
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The crucial role of silver(I)-salts as additives in C–H activation reactions: overall analysis of their versatility and applicability
Renato L.
de Carvalho
 a,
Emilay B. T.
Diogo
a,
Simon L.
Homölle
b,
Suman
Dana
b,
Eufrânio N.
da Silva Júnior
*a and
Lutz
Ackermann
*b
a,
Emilay B. T.
Diogo
a,
Simon L.
Homölle
b,
Suman
Dana
b,
Eufrânio N.
da Silva Júnior
*a and
Lutz
Ackermann
*b
aInstitute of Exact Sciences, Department of Chemistry, Federal University of Minas Gerais-UFMG, 31270-901, Belo Horizonte, MG, Brazil. E-mail: eufranio@ufmg.br; Web: https://www.eufraniolab.com
bInstitut für Organische und Biomolekulare Chemie and Wöhler Research Institute for Sustainable Chemistry, Georg-August-Universität Göttingen, Tammannstrasse 2, 37077 Göttingen, Germany. E-mail: Lutz.Ackermann@chemie.uni-goettingen.de; Web: https://www.ackermann.chemie.uni-goettingen.de/ Web: https://wisch.chemie.uni-goettingen.de/
First published on 1st September 2023
Abstract
Transition-metal catalyzed C–H activation reactions have been proven to be useful methodologies for the assembly of synthetically meaningful molecules. This approach bears intrinsic peculiarities that are important to be studied and comprehended in order to achieve its best performance. One example is the use of additives for the in situ generation of catalytically active species. This strategy varies according to the type of additive and the nature of the pre-catalyst that is being used. Thus, silver(I)-salts have proven to play an important role, due to the resulting high reactivity derived from the pre-catalysts of the main transition metals used so far. While being powerful and versatile, the use of silver-based additives can raise concerns, since superstoichiometric amounts of silver(I)-salts are typically required. Therefore, it is crucial to first understand the role of silver(I) salts as additives, in order to wisely overcome this barrier and shift towards silver-free systems.
 Renato L. de Carvalho | Renato Lúcio de Carvalho concluded his doctorate in 2022 at the Federal University of Minas Gerais (UFMG), after developing his research under the supervision of Prof. Dr Eufrânio N. da Silva Júnior, towards the development of new methods involving C–H annulation reactions for achieving new quinoidal bioactive compounds. At present, he is a postdoctoral fellow at the University of Liverpool, United Kingdom. He has experience in synthetic methods for organic compounds, for example, C–C coupling, C–H activation, synthesis and application of arynes, reductive/oxidative methods, chemistry of carbonylated compounds and 1,3-dipolar cycloadditions. His research interests extend to biomedicinal chemistry, catalysis in general, and studies related to potential pharmacological molecules, on both new anticancer and trypanocidal compounds. |
 Emilay B. T. Diogo | Emilay Baessa Teixeira Diogo received her degree in Chemistry in 2015. In 2021, she completed her MSc degree at the Federal Center for Technological Education of Minas Gerais (CEFET-MG) under the supervision of Prof. Dr Emerson Fernandes Pedroso. In the same year, she started her PhD studies at the Federal University of Minas Gerais (UFMG) under the supervision of Prof. Dr Eufrânio N. da Silva Júnior and Prof. Dr Cynthia L. M. Pereira, working with methods involving C–H bond activation and electroorganic chemistry for the synthesis of new bioactive compounds. Her research interests are focused on C–H bond activation reactions, mechanistic investigations, electrosynthesis and synthesis of naphthoquinoidal bioactive compounds. |
 Simon L. Homölle | Simon L. Homölle studied chemistry at the Georg-August-University Göttingen and obtained his MSc degree in 2021 under the supervision of Prof. Dr Lutz Ackermann. During his studies he joined the group of Prof. Dr W. Hell at the MPI for a research stay in biophysical chemistry. He presented his Master's thesis at Symrise AG in Holzminden in the group of Dr L. Zygalski working on the synthesis of long-chain polyunsaturated aldehydes. In the same year, he started his doctoral studies in the group of Prof. Dr Lutz Ackermann. His research subject is synthetic organic electrochemistry, with focus on transition-metal-catalyzed C–H activation and electrocatalysis. Besides his PhD he is the spokesperson of the Göttinger GDCh-JCF and was responsible member for the organisation of the annual FJS 2022 in Hannover. |
 Suman Dana | Suman Dana was born in 1992, in West Bengal, India. He obtained his bachelor's degree in Chemistry (Honours) from Bankura Christian College, India, and Master's degree (Gold Medallist) from the School of Chemistry, University of Hyderabad, India. He received his PhD degree in April 2021 from Indian Institute of Technology, Madras, under the supervision of Dr Mahiuddin Baidya's. In 2021, he joined the research group of Prof. Dr Lutz Ackermann at the Georg-August-Universität Göttingen as a post-doctoral researcher, where he is involved in developing new asymmetric transformations using metallaelectrocatalyzed C–H bond activation reactions. |
 Eufrânio N. da Silva Júnior | Eufrânio N. da Silva Júnior received his PhD from the University of Brasilia (UnB) in 2009. In 2010, he started his independent career as an adjunct professor at the Federal University of Minas Gerais, where he was promoted to Associate Professor in 2018. He has received the RSC/BMOS Young Investigator Award (2015), Jones Travelling Fellowship (2015), MedChemComm New Talent: Americas (2017), Themed collection’Contributors to the emerging investigators issue 2018’ of ChemSocRev and CNPq-SBQ-RSC Brazilian Chemistry Certificate of Distinction (2019). In 2018, he received the Capes-Humboldt Research Fellowship for experienced researchers, spending a year as visiting professor at Georg-August-Universität Göttingen hosted by Prof. Ackermann. He is a member of the Brazilian Academy of Science and Fellow of the Royal Society of Chemistry. His research interests are focused on C–H bond activation reactions and synthesis of heterocyclic and naphthoquinoidal bioactive and fluorescent compounds. |
 Lutz Ackermann | Lutz Ackermann studied chemistry at Kiel University (Germany), and undertook his PhD with Prof. Alois Fürstner at the Max-Plank-Institut für Kohlenforschung (Mülheim an der Ruhr, 2001). After a postdoctoral stay at the University of California, Berkeley, with Prof. Robert G. Bergman, he initiated his independent research career in 2003 at the Ludwig-Maximilians-Universität München. In 2007, he became a Full Professor (W3) at Georg-August-Universität Göttingen. His recent awards and distinctions include an AstraZeneca Excellence in Chemistry Award (2011), an ERC consolidator and Advance Grants (2012, 2021) and a Gottfried Wilhelm Leibniz-Preis (2017). The development and application of novel concepts for sustainable catalysis are his major current research interests, with a topical focus on C–H activation, mechanistic studies, Earth-abundant 3d transition metal catalysis and electrochemistry. He has recently reached over 500 publications on the subject of C–H activation. |
1. Introduction
In the past three decades, direct transition-metal catalyzed organometallic functionalization of C–H bonds, typically termed “C–H activation”, has exhibited tremendous potential in synthetic organic chemistry.1 This process exploits ubiquitous C–H bonds as a staple to functionalize organic molecules, enhancing the step- and atom-economies of the process. The inception and growth of C–H activation reactions facilitated the rapid construction of numerous synthetically challenging targets and thus, they have been extensively used as a potential approach for the late-stage diversification of bioactive molecules.2 Notably, this innovative approach somewhat depends on the nature of the applied catalyst, the use (or not) of removable directing groups, and the structure of the substrates (Scheme 1).1,3–6 Furthermore, specific additives play a decisive role in the process and determine the catalytic efficacy as well as turnover.5 In this context, the use of Ag(I)-additives is common in numerous C–H activation reactions in combination with different transition-metal catalysts, such as Ir, Rh, Ru, Co, Pd, etc.1 Even, the presence of these additives sometimes decides the fate of the transformation, making them irreplaceable in this domain.5b–d These additives usually help the in situ generation of catalytically active species, originating usually from dimeric ruthenium-(p-cymene),1i–l rhodium-Cp*,1b,f iridium-Cp*1b,c and cobalt-Cp*1b half-sandwich pre-catalysts (Scheme 1). In contrast, for palladium catalysis, the role of the additive is shifted towards the formation of heterodimeric Pd–Ag complexes (Scheme 1).5b–d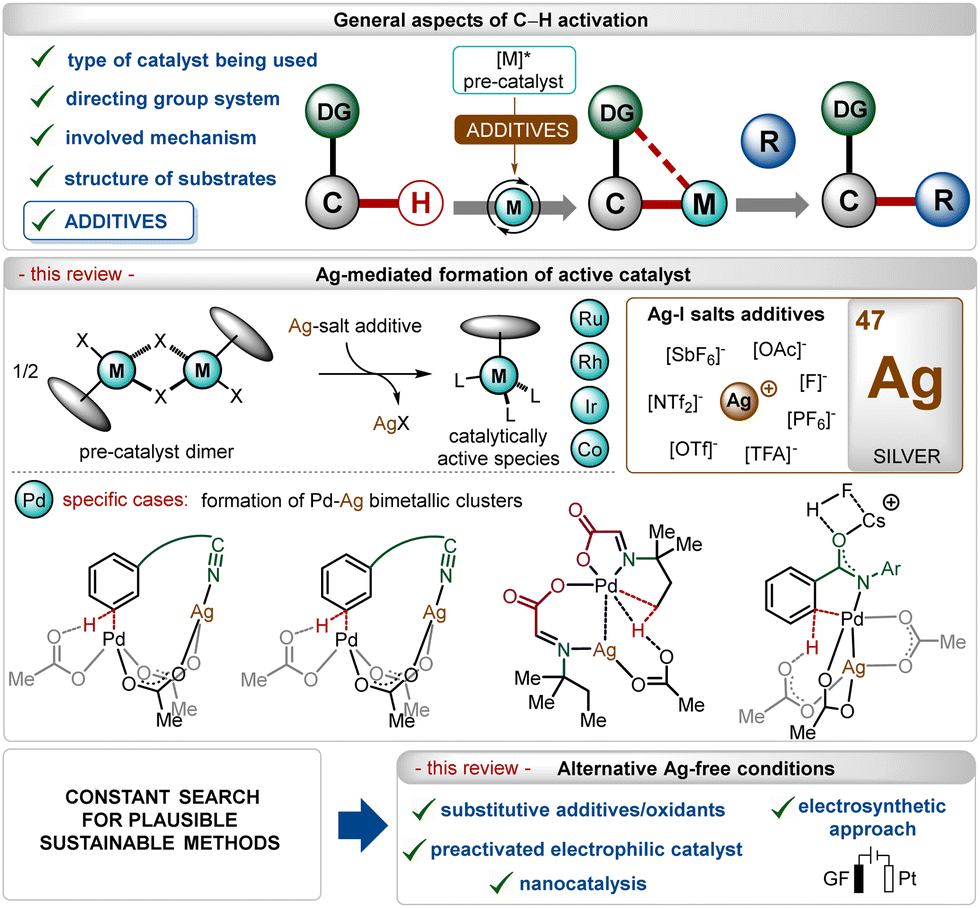 | ||
| Scheme 1 Overview. | ||
In this field, among other common additives such as carboxylates, carbonates, carboxylic acids,7 and amine derivates,8 silver(I)-salts have turned out as an essential additive owing to several reasons.5b–d,9,10 First of all, the quick and effective interaction between the Ag(I)-cation and a halide ion (X−) led to the formation of AgX and this halophilicity of Ag(I)-salts stands out from other common metallic additives. Furthermore, Ag(I)-salts are recognized for their ease of forming bimetallic species bridged by carboxylate ligands, which can effectuate many transition-metal catalyzed transformations. Often, the oxidizing and Lewis-acidic properties of Ag(I)-salts also facilitate their use in C–H activation reactions.
Previously published reviews on C–H activation focused on different aspects,4b–f including the nature of the directing groups,4g–j transition-metal catalysis,4k–n even more specifically related to 3d-metals,4o–q but only a limited number rationalized the use of additives,5a,7a and only fewer discussed the key role of silver(I)-based additives.5d,15 In this review, we have summarized discussions about general and specific applications of Ag(I)-salts as a pre-catalyst activator on different transition-metal catalyzed C–H activation reactions, which has not been specifically elaborated earlier. The review excludes the discussion on strategies involving Ag(I)-salts as oxidants and specifically focuses on their utility as a critical additive in the transformation. Owing to the enormous number of reports available in this regime, specific examples have been selected and analyzed. The following sections will highlight a precise overview of the role of Ag(I)-salt additives along with further discussion about the replacement of these salts with other materials and activating methods. Notably, on many occasions, an excess of inherently toxic silver(I)-salts is required to promote the transformation.10b–e Thus, there is an intrinsic urge to shift the methodologies towards Ag-free conditions, improving the sustainable features of these reactions.11 This aspect has also been covered briefly through a precise discussion on enabling alternatives recognized to execute C–H activation reactions without Ag(I)-additives (Scheme 1).
2. Ruthenium catalysis with silver additives
Since the pioneering contribution by Murai and co-workers,12 ruthenium-catalyzed C–H activation reactions have flourished significantly in the past two decades with major contributions from Ackermann,1j–m,13 Dixneuf,14 and Miura,15 among others.1j–m,16,17In this regime, the most used catalyst to enable C–H activation reactions is inexpensive, bench-stable, and commercially available [Ru(p-cymene)Cl2]2.1j–m This classical half-sandwich pre-catalyst has proven to be significantly reactive in combination with large varieties of strongly as well as weakly coordinating directing groups. However, in order to obtain superior interaction with the substrates, it must be converted to its respective monomeric cationic complex. In their early findings, Ackermann validated the efficacy of the cationic Ru(II)-complex through oxidative annulation reactions with alkynes.13h,i In this context, for the in situ generation of the cationic Ru(II)-complex, silver-based additives have emerged as a general candidate, which facilitate the formation of the cationic Ru(II)-complex through the precipitation of AgCl. Generally, this process is combined with an additional carboxylate ligand, such as acetate,1j–m which coordinates with the electron-deficient Ru(II)-center with η3-coordination. The carboxylate ligand plays a crucial role in the C–H activation step through concerted metalation deprotonation (CMD) or more often a base-assisted electrophilic substitution (BIES) pathway. Notably, the electron-deficient Ru(II)-cationic complex exhibits enhanced interactions with the weakly coordinating O-centered directing groups and furthers the electrophilic C–H activation strategy through decreasing the energy of LUMO of the Ru-catalyst.18
Amongst several possible silver salts, silver hexafluoroantimonate (AgSbF6) is one of the commonly used additives. It has found widespread application in alkenylation,13j,h,15d,19 alkylation,13a,20 alkynylation,21 allylation,22 arylation,23 amidation,24 and annulation13c,i,25 reactions (Scheme 2). In this realm, the Ackermann group has a contribution, where their initial findings disclosed the oxidative olefination of carboxylic acid esters and carbamates, exploiting weakly coordinating O-centered directing groups.13j,h
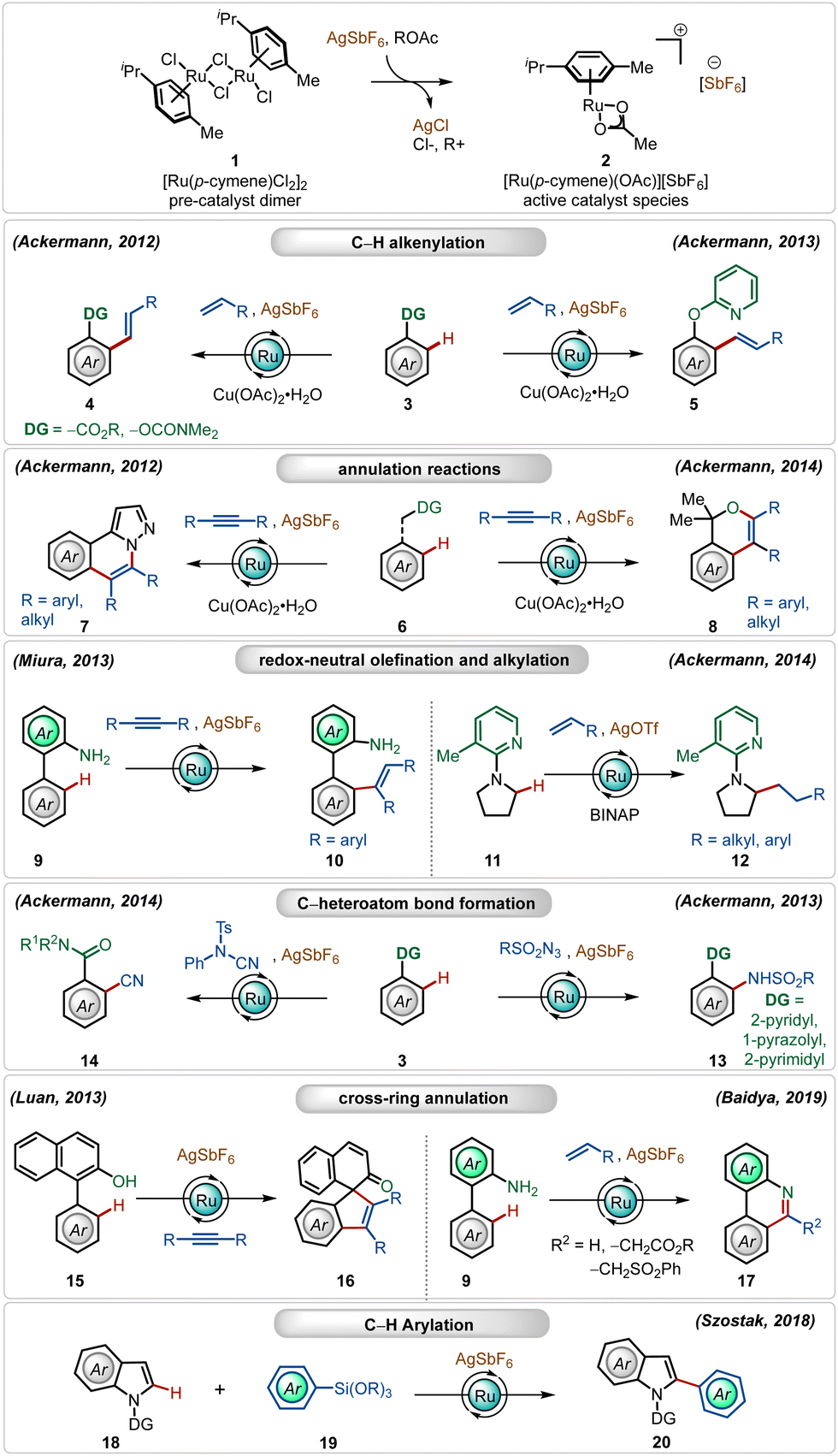 | ||
| Scheme 2 Overview of Ag-assisted [Ru]-catalyzed C–H activations. | ||
Later, they also described ortho-C–H olefination of phenols using a removable 2-pyridyl directing group.13e They further adopted this cationic Ru(II)-catalyst promoted approach for oxidative annulation reactions with internal alkynes, constructing valuable heterocyclic architectures from substituted 1H-pyrazoles.13i This strategy was also successful executing redox-neutral C–H activation reactions, where Miura reported an ortho-olefination of arenes through alkyne hydroarylation.15c
Ackermann and co-workers further found that the cationic Ru(II)-complex could execute challenging redox-neutral alkylation of C(sp3)–H bonds of pyrrolidines using olefins as the alkyl source, where the AgOTf additive was found to be optimal in combination with the RuCl2(PPh3)3 catalyst and the BINAP ligand.13b In this context, it is worth noting that Ru(II)-catalyzed C(sp3)–H bond functionalization reactions are rare in the literature. Simultaneously, they also elaborated on challenging amidation24 and cyanation13d reactions using the bench-stable Ru(II)-catalyst along with AgSbF6. In all these transformations, the Ag(I)-additive played a pivotal role, the absence of which offered deteriorated outcome. Even cross-ring annulation reactions with alkynes and activated olefins25 and C-2 arylation of indoles23 were also feasible, providing modular access to a large variety of functionalized heterocycles.
Among their other significant contribution in this domain, in 2018, the Ackermann group developed the first example of a weak coordination assisted distal C(sp2)–H functionalization of aryl acetamide analogues (Scheme 3).26 The oxidative olefination reaction tolerated diverse aryl acetamides with ease and consisted a broad scope. The reaction was executed using a cationic Ru-complex, formed in situ in the presence of AgSbF6 and the transformation failed in the absence of AgSbF6. The plausible mechanism involved a base-assisted internal electrophilic type substitution (BIES) pathway for the C–H activation step, forming the Ruthenacycle intermediate 23-A, which on exposure to the olefin underwent migratory insertion followed by β-hydride elimination to generate the olefinated product. The Ru(II)-catalyst was regenerated by the Cu(II) oxidant. Furthermore, this catalytic strategy was also applicable for redox-neutral hydroarylation of alkynes (25), albeit in this case adamantane carboxylic acid additive was necessary instead of Cu(II)-salt.
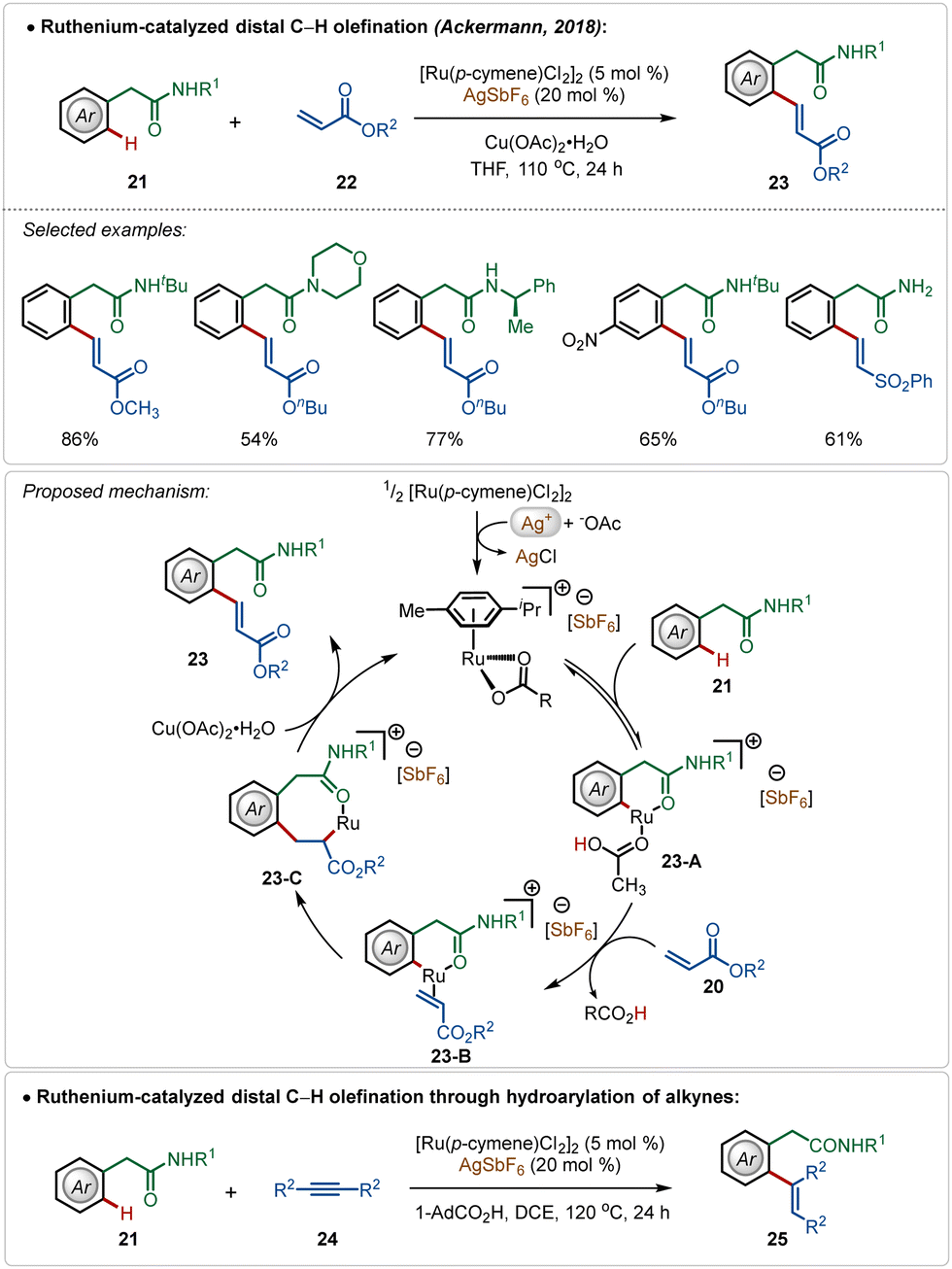 | ||
| Scheme 3 Ruthenium-catalyzed distal C–H alkenylation. | ||
In 2020, Chatani and co-workers described an interesting example of directed ruthenium-catalyzed ortho-C–H acyloxylation on 2-aroyl-imidazoles, using simple carboxylic acids as reaction partners (Scheme 4).27 Using this method, varieties of phenyl ester derivatives were synthesized in moderate to good yields. The presence of radical quencher TEMPO did not prevent the reaction, excluding the involvement of radical intermediates. Based on the plausible mechanistic proposal, the reaction starts with the usual formation of the active catalyst from the pre-catalyst in the presence of silver(I)-carbonate and carboxylic acid. The active catalyst promoted a reversible C–H activation reaction to form intermediate 28-B, which upon reductive elimination afforded the desired product along with the regeneration of the active ruthenium-catalyst.
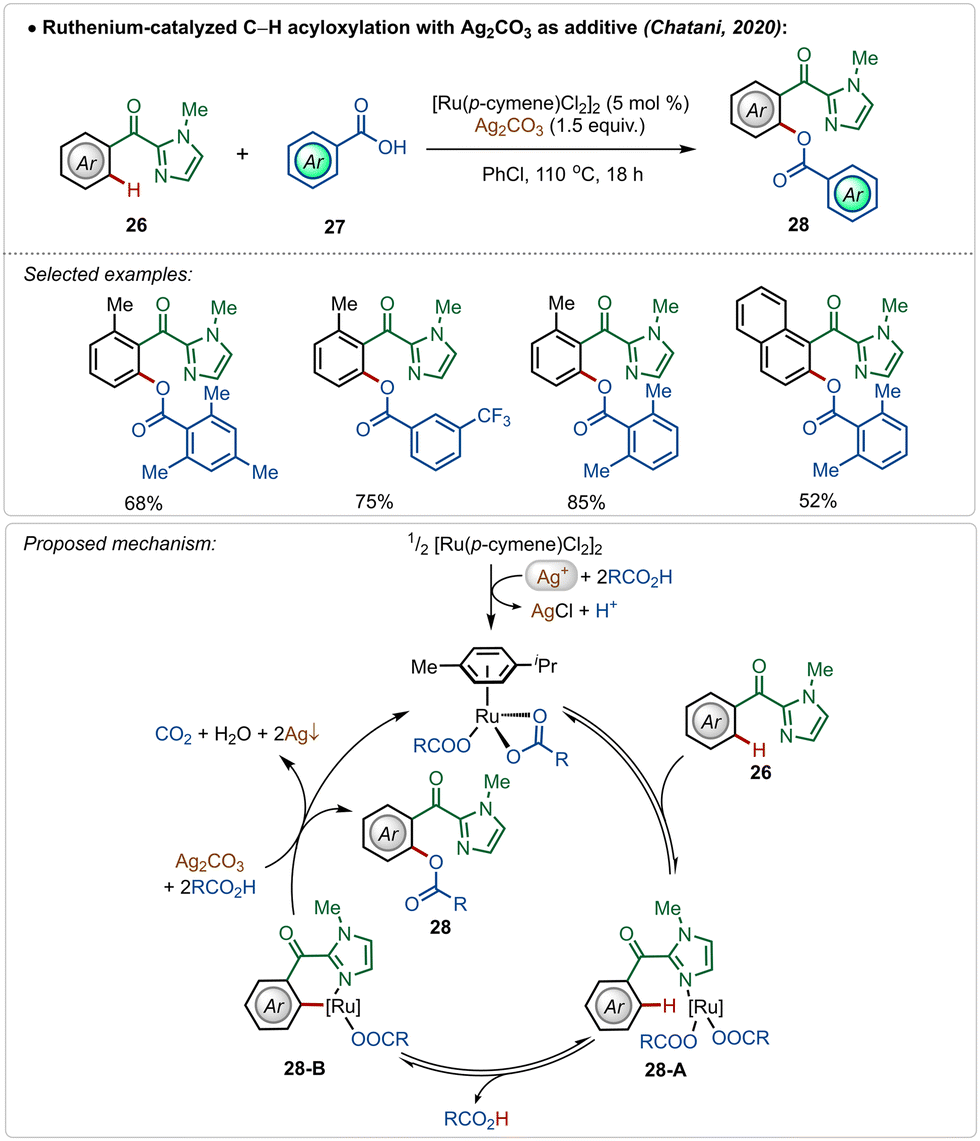 | ||
| Scheme 4 Ruthenium-catalyzed C–H acyloxylation. | ||
Very recently, the Ackermann group has described an interesting example of ruthenium-catalyzed ortho-C–H alkylation of indole derivatives where both point and axial chirality were generated consequently. Easily accessible chiral imidazolidine carboxylic acids were used for the chiral induction (Scheme 5).13a The strategy successfully involved diverse allyl arenes and vinyl silanes as the coupling partners in combination with different indole analogues. The chiral carboxylic acid played a decisive role in both the atroposelective C–H activation (intermediates 31-A and 31-B) in the C-2 position of indole and in the migratory insertion of the olefin (31-C–31-F) to generate the chiral molecules.
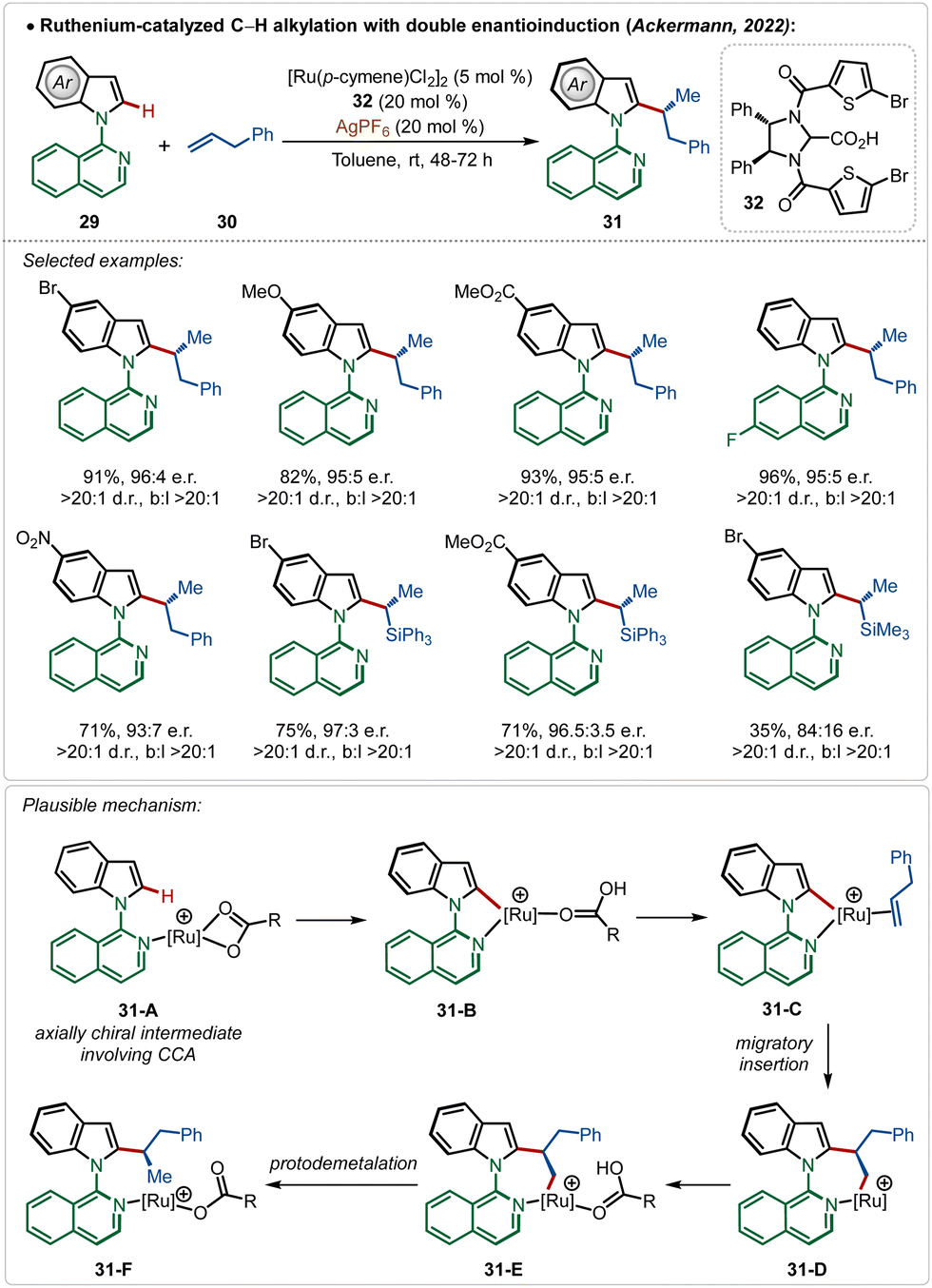 | ||
| Scheme 5 Ruthenium-catalyzed C–H alkylation with double enantio-induction. | ||
3. Rhodium catalysis with silver additives
In this expanse, rhodium-catalyzed C–H activation reactions have also exhibited tremendous potential owing to the high reactivity and versatility.1b,f,28 The most popular half-sandwich catalyst used in these transformations is the rhodium pentamethylcyclopentadienyl chloride dimer ([Cp*RhCl2]2). Similar to the reactivity of the ruthenium catalyst, already discussed in the preceding section, this dimer is also a pre-catalyst, which requires Ag-mediated activation to form its monomeric cationic variant.1b,f In these cases, silver hexafluoroantimonate (AgSbF6), silver triflimide (AgNTf2), silver carbonate (Ag2CO3), silver tetrafluoroborate (AgBF4), silver acetate (AgOAc) or silver trifluoroacetate (AgTFA) are used as common additives in combination with additional carboxylate sources. These silver salts, when combined with the Rh(III)-catalyst, mediate different C–H functionalization reactions, such as C–H alkenylation,29 C–H alkylation,30 C–H alkynylation,31 C–H allylation,32 C–H arylation,33etc. (Scheme 6).1b,f,28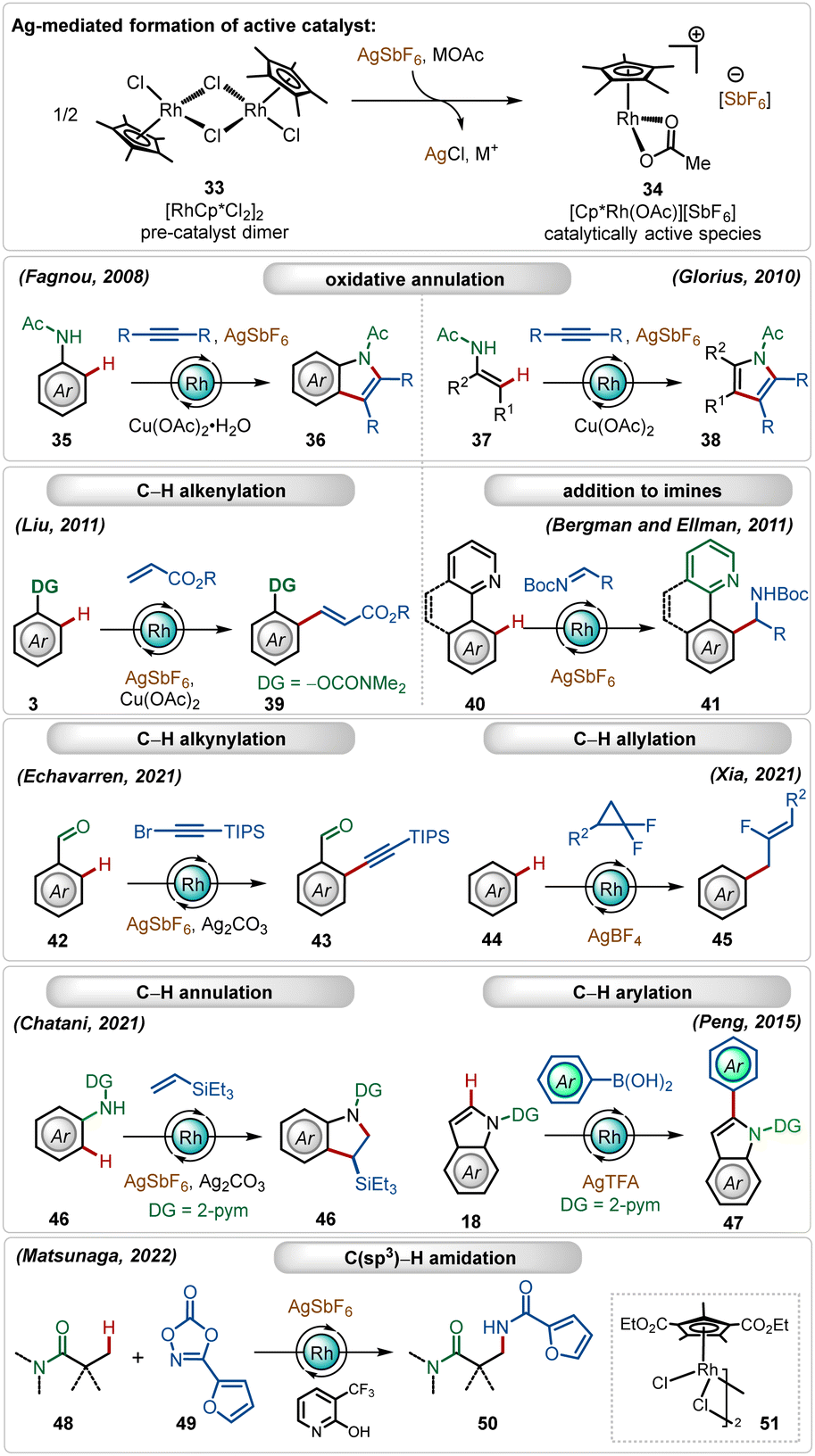 | ||
| Scheme 6 Overview of Ag-assisted [Rh]-catalyzed C–H activations. | ||
Early examples of the efficiency of cationic Rh(III)-complexes were demonstrated by Fagnou and co-workers through an oxidative C–H/N–H annulation of anilides with internal alkynes.34 Later, Glorius reported a similar Rh(III)-catalyzed C–H/N–H annulation of enamides with internal alkynes.35 An important example of in situ generated cationic Rh(III)-catalyzed oxidative olefination of phenols were illustrated by Liu.36 Ellman and Bergman showed that the combination of the [Cp*RhCl2]2 catalyst with AgSbF6 could also promote a Grignard like addition of C–H bonds to aldimines.37 Recently, Echavarren delineated alkynylation of aldehydes using alkynyl bromides as the reaction partner.31 In a similar trend, Xia and co-workers described the direct addition of C–H bonds to difluorocyclopropanes forming fluorinated allyl benzenes, where AgBF4 was utilized as a suitable additive.32 This Ag(I)-mediated cationic Rh(III)-catalyzed approach was also garnered for annulation with olefins38 and C-2 arylation of indoles.33 This catalytic system is also suitable for C(sp3)–H bond activation reactions. Among other important findings,39 Matsunaga and co-workers described a C(sp3)–H amidation of aliphatic amides using dioxazolones as the amidating agent, where electron-deficient Rh(III)-pre-catalyst and pyridone ligands were an imperative part of the transformation.39a
Sun and co-workers reported an elegant example of rhodium-catalyzed C–H alkylation of modified ferrocenes, where various alkylated ferrocenes were obtained in good yields (Scheme 7).29 In this work, AgNTf2 was used as the additive in combination with the KOAc co-additive. This combination successfully led to the in situ formation of the crucial catalytically active cationic [Rh]-species. Diazo-salts were used as the suitable alkyl source, where the reaction proceeded via the formation of Rh-carbenoid intermediates. An amide directed regioselective insertion of these Rh-carbenoid intermediates afforded the alkylated products.
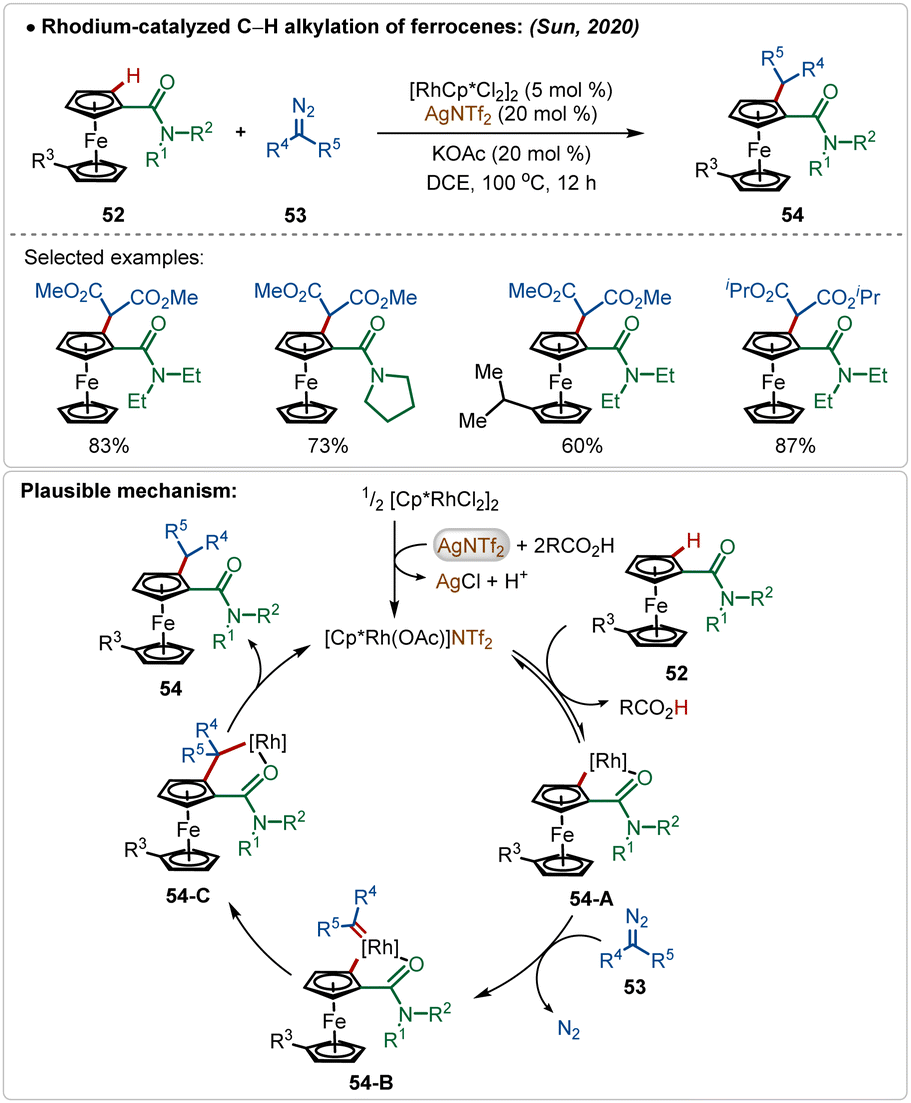 | ||
| Scheme 7 Rhodium-catalyzed C–H alkylation. | ||
Another interesting Rh(III)-catalyzed C–H activation reaction was recently described by Wei, Shi, and co-workers,40 in which novel isoquinoline derivatives were successfully fabricated in moderate to good yields (Scheme 8). In this process, AgSbF6 played a dual role. First of all, the Ag(I)-salt acted as a Lewis acid to activate the internal alkyne to mediate an intramolecular cyclization with oxime ether functionality, constructing the corresponding isoquinoline analogue 56-C. Then the isoquinoline directed ortho-C–H bond activation of the arene took place (56-D) followed by the formation of an Rh-carbenoid species 56-F after a loss of DMSO. Next, the respective intermediate underwent a migratory insertion to generate 56-G, protodemetalation, which led to the formation of the desired product (56). In the latter case, Ag(I)-salt assisted the formation of catalytically active cationic Rh(III)-catalyst, which enabled the C–H activation.
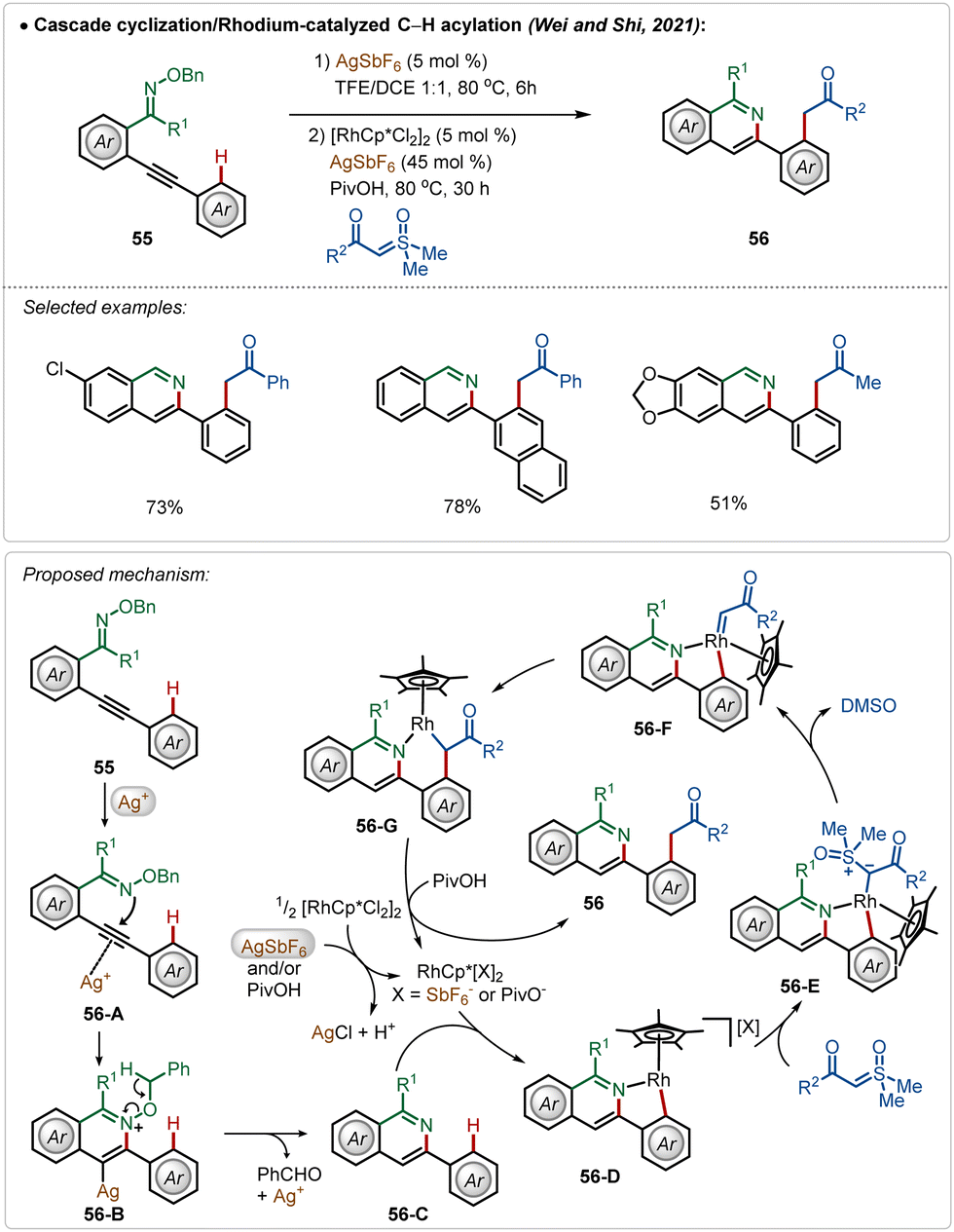 | ||
| Scheme 8 Rhodium-catalyzed C–H activation with bifocal use of AgSbF6. | ||
4. Iridium catalysis with silver additives
Cationic iridium(III)-catalysts have also found widespread application in many C–H activation reactions.1b,c,41 One of the main half-sandwich iridium-catalysts is the iridium pentamethylcyclopentadienyl chloride dimer ([Cp*IrCl2]2), which is similar to the above-mentioned rhodium catalyst that must be converted into its monomeric form using Ag(I)-additives.1b,c,41 In this case, usually used silver salt additives are silver hexafluoroantimonate (AgSbF6), silver carbonate (Ag2CO3), silver triflimide (AgNTf2), or silver fluoride (AgF).1c,41 This Ir(III)-catalyzed strategy has been successful for a large variety of direct C–H activation reactions (Scheme 9).41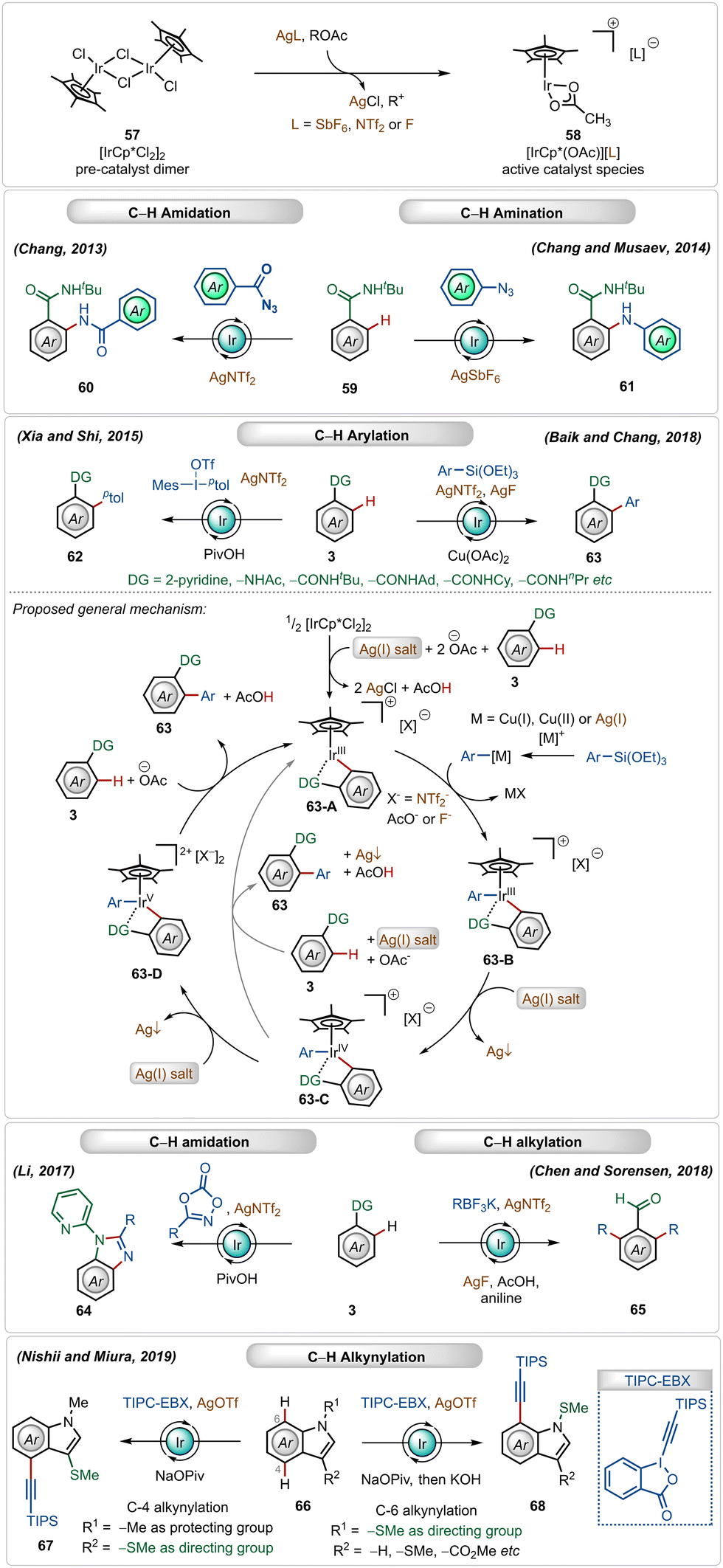 | ||
| Scheme 9 Overview of Ag-assisted [Ir]-catalyzed C–H activations. | ||
In 2013, Chang utilized the bench-stable [Cp*IrCl2]2 catalyst in combination with the AgNTf2 additive to perform ortho-C–H amidation of benzamides with acyl azides.42a Later, in 2014, they also established cationic Ir(III)-catalyzed direct C–H amination with sulfonyl azides and aryl azides as amidating agents.42b,43 The Ir(III)-catalyst was also suitable for C–H arylation of arenes using arylsiloxanes44a and diaryliodonium salts as the aryl source.44c Recently, Chang and Baik also used aryl siloxanes as arylating agents for C–H arylation reactions under Ir(III)-catalysis.44b In both reactions, the presence of the AgNTf2 additive was crucial.
In 2018, Chen and Sorensen reported ortho-C–H alkylation of arenes utilizing alkyl trifluoroborates as the alkylating agent, where the presence of AgNTf2 and AgF was necessary for the optimal outcome.45 Nishi and Miura discovered an interesting example of an in situ generated cationic Ir(III)-catalyzed regioselective alkynylation strategy, achieving C-4 and C-7 selective alkynylation of indoles.46
In a fascinating transformation, Chatani and co-workers disclosed an iridium-catalyzed C–H olefination strategy for the synthesis of 1,1-diaryl olefins, where both AgSbF6 and Ag2CO3 were used as additives (Scheme 10).47 Here, aniline derivatives were used as suitable substrates employing a pyrimidine-directing group, which provided access to several styrene derivatives in moderate to good yields. Unlike typical C–H olefination reactions, this process leads to branched olefins exclusively.
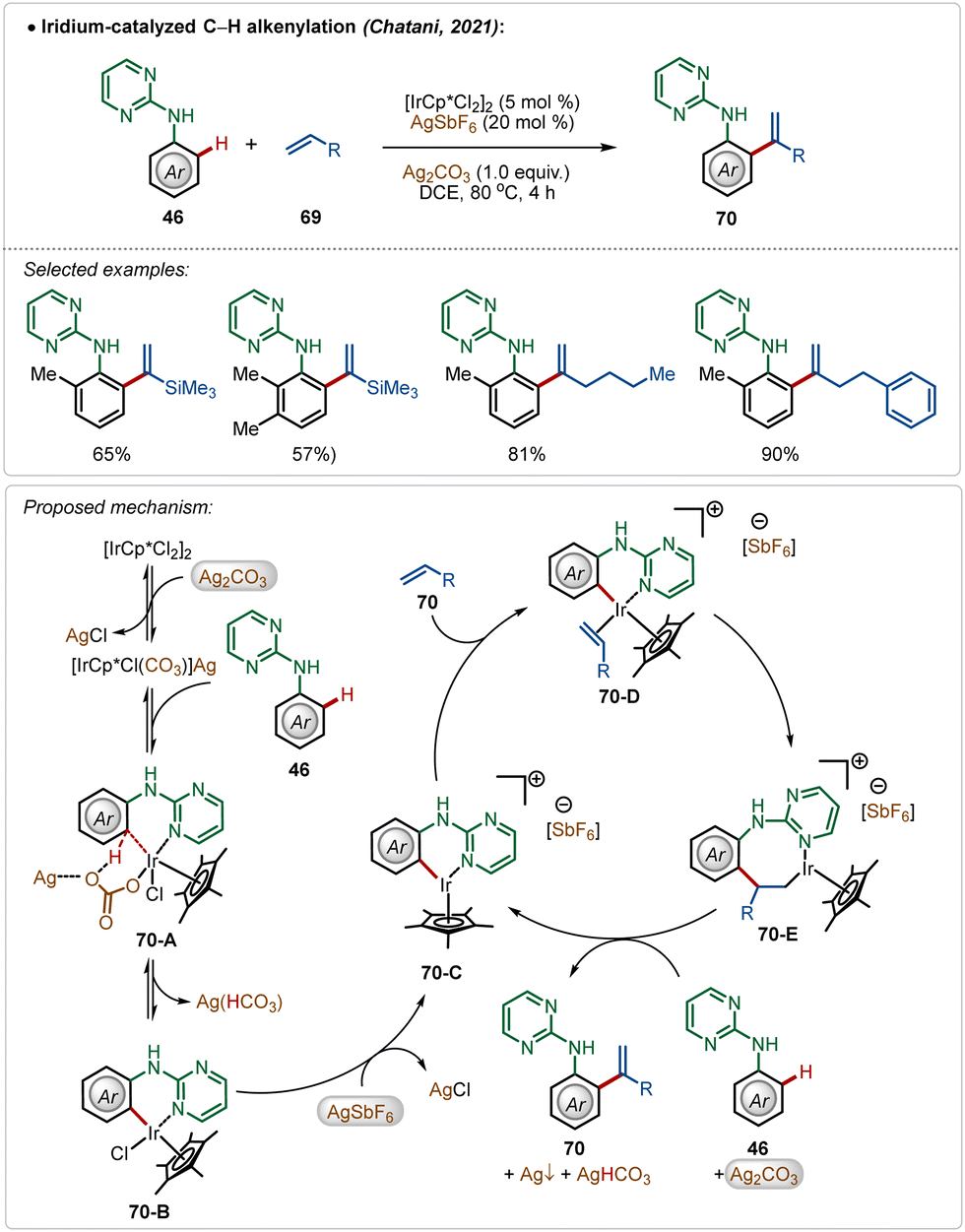 | ||
| Scheme 10 Iridium-catalyzed C–H alkenylation. | ||
Based on the experimental results, DFT calculations, and literature precedents, the plausible mechanism was proposed. First, the reaction of pre-catalyst [Cp*IrCl2]2 with additive Ag2CO3 generated the intermediate [Cp*IrCl(CO3)](Ag). This species executed the desired C–H activation after an interaction with the substrate. The addition of AgSbF6 promoted the reaction by removing the other residual chloride, forming AgCl and the active species 70-C, which actively participated in the catalytic transformation. The cationic intermediate 70-C interacted with the olefin, forming intermediate 70-E after a migratory insertion, which upon β-hydride elimination gave the olefinated product (70).
In 2015, Chang and co-workers depicted the divergent reactivity of the Ir(III)-catalyst through the olefination and alkylation of arenes with activated olefins, where the product selectivity was largely dependent upon the directing group (Scheme 11).48 Strongly coordinating pyridyl, pyrimidyl, and pyrazolyl groups led to alkylated products, whereas weakly coordinating ketone, anilide, and amide generated the olefinated products. In these transformations, AgNTf2 played a critical role in forming the active cationic iridium catalyst, which was responsible for the product formation.
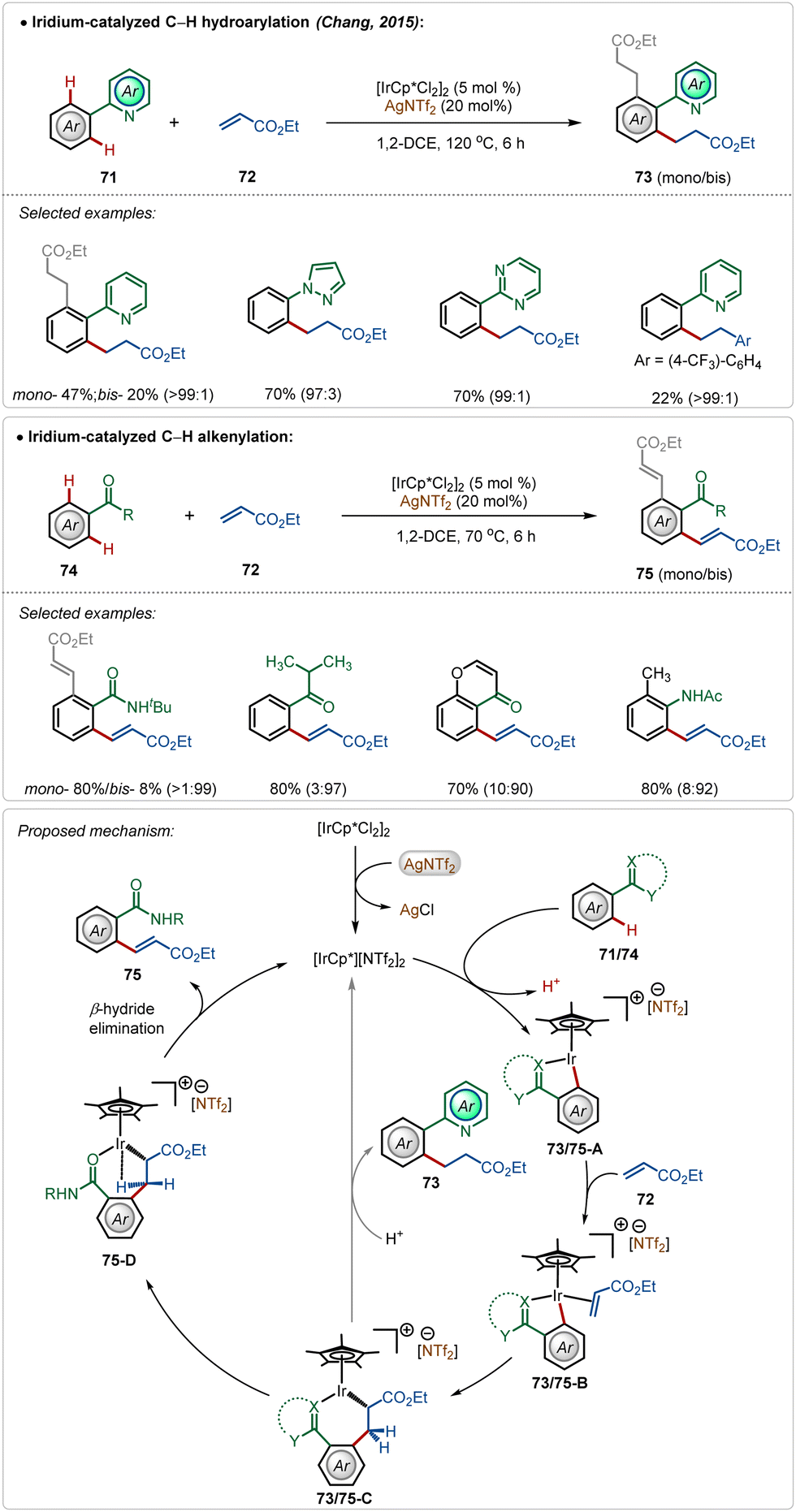 | ||
| Scheme 11 Iridium-catalyzed C–H alkenylation vs alkylation guided by directing groups. | ||
Initial steps of the catalytic cycles, such as C–H activation, olefin coordination, and migratory insertion were the same for both N-centered and O-centered directing groups. The directing group played a decisive role in chemoselectivity. The product was formed after a structural modification of the key intermediate 73/75-C. Now the strong interaction between strongly Lewis basic N-centre with the Lewis acidic Ir(III)-centre restricted the change in the orientation owing to the high rigidity of the iridacycle (73/75-C), which led to alkylation. The weakly Lewis basic O-centered directing group had a weaker interaction with the Ir(III)-centre leading to a feasible change in the structural orientation, which underwent β-hydride elimination through agostic interaction to deliver the olefinated product (Scheme 11).
The Chang group further demonstrated an iridium catalyzed amination using alkyl amines as the amine source.49 Similar to earlier examples, this reaction was also promoted by a cationic Ir(III)-catalyst, generated through the reaction between the bench-stable Ir(III)-dimeric catalyst and AgNTf2. The interesting feature of this transformation was the involvement of an oxidation-induced reductive elimination process, which involved an Ir(V)-intermediate consisting of an amido ligand. The insertion on the amido-Ir(V) intermediate (78-D) followed by protodemetalation (78-E) produced the aminated product. Poor reactivity of secondary amines in this transformation further justified the involvement of an oxidation-induced reductive elimination pathway instead of a direct reductive elimination from the Ir(III)-intermediate (Scheme 12).
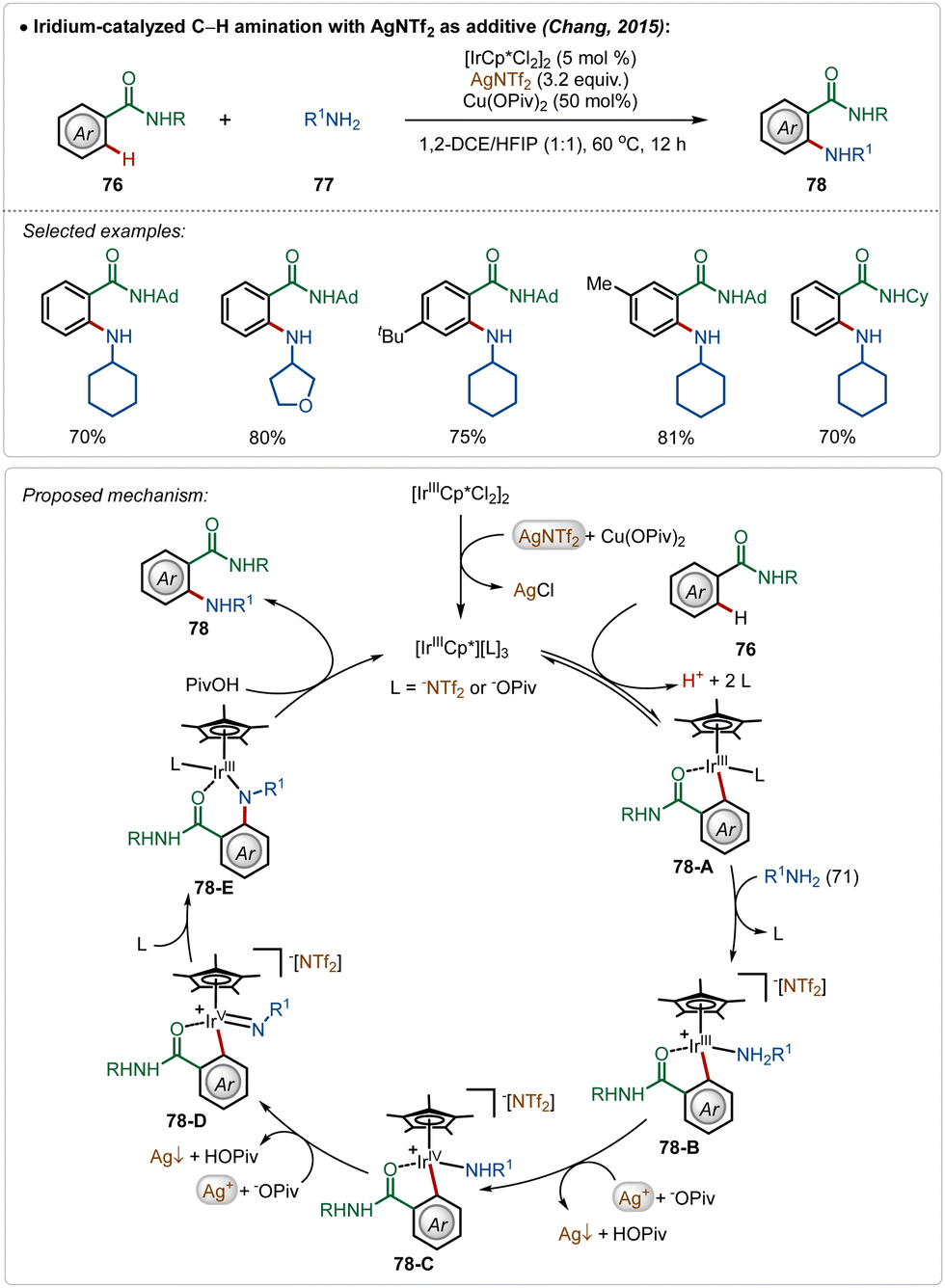 | ||
| Scheme 12 Iridium-catalyzed amination using aliphatic primary amines. | ||
A unique dihydroquinolinone synthesis was depicted by Pan, Yu and co-workers through an iridium-catalyzed C–H amidation followed by an intramolecular aza-Michael addition strategy.50 Starting from a large variety of chalcones, through this one-pot procedure different cyclic dihydroquinolinone derivatives were accessed in good yields, where two new C–N bonds were successfully formed (Scheme 13). The proposed mechanism played a similar role to AgSbF6, which in combination with pivalic acid, led to the formation of the active iridium-species. After C–H activation followed by the nitrene insertion, the amidated product 81-D was formed, which underwent a facile Michael addition to the α,β-unsaturated ketone moiety to form the heterocyclic dihydroquinolinone product.
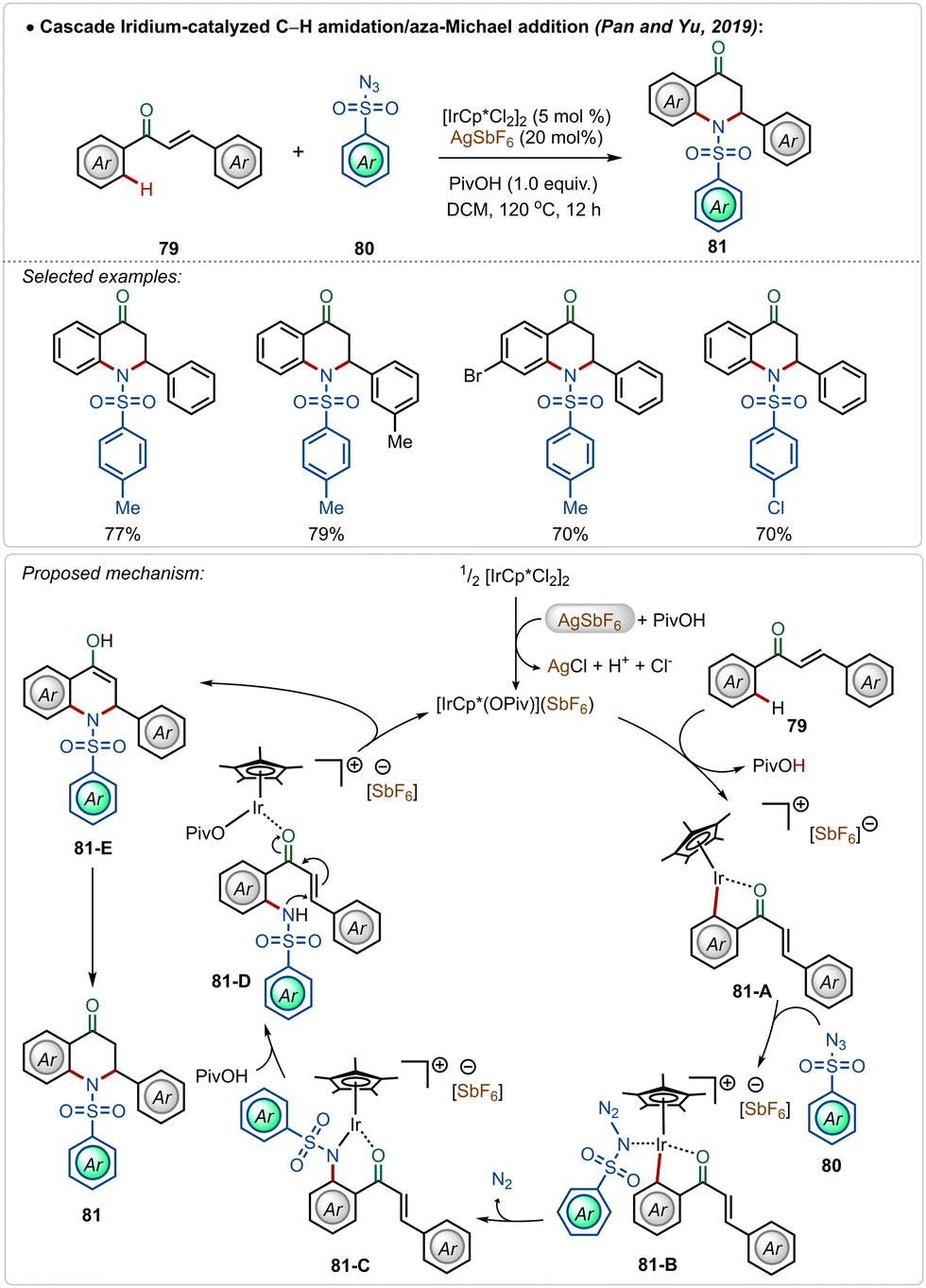 | ||
| Scheme 13 Iridium-catalyzed C–H amidation/aza-Michael addition cascade. | ||
5. Palladium catalysis with silver additives
In the C–H activation regime, palladium-catalysis remains at the forefront owing to its diverse applicability in proximal and distal C(sp2)–H and C(sp3)–H bond functionalization.51 Notably, many of these transformations require Ag-additives to obtain the optimal outcome.5b–d Typically, these additives play a crucial role as a terminal oxidant to enable the catalytic turnover.51However, recently, Larrosa has depicted that phosphine ligated Ag(I)-carboxylates could also promote C–H activation of electron-deficient arenes forming Ag-aryl intermediates, which then realized transmetalation with a Pd-intermediate to accomplish the arylation with aryl iodides.52 Recent findings have further reported some interesting insights into Pd-catalyzed C–H activation reactions promoted by Ag-additives, which divulges the critical role of a Pd–Ag heterobimetallic intermediate species in these transformations.5b–d For example, Pd(II)-catalyzed ortho-amination reaction of benzamides, which follows a Pd(II)/Pd(IV) pathway, does not necessarily need an additional Ag-oxidant for product formation.9a However, experimental findings suggested an indispensable role of AgOAc in this reaction. Theoretical studies by Sunoj and Schafer unveiled the formation of an energetically favourable Pd–Ag heterometallic intermediate during the process, which necessitated the use of Ag(I)-salt in this transformation (Scheme 14a).9
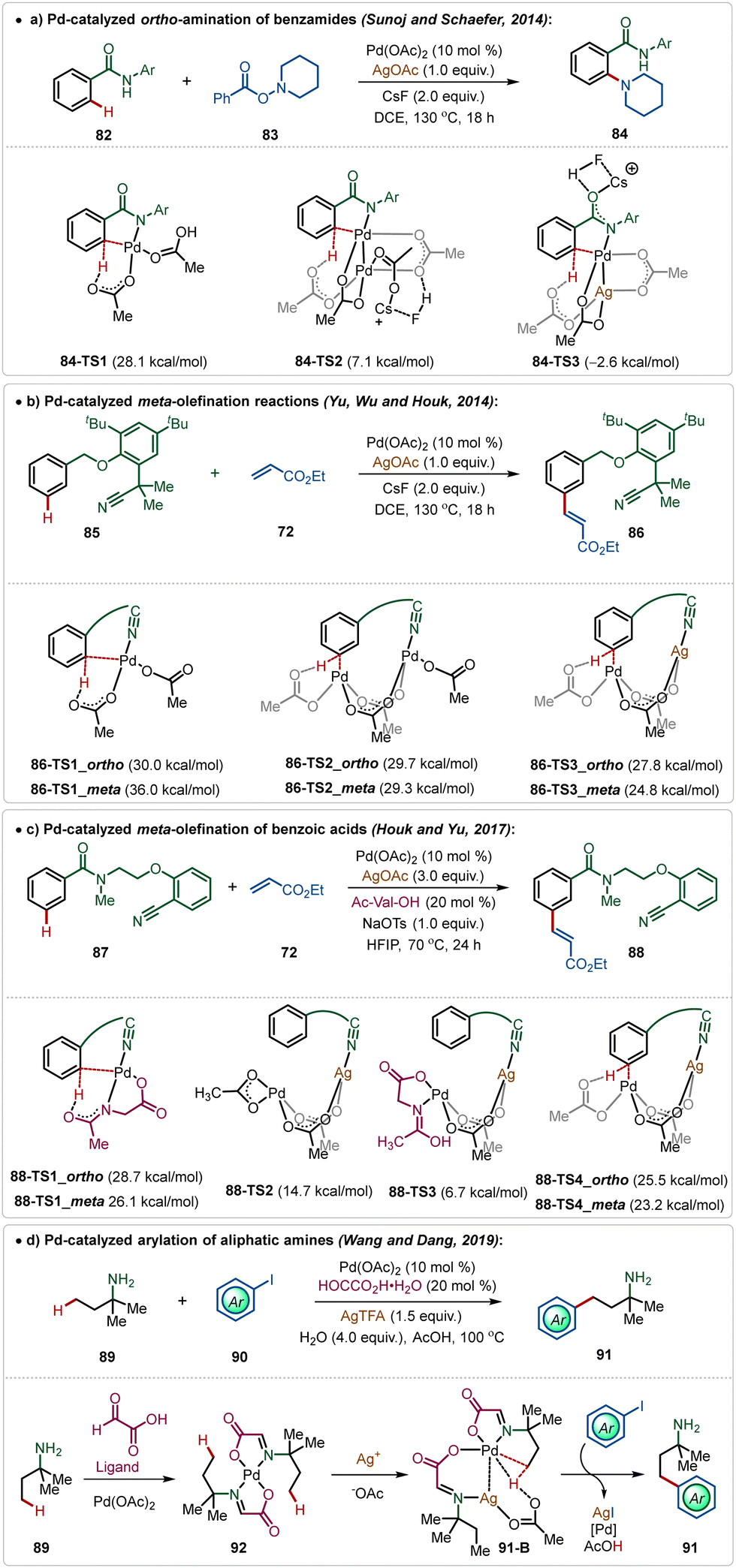 | ||
| Scheme 14 Overview of Ag-assisted [Pd]-catalyzed C–H activations. | ||
Similarly, Houk and Yu disclosed that the Pd-catalyzed distal C(sp2)–H olefination reaction was enabled by the formation of critical Pd–Ag heterometallic intermediates.53 Based on their findings, it was apparent that the Pd-monomeric complex generated exclusive ortho-selectivity, while the Pd-dimeric complex led to a ∼3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 meta/ortho regio-isomeric mixture. Interestingly, the incorporation of an isoelectronic Ag(I)-species instead of Pd(II)-species stabilized the transition states and elevated the meta-selectivity. The direct coordination of Ag(I)-species with the nitrile functionality of the directing group was responsible for the superior meta-selectivity (Scheme 14b).
:1 meta/ortho regio-isomeric mixture. Interestingly, the incorporation of an isoelectronic Ag(I)-species instead of Pd(II)-species stabilized the transition states and elevated the meta-selectivity. The direct coordination of Ag(I)-species with the nitrile functionality of the directing group was responsible for the superior meta-selectivity (Scheme 14b).
This conceptual basis was also applicable for the template-assisted meta-olefination of benzoic acid derivatives.54 Though the transformation was ineffective in the absence of mono-protected amino acid (MPAA) ligands, computational studies supported the formation of a Pd–Ag bimetallic transition state and MPAA was found to stabilize this species (Scheme 14c).
The formation of Pd–Ag bimetallic clusters was also found in Pd-catalyzed C(sp3)–H bond activation reactions. Dang and Wang showed a Pd-catalyzed C(sp3)–H arylation of aliphatic amines, which was enabled by the formation of Pd–Ag heterodimeric species consisting of carboxylate bridges (Scheme 14d).55
Maiti also reported the meta-alkynylation of arenes, where the formation of a Pd–Ag bimetallic cluster was the key for product formation.56 Noticeably, in this case the directing group was directly coordinated to the Pd(II)-species, which was connected to the Ag(I)-centre through an acetate bridge. The halophilic Ag(I)-species assisted bromide removal from the alkynyl bromide in the transition-state (95-A) and thus influenced product formation (Scheme 15).
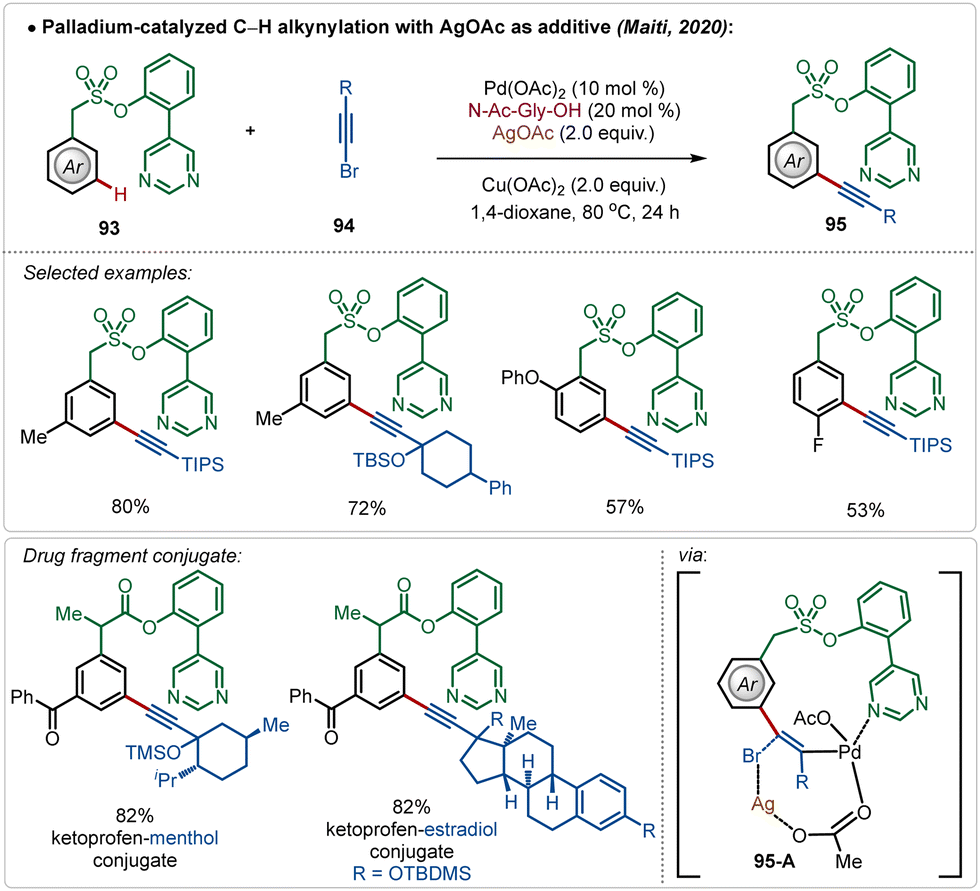 | ||
| Scheme 15 Ag-assisted [Pd]-catalyzed C–H alkynylation. | ||
6. Cobalt catalysis with silver additives
In recent years, there is a surge in interest towards the development of more practical ways to perform C–H activation reactions reducing the undesired barrier between the academic and industrial research.1 This can be achieved by replacing expensive ruthenium-, rhodium-, iridium- and palladium-catalysts with cheap, comparatively less toxic, and Earth-abundant 3d transition metal catalysts.1eIn this domain, among all the possible 3d transition metals, currently the most used transition-metal catalyst is cobalt.57,58 The most common cobalt catalyst used in C–H activation reactions is the half-sandwich complex [Cp*Co(CO)I2].57,58 Although it is already a monomeric catalyst, it is less reactive and mainly acts a pre-catalyst. Therefore, it usually requires the presence of Ag-based additives to exchange its iodide ligands for the formation of reactive cationic species.1b In this case, silver carbonate, silver hexafluoroantimonate (AgSbF6), silver acetate (AgOAc), or silver triflate (AgOTf) are common additives. This combination under the optimal reaction conditions has been applied to a diversity of transformation, such as C–H alkylation,59 C–H carboamination,60 C–H alkenylation,61 C–H alkynylation,62 C–H allylation,63 and C–H annulation64 (Scheme 16). A notable contribution has been documented by the Ackermann group,65 where they described a versatile Co(III)-catalyzed oxidative annulation with internal alkynes with O-acetyl oximes65l and nitrones65i without using external oxidants. They also demonstrated an effective application of the in situ generated cationic Co(III)-catalyst for a selective synthesis of Z-olefins, where vinyl cyclopropanes were used as the reaction partners.65g This catalytic manifold was also suitable for C-2 alkynylation of indoles with alkynyl bromides. Among other notable findings, Ackermann and co-workers also established the C–H/N–H annulation reaction involving α-diazocarbonyl compounds as the carbene precursor65h and C–H amidation with dioxazolones as the nitrene source.65f They also discovered a versatile C-2 alkylation of indoles with full selectivity control.59e The selectivity was governed by the carboxylic acid additive. In the presence of acid, branched alkylation was observed under mild conditions, whereas in the absence of the carboxylic acid linear alkylation was the exclusive reaction. In both cases, the AgSbF6 additive played a critical role in generating the catalytically active Co(III)-catalyst. Later, the Ackermann group also demonstrated a versatile ortho-methylation strategy using a Co(III)-sandwich complex [Cp*Co(C6H6)](PF6)2.59a This Co(III)-catalyzed methylation strategy enabled the methylation of a large variety of arene analogues, tolerating different weakly as well as strongly coordinating directing groups. Furthermore, the catalytic conditions smoothly executed late-stage methylation of several structurally complex natural products and drug molecules. Sundararaju and co-workers reported an early example of Co(III)-catalyzed C(sp3)–H olefination through hydroarylation of alkynes.61 Another attractive allene hydroarylation strategy was unveiled by Ackermann, offering modular access to alkenylated arenes.65e
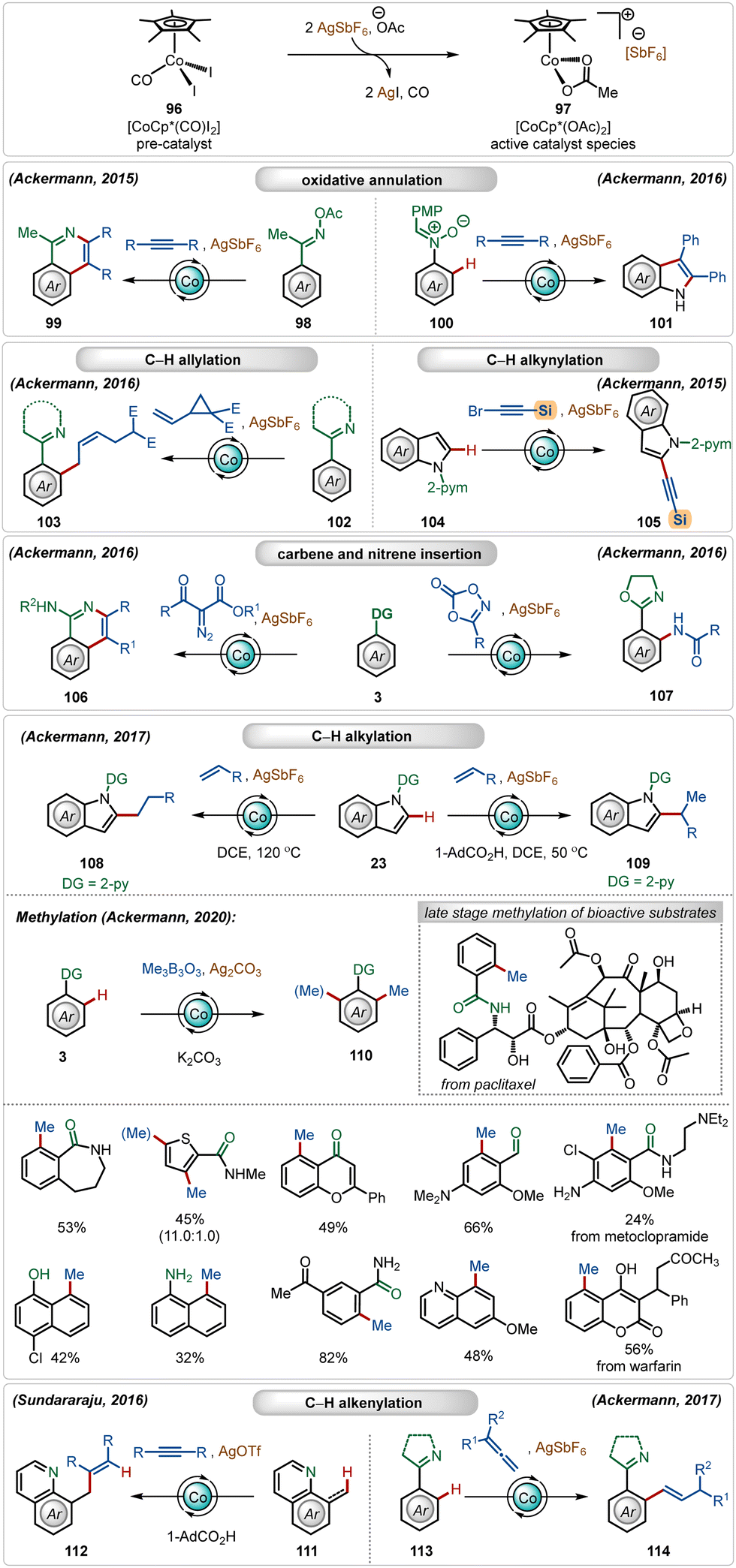 | ||
| Scheme 16 Overview of Ag-assisted [Co]-catalyzed C–H activations. | ||
Clavier and co-workers described a cobalt-catalyzed C–H allylation of arenes with vinyl aziridines (Scheme 17).66 A separable mixture of (E/Z)-alkenes was achieved in a proportion close to 1:1 in most cases.
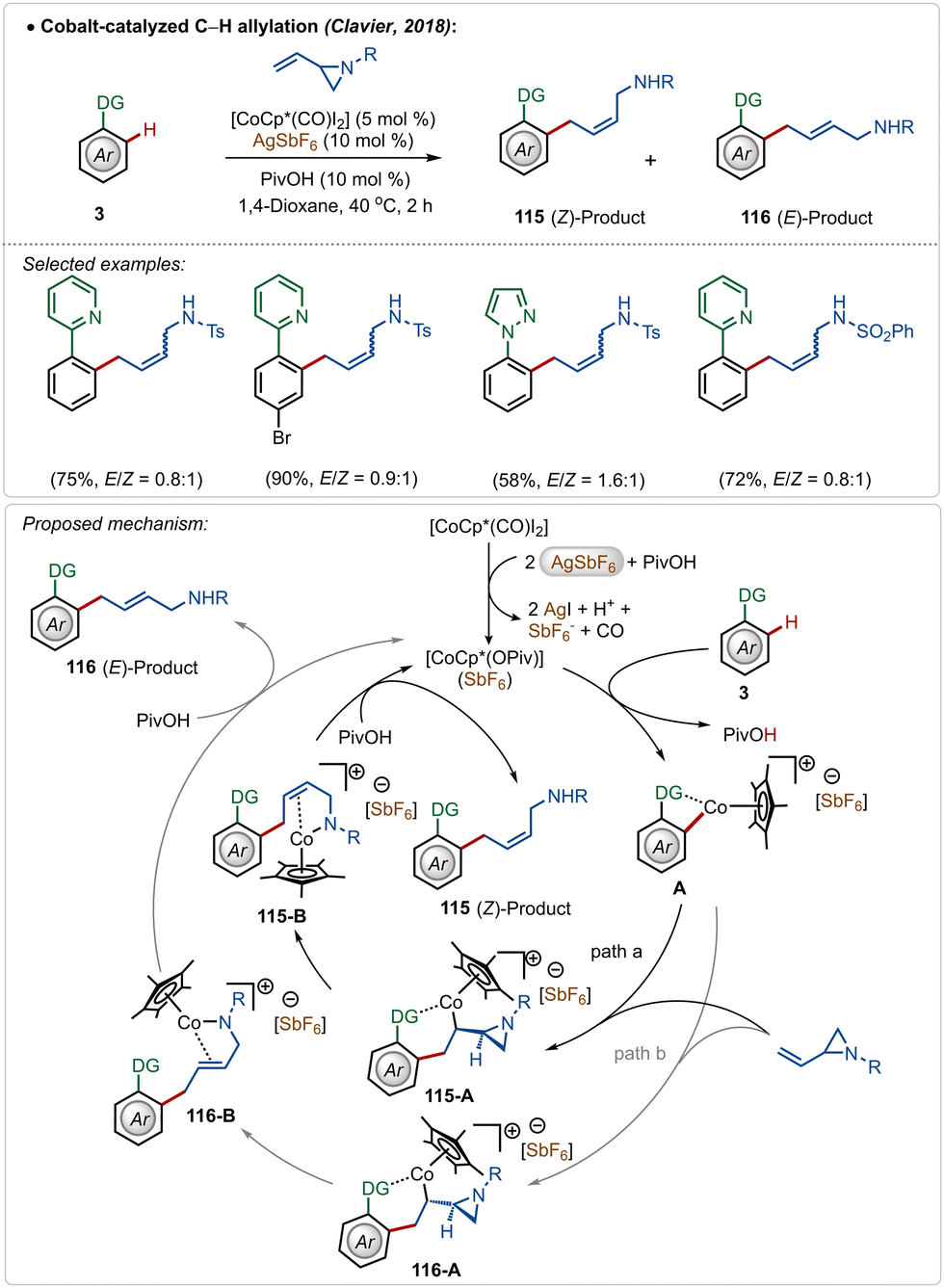 | ||
| Scheme 17 Cobalt-catalyzed C–H allylation with vinylaziridines. | ||
Although the method presented a low selectivity, this strategy worked efficiently on different substrates with strongly coordinating 2-pyridyl and 1-pyrazolyl directing groups. It was initiated with the known modification of the pre-catalyst [Cp*Co(CO)I2] to its active cationic form [Cp*Co(OPiv)](SbF6) with the aid of AgSbF6. Then the active species interacted with the substrate to form metallacycle intermediate A through C–H activation. It then realized olefin-insertion on both possible faces of the coupling partner, and this difference led the mechanism to slip into path “a” and “b”. Usually there was no preferential pathway, which led to the formation of both the isomers in a low selectivity. A subsequential β-N-elimination and ligand exchange governed the final (Z)- and (E)-products respectively and regenerated the active cobalt-catalyst.
In 2015, Chang and co-workers delineated Co(III)-catalyzed amidation of 2-arylpyridines (Scheme 18).67 The reaction was enabled by a Co(III)-cationic complex, formed in situ with the aid of AgSbF6. The active cationic complex on the exposure of 2-arylpyridines generated the cyclometalated intermediate 118-A, which then delivered the desired product by reacting with the amidating agent through intermediates 118-B–118-D. The amidation step plausibly involved a Co(III)-amido intermediate, while the possible involvement of a Co(V)-nitrenoid intermediate in the transformation could not be overlooked.
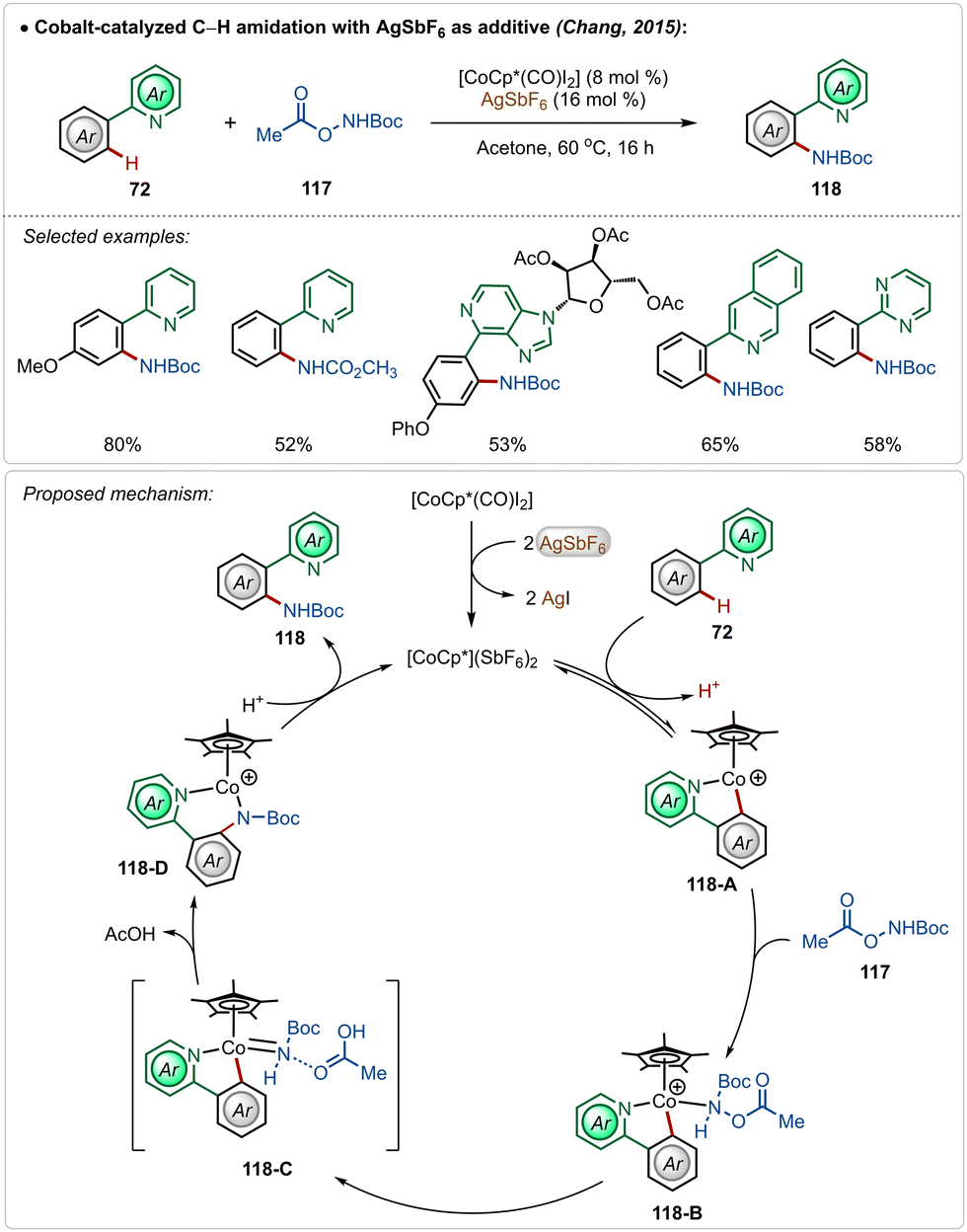 | ||
| Scheme 18 Cobalt-catalyzed C–H amidation. | ||
In 2018, Ackermann and co-workers described the first example of high-valent cobalt-catalyzed enantioselective ortho-C–H alkylation of indoles (Scheme 19).59d The enantioselectivity was realized using easily accessible imidazolidine carboxylic acid analogue, which was induced during the insertion and protodemetalation through the direct coordination of the chiral carboxylic acid (104-B and 104-C). Even in this transformation the role of AgSbF6 was critical to form the catalytically active species. This enantioselective alkylation strategy involved diverse indole and allyl arene analogues to construct the corresponding alkylated products in good yields and selectivity. The pyridyl directing group was also removed without affecting the enantiomeric excess after the Co-catalyzed transformation, obtaining free indoles in high yields.
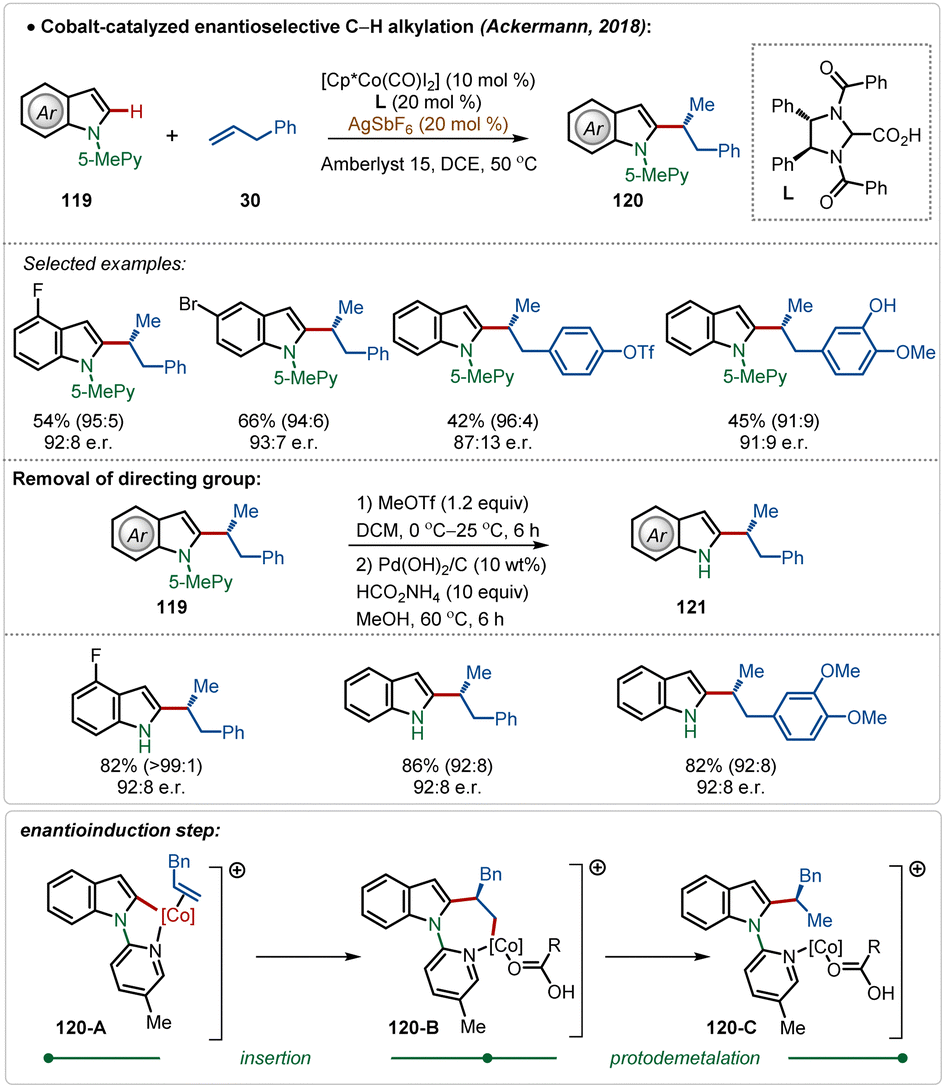 | ||
| Scheme 19 Cobalt-catalyzed enantioselective C–H alkylation. | ||
7. Ag-additive free C–H activation strategies
Modern organic synthesis focuses on more practical and sustainable means to execute synthetic transformations. Avoiding the necessary additives from these transformations can be extremely effective, as it makes the approach more atom economical and reduces the trace-metal contamination on the final product.68 In this context, Ackermann and co-workers have disclosed several strategies using Ru,69 Rh,70 and Ir71 catalysts, where the need for otherwise indispensable Ag-additives can be obviated by harvesting renewable electricity.68c–fIn their pioneering findings, Ackermann described that the need for the AgSbF6 salt could be avoided when the C–H activation was performed electrochemically.69c The reaction did not involve a cationic Ru(II)-catalyst and was enabled in an aqueous medium. Anodic oxidation of Ru(0)-intermediate 123-D regenerated the active Ru(II)-catalyst and the cathodic reduction of protons completed the electrochemical process with the generation of hydrogen gas. This electrochemical approach was possible with both arene carboxylic acids as well as amides and possessed high functional group tolerance leading to a broad scope (Scheme 20).
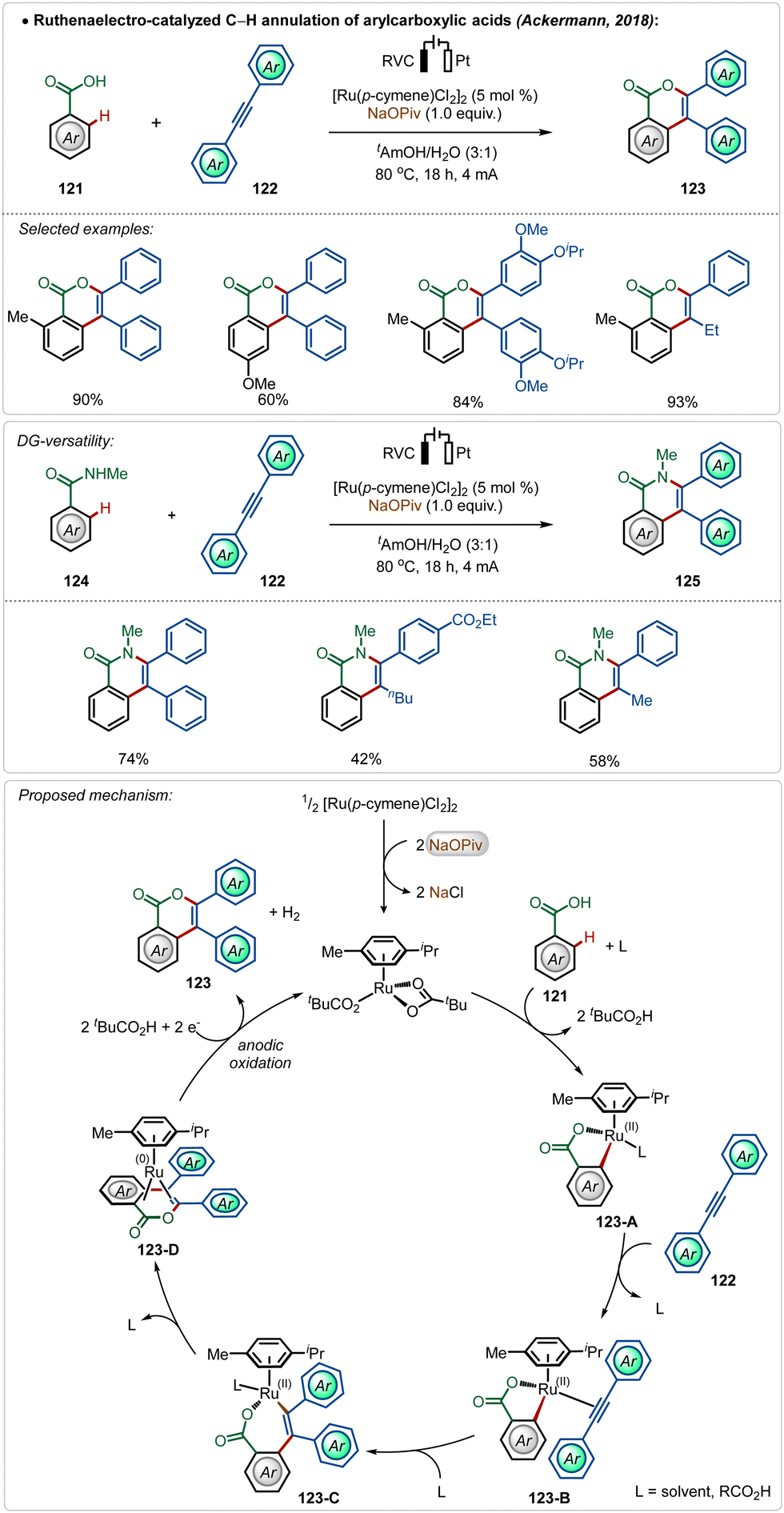 | ||
| Scheme 20 Electrooxidative ruthenium-catalyzed annulation. | ||
Ackermann and co-workers further reported the first Rh(III)-catalyzed oxidative olefination/annulation strategy with weakly coordinating carboxylic acids and benzamides.70c In contrast to the prior Ru(II)-catalyzed processes, this transformation underwent smoothly without otherwise necessary Ag(I)-salt additives. The reaction was achievable in an aqueous medium and coupled with the hydrogen evolution reaction as the cathodic half-reaction. The strategy was versatile and accommodated a diverse range of arenes in this transformation. Even, C-2 olefination of indole was also demonstrated utilizing similar electrochemical reaction conditions. The key step enabled by electricity was the anodic oxidation of the Rh(I)-intermediate (126-D), formed in situ after β-hydride elimination (Scheme 21).
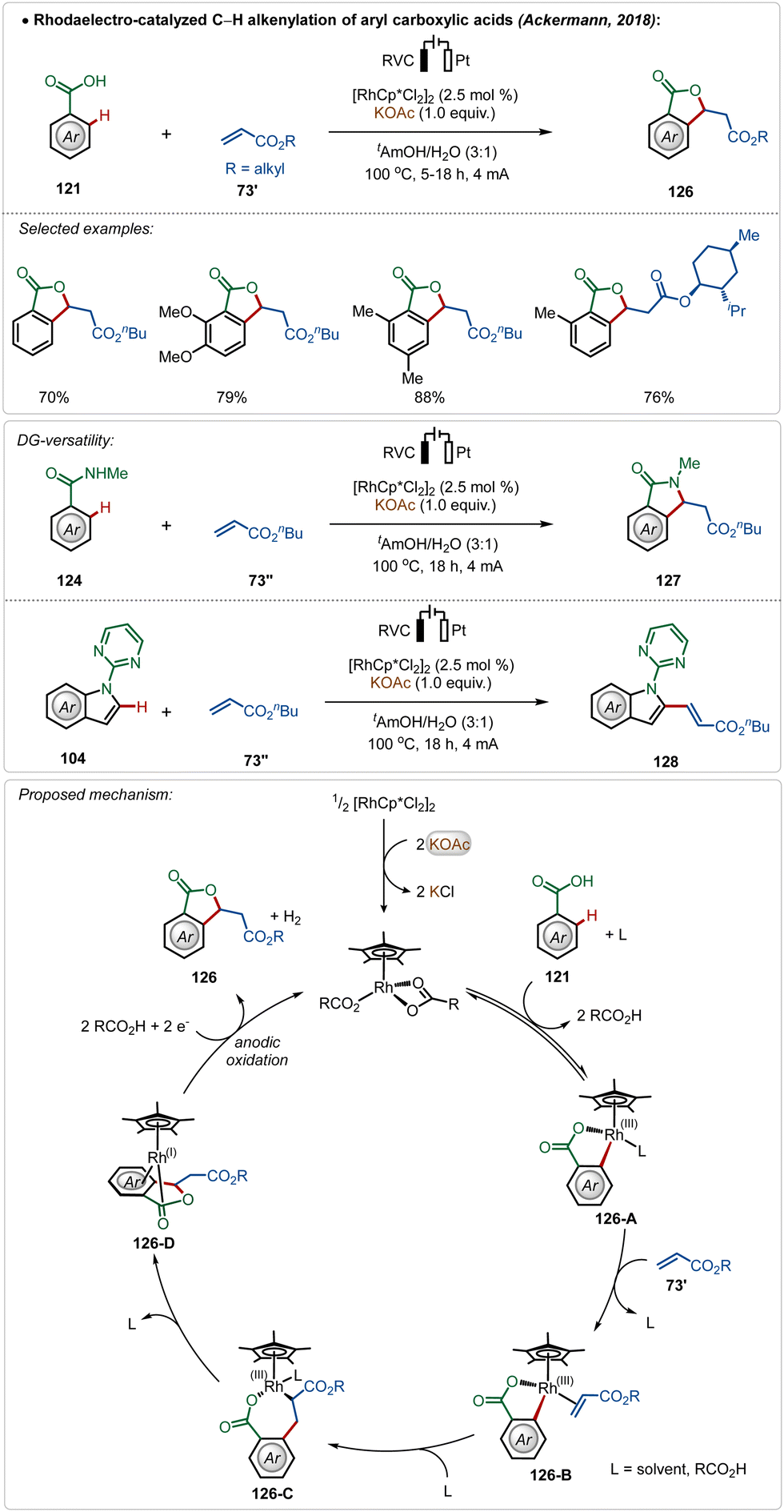 | ||
| Scheme 21 Rhodaelectro-catalyzed Ag-free C–H alkenylation of arylcarboxylic acids. | ||
Later, they also detailed an elegant rhodaelectro-catalyzed C–H alkenylation at the C-8 position of naphthol with styrene-derivatives.70b This methodology offered direct access to a broad range of C-8 olefinated naphthols in good to excellent yields. Interestingly, 5-hydroxyquinoline under electrochemical conditions furnished annulated products in moderate to good yields, displaying the versatility of the approach. Based on DFT calculations a mechanism was proposed starting with the activation of the pre-catalyst, [Cp*RhCl2]2, mediated by the additive potassium pivalate. The coordination of the hydroxy group guided the C–H activation at the C-8 position of the substrate, resulting in intermediate 131-A. A sequence of coordination of the coupling partner and migratory insertion led to the respective cyclometalated intermediate, which suffered an oxidatively induced β-hydride elimination from Rh(IV)-intermediate 131-C, leading to the product 131 and a Rh(II) species. Anodic oxidation regenerated the active Rh(III)-catalyst (Scheme 22). In previous examples, such reactions were performed in the presence of silver(I) salts along with an additional oxidant.72 Here renewable electricity was used as the terminal oxidant in the absence of Ag-salt additives, which makes these processes more general, sustainable, and resource-economic.
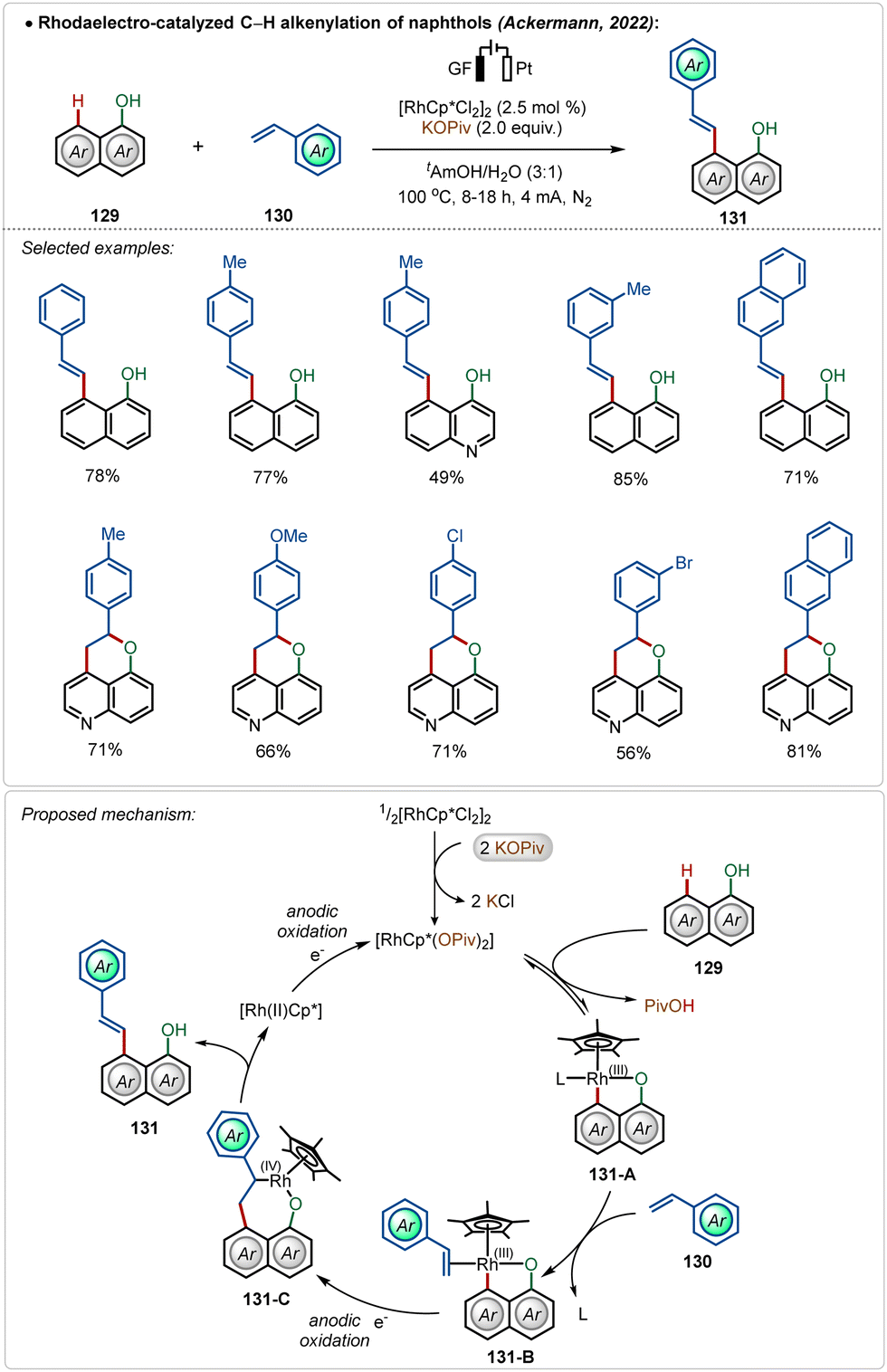 | ||
| Scheme 22 Rhodaelectro-catalyzed Ag-free C–H alkenylation of naphthols. | ||
Another convenient approach for silver-additive free C–H activation is the use of a preactivated electrophilic catalyst consisting of ligands that are labile enough to facilitate the interaction between the substrate and the metal centre,73 which was well exemplified by the work by Nan and co-workers.73a They delineated a successful rhodium-catalyzed C–H annulation of anilines for the formation of quinoline derivatives in moderate to good yields (Scheme 23). This strategy was empowered by the cationic [Cp*Rh(MeCN)3](SbF6)2 catalyst. The plausible mechanism involved an aryl-rhodium intermediate 135, probably achieved through a direct electrophilic metalation with the substrate.
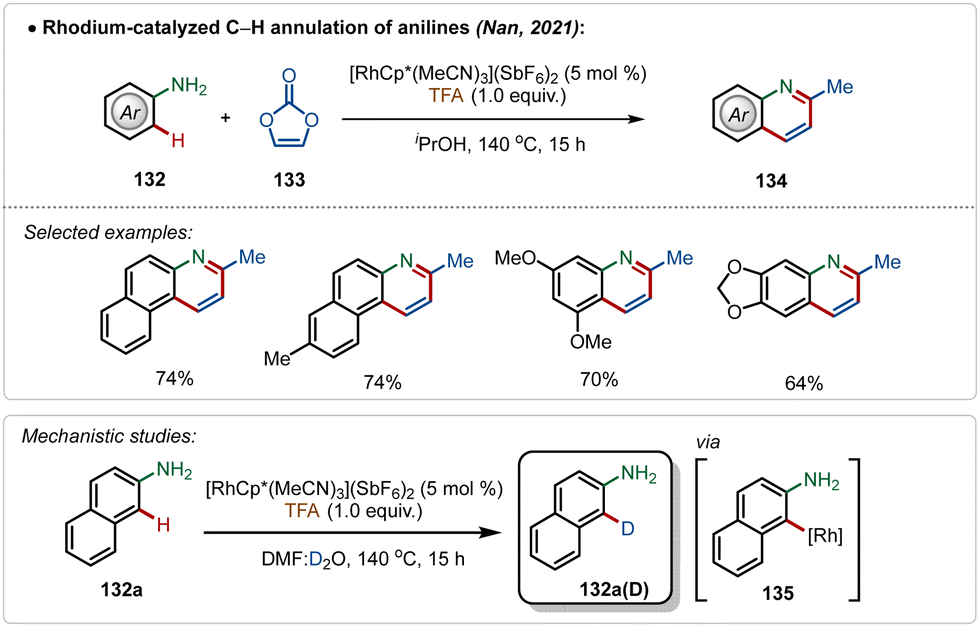 | ||
| Scheme 23 Rhodium-catalyzed Ag-free C–H annulation of anilines. | ||
In fact, Co(III)-catalyzed C–H activation is also viable in the absence of Ag(I)-additives.73g–k In 2014, Matsunaga and Kanai depicted an interesting cobalt-catalyzed C–H alkenylation of indoles with alkynes, constructing C-2 olefinated indoles in good yields (Scheme 24).74 This olefination reaction avoided the use of an additional Ag(I)-additive and used the preactivated cationic [Cp*Co(C6H6)](PF6)2 catalyst, which in the presence of KOAc smoothly executed the transformation. Plausibly, an active cationic [Cp*Co(OAc)]+ species was responsible for the transformation. A sequential annulation was also realized with morpholine derived amide directing group, where the catalytic conditions successfully engendered pyrroloindolone derivatives in good yields (Scheme 24).
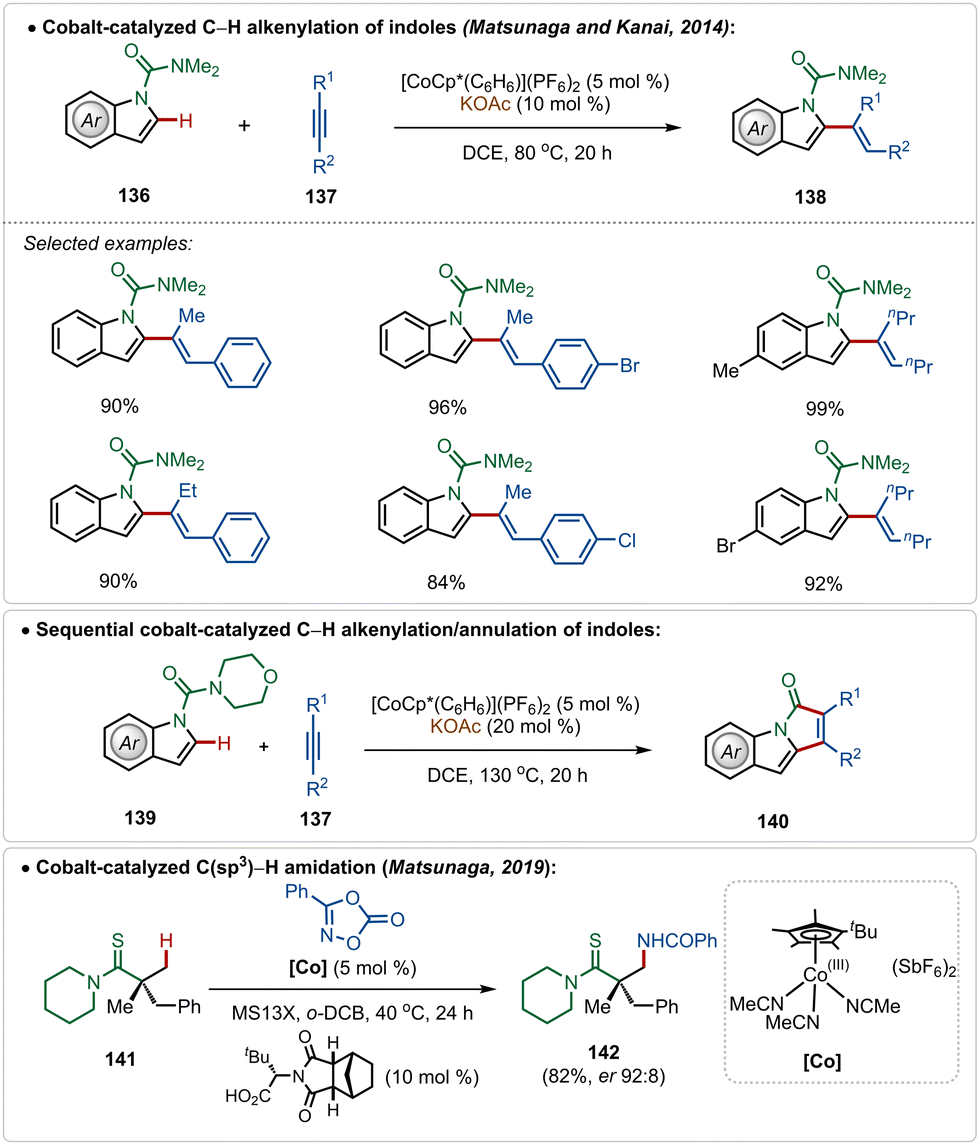 | ||
| Scheme 24 Cobalt-catalyzed Ag-free C–H alkenylation/annulation of indoles. | ||
Later, in 2019, they also revealed that this silver free approach was also feasible to perform enantioselective C(sp3)–H amidation reaction, where the chiral carboxylic acid was involved for chiral induction.75 With their continuous effort on 3d transition-metal catalysis, the Ackermann group has also contributed in Cp*Co(III)-catalyzed reactions under Ag(I)-additive free conditions. In 2016, they established a Co(III)-catalyzed ortho-C–H amidation of arenes, where various N-heterocycle susbtitutions were tolerated under the catalytic conditions and the functionalization was enabled selectively by the imidate directing group (Scheme 25).76 Different quinazoline derivatives were easily accessed in good to excellent yields through this approach. Easily accessible dioxazolones were used as the amidating agents, which after insertion to the cobaltacycle intermediate 144-A delivered the ortho-amidated intermediate. Intermediate 144-C after intramolecular condensation with the imidate formed the quinazoline products.
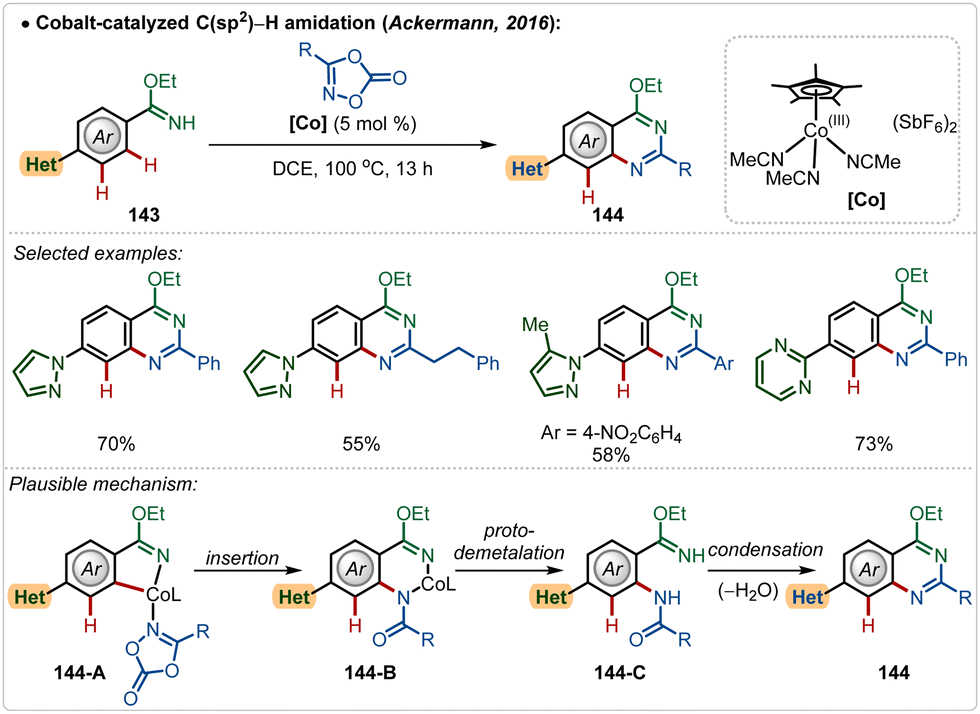 | ||
| Scheme 25 Overcoming the limitation of strongly coordinating N-heterocyles through Co(III)-catalysis. | ||
Recently, Ackermann has also explored Ag(I)-additive free Co(III)-catalyzed protocol for the late-stage modification of peptides (Scheme 26).77 Tryptophan-derived peptides were easily allylated with allyl acetate using the electrophilic Co(III)-catalyst. The reaction conditions were amenable to stitching amino acid derived terminal alkyne generating olefinated products (146). Interestingly, the reaction conditions tolerated olefin functionality in the substrate. Thus, the allylated products were easily manipulated into various cyclic peptides having modular ring sizes.
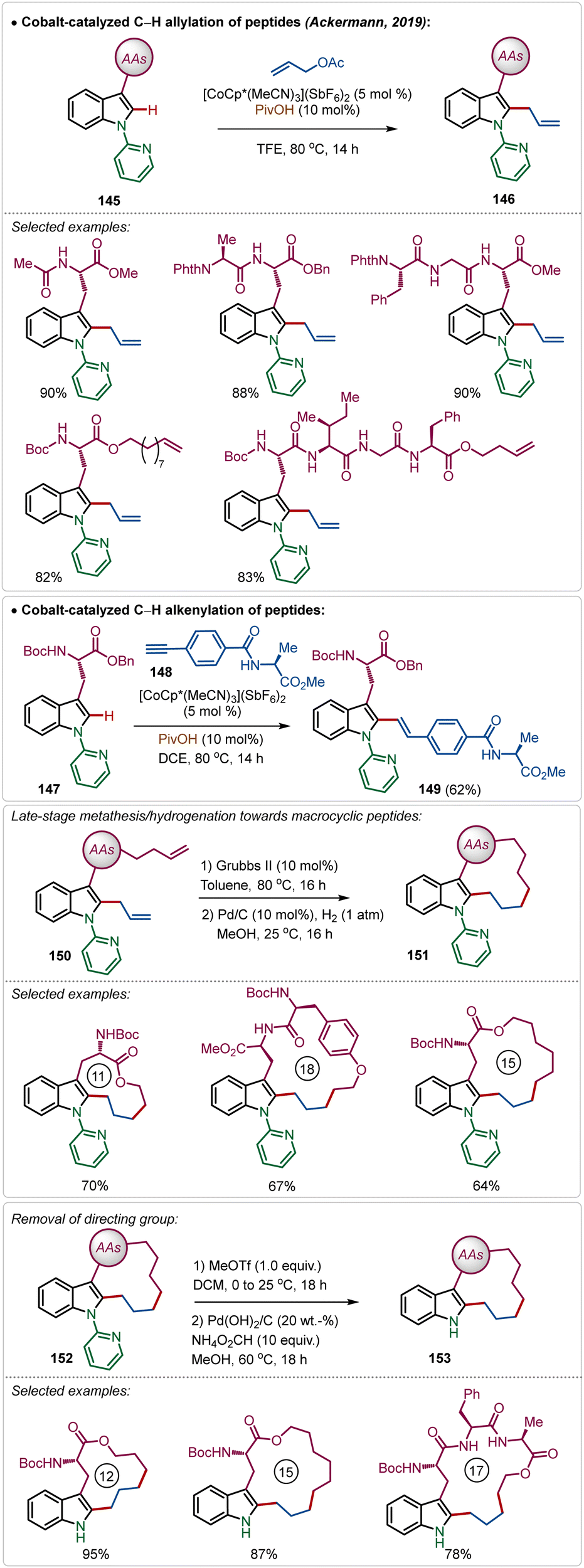 | ||
| Scheme 26 Late-stage functionalization of peptides with Co(III)-catalysis. | ||
An alternative useful way to avoid silver additives in C–H activation reactions is nanocatalysis.78 Such approaches are elusive and require superficial availability of the transition-metal containing nano-catalyst supported on the surface to mediate C–H activation. In 2021, Kilic, Metin and co-workers depicted an example78a of C–H arylation of imidazopyridine with bromo or iodobenzene using a Ni-Pd based nanocatalyst (Scheme 27). In this study, a nanocatalyst was designed using a nickel–palladium alloy nanoparticles, supported on a layer of graphene oxide previously modified with cobalt ferrite (CoFe2O4-rGO/Ni Pd NPs). This modification made it possible to magnetically separate the catalyst from the reaction media and reuse it on five consecutive cycles, without significantly compromising the achieved yield. Using this approach various arylated imidazopyridine derivatives were successfully accessed in good to excellent yields.
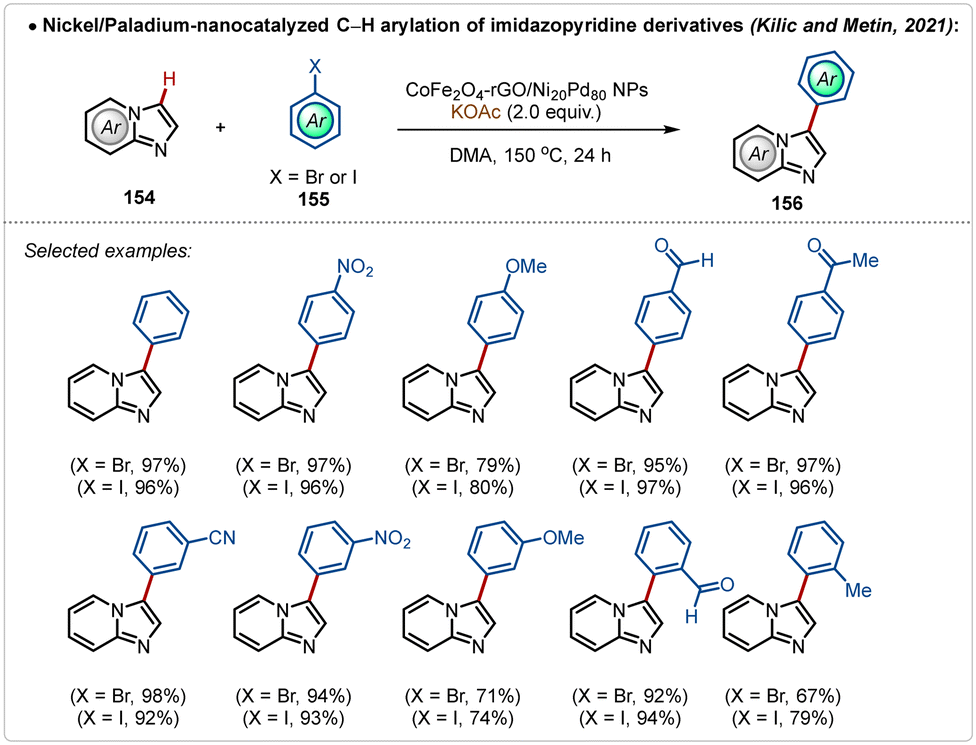 | ||
| Scheme 27 Nickel/paladium-nanocatalyzed Ag-free C–H arylation of imidazopyridine derivatives. | ||
8. Conclusions
Silver(I)-salts, being one of the most used additives in C–H activation reactions, have been investigated for detailed understanding of their role in different transition-metal catalysis. The preceding sections briefly elaborated on the crucial role of Ag(I)-salts in the generation of catalytically active species typically from dimeric half sandwiches, acting as a halide scavenger. This type of activation process is the common basis in ruthenium, rhodium, iridium, and cobalt catalysis, which makes it an indispensable additive for such transformations. In Pd(II)-catalyzed reactions, the Ag(I)-salt can act as a key metal to form bimetallic intermediates, which are decisive for product formation and often necessary to govern the selectivity. Herein, we have summarized representative examples of C–H activations involving Ag(I)-additives, along with a mechanistic analysis. Understanding these insights can influence the impending studies and serve as a guideline for the development of new strategies in and beyond organometallic C–H activation chemistry. Furthermore, the full understanding of the role of silver in C–H activation reactions is a crucial step towards sustainable silver-free approaches towards resource-economic electrochemical approaches. This coherent summary of the role of Ag(I)-additives in C–H activation reactions is expected to guide research in C–H activation to major advances, especially for the development of heterobimetallic catalysis and Ag-free strategies in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
E. N. da Silva Júnior acknowledges funding from CNPq (PQ 309774/2020-9 and Universal Project 405052/2021-9), FAPEMIG (PPM-00635-18 and TEC-RED-00282-16), INCT-Catálise/CNPq/FAPESC, the Return Fellowship of the Alexander von Humboldt Foundation (AvH), and CAPES Finance Code 001 and PROBRAL (88881.627934/2021-01). LA acknowledges the support from the ERC Advanced grant no. 101021358 and the DFG Gottfried-Wilhelm-Leibniz award.Notes and references
- (a) T. Rogge, N. Kaplaneris, N. Chatani, J. Kim, S. Chang, B. Punji, L. L. Schafer, D. G. Musaev, J. Wencel-Delord, C. A. Roberts, R. Sarpong, Z. E. Wilson, M. A. Brimble, M. J. Johansson and L. Ackermann, Nature Rev. Meth. Primers, 2021, 1, 43 CrossRef CAS; (b) T. Yoshino and S. Matsunaga, ACS Catal., 2021, 11, 6455–6466 CrossRef CAS; (c) P. Gandeepan, L. H. Finger, T. H. Meyer and L. Ackermann, Chem. Soc. Rev., 2020, 49, 4254–4272 RSC; (d) X. Li, W. Ouyang, J. Nie, S. Ji, Q. Chen and Y. Huo, ChemCatChem, 2020, 12, 2358–2384 CrossRef CAS; (e) P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS PubMed; (f) W. Liu and L. Ackermann, ACS Catal., 2016, 6, 3743–3752 CrossRef CAS; (g) S. S. Li, L. Qin and L. Dong, Org. Biomol. Chem., 2016, 14, 4554–4570 RSC; (h) F. F. Khan, S. K. Sinha, G. K. Lahiri and D. Maiti, Chem. Asian J., 2018, 13, 2243–2256 CrossRef CAS PubMed; (i) M. Moselage, J. Li and L. Ackermann, ACS Catal., 2016, 6, 498–525 CrossRef CAS; (j) L. Ackermann, Acc. Chem. Res., 2014, 47, 281–295 CrossRef CAS PubMed; (k) S. De Sarkar, W. Liu, S. I. Kozhushkov and L. Ackermann, Adv. Synth. Catal., 2014, 356, 1461–1479 CrossRef CAS; (l) L. Ackermann, Acc. Chem. Res., 2014, 47, 281–295 CrossRef CAS PubMed; (m) S. I. Kozhushkov and L. Ackermann, Chem. Sci., 2013, 4, 886–896 RSC; (n) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236–10254 CrossRef CAS PubMed; (o) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed; (p) L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed; (q) A. Deb, S. Manna, A. Modak, T. Patra, S. Maity and D. Maiti, Angew. Chem., Int. Ed., 2013, 52, 9747–9750 CrossRef CAS PubMed; (r) S. Maity, S. Manna, S. Rana, T. Naveen, A. Mallick and D. Maiti, J. Am. Chem. Soc., 2013, 135, 3355–3358 CrossRef CAS PubMed; (s) A. Maji, A. Hazra and D. Maiti, Org. Lett., 2014, 16, 4524–4527 CrossRef CAS PubMed.
- (a) L. Guillemard, N. Kaplaneris, L. Ackermann and M. J. Johansson, Nat. Rev. Chem., 2021, 5, 522–545 CrossRef CAS PubMed; (b) R. L. de Carvalho, A. S. de Miranda, M. P. Nunes, R. S. Gomes, G. A. M. Jardim and E. N. da Silva Júnior, Beilstein J. Org. Chem., 2021, 17, 1849–1938 CrossRef PubMed; (c) A. Dey, S. Maity and S. Maiti, Chem. Commun., 2016, 52, 12398–12414 RSC; (d) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651–3678 RSC.
- (a) K. Murali, L. A. Machado, R. L. de Carvalho, L. F. Pedrosa, R. Mukherjee, E. N. da Silva Júnior and D. Maiti, Chem. – Eur. J., 2021, 27, 12453–12508 CrossRef CAS PubMed; (b) R. L. de Carvalho, R. G. Almeida, K. Murali, L. A. Machado, L. F. Pedrosa, P. Dolui, D. Maiti and E. N. da Silva Júnior, Org. Biomol. Chem., 2021, 19, 525–547 RSC.
- (a) X.-S. Xue, P. Ji, B. Zhou and J.-P. Cheng, Chem. Rev., 2017, 117, 8622–8648 CrossRef CAS PubMed; (b) J. R. Hummel, J. A. Boerth and J. A. Ellman, Chem. Rev., 2017, 117, 9163–9227 CrossRef CAS PubMed; (c) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754–8786 CrossRef CAS PubMed; (d) T. Piou and T. Rovis, Acc. Chem. Res., 2018, 51, 170–180 CrossRef CAS PubMed; (e) S. Vásquez-Céspedes, X. Wang and F. Glorius, ACS Catal., 2018, 8, 242–257 CrossRef; (f) M.-Z. Lu, J. Goh, M. Maraswami, Z. Jia, J.-S. Tian and T.-P. Loh, Chem. Rev., 2022, 122, 17479–17646 CrossRef CAS PubMed; (g) S. Rej, Y. Ano and N. Chatani, Chem. Rev., 2020, 120, 1788–1887 CrossRef CAS PubMed; (h) A. Dey, S. K. Sinha, T. K. Achar and D. Maiti, Angew. Chem., Int. Ed., 2018, 58, 10820–10843 CrossRef PubMed; (i) Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107–1295 RSC; (j) R.-Y. Zhu, M. E. Farmer, Y.-Q. Chen and J.-Q. Yu, Angew. Chem., Int. Ed., 2016, 55, 10578–10599 CrossRef CAS PubMed; (k) J. M. Zakis, T. Smejkal and J. Wencel-Delord, Chem. Commun., 2022, 58, 483–490 RSC; (l) C. Haldar, M. E. Hoque, R. Bisht and B. Chattopadhyay, Tetrahedron Lett., 2018, 59, 1269–1277 CrossRef CAS; (m) O. Baudoin, Acc. Chem. Res., 2017, 50, 1114–1123 CrossRef CAS PubMed; (n) Y. Minami and T. Hiyama, Acc. Chem. Res., 2016, 49, 67–77 CrossRef CAS PubMed; (o) Ł. Woźniak and N. Cramer, Trends. Chem., 2019, 1, 471–484 CrossRef; (p) L. Ackermann, Acc. Chem. Res., 2020, 53, 84–104 CrossRef CAS PubMed; (q) J. Loup, U. Dhawa, F. Pesciaioli, J. Wencel-Delord and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 12803–12818 CrossRef CAS PubMed.
- (a) S. Ghosh, S. Shilpa, C. Athira and R. B. Sunoj, Topics in Catal., 2022, 65, 141–164 CrossRef CAS; (b) T. Bhattacharya, S. Dutta and D. Maiti, ACS Catal., 2021, 11, 9702–9714 CrossRef CAS; (c) Á. L. Mudarra, S. Martínez De Salinas and M. H. Pérez-Temprano, Org. Biomol. Chem., 2019, 17, 1655–1667 RSC; (d) K. L. Bay, Y.-F. Yang and K. N. Houk, J. Organomet. Catal., 2018, 864, 19–25 CrossRef CAS.
- R. L. de Carvalho, G. G. Dias, C. L. M. Pereira, P. Ghosh, D. Maiti and E. N. da Silva Júnior, J. Braz. Chem. Soc., 2021, 32, 917–952 Search PubMed.
- (a) H. Jiang and T. Y. Sun, Molecules, 2021, 26, 4083 CrossRef CAS PubMed; (b) J. Becica and G. E. Dobereiner, Org. Biomol. Chem., 2019, 17, 2055–2069 RSC; (c) D. L. Davies, S. A. Macgregor and C. L. McMullin, Chem. Rev., 2017, 117, 8649–8709 CrossRef CAS PubMed.
- J. Loup, D. Zell, J. C. A. Oliveira, H. Keil, D. Stalke and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 14197–14201 CrossRef CAS PubMed.
- (a) M. Anand, R. B. Sunoj and H. F. Schaefer, J. Am. Chem. Soc., 2014, 136, 5535–5538 CrossRef CAS PubMed; (b) B. Bhaskararao, S. Singh, M. Anand, P. Verma, P. Prakash, A. C. S. Malakar, H. F. Schaefer and R. B. Sunoj, Chem. Sci., 2020, 11, 208–216 RSC.
- For selected recent examples, see: (a) Y. L. Kim., S. Park and J. H. Kim, Eur. J. Org. Chem., 2020, 4026–4030 CrossRef CAS; (b) R. J. Harris, J. Park, T. A. F. Nelson, N. Iqbal, D. C. Salgueiro, J. Bacsa, C. E. MacBeth, M.-H. Baik and S. B. Blakey, J. Am. Chem. Soc., 2020, 142, 5842–5851 CrossRef CAS PubMed; (c) A. Mondal, H. Chen, L. Flämig, P. Wedi and M. van Gemmeren, J. Am. Chem. Soc., 2019, 141, 18662–18667 CrossRef CAS PubMed; (d) X.-L. Wu and L. Dong, Org. Lett., 2018, 20, 6990–6993 CrossRef CAS PubMed; (e) H. Kim, K. Shin and S. Chang, J. Am. Chem. Soc., 2014, 136, 5904–5907 CrossRef CAS PubMed.
- A. Mondal and M. van Gemmeren, Angew. Chem., Int. Ed., 2022, 61, e202210825 CrossRef CAS PubMed.
- S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529–531 CrossRef CAS.
- For selected examples, see: (a) Y. Li, Y.-C. Liou, J. C. A. Oliveira and L. Ackermann, Angew. Chem., Int. Ed., 2022, 61, e202212595 CrossRef CAS PubMed; (b) M. Schinkel, L. Wang, K. Bielefeld and L. Ackermann, Org. Lett., 2014, 16, 1876–1879 CrossRef CAS PubMed; (c) S. Nakanowatari and L. Ackermann, Chem. – Eur. J., 2014, 20, 5409–5413 CrossRef CAS PubMed; (d) W. Liu and L. Ackermann, Chem. Commun., 2014, 50, 1878–1881 RSC; (e) W. Ma and L. Ackermann, Chem. – Eur. J., 2013, 19, 13925–13928 CrossRef CAS PubMed; (f) L. Ackermann and A. V. Lygin, Org. Lett., 2012, 14, 764–767 CrossRef CAS PubMed; (g) C. Kornhaaß, J. Li and L. Ackermann, J. Org. Chem., 2012, 77, 9190–9198 CrossRef PubMed; (h) K. Graczyk, W. Ma and L. Ackermann, Org. Lett., 2012, 14, 4110–4113 CrossRef CAS PubMed; (i) W. Ma, K. Graczyk and L. Ackermann, Org. Lett., 2012, 14, 6318–6321 CrossRef CAS PubMed; (j) J. Li, C. Kornhaaß and L. Ackermann, Chem. Commun., 2012, 48, 11343–11345 RSC; (k) I. Choi, A. M. Messinis and L. Ackermann, Angew. Chem., Int. Ed., 2020, 59, 12534–12540 CrossRef CAS PubMed; (l) E. N. Silva Júnior, R. L. Carvalho, R. G. Almeida, L. G. Rosa, F. Fantuzzi, T. Rogge, P. M. S. Costa, C. Pessoa, C. Jacob and L. Ackermann, Chem. – Eur. J., 2020, 26, 10981–10986 CrossRef PubMed; (m) W. Ma, Z. Weng, X. Fang, L. Gu, Y. Song and L. Ackermann, Eur. J. Org. Chem., 2019, 41–45 CrossRef CAS; (n) W. Ma, Z. Weng, T. Rogge, L. Gu, J. Lin, A. Peng, X. Luo, X. Gou and L. Ackermann, Adv. Synth. Catal., 2018, 360, 704–710 CrossRef CAS.
- (a) Q. Lin, Z. Lin, M. Pan, Q. Zheng, H. Li, X. Chen, C. Darcel, P. H. Dixneuf and B. Li, Org. Chem. Front., 2021, 8, 514–521 RSC; (b) K. S. Singh, S. G. Sawant and P. H. Dixneuf, ChemCatChem, 2016, 6, 1046–1050 CrossRef; (c) B. Li, C. Darcel and P. H. Dixneuf, Chem. Commun., 2014, 50, 5970–5972 RSC.
- For selected examples, see: (a) T. Okada, K. Nobushige, T. Satoh and M. Miura, Org. Lett., 2016, 18, 1150–1153 CrossRef CAS PubMed; (b) Y. Hashimoto, K. Hirano, T. Satoh, F. Kakiuchi and M. Miura, Org. Lett., 2012, 14, 2058–2061 CrossRef CAS PubMed; (c) C. Suzuki, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2013, 15, 3990–3993 CrossRef CAS PubMed; (d) Y. Hashimoto, T. Ortloff, K. Hirano, T. Satoh, C. Bolm and M. Miura, Chem. Lett., 2012, 41, 151–153 CrossRef CAS.
- A. Biafora, T. Krause, D. Hackenberger, F. Belitz and L. J. Gooßen, Angew. Chem., Int. Ed., 2016, 55, 14752–14755 CrossRef CAS PubMed.
- (a) A. Hogg, M. Wheatley, P. Domingo-Legarda, A. Carral-Menoyo, N. Cottam and I. Larrosa, J. Am. Chem. Soc., 2022, 11, 2529–2538 Search PubMed; (b) G.-W. Wang, M. Wheatley, M. Simonetti, D. M. Cannas and I. Larrosa, Chem, 2020, 6, 1459–1468 CrossRef CAS; (c) M. Simonetti, D. M. Cannas, A. Panigrahi, S. Kujawa, M. Kryjewski, P. Xie and I. Larrosa, Chem. – Eur. J., 2017, 23, 549–553 CrossRef CAS PubMed; (d) M. Zheng, J. Zhou, F. Fang, Z. Xu, F. Zheng, Z. Jiang, T. Liu, H. Liu, L. Zhao and Y. Zhou, Org. Chem. Front., 2023, 10, 62–67 RSC.
- (a) F. Roudesly, J. Oble and G. Poli, J. Mol. Catal. A Chem., 2017, 426, 275–296 CrossRef CAS; (b) D. L. Davies, S. M. A. Donald, O. Al-duaij and S. A. Macgregor, J. Am. Chem. Soc., 2006, 128, 4210–4211 CrossRef CAS PubMed.
- For selective examples, see: (a) G. G. Dias, T. A. do Nascimento, A. K. A. de Almeida, A. C. S. Bombaça, R. F. S. Menna-Barreto, C. Jacob, E. N. da Silva Júnior and L. Ackermann, Eur. J. Org. Chem., 2019, 2344–2353 CrossRef CAS; (b) X. Lu, C. Shen, K. Meng, L. Zhao, T. Li, Y. Sun, J. Zhang and G. Zhong, Chem. Commun., 2019, 55, 826–829 RSC; (c) Q. Bu, T. Rogge, V. Kotek and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 765–768 CrossRef CAS PubMed; (d) R. Manoharan and M. Jeganmohan, Eur. J. Org. Chem., 2016, 4013–4019 CrossRef CAS.
- For selective examples, see: (a) U. Dhawa, R. Connon, J. C. A. Oliveira, R. Steinbock and L. Ackermann, Org. Lett., 2021, 23, 2760–2765 CrossRef CAS PubMed; (b) C.-S. Wang, P. H. Dixneuf and J.-F. Soulé, ChemCatChem, 2017, 9, 3117–3120 CrossRef CAS.
- Y. Liu, F. Chang, Q. Jiang, Z. Ma and C. Liu, Synlett, 2018, 658–662 CrossRef.
- For selective examples, see: (a) G. Bairy, A. Nandi, K. Manna and R. Jana, Synthesis, 2019, 2523–2531 CAS; (b) G. S. Kumar and M. Kapur, Org. Lett., 2016, 18, 1112–1115 CrossRef CAS PubMed.
- For selective examples, see: P. Nareddy, F. Jordan and M. Szostak, Org. Lett., 2018, 20, 341–344 CrossRef CAS PubMed.
- V. S. Thirunavukkarasu, K. Raghuvanshi and L. Ackermann, Org. Lett., 2013, 15, 3286–3289 CrossRef CAS PubMed.
- (a) D. Chowdhury, S. Dana, A. Mandal and M. Baidya, Chem. Commun., 2019, 55, 11908 RSC; (b) J. Nan, Z. Zuo, L. Luo, L. Bai, H. Zheng, Y. Yuan, J. Liu, X. Luan and Y. Wang, J. Am. Chem. Soc., 2013, 135, 17306–17309 CrossRef CAS PubMed.
- Q. Bu, T. Rogge, V. Kotek and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 765–768 CrossRef CAS PubMed.
- C.-A. Wang and N. Chatani, Org. Chem. Front., 2020, 7, 2955–2959 RSC.
- For selective examples, see: (a) Y. Yuan, J. Zhu, Z. Yang, S.-F. Ni, Q. Huang and L. Ackermann, CCS Chem., 2022, 4, 1858–1870 CrossRef CAS; (b) A. Das and N. Chatani, Chem. Sci., 2021, 12, 3202–3209 RSC; (c) N. Ohara, S. Rej and N. Chatani, Chem. Lett., 2020, 49, 1053–1057 CrossRef CAS; (d) S.-S. Li, L. Qin and L. Dong, Org. Biomol. Chem., 2016, 14, 4554–4570 RSC; (e) Y. Yokoyama, Y. Unoh, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2014, 79, 7649–7655 CrossRef CAS PubMed; (f) X. Yang, S. Li, Y. He, D. Dai, M. Bao, Z. Luo, X. Liu, Y. Geng and L. Fan, Org. Biomol. Chem., 2023, 21, 797–806 RSC; (g) A. S. Tsai, M. Brasse, R. G. Bergman and J. A. Ellman, Org. Lett., 2011, 13, 540–542 CrossRef CAS PubMed; (h) K. Ueura, T. Satoh and M. Miura, Org. Lett., 2007, 9, 1407–1409 CrossRef CAS PubMed; (i) T. Uto, M. Shimizu, K. Ueura, H. Tsurugi, T. Satoh and M. Miura, J. Org. Chem., 2008, 73, 298–300 CrossRef CAS PubMed.
- Selective example: S. Hazra, K. Hirano and M. Miura, Asian J. Org. Chem., 2019, 8, 1097–1101 CrossRef CAS.
- Selective example: L. Zhang, J. Zhao, Q. Mou, D. Teng, X. Meng and B. Sun, Adv. Synth. Catal., 2020, 362, 955–959 CrossRef CAS.
- Selective example: E. Tan, L. J. Nannini, O. Stoica and A. M. Echavarren, Org. Lett., 2021, 23, 1263–1268 CrossRef CAS PubMed.
- Z. T. Jiang, J. Huang, Y. Zeng, F. Hu and Y. Xia, Angew. Chem., Int. Ed., 2021, 60, 10626–10631 CrossRef CAS PubMed.
- L. Wang, X. Qu, Z. Li and W.-M. Peng, Tetrahedron Lett., 2015, 56, 3754–3757 CrossRef CAS.
- D. R. Stuart, M. Bertrand-Laperle, K. M. N. Burgess and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 16474–16475 CrossRef CAS PubMed.
- S. Rakshit, F. W. Patureau and F. Glorius, J. Am. Chem. Soc., 2010, 132, 9585–9587 CrossRef CAS PubMed.
- T.-J. Gong, B. Xiao, Z.-J. Liu, J. Wan, J. Xu, D.-F. Luo, Y. Fu and L. Liu, Org. Lett., 2011, 13, 3235–3237 CrossRef CAS PubMed.
- A. S. Tsai, M. E. Tauchert, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2011, 133, 1248–1250 CrossRef CAS PubMed.
- S. M. Khake and N. Chatani, ACS Catal., 2021, 11, 12375–12383 CrossRef CAS.
- (a) T. Wakikawa, D. Sekine, Y. Murata, Y. Bunno, M. Kojima, Y. Nagashima, K. Tanaka, T. Yoshino and S. Matsunaga, Angew. Chem., Int. Ed., 2022, 61, e202213659 CrossRef CAS PubMed; (b) Y. Kato, L. Lin, M. Kojima, T. Yoshino and S. Matsunaga, ACS Catal., 2021, 11, 4271–4277 CrossRef CAS; (c) S. Fukagawa, M. Kojima, T. Yoshino and S. Matsunaga, Angew. Chem., Int. Ed., 2019, 58, 18154–18158 CrossRef CAS PubMed.
- Q. Li, R. Liu, Y. Wei and M. Shi, Adv. Synth. Catal., 2021, 363, 2664–2669 CrossRef CAS.
- For selective examples, see: (a) R. Yabe, Y. Ebe and T. Nishimura, Synthesis, 2021, 3051–3056 CAS; (b) A. T. Tran and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 10530–10534 CrossRef CAS PubMed; (c) S. Kathiravan and I. A. Nicholls, Chem. – Eur. J., 2017, 23, 7031–7036 CrossRef CAS PubMed; (d) C. Pan, N. Jin, H. Zhang, J. Han and C. Zhu, J. Org. Chem., 2014, 79, 9427–9432 CrossRef CAS PubMed; (e) M. Nagamoto and T. Nishimura, Chem. Commun., 2014, 50, 6274–6277 RSC; (f) Z. Liu, Q. Li, C. Yang, X. Zheng, D. Wu, G. Gao and J. Lan, Chem. Commun., 2022, 58, 7042–7045 RSC; (g) J. Xia, X. Yang, Y. Li and X. Li, Org. Lett., 2017, 19, 3243–3246 CrossRef CAS PubMed.
- (a) J. Ryu, J. Kwak, K. Shin, D. Lee and S. Chang, J. Am. Chem. Soc., 2013, 135(34), 12861 CrossRef CAS PubMed; (b) J. Kim and S. Chang, Angew. Chem., Int. Ed., 2014, 53, 2203–2207 CrossRef CAS PubMed.
- T. M. Figg, S. Park, J. Park, S. Chang and D. G. Musaev, Organometallics, 2014, 33, 4076–4085 CrossRef CAS.
- (a) W. Sarkar, A. Bhowmik, S. Das, A. B. Sulekha, A. Mishra and I. Deb, Org. Biomol. Chem., 2020, 18, 7074–7078 RSC; (b) K. Shin, Y. Park, M.-H. Baik and S. Chang, Nat. Chem., 2018, 10, 218–224 CrossRef CAS PubMed; (c) P. Gao, W. Guo, J. Xue, Y. Zhao, Y. Yuan, Y. Xia and Z. Shi, J. Am. Chem. Soc., 2015, 137, 12231–12240 CrossRef CAS PubMed.
- X.-Y. Chen and E. J. Sorensen, Chem. Sci., 2018, 9, 8951–8956 RSC.
- C. N. Kona, Y. Nishii and M. Miura, Angew. Chem., Int. Ed., 2019, 58, 9856–9860 CrossRef CAS PubMed.
- S. M. Khake, K. Yamazaki, Y. Ano and N. Chatani, ACS Catal., 2021, 11, 5463–5471 CrossRef CAS.
- J. Kim, S.-W. Park, M.-H. Baik and S. Chang, J. Am. Chem. Soc., 2015, 137, 13448–13451 CrossRef CAS PubMed.
- H. Kim and S. Chang, ACS Catal., 2015, 5, 6665–6669 CrossRef CAS.
- C. Pan, Z. Yang, H. Xiong, J. Teng, Y. Wang and J.-T. Yu, Chem. Commun., 2019, 55, 1915–1918 RSC.
- (a) H. S. Park, Z. Fan, R.-Y. Zhu and J.-Q. Yu, Angew. Chem., Int. Ed., 2020, 59, 12853–12859 CrossRef CAS PubMed; (b) Z. Zhuang and J.-Q. Yu, J. Am. Chem. Soc., 2020, 142, 12015–12019 CrossRef CAS PubMed; (c) S. Pimparkar, T. Bhattacharya, A. Maji, A. Saha, R. Jayarajan, U. Dutta, G. Lu, D. W. Lupton and D. Maiti, Chem. – Eur. J., 2020, 26, 11558–11564 CrossRef CAS PubMed; (d) E. T. Nadres, G. I. F. Santos, D. Shabashov and O. Daugulis, J. Org. Chem., 2013, 78, 9689–9714 CrossRef CAS PubMed; (e) S. Ueda, T. Okada and H. Nagasawa, Chem. Commun., 2010, 46, 2462–2464 RSC; (f) U. Dhawa, T. Wdowik, X. Hou, B. Yuan, J. C. A. Oliveira and L. Ackermann, Chem. Sci., 2021, 12, 14182–14188 RSC; (g) S. Agasti, B. Mondal, T. K. Achar, S. K. Sinha, A. S. Suseelan, K. J. Szabo, F. Schoenebeck and D. Maiti, ACS Catal., 2019, 9, 9606–9613 CrossRef CAS; (h) X. Bao, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2017, 56, 9577–9581 CrossRef CAS PubMed; (i) X. Ye, C. Xu, L. Wojtas, N. G. Akhmedov, H. Chen and X. Shi, Org. Lett., 2016, 18, 2970–2973 CrossRef CAS PubMed; (j) T. Yamada, Y. Hashimoto, K. Tanaka, N. Morita and O. Tamura, RSC Adv., 2022, 12, 21548–21557 RSC; (k) M. Bera, A. Maji, S. K. Sahoo and D. Maiti, Angew. Chem., Int. Ed., 2015, 54, 8515–8519 CrossRef CAS PubMed; (l) N. Goswami, S. K. Sinha, P. Mondal, S. Adhya, A. Datta and D. Maiti, Chem, 2023, 9, 989–1003 CrossRef CAS; (m) M. Bera, A. Modak, T. Patra, A. Maji and D. Maiti, Org. Lett., 2014, 16, 5760–5763 CrossRef CAS PubMed; (n) M. Bera, S. K. Sahoo and D. Maiti, ACS Catal., 2016, 6, 3575–3579 CrossRef CAS; (o) S. Bag, R. Jayarajan, R. Mondal and D. Maiti, Angew. Chem., Int. Ed., 2017, 56, 3182–3186 CrossRef PubMed; (p) S. Bag, R. Jayarajan, U. Dutta, R. Chowdhury, R. Mondal and D. Maiti, Angew. Chem., Int. Ed., 2017, 56, 12538–12542 CrossRef CAS PubMed; (q) R. Jayarajan, J. Das, S. Bag, R. Chowdhury and D. Maiti, Angew. Chem., Int. Ed., 2018, 57, 7659–7663 CrossRef CAS PubMed; (r) S. Bag, S. Jana, S. Pradhan, S. Bhowmick, N. Goswami, S. K. Sinha and D. Maiti, Nat. Commun., 2021, 12, 1393 CrossRef CAS PubMed.
- D. Whitaker, J. Burés and I. Larossa, J. Am. Chem. Soc., 2016, 138, 8384–8387 CrossRef CAS PubMed.
- Y.-F. Yang, G.-J. Cheng, P. Liu, D. Leow, T.-Y. Sun, P. Chen, X. Zhang, J.-Q. Yu, Y.-D. Wu and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 344–355 CrossRef CAS.
- L. Fang, T. G. Saint-Denis, B. L. H. Taylor, S. Ahlquist, K. Hong, S. Liu, L. Han, K. N. Houk and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 10702–10714 CrossRef CAS PubMed.
- W. Feng, T. Wang, D. Liu, X. Wang and Y. Dang, ACS Catal., 2019, 9, 6672–6680 CrossRef CAS.
- S. Porey, X. Zhang, S. Bhowmick, V. K. Singh, S. Guin, R. S. Paton and D. Maiti, J. Am. Chem. Soc., 2020, 142, 3762–3774 CrossRef CAS PubMed.
- (a) C. Tian, T. H. Meyer, M. Stangier, U. Dhawa, K. Rauch, L. H. Finger and L. Ackermann, Nat. Protoc., 2020, 15, 1760–1774 CrossRef CAS PubMed; (b) R. Mei, U. Dhawa, R. C. Samanta, W. Ma, J. Wencel-Delord and L. Ackermann, ChemSusChem, 2020, 13, 3306–3356 CrossRef CAS PubMed; (c) N. Sauermann, T. H. Meyer and L. Ackermann, Chem. – Eur. J., 2018, 24, 16209–16217 CrossRef CAS PubMed.
- For selective examples, see: (a) Q.-J. Yao, F.-R. Huang, J.-H. Chen, M.-Y. Zhong and B.-F. Shi, Angew. Chem., Int. Ed., 2023, 62, e202218533 CrossRef CAS PubMed; (b) P. Dhillon, P. Anaspure, J. G. Wiklander, S. Kathiravan and I. A. Nicholls, Org. Biomol. Chem., 2023, 21, 1942–1951 RSC; (c) T. V. Münchow, S. Dana, Y. Xu, B. Yuan and L. Ackermann, Science, 2023, 379, 1036–1042 CrossRef PubMed; (d) Y.-H. Huang and L. Dong, Adv. Synth. Catal., 2022, 365, 23–30 CrossRef; (e) S. Saha and M. S. Maji, Chem. Commun., 2022, 58, 10865–10868 RSC; (f) T. Yoshino, H. Ikemoto, S. Matsunaga and M. Kanai, Angew. Chem., Int. Ed., 2013, 52, 2207–2211 CrossRef CAS PubMed; (g) T. Yoshino, H. Ikemoto, S. Matsunaga and M. Kanai, Chem. – Eur. J., 2013, 19, 9142–9146 CrossRef CAS PubMed.
- For selective examples, see: (a) S. D. Friis, M. J. Johansson and L. Ackermann, Nat. Chem., 2020, 12, 511–519 CrossRef CAS PubMed; (b) Y.-H. Liu, P.-P. Xie, L. Liu, J. Fan, Z.-Z. Zhang, X. Hong and B.-F. Shi, J. Am. Chem. Soc., 2021, 143, 19112–19120 CrossRef CAS PubMed; (c) T. Li, C. Shen, Y. Sun, J. Zhang, P. Xiang, X. Lu and G. Zhong, Org. Lett., 2019, 21, 7772–7777 CrossRef CAS PubMed; (d) F. Pesciaioli, U. Dutta, J. C. A. Oliveira, R. Yin, M. John and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 15425–15429 CrossRef CAS PubMed; (e) D. Zell, M. Bursch, V. Müller, S. Grimme and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 10378–10382 CrossRef CAS PubMed.
- K. Ozols, S. Onodera, Ł. Woźniak and N. Cramer, Angew. Chem., Int. Ed., 2021, 60, 655–659 CrossRef CAS PubMed.
- M. Sen, B. Emayavaramban, N. Barsu, J. R. Premkumar and B. Sundararaju, ACS Catal., 2016, 6, 2792–2796 CrossRef CAS.
- N. Sauermann, M. J. González and L. Ackermann, Org. Lett., 2015, 17, 5316–5319 CrossRef CAS PubMed.
- K. Ramachandran and P. Anbarasan, Eur. J. Org. Chem., 2017, 3965–3968 CrossRef CAS.
- M. Liu, J.-L. Niu, D. Yang and M.-P. Song, J. Org. Chem., 2020, 85, 4067–4078 CrossRef CAS PubMed.
- (a) E. Detmar, V. Müller, D. Zell, L. Ackermann and M. Breugst, Beilstein J. Org. Chem., 2018, 14, 1537–1545 CrossRef CAS PubMed; (b) Z. Zhang, S. Han, M. Tang, L. Ackermann and J. Li, Org. Lett., 2017, 19, 3315–3318 CrossRef CAS PubMed; (c) H. Wang, M. M. Lorion and L. Ackermann, ACS Catal., 2017, 7, 3430–3433 CrossRef CAS; (d) D. Zell, V. Müller, U. Dhawa, M. Bursch, R. R. Presa, S. Grimme and L. Ackermann, Chem. – Eur. J., 2017, 23, 12145–12148 CrossRef CAS PubMed; (e) S. Nakanowatari, R. Mei, M. Feldt and L. Ackermann, ACS Catal., 2017, 7, 2511–2515 CrossRef CAS; (f) R. Mei, J. Loup and L. Ackermann, ACS Catal., 2016, 6, 793–797 CrossRef CAS; (g) D. Zell, Q. Bu, M. Feldt and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 7408–7412 CrossRef CAS PubMed; (h) J. Li, M. Tang, L. Zang, X. Zhang, Z. Zhang and L. Ackermann, Org. Lett., 2016, 18, 2742–2745 CrossRef CAS; (i) H. Wang, M. Moselage, M. J. González and L. Ackermann, ACS Catal., 2016, 6, 2705–2709 CrossRef CAS; (j) N. Sauermann, M. J. González and L. Ackermann, Org. Lett., 2015, 17, 5316–5319 CrossRef CAS PubMed; (k) J. Li and L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 8551–8554 CrossRef CAS; (l) H. Wang, J. Koeller, W. Liu and L. Ackermann, Chem. – Eur. J., 2015, 21, 15525–15528 CrossRef CAS PubMed; (m) M. Moselage, N. Sauermann, J. Koeller, W. Liu, D. Gelman and L. Ackermann, Synlett, 2015, 1596–1600 CAS.
- L. Kong, B. Biletskyi, D. Nuel and H. Clavier, Org. Chem. Front., 2018, 5, 1600–1603 RSC.
- P. Patel and S. Chang, ACS Catal., 2015, 5, 853–858 CrossRef CAS.
- (a) C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7, 415–431 CrossRef CAS PubMed; (b) T. H. Meyer, I. Choi, C. Tian and L. Ackermann, Chem, 2020, 6, 2484–2496 CrossRef CAS for further electrochemistry examples, see: ; (c) C.-Y. Cai, X.-L. Lai, Y. Wang, H.-H. Hu, J. Song, Y. Yang, C. Wang and H.-C. Xu, Nat. Catal., 2022, 5, 943–951 CrossRef CAS; (d) H. Long, T.-S. Chen, J. Song, S. Zhu and H.-C. Xu, Nat. Commun., 2022, 13, 3945 CrossRef CAS PubMed; (e) H. Long, C. Huang, Y.-T. Zheng, Z.-Y. Li, L.-H. Jie, J. Song, S. Zhu and H.-C. Xu, Nat. Commun., 2021, 12, 6629 CrossRef CAS PubMed; (f) M. Chen, Z.-J. Wu, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2022, 61, e202115954 CrossRef CAS PubMed.
- (a) Y. Wang, H. Simon, X. Chen, Z. Lin, S. Chen and L. Ackermann, Angew. Chem., Int. Ed., 2022, 61, e202201595 CrossRef CAS PubMed; (b) X. Hou, N. Kaplaneris, B. Yuan, J. Frey, T. Ohyama, A. M. Messinis and L. Ackermann, Chem. Sci., 2022, 13, 3461–3467 RSC; (c) Y. Qiu, C. Tian, L. Massignan, T. Rogge and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 5818–5822 CrossRef CAS PubMed; (d) G. G. Dias, T. Rogge, R. Kuniyl, C. Jacob, R. F. S. Menna-Barreto, E. N. da silva Júnior and L. Ackermann, Chem. Commun., 2018, 54, 12840–12843 RSC.
- (a) B. Sadowski, B. Yuan, Z. Lin and L. Ackermann, Angew. Chem., Int. Ed., 2022, 61, e202117188 CrossRef CAS PubMed; (b) W. Wei, A. Scheremetjew and L. Ackermann, Chem. Sci., 2022, 13, 2783–2788 RSC; (c) Y. Qiu, W.-J. Kong, J. Struwe, N. Sauermann, T. Rogge, A. Scheremetjew and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 5828–5832 CrossRef CAS PubMed.
- (a) B. Sadowski, B. Yuan, Z. Lin and L. Ackermann, Angew. Chem., Int. Ed., 2022, 61, e202117188 CrossRef CAS PubMed; (b) W. Wei, A. Scheremetjew and L. Ackermann, Chem. Sci., 2022, 13, 2783–2788 RSC; (c) Y. Qiu, W.-J. Kong, J. Struwe, N. Sauermann, T. Rogge, A. Scheremetjew and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 5828–5832 CrossRef CAS PubMed.
- X. W. Wang, X. T. Li, J. Xiao, Y. Jiang and X. W. Li, Synlett, 2012, 1649–1652 CAS.
- (a) J. Nan, J. Yin, X. Gong, Y. Hu and Y. Ma, Org. Lett., 2021, 23, 8910–8915 CrossRef CAS PubMed; (b) N. Aravindan and M. Jeganmohan, J. Org. Chem., 2021, 86, 14826–14843 CrossRef CAS PubMed; (c) K. Korvorapun, J. Struwe, R. Kuniyil, A. Zangarelli, A. Casnati, M. Waeterschoot and L. Ackermann, Angew. Chem., Int. Ed., 2020, 59, 18103–18109 CrossRef CAS PubMed; (d) D. Doiuchi, T. Nakamura, H. Hayashi and T. Uchida, Chem. Asian J., 2020, 15, 762–765 CrossRef CAS PubMed; (e) X. Yan, P. Zhao, H. Liang, H. Xie, J. Jiang, S. Gou and J. Wang, Org. Lett., 2020, 22, 3219–3223 CrossRef CAS PubMed; (f) M. R. Jones, C. D. Fast and N. D. Schley, J. Am. Chem. Soc., 2020, 142, 6488–6492 CrossRef CAS PubMed; (g) G. Zhou, J.-H. Chen, Q.-J. Yao, F.-R. Huang, Z.-K. Wang and B. F. Shi, Angew. Chem., Int. Ed., 2023, 62, e202302964 CrossRef CAS PubMed; (h) Y.-B. Zhou, T. Zhou, P.-F. Qian, J.-Y. Li and B.-F. Shi, ACS Catal., 2022, 12, 9806–9811 CrossRef CAS; (i) L.-B. Zhang, X.-Q. Hao, S.-K. Zhang, Z.-J. Liu, X.-X. Zheng, J.-F. Gong, J.-L. Niu and M.-P. Song, Angew. Chem., Int. Ed., 2015, 54, 272–275 CrossRef CAS PubMed; (j) D. Yang, X. Zhang, X. Wang, X.-J. Si, J. Wang, D. Wei, M.-P. Song and J.-L. Niu, ACS Catal., 2023, 13, 4250–4260 CrossRef CAS; (k) X.-J. Si, X. Zhao, J. Wang, X. Wang, Y. Zhang, D. Yang, M.-P. Song and J.-L. Niu, Chem. Sci., 2023, 14, 7291–7303 RSC.
- H. Ikemoto, T. Yoshino, K. Sakata, S. Matsunaga and M. Kanai, J. Am. Chem. Soc., 2014, 136, 5424–5431 CrossRef CAS PubMed.
- S. Fukagawa, Y. Kato, R. Tanaka, M. Kojima, T. Yoshino and S. Matsunaga, Angew. Chem., Int. Ed., 2019, 58, 1153–1157 CrossRef CAS PubMed.
- H. Wang, M. M. Lorion and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 10386–10390 CrossRef CAS PubMed.
- M. M. Lorion, N. Kaplaneris, J. Son, R. Kuniyil and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 1684–1688 CrossRef CAS PubMed.
- (a) Y. Zozik, M. Sevim, F. Lafzi, H. Kilic and Ö. Metin, Dalton Trans., 2021, 50, 17515–17523 RSC; (b) K. Kim, Y. Jung, S. Lee, M. Kim, D. Shin, H. Byun, S. J. Cho, H. Song and H. Kim, Angew. Chem., Int. Ed., 2017, 56, 6952–6956 CrossRef CAS PubMed; (c) V. Pascanu, F. Carson, M. V. Solano, J. Su, X. Zou, M. J. Johansson and B. Matín-Matute, Chem. – Eur. J., 2016, 22, 3729–3737 CrossRef PubMed; (d) H. Woo, J. Park, J. C. Park, S. Park, J. M. Lee and K. H. Park, RSC Adv., 2016, 6, 36211–36217 RSC; (e) M. Jafarpour, H. Kargar and A. Rezaeifard, RSC Adv., 2016, 6, 25034–25046 RSC; (f) C. Taglang, L. M. Martínez-Prieto, I. del Rosal, L. Maron, R. Poteau, K. Philippot, B. Chaudret, S. Perato, A. S. Lone, C. Puente, C. Dugave, B. Rousseau and G. Pieters, Angew. Chem., Int. Ed., 2015, 54, 10474–10477 CrossRef CAS PubMed; (g) A. Paul, D. Chatterjee, S. Banerjee and S. Yadav, Beilstein J. Org. Chem., 2020, 16, 140–148 CrossRef CAS PubMed; (h) P. Bora, D. Konwar, A. Dewan, M. R. Das and U. Bora, New J. Chem., 2022, 46, 12551–12557 RSC; (i) R. Xie, F. Xie, C.-J. Zhou, H.-F. Jiang and M. Zhang, J. Catal., 2019, 377, 449–454 CrossRef CAS; (j) N. Abeadi, R. Zhiani, A. Motavalizadehkakhky, M. Omidvar and M. S. Hosseiny, Inorg. Chem. Commun., 2021, 123, 108356 CrossRef CAS; (k) P. Rana, R. Gaur, B. Kaushik, P. Rana, S. Yadav, P. Yadav, P. Sharma, M. B. Gawande and R. K. Sharma, J. Catal., 2021, 401, 297–308 CrossRef CAS; (l) P. Rana, R. Gaur, R. Gupta, G. Arora, A. Jayashree and R. K. Sharma, Chem. Commun., 2019, 55, 7402–7405 RSC.
| This journal is © The Royal Society of Chemistry 2023 |