
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAcid-base-induced fac → mer isomerization of luminescent iridium(III) complexes†
Anastasia
Yu. Gitlina
 a,
Farzaneh
Fadaei-Tirani
a,
Albert
Ruggi
b,
Carolina
Plaice
a and
Kay
Severin
*a
a,
Farzaneh
Fadaei-Tirani
a,
Albert
Ruggi
b,
Carolina
Plaice
a and
Kay
Severin
*a
aInstitut des Sciences et Ingénierie Chimiques, École Polytechnique Fédérale de Lausanne (EPFL), 1015 Lausanne, Switzerland. E-mail: kay.severin@epfl.ch
bDépartement de Chimie, Université de Fribourg, 1700 Fribourg, Switzerland
First published on 22nd August 2022
Abstract
Luminescent Ir(C^N)3 complexes (C^N = cyclometalated arylpyridine ligand) exist in the form of two stable isomers with distinct photophysical and electrochemical properties: fac and mer. Herein, we show that fac-Ir(C^N)3 complexes can be converted into the thermodynamically less stable mer forms by a consecutive reaction with first acid and then base. The chemically induced isomerization is fast, quantitative, and stereoselective, and it can be inversed by light. The new isomerization process opens the possibility to use highly luminescent Ir(C^N)3 complexes as molecular switches.
Introduction
Iridium(III) complexes with cyclometalated arylpyridine ligands (C^N ligands) display intriguing photophysical and chemical properties. Noteworthy characteristics include high photoluminescence quantum yields, reversible redox transitions, a pronounced chemical and thermal stability, and the possibility to tune the emission color, the phosphorescence lifetime, and the redox properties by structural modifications.1 The unique properties of Ir(C^N)3 complexes have led to diverse applications.1–10 For example, Ir(C^N)3 complexes have been used as emitters in organic light-emitting diodes (OLEDs),2,3 as photoredox catalysts,4,5 as chemical or biological probes,6–8 and as building blocks for supramolecular assemblies.9,10Ir(C^N)3 complexes form two stable isomers with distinct photophysical and electrochemical properties: fac and mer.11 The mer isomers are thermodynamically less stable than the fac isomers. By adjusting the reaction conditions, it is possible to obtain the mer isomer in a kinetically controlled reaction.11,12 A mer → fac isomerization can be achieved at high temperature or by irradiation.11,13–15 The inverse fac → mer isomerization, which is thermodynamically uphill, has not been reported so far.
Herein, we show that a fac → mer isomerization can be achieved by a consecutive reaction with first acid and then base. The chemically induced isomerization allows using highly luminescent Ir(C^N)3 complexes as molecular switches.
Results and discussion
While studying the chemistry of fac-Ir(ppy)3 (ppy = metalated 2-phenylpyridine), we made a surprising observation: when a solution of fac-Ir(ppy)3 in dichloromethane was treated in a successive fashion with first trifluoroacetic acid (TFA, 10 equiv.) and then with triethylamine (10.5 equiv.), a quantitative conversion into the mer isomer was observed (Scheme 1).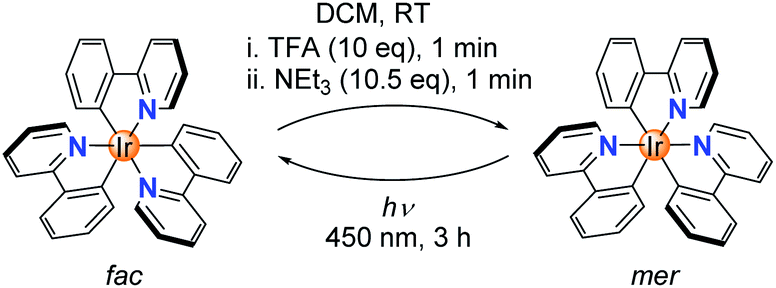 | ||
| Scheme 1 Interconversion of fac-Ir(ppy)3 and mer-Ir(ppy)3. | ||
The chemically induced transformation of fac-Ir(ppy)3 into mer-Ir(ppy)3 was followed by 1H NMR spectroscopy using CD2Cl2 as solvent. The TFA treatment resulted in the rapid (<1 min) formation of a defined complex of low symmetry, with three sets of signals for the three ligands (Fig. 1a). Furthermore, the NMR data indicated that protonation had occurred at one of the phenyl groups, thereby converting a chelating, anionic C^N ligand into a monodentate, neutral N-donor ligand.
 | ||
| Fig. 1 (a) 1H NMR spectra (400 MHz, CD2Cl2, aromatic region) for the conversion of fac-Ir(ppy)3 into mer-Ir(ppy)3 and back. (b) Molecular structure of [Ir(ppy)2(Hppy)](NTf2) in the crystal with thermal ellipsoids at 50% probability; the anion is not shown for clarity. (c) Photos of solutions containing fac-Ir(ppy)3, [Ir(ppy)2(Hppy)](O2CCF3) (‘open form’), and mer-Ir(ppy)3 under UV-light (λex = 366 nm). | ||
Attempts to characterize the TFA adduct by single crystal X-ray crystallography were not successful. However, when using bistriflimidic acid (HNTf2) instead of TFA, we were able to crystallize the ‘open form’. The result of a crystallographic analysis is depicted in Fig. 1b. In line with the NMR data, the cationic Ir complex features two orthometalated 2-phenylpyridine ligands and one neutral 2-phenylpyridine ligand, which is bound via the pyridine group to the metal. One C–H group of the phenyl ring forms an agostic interaction16 with the cationic Ir center (Ir⋯H ∼ 1.97 Å; Ir–H–C ∼ 115°). When the agostic interaction is taken into account, the coordination environment around Ir can be described as distorted octahedral. The open form has a meridional arrangement of the three N-donor atoms, and a base-induced metalation is primed to give the mer isomer of Ir(ppy)3, as found experimentally. Within one minute after the addition of NEt3, a quantitative formation of mer-Ir(ppy)3 was observed (Fig. 1a). As described in the literature,11–15 it is possible to convert the mer isomer back into the fac isomer by irradiation. For this purpose, we have used a 90 W LED lamp with an emission maximum at 450 nm. A clean back-isomerization was achieved within 3 h, as evidenced again by 1H NMR spectroscopy (Fig. 1a). The fac and the mer isomer of Ir(ppy)3 show distinct emission colours and intensities, with a weaker, red-shifted emission for the mer form (Fig. 1c).11 The open form [Ir(ppy)2(Hppy)](O2CCF3) is largely non-luminescent.
The acid-base reaction accompanying the transformation of fac-Ir(ppy)3 to mer-Ir(ppy)3 results in the formation of (HNEt3)(O2CCF3) (Fig. 2a). In order to examine if the presence of this salt would compromise subsequent isomerization steps, we have performed multiple fac → mer → fac cycles in the same reaction flask without separation/purification steps. The conversion efficiency was monitored by recording the emission spectra after each isomerization. The results indicate that for ten cycles, neither the chemically induced fac → mer isomerization nor the photochemical mer → fac isomerization is compromised to a significant extent by the presence of accumulating amounts of salt (ESI, Fig. S37†). The robustness of the switching process was substantiated by 1H NMR studies in CD2Cl2. After three fac → mer → fac cycles, only small amounts of side products could be detected by 1H NMR spectroscopy (ESI, Fig. S2–4†).
 | ||
| Fig. 2 Acid-base treatment followed by irradiation allows repetitive cycling between fac- and mer-Ir(ppy)3. The interference of accumulating (HNEt3)(O2CCF3) is low for the first ten cycles, as evidenced by spectroscopic monitoring. | ||
Next, we examined if other Ir(C^N)3 complexes would also undergo acid-base-induced fac → mer isomerizations. Complexes with orthometalated p-tolylpyridine (tpy), 2-(4-methoxyphenyl)pyridine (meppy) and 2-(4-tert-butylphenyl)pyridine (buppy) ligands behaved in a similar fashion as Ir(ppy)3: a clean and fast fac → mer conversion could be induced by consecutive addition of TFA (5–10 equiv.) and NEt3 (5.5–10.5 equiv.) (Scheme 2). A quantitative back-isomerization was achieved by irradiation with blue light (ESI, Fig. S5–10, S17†). For complexes based on 2-(4-fluorophenyl)pyridine (Hfppy) and 2-(2,4-difluorophenyl)pyridine (Hdfppy), on the other hand, TFA addition did not result in the formation of an open form. We hypothesized that the reduced basicity of the fluorinated phenyl group was responsible for the lack of reactivity. Indeed, the fac → mer isomerization of fac-Ir(fppy)3 and fac-Ir(dfppy)3 was achieved with the stronger acid HNTf2 (2 and 5 equiv., respectively) (Fig. S11–16 and S18†).
 | ||
| Scheme 2 Chemically induced fac → mer isomerizations are possible for different Ir(C^N)3 complexes using either TFA or HNTf2 for the protonation step. | ||
The protonated complexes [Ir(C^N)2(HC^N)]+ feature a monodentate N-donor ligand. This ligand is expected to be more labile than the chelating C^N ligands. It was therefore conceivable that ligand exchange processes can occur during isomerization at the stage of the ‘open form’. In order to evaluate if ligand scrambling can happen, we have examined the fac → mer → fac isomerization of an equimolar mixture of fac-Ir(ppy)3 and fac-Ir(tpy)3 in CD2Cl2. Analysis of the mixture after TFA/NEt3 treatment by 1H NMR spectroscopy showed the clean formation of the homoleptic complexes mer-Ir(ppy)3 and mer-Ir(tpy)3 (Scheme 3 and ESI, Fig. S39†). The results of the NMR analysis were corroborated by high-resolution mass spectrometry: signals of the heteroleptic complexes mer-Ir(ppy)2(tpy) and mer-Ir(ppy)(tpy)2 were not detected (ESI, Fig. S40†). In line with literature reports,11a the photochemical mer → fac back-isomerization also proceeded without ligand scrambling.
 | ||
| Scheme 3 A fac → mer → fac isomerization sequence with a mixture of Ir(ppy)3 and Ir(tpy)3 does not lead to ligand scrambling. | ||
Compounds, which can be cycled between two (meta)stable states A and B, are often referred to as ‘molecular switches’.17 Some applications of molecular switches rely on the geometric differences between the forms A and B. In order to demonstrate that large structural changes can be achieved with fac → mer isomerizations, we have prepared an Ir(C^N)3 complex with a long 4-(2-pyridyl)-p-terphenyl ligand (tppy). The free ligand Htppy was obtained in 85% yield by Pd-catalyzed cross-coupling of 2-(4-bromophenyl)pyridine with 4-biphenylboronic acid. In analogy to a reported procedure,18 we then synthesized the chloro-bridged dimer [Ir(tppy)2(μ-Cl)]2 by reaction of Htppy with IrCl3(H2O)3 in 2-ethoxyethanol-water (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) using microwave heating (150 °C, 20 min, yield: 82%, for details, see the ESI†). The homoleptic complex mer-Ir(tppy)3 was obtained in 67% yield by reaction of [Ir(tppy)2(μ-Cl)]2 with Htppy and K2CO3 in glycerol at 200 °C (Scheme 4). Quantitative isomerization into fac-Ir(tppy)3 was achieved by heating of a phenol solution of mer-Ir(tppy)3 to 185 °C for 20 h.
:1) using microwave heating (150 °C, 20 min, yield: 82%, for details, see the ESI†). The homoleptic complex mer-Ir(tppy)3 was obtained in 67% yield by reaction of [Ir(tppy)2(μ-Cl)]2 with Htppy and K2CO3 in glycerol at 200 °C (Scheme 4). Quantitative isomerization into fac-Ir(tppy)3 was achieved by heating of a phenol solution of mer-Ir(tppy)3 to 185 °C for 20 h.
 | ||
| Scheme 4 Synthesis of fac- and mer-Ir(tppy)3. The structures of the products are based on crystallographic analyses, with thermal ellipsoids at the 50% probability level. Hydrogen atoms are not shown for clarity. | ||
Complex fac-Ir(tppy)3 can be converted cleanly into the mer form using an acid-base treatment with TFA and NEt3. Crystallographic analyses of the two isomers show the pronounced differences in geometry upon isomerization (Scheme 4). It is worth noting that a conversion of mer-Ir(tppy)3 into fac-Ir(tppy)3 was only achieved thermally, and not with light. Apparently, the bulky ligands inhibit the photochemical isomerization, which was suggested to involve the rupture of one Ir–N bond.13,14
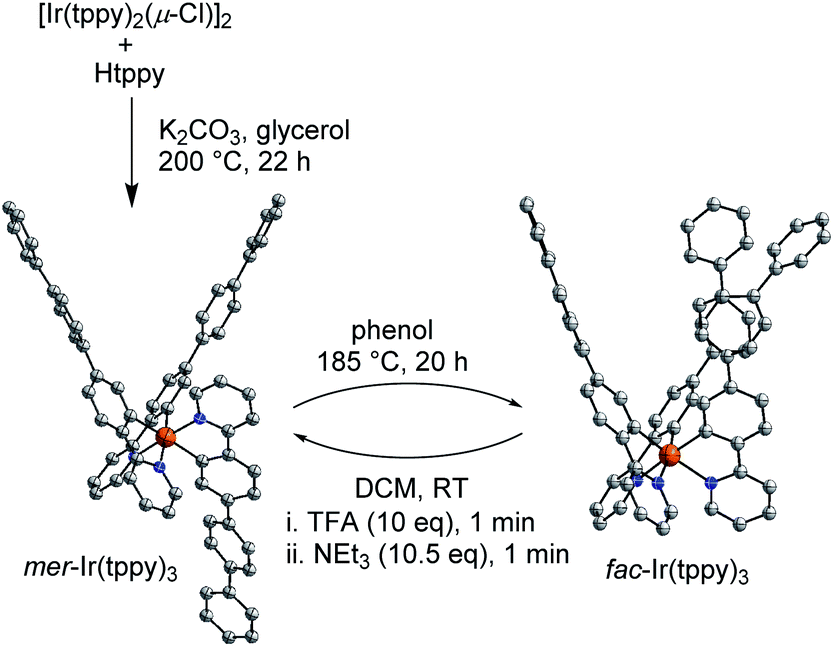
To study the stereoselectivity of the acid-base-induced fac → mer isomerization, we have prepared enantio-enriched fac-Ir(ppy)3 by chromatographic separation (HPLC, Chiralpak IA column, for details, see the ESI†).19 Analysis of the sample before and after the TFA/NEt3 treatment showed the same enantiomeric ratio of Δ:Λ = 83:17, implying that the fac → mer isomerization proceeds without racemization (Fig. 3a).20
 | ||
| Fig. 3 (a) HPLC profile of a sample containing enantio-enriched fac-Ir(ppy)3 before (top) and after (bottom) acid-base-induced fac → mer isomerization. (b) Plausible mechanism for the isomerization involving an in-plane movement of the protonated Hppy ligand and a 180° rotation around the Ir–N bond. A sequential movement–rotation is shown for illustrative purposes. | ||
The high stereoselectivity indicates that the isomerization involves a selective reorientation of one of the three ppy ligands. Most likely, this reorientation occurs upon protonation. The protonated Hppy ligand needs to undergo an in-plane movement along with a 180° rotation around the Ir–N bond to give to pre-meridional form I (Fig. 3b). Base-induced Ir–C bond formation could then occur with minor structural rearrangements. This proposition is supported by the crystallographic analysis of the protonated complex [Ir(ppy)2(Hppy)](NTf2), which shows a pseudo-mer orientation of the three ligands and an agostic interaction between the aromatic C–H group and the cationic Ir center (Fig. 1b). It is worth noting that the photochemical mer → fac back-isomerization proceeds via a different pathway, namely via Ir–N bond rupture14 rather than Ir–C bond rupture. Therefore, Ir(C^N)3 complexes could be regarded as molecular motors performing a complex molecular motion.21,22
Having established that homoleptic fac-Ir(C^N)3 complexes can be isomerized chemically, we turned our attention to a heteroleptic complex, fac-Ir(dfppy)2(tpy).23 In principle, three different complexes could form during the chemically induced fac → mer isomerization (Scheme 5). Switching the orientation of the tpy ligand would give isomer A, whereas switching the orientation of one of the dfppy ligands would give isomer B or C. In view of the lower basicity of the fluorinated dfppy ligands, we anticipated that protonation of tpy would be preferred. Indeed, 1H NMR spectroscopic analysis of a mixture of fac-Ir(dfppy)2(tpy) and TFA (100 equiv.) in CD2Cl2 revealed that the more basic tpy ligand was protonated selectively (ESI, pages S88 and S89†). Subsequent ring closure with NEt3 gave a single isomer, and a crystallographic analysis of the product revealed that isomer A had formed.
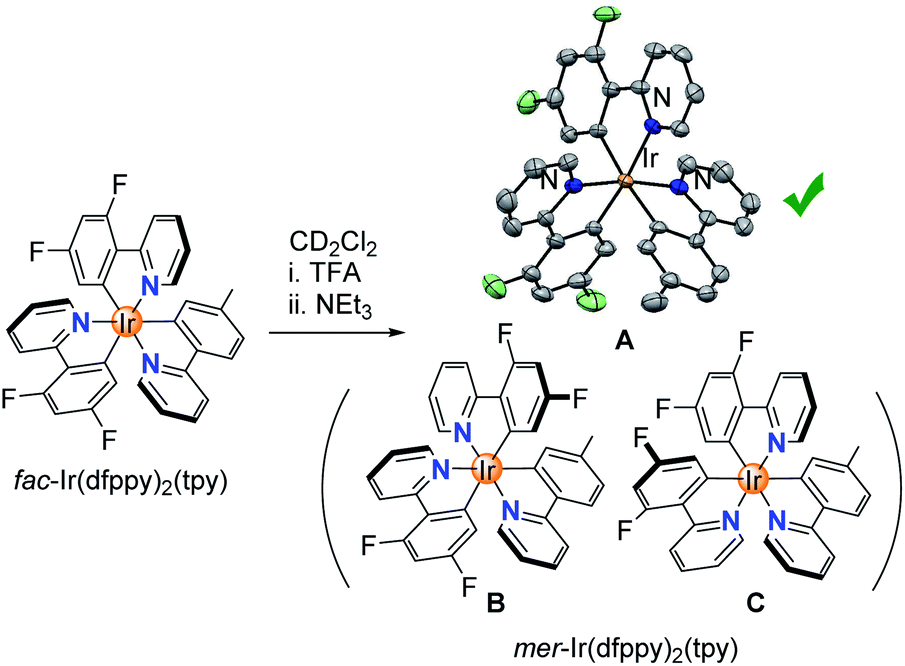 | ||
| Scheme 5 Isomerization of fac-Ir(dfppy)2(tpy) gives selectively complex A. The structure of the product is based on a crystallographic analysis, with thermal ellipsoids at the 50% probability level. Hydrogen atoms are not shown for clarity. | ||
The possibility to interconvert fac- and mer-Ir(C^N)3 complexes provides the opportunity to use these highly luminescent complexes for rewritable data storage devices.24 As a proof-of-concept, we have used a well plate filled with o-dichlorobenzene solutions of fac-Ir(ppy)3 as a luminescent display. The text ‘Iridium’ was written chemically by addition of TFA (green-black contrast) and then NEt3 (green–orange contrast), and reset was achieved by light (Fig. 4).
 | ||
| Fig. 4 Photos of a well plate filled with solutions of (a) fac-Ir(ppy)3 in o-dichlorobenzene, (b) after addition of TFA to selected wells, (c) after addition of NEt3 to the same wells, and (d) after irradiation with blue light. Details of the procedure are given in the ESI.† | ||
Conclusions
In conclusion, we have demonstrated that fac-Ir(C^N)3 complexes can be converted cleanly into the thermodynamically less stable mer isomers by sequential treatment with first acid and then base. Spectroscopic and structural data indicate that the acid protonates a phenyl group of one ligand, resulting in Ir–C bond rupture. The intermediate protonated complex displays a mer arrangement of the N-donor atoms, priming the system for the kinetically controlled formation of mer-Ir(C^N)3 in the presence of base. Notably, the structural rearrangement of the protonated HC^N ligand does not change the relative orientation of the two other C^N ligands. As a consequence, the isomerization is completely stereoselective, i.e. we observe conversion of fac-Δ into mer-Δ, and of fac-Λ into mer-Λ. The mer isomers can be converted back into the fac isomers by irradiation with light, and it is possible to perform multiple fac → mer → fac cycles with minor decomposition of the complexes.fac-Ir(C^N)3 and mer-Ir(C^N)3 complexes are thermally highly stable, and the possibility to switch between these two stable forms opens up potential applications. One possible direction is rewritable data storage devices, and the luminescent display shown in Fig. 4 is a first proof-of-principle that such devices are feasible. Another possibility of the utilization of Ir(C^N)3 complexes as geometric switches is in more complex nanostructures. The results obtained for Ir(tppy)3 are evidence that large structural changes can be realized when converting fac isomers into mer isomers, and vice versa.
Finally, the results are also of interest from a synthetic point of view. Our new method allows preparing mer isomers of Ir(C^N)3 complexes in a clean and fast fashion from the corresponding fac isomers. Notably, it is possible to access mer isomers, which are difficult to prepare otherwise. For example, the standard procedure to prepare the heteroleptic complex mer-Ir(dfppy)2(tpy) gives isomer C (Scheme 5) rather than isomer A.11a,23
Data availability
Raw data for this publication are available at Zenodo at: https://zenodo.org/record/7002606#.YvympnaxUQ.Author contributions
A. Y. G. and K. S. initiated the study, A. Y. G. performed the experiments and analyzed the data, F. F.-T. collected and processed the X-ray data, A. R. performed the photophysical measurements, C. P. helped with the synthesis and the analysis of the heteroleptic Ir(dfppy)2(tpy) complexes, and A. Y. G. and K. S. co-wrote the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the Swiss National Science Foundation and by the École Polytechnique Fédérale de Lausanne (EPFL). We thank Dr Łukasz Woźniak for help with the HPLC measurements, and the Protein Production and Structure Core Facility of the EPFL for support regarding CD spectroscopy measurements.References
-
E. Zysman-Colman, Iridium(III) in Optoelectronic and Photonics Application, John Wiley & Sons, Chichester, West Sussex, 2017 Search PubMed
.
- A. R. B. M. Yusoff, A. J. Huckaba and M. K. Nazeeruddin, Top. Curr. Chem., 2017, 375, 39 CrossRef PubMed
- P.-T. Chou and Y. Chi, Chem. - Eur. J., 2007, 13, 380–395 CrossRef CAS PubMed
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed
- R. C. McAtee, E. J. McClain and C. R. J. Stephenson, Trends Chem., 2019, 1, 111–125 CrossRef CAS
- H. Shi, Y. Wang, S. Lin, J. Lou and Q. Zhang, Dalton Trans., 2021, 50, 6410–6417 RSC
- P.-Y. Ho, C.-L. Ho and W.-Y. Wong, Coord. Chem. Rev., 2020, 413, 213267 CrossRef CAS
- D.-L. Ma, V. P.-Y. Ma, D. S.-H. Chan, K.-H. Leung, H.-Z. He and C.-H. Leung, Coord. Chem. Rev., 2012, 256, 3087–3113 CrossRef CAS
- D. R. Martir and E. Zysman-Colman, Chem. Commun., 2018, 55, 139–158 RSC
- D. R. Martir and E. Zysman-Colman, Coord. Chem. Rev., 2018, 364, 86–117 CrossRef
-
(a) A. R. McDonald, M. Lutz, L. S. von Chrzanowski, G. P. M. van Klink, A. L. Spek and G. van Koten, Inorg. Chem., 2008, 47, 6681–6691 CrossRef CAS PubMed
- For the targeted synthesis of mer Ir(C^N)3 complexes, see:
(a) A. Maity, B. L. Anderson, N. Deligonul and T. G. Gray, Chem. Sci., 2013, 4, 1175–1181 RSC
- D. Escudero, ChemPhotoChem, 2019, 3, 697–701 CrossRef CAS
- K. Tsuchiya, E. Ito, S. Yagai, A. Kitamura and T. Karatsu, Eur. J. Inorg. Chem., 2009, 14, 2104–2109 CrossRef
- T. Karatsu, T. Nakamura, S. Yagai, A. Kitamura, K. Yamaguchi, Y. Matsushima, T. Iwata, Y. Hori and T. Hagiwara, Chem. Lett., 2003, 32, 886–887 CrossRef CAS
- M. Brookhart, M. L. H. Green and G. Parkin, Proc. Natl. Acad. Sci., 2007, 104, 6908–6914 CrossRef CAS
-
(a) J. D. Harris, M. J. Moran and I. Aprahamian, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9414–9422 CrossRef CAS
- B. Orwat, M. J. Oh, M. Zaranek, M. Kubicki, R. Januszewski and I. Kownacki, Inorg. Chem., 2020, 59, 9163–9176 CrossRef CAS PubMed
- For the chiral separation of Ir(C^N)3 complexes, see:
(a) T.-Y. Li, Y.-M. Jing, X. Liu, Y. Zhao, L. Shi, Z. Tang, Y.-X. Zheng and J.-L. Zuo, Sci. Rep., 2015, 5, 14912 CrossRef CAS PubMed
- The photochemical mer → fac isomerization of Ir(C^N)3 complexes was reported to lead to partial racemization. See ref. 14.
- For a discussion of the differences between molecular switches and molecular motors, see:
(a) D. Datler, G. Fuks, J. Heiser, E. Moulin, A. Perrot, X. Yao and N. Giuseppone, Chem. Rev., 2020, 120, 310–433 CrossRef PubMed
- For examples of chemically powered molecular motors, see:
(a) S. Erbas-Cakmak, S. D. P. Fielden, U. Karaca, D. A. Leigh, C. T. McTernan, D. J. Tetlow and M. R. Wilson, Science, 2017, 358, 340–343 CrossRef CAS PubMed
-
(a) J. Haribabu, Y. Tamura, K. Yokoi, C. Balachandran, M. Umezawa, K. Tsuchiya, Y. Yamada, R. Karvembu and S. Aoki, Eur. J. Inorg. Chem., 2021, 18, 1796–1814 CrossRef
-
(a) H. Wang, X. Ji, Z. A. Page and J. L. Sessler, Mater. Chem. Front., 2020, 4, 1024–1039 RSC
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2101920, 2102440, 2096259, 2102439, 2118152 and 2122690. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc02808e |
| This journal is © The Royal Society of Chemistry 2022 |