267–282 CrossRef CAS;
(j) V. Fasano and M. J. Ingleson, Synthesis, 2018, 1783–1795 CAS;
(k) H. Wang, Y. Zheng, Z. Pan, H. Fu, F. Ling and W. Zhong, Chin. J. Org. Chem., 2017, 37, 301–313 CrossRef CAS;
(l) N. Li and W.-X. Zhang, Chin. J. Chem., 2020, 38, 1360–1370 CrossRef CAS;
(m) J. Lam, K. M. Szkop, E. Mosaferi and D. W. Stephan, Chem. Soc. Rev., 2019, 48, 3592–3612 RSC;
(n) D. W. Stephan, J. Am. Chem. Soc., 2021, 143, 20002–20014 CrossRef CAS PubMed.
- D. Chen and J. Klankermayer, Chem. Commun., 2008, 2130–2131 RSC.
- For leading reviews, see:
(a) Y. Liu and H. Du, Huaxue Xuebao, 2014, 72, 771–777 CrossRef CAS;
(b) X. Feng and H. Du, Tetrahedron Lett., 2014, 55, 6959–6964 CrossRef CAS;
(c) L. Shi and Y.-G. Zhou, ChemCatChem, 2015, 7, 54–56 CrossRef CAS;
(d) L. C. Wilkins and R. L. Melen, Coord. Chem. Rev., 2016, 324, 123–129 CrossRef CAS;
(e) J. Paradies, Top. Organomet. Chem., 2017, 62, 193–216 CrossRef;
(f) W. Meng, X. Feng and H. Du, Acc. Chem. Res., 2018, 51, 191–201 CrossRef CAS PubMed;
(g)
X. Feng, W. Meng and H. Du, Frustrated Lewis Pair Catalyzed Asymmetric Reactions. In Frustrated Lewis Pairs; Molecular Catalysis, ed. J. C. Slootweg and A. R. Jupp, Springer Nature Switzerland AG, Cham, Switzerland, 2020, vol 2, pp. 29–86 Search PubMed;
(h) X. Feng, W. Meng and H. Du, Chin. J. Chem., 2022, 40, 1109–1116 CrossRef CAS;
(i) Z.-Y. Yang, M. Zhang and X.-C. Wang, Synthesis, 2022, 1527–1536 CAS.
- D. Chen, Y. Wang and J. Klankermayer, Angew. Chem., Int. Ed., 2010, 49, 9475–9478 CrossRef CAS PubMed.
- G. Ghattas, D. Chen, F. Pan and J. Klankermayer, Dalton Trans., 2012, 41, 9026–9028 RSC.
- A. Hamza, K. Sorochkina, B. Kótai, K. Chernichenko, D. Berta, M. Bolte, M. Nieger, T. Repo and I. Pápai, ACS Catal., 2020, 10, 14290–14301 CrossRef CAS.
- X. Wang, G. Kehr, C. G. Daniliuc and G. Erker, J. Am. Chem. Soc., 2014, 136, 3293–3303 CrossRef CAS PubMed.
- K.-Y. Ye, X. Wang, C. G. Daniliuc, G. Kehr and G. Erker, Eur. J. Inorg. Chem., 2017, 368–371 CrossRef.
- V. Sumerin, K. Chernichenko, M. Nieger, M. Leskelä, B. Rieger and T. Repo, Adv. Synth. Catal., 2011, 353, 2093–2110 CrossRef CAS.
- M. Lindqvist, K. Borre, K. Axenov, B. Kotai, M. Nieger, M. Leskela, I. Papai and T. Repo, J. Am. Chem. Soc., 2015, 137, 4038–4041 CrossRef CAS PubMed.
-
(a) D. J. Parks, R. E. v H. Spence and W. E. Piers, Angew. Chem., Int. Ed. Engl., 1995, 34, 809–811 CrossRef CAS;
(b) D. J. Parks, W. E. Piers and G. P. A. Yap, Organometallics, 1998, 17, 5492–5503 CrossRef CAS.
- Z. Cao and H. Du, Org. Lett., 2010, 12, 2602–2605 CrossRef CAS PubMed.
- Y. Liu and H. Du, J. Am. Chem. Soc., 2013, 135, 6810–6813 CrossRef CAS PubMed.
- X. Liu, T. Liu, W. Meng and H. Du, Org. Biomol. Chem., 2018, 16, 8686–8689 RSC.
- X.-S. Tu, N.-N. Zeng, R.-Y. Li, Y.-Q. Zhao, D.-Z. Xie, Q. Peng and X.-C. Wang, Angew. Chem., Int. Ed., 2018, 57, 15096–15100 CrossRef CAS PubMed.
- J. Lam, B. A. R. Günther, J. M. Farrell, P. Eisenberger, B. P. Bestvater, P. D. Newman, R. L. Melen, C. M. Crudden and D. W. Stephan, Dalton Trans., 2016, 45, 15303–15316 RSC.
- D. M. Mercea, M. G. Howlett, A. D. Piascik, D. J. Scott, A. Steven, A. E. Ashley and M. J. Fuchter, Chem. Commun., 2019, 55, 7077–7080 RSC.
- D. W. Stephan, S. Greenberg, T. W. Graham, P. Chase, J. J. Hastie, S. J. Geier, J. M. Farrell, C. C. Brown, Z. M. Heiden, G. C. Welch and M. Ullrich, Inorg. Chem., 2011, 50, 12338–12348 CrossRef CAS PubMed.
- Y. Zhang, S. Chen, A. M. Al-Enizi, A. Nafady, Z. Tang and S. Ma, Angew. Chem., Int. Ed., 2023, 62, e202213399 CrossRef CAS PubMed.
- X. Zhu and H. Du, Org. Lett., 2015, 17, 3106–3109 CrossRef CAS PubMed.
- S. Wei, X. Feng and H. Du, Org. Biomol. Chem., 2016, 14, 8026–8029 RSC.
-
(a) J. Mohr and M. Oestreich, Angew. Chem., Int. Ed., 2014, 53, 13278–13281 CrossRef CAS PubMed;
(b) J. Mohr, D. Porwal, I. Chatterjee and M. Oestreich, Chem. – Eur. J., 2015, 21, 17583–17586 CrossRef CAS PubMed.
- K. Yu, X. Feng and H. Du, Org. Biomol. Chem., 2022, 20, 3708–3711 RSC.
- K. Yu, W. Meng, X. Feng and H. Du, Org. Lett., 2023, 25, 3607–3610 CrossRef CAS PubMed.
- Z. Zhang and H. Du, Angew. Chem., Int. Ed., 2015, 54, 623–626 CrossRef CAS PubMed.
- Z. Zhang and H. Du, Org. Lett., 2015, 17, 2816–2819 CrossRef CAS PubMed.
- Z. Zhang and H. Du, Org. Lett., 2015, 17, 6266–6269 CrossRef CAS PubMed.
- C. Han, E. Zhang, X. Feng, S. Wang and H. Du, Tetrahedron Lett., 2018, 59, 1400–1403 CrossRef CAS.
- X. Li, J.-J. Tian, N. Liu, X.-S. Tu, N.-N. Zeng and X.-C. Wang, Angew. Chem., Int. Ed., 2019, 58, 4664–4668 CrossRef CAS PubMed.
- W. Wang, X. Feng and H. Du, Org. Biomol. Chem., 2016, 14, 6683–6686 RSC.
- Y. Liu and H. Du, J. Am. Chem. Soc., 2013, 135, 12968–12971 CrossRef CAS PubMed.
- H. Wang, R. Fröhlich, G. Kehr and G. Erker, Chem. Commun., 2008, 5966–5968 RSC.
- L. Greb, P. Oña-Burgos, A. Kubas, F. C. Falk, F. Breher, K. Fink and J. Paradies, Dalton Trans., 2012, 41, 9056–9060 RSC.
- S. Wei and H. Du, J. Am. Chem. Soc., 2014, 136, 12261–12264 CrossRef CAS PubMed.
- X. Ren, G. Li, S. Wei and H. Du, Org. Lett., 2015, 17, 990–993 CrossRef CAS PubMed.
-
(a) T. Mahdi and D. W. Stephan, J. Am. Chem. Soc., 2014, 136, 15809–15812 CrossRef CAS PubMed;
(b) T. Mahdi and D. W. Stephan, Angew. Chem., Int. Ed., 2015, 54, 8511–8514 CrossRef CAS PubMed.
-
(a) D. J. Scott, M. J. Fuchter and A. E. Ashley, J. Am. Chem. Soc., 2014, 136, 15813–15816 CrossRef CAS PubMed;
(b) Á. Gyömöre, M. Bakos, T. Földes, I. Pápai, A. Domján and T. Soós, ACS Catal., 2015, 5, 5366–5372 CrossRef;
(c) D. J. Scott, T. R. Simmons, E. J. Lawrence, G. G. Wildgoose, M. J. Fuchter and A. E. Ashley, ACS Catal., 2015, 5, 5540–5544 CrossRef CAS PubMed.
- B. Gao, X. Feng, W. Meng and H. Du, Angew. Chem., Int. Ed., 2020, 59, 4498–4504 CrossRef CAS PubMed.
- Y. Dai, W. Meng, X. Feng and H. Du, Chem. Commun., 2022, 58, 1558–1560 RSC.
- J. Chen, B. Gao, X. Feng, W. Meng and H. Du, Org. Lett., 2021, 23, 8565–8569 CrossRef CAS PubMed.
- J. Chen, W. Meng, X. Feng and H. Du, J. Org. Chem., 2022, 87, 10544–10549 CrossRef CAS PubMed.
- D. J. Parks and W. E. Piers, J. Am. Chem. Soc., 1996, 118, 9440–9441 CrossRef CAS.
- D. T. Hog and M. Oestreich, Eur. J. Org. Chem., 2009, 5047–5056 CrossRef CAS.
- D. Chen, V. Leich, F. Pan and J. Klankermayer, Chem. – Eur. J., 2012, 18, 5184–5187 CrossRef CAS PubMed.
- M. Mewald and M. Oestreich, Chem. – Eur. J., 2012, 18, 14079–14084 CrossRef CAS PubMed.
- X. Zhu and H. Du, Org. Biomol. Chem., 2015, 13, 1013–1016 RSC.
- T. Liu, X. Feng and H. Du, Tetrahedron Lett., 2022, 111, 154202 CrossRef CAS.
- S. Rendler and M. Oestreich, Angew. Chem., Int. Ed., 2008, 47, 5997–6000 CrossRef CAS PubMed.
- X. Ren and H. Du, J. Am. Chem. Soc., 2016, 138, 810–813 CrossRef CAS PubMed.
- L. Süsse, J. Hermeke and M. Oestreich, J. Am. Chem. Soc., 2016, 138, 6940–6943 CrossRef PubMed.
- X. Liu, Q. Wang, C. Han, X. Feng and H. Du, Chin. J. Chem., 2019, 37, 663–666 CrossRef CAS.
- Q. Wang, C. Han, X. Feng and H. Du, Chin. J. Org. Chem., 2019, 39, 2257–2263 CrossRef CAS.
- X. Ren, C. Han, X. Feng and H. Du, Synlett, 2017, 2421–2424 Search PubMed.
- Z. Luo, Z. Li, H. Zhao, J. Yang, L. Xu, M. Lei, Q. Fan and P. J. Walsh, Angew. Chem., Int. Ed., 2023, e202305449 CAS.
- S. Li, G. Li, W. Meng and H. Du, J. Am. Chem. Soc., 2016, 138, 12956–12962 CrossRef CAS PubMed.
- W. Zhao, X. Feng, J. Yang and H. Du, Tetrahedron Lett., 2019, 60, 1193–1196 CrossRef CAS.
-
(a) Q. Zhou, W. Meng, J. Yang and H. Du, Angew. Chem., Int. Ed., 2018, 57, 12111–12115 CrossRef CAS PubMed;
(b) Q. Zhou, X. Feng, J. Yang and H. Du, Chin. J. Org. Chem., 2019, 39, 2188–2195 CrossRef CAS.
- S. Li, W. Meng and H. Du, Org. Lett., 2017, 19, 2604–2606 CrossRef CAS PubMed.
- W. Zhao, Z. Zhang, X. Feng, J. Yang and H. Du, Org. Lett., 2020, 22, 5850–5854 CrossRef CAS PubMed.
- Q. Wang, J. Chen, X. Feng and H. Du, Org. Biomol. Chem., 2018, 16, 1448–1451 RSC.
- For selected examples, see:
(a) Q.-A. Chen, K. Gao, Y. Duan, Z.-S. Ye, L. Shi, Y. Yang and Y.-G. Zhou, J. Am. Chem. Soc., 2012, 134, 2442–2448 CrossRef CAS PubMed;
(b) Z.-P. Chen, M.-W. Chen, R.-N. Guo and Y.-G. Zhou, Org. Lett., 2014, 16, 1406–1409 CrossRef CAS PubMed;
(c) M.-W. Chen, B. Wu, Z.-P. Chen, L. Shi and Y.-G. Zhou, Org. Lett., 2016, 18, 4650–4653 CrossRef CAS PubMed;
(d) Z.-B. Zhao, X. Li, M.-W. Chen, B. Wu and Y.-G. Zhou, Chin. J. Chem., 2020, 38, 1691–1695 CrossRef CAS.
-
(a) L.-Q. Lu, Y. Li, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 8382–8386 CrossRef CAS PubMed;
(b) L.-Q. Lu, Y. Li, K. Junge and M. Beller, J. Am. Chem. Soc., 2015, 137, 2763–2768 CrossRef CAS PubMed.
- B. Gao, W. Meng, X. Feng and H. Du, Org. Lett., 2022, 24, 3955–3959 CrossRef CAS PubMed.
- B. Gao, Z. Han, W. Meng, X. Feng and H. Du, J. Org. Chem., 2023, 88, 3335–3339 CrossRef CAS PubMed.
- J.-J. Tian, Z.-Y. Yang, X.-S. Liang, N. Liu, C.-Y. Hu, X.-S. Tu, X. Li and X.-C. Wang, Angew. Chem., Int. Ed., 2020, 59, 18452–18456 CrossRef CAS PubMed.
- C. Han, X. Feng and H. Du, Org. Lett., 2021, 23, 7325–7329 CrossRef CAS PubMed.
- C. Han, W. Meng, X. Feng and H. Du, Angew. Chem., Int. Ed., 2022, e202200100 CAS.
- M. Shang, X. Wang, S. M. Koo, J. Youn, J. Z. Chan, W. Yao, B. T. Hastings and M. Wasa, J. Am. Chem. Soc., 2017, 139, 95–98 CrossRef CAS PubMed.
- M. Shang, M. Cao, Q. Wang and M. Wasa, Angew. Chem., Int. Ed., 2017, 56, 13338–13341 CrossRef CAS PubMed.
- J.-J. Tian, N. Liu, Q.-F. Liu, W. Sun and X.-C. Wang, J. Am. Chem. Soc., 2021, 143, 3054–3059 CrossRef CAS PubMed.
- Q. Zhang, Y. Li, L. Zhang and S. Luo, Angew. Chem., Int. Ed., 2021, 60, 10971–10976 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Wei
Meng
ab,
Wei
Meng