
Nanoscale probing and imaging of HIV-1 RNA in cells with a chimeric LNA–DNA sensor†
Alessia
Amodio‡
 ab,
Marco
Cassani‡
ac,
Liviana
Mummolo
a,
Christina
Cortez-Jugo
a,
Sukhvir Kaur
Bhangu
d,
Jori
Symons
e,
Chantelle L.
Ahlenstiel
f,
Giancarlo
Forte
c,
Francesco
Ricci
b,
Anthony D.
Kelleher
f,
Sharon R.
Lewin
egh,
Francesca
Cavalieri
*bd and
Frank
Caruso
*a
ab,
Marco
Cassani‡
ac,
Liviana
Mummolo
a,
Christina
Cortez-Jugo
a,
Sukhvir Kaur
Bhangu
d,
Jori
Symons
e,
Chantelle L.
Ahlenstiel
f,
Giancarlo
Forte
c,
Francesco
Ricci
b,
Anthony D.
Kelleher
f,
Sharon R.
Lewin
egh,
Francesca
Cavalieri
*bd and
Frank
Caruso
*a
aDepartment of Chemical Engineering, The University of Melbourne, Parkville, Victoria 3010, Australia. E-mail: fcaruso@unimelb.edu.au
bDipartimento di Scienze e Tecnologie Chimiche, Università degli Studi di Roma Tor Vergata, Via della Ricerca Scientifica 1, 00133 Rome, Italy
cInternational Clinical Research Center (ICRC), St Anne's University Hospital, CZ-65691 Brno, Czech Republic
dSchool of Science, RMIT University, Victoria 3000, Australia. E-mail: francesca.cavalieri@rmit.edu.au
eDepartment of Infectious Diseases, The University of Melbourne at the Peter Doherty Institute for Infection and Immunity, Melbourne, Victoria 3000, Australia
fKirby Institute, University of New South Wales, New South Wales 2052, Australia
gVictorian Infectious Diseases Service, Royal Melbourne Hospital at the Peter Doherty Institute for Infection and Immunity, Melbourne, Victoria 3000, Australia
hDepartment of Infectious Diseases, Alfred Hospital and Monash University, Melbourne, Victoria 3004, Australia
First published on 31st January 2022
Abstract
Real-time detection and nanoscale imaging of human immunodeficiency virus type 1 ribonucleic acid (HIV-1 RNA) in latently infected cells that persist in people living with HIV-1 on antiretroviral therapy in blood and tissue may reveal new insights needed to cure HIV-1 infection. Herein, we develop a strategy combining DNA nanotechnology and super-resolution expansion microscopy (ExM) to detect and image a 22 base sequence transcribed from the HIV-1 promoter in model live and fixed cells. We engineer a chimeric locked nucleic acid (LNA)–DNA sensor via hybridization chain reaction to probe HIV-1 RNA in the U3 region of the HIV-1 long terminal repeat (LTR) by signal amplification in live cells. We find that the viral RNA transcript of the U3 region of the HIV-1 LTR, namely PromA, is a valid and specific biomarker to detect infected live cells. The efficiency and selectivity of the LNA–DNA sensor are evaluated in combination with ExM. Unlike standard ExM methods, which rely on additional custom linkers to anchor and immobilize RNA molecules in the intracellular polymeric network, in the current strategy, we probe and image the HIV-1 RNA target at nanoscale resolution, without resorting to chemical linkers or additional preparation steps. This is achieved by physical entrapment of the HIV-1 viral transcripts in the cells post-expansion by finely tuning the mesh size of the intracellular polymeric network.
Introduction
Nanoscale-resolution imaging and quantification of proteins and nucleic acids in cells and tissues is key to understanding cellular processes and for defining cell phenotypes.1–3 Probing ribonucleic acid (RNA) molecules in live cells and tissues by pairing in situ hybridization and amplification techniques with super-resolution imaging technologies would be useful for defining cell types and states in normal and pathological biological settings. For instance, biosensing and imaging techniques that are suited to identifying infected cells that harbor productive or latent human immunodeficiency virus type 1 (HIV-1) in vivo are of high interest. HIV-1 establishes latent infection in resting and proliferating CD4+ T-cells, and these cells can persist in people living with HIV-1 despite prolonged antiretroviral therapy (ART).4 Simple methods for real-time probing of HIV-1-infected cells that persist during ART would enable a better understanding of viral latency, reservoir dynamics and reactivation. Significant research has focused on developing strategies for detecting HIV-1 in plasma, including electrochemical-, optical-, and mechanical-based assays, as well as the use of miniaturized platforms.5 Current available methods to detect HIV-1 rely on the quantification of proviral DNA and RNA transcripts, including a viral outgrowth assay,6in situ hybridization RNAscope and DNAscope,7 and polymerase chain reaction.8,9 Although highly specific and sensitive (Table S1†), these techniques are labor-intensive and require a number of reagents and purifications steps, thus increasing the cost and time of analysis. In addition, these techniques are performed on formalin-fixed cells and paraffin-embedded tissues, hence precluding the real-time detection of HIV-1 DNA and RNA in live cells.In the present study, a chimeric locked nucleic acid (LNA)–DNA sensor was designed to enable hybridization chain reaction (HCR) for the efficient detection of HIV-1 RNA transcripts in fixed and live cells. Previous studies have indicated that a small-interfering RNA (siRNA) siPromA (Fig. 1a) is a potent siRNA transcriptional suppressor of HIV-1 in vitro and in vivo.10 siPromA targets the tandem nuclear factor kappa B (NF-κB) binding sites in the HIV-1 long terminal repeat (LTR). We postulate that the PromA target sequence in the viral RNA transcripts of the U3 region of the HIV-1 LTR can be a valid specific biomarker of infected cells as it is highly conserved in different HIV-1 strains and is unique to the HIV-1 genome, with little homogeneity to the human/host genome.
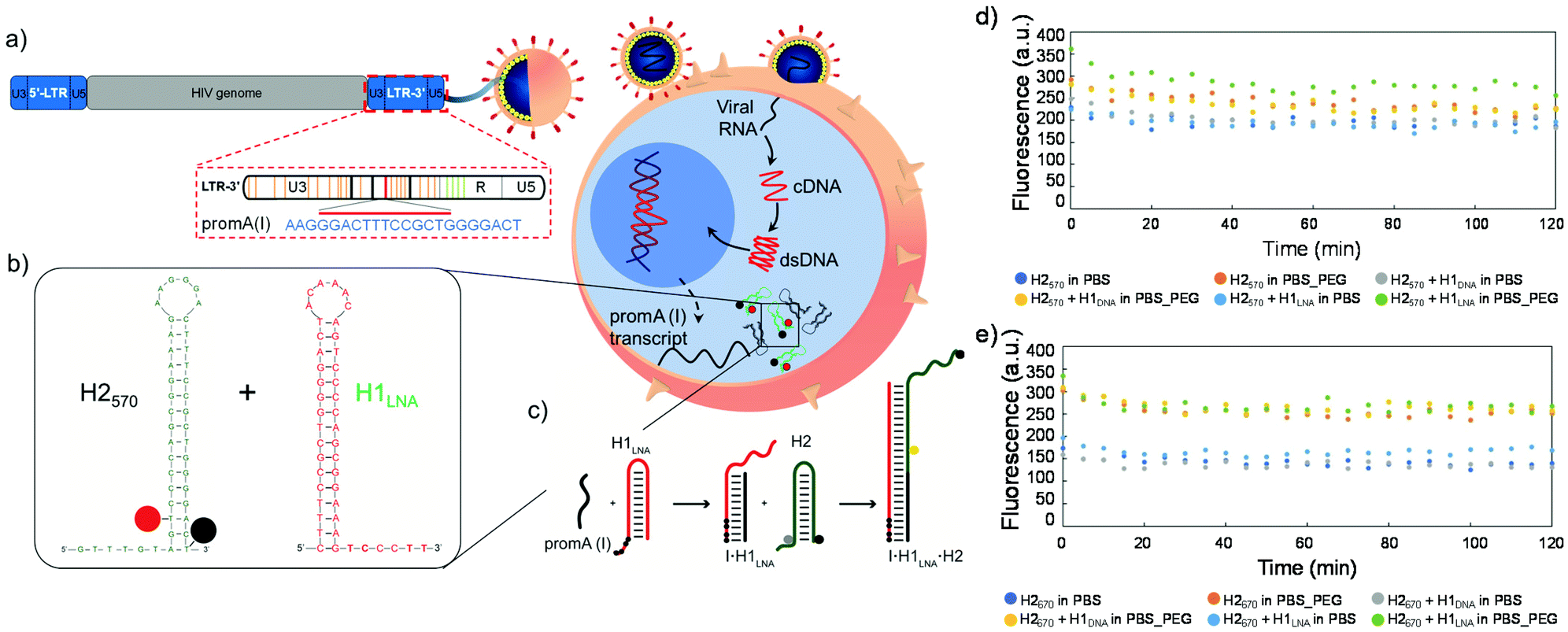 | ||
| Fig. 1 Schematic representation of an infected cell and mechanism of action of hairpins. Upon cell infection with HIV-1, (a) the viral genome containing the 5′ and 3′LTR regions with the PromA target sequence is integrated in the genome. The 5′LTR functions as the virus promoter and the 3′LTR acts to cap viral transcripts and is incorporated into all HIV-1 gene products. (b) Sequence of the H2 signal element and H1LNA or H1DNA detection element. (c) Hairpins H1 and H2 are stable in the absence of the mRNAPromA initiator I. Initiator I can nucleate at the sticky end of H1 and undergo a strand displacement reaction to open the hairpin. The newly exposed sticky end of H1 binds to the sticky end of H2 and opens the hairpin to expose a sticky end on H2 that is identical in sequence to I. cDNA, complementary DNA; dsDNA, double-stranded DNA. Black circle represents the quencher, grey and yellow circles represent the fluorophore. The black circles in the sticky end region of H1 represent the LNA bases. (d and e) Stability of H2570 and H2670 in the presence of H1: fluorescence signal as a function of time of H2570, H2570 + H1DNA, or H2570 + H1LNA in PBS or PBS + 30% w/v PEG3350 (PBS_PEG) (d) and fluorescence signal as a function of time of H2670, H2670 + H1DNA, or H2670 + H1LNA in PBS or PBS_PEG (e). The concentration of the hairpins (H1 and H2) in the reaction mixture was constant (100 nM). The reaction kinetics was followed for 2 h at 37 °C. | ||
HCR is an efficient enzyme-free amplification biosensing technique based on single-stranded DNA hairpins H1 and H2.11 Recent studies have reported the imaging and detection of RNA and DNA by HCR in cells from different organisms,12,13 including tumor cells and living mice.14 Briefly, the two hairpins H1 and H2 are kinetically trapped in metastable states in the absence of a nucleic acid initiator, I. The introduction of I opens H1 via toehold-mediated strand displacement to expose a domain that opens H2 by hybridization to its sticky end. Upon opening, H2 exposes a domain identical to the sequence of I that triggers a chain reaction of alternating H1 and H2 polymerization steps, ultimately leading to the formation of a nicked double-stranded polymeric DNA. Using this approach, viral RNA transcripts can serve as the initiator and readily trigger signal transduction in fixed cells and tissues. However, to detect intracellular HIV-1 RNA targets in live cells (Fig. 1b), the molecular probes H1 and H2 need to overcome several barriers including transfection efficiency, intracellular trafficking, and endosomal escape. These barriers typically limit the uptake and HCR efficiency of the H1 and H2 hairpins in live cells, resulting in poor signal output and high interference due to the intracellular nuclease-mediated degradation of the probes. Hence, the engineering of a highly efficient and specific LNA–DNA chimera probe is key to improving the fluorescence signal or readout provided by HCR in live cells.15
In combination with the advanced design of the chimeric LNA–DNA probe for live cell imaging of viral transcripts, we employed super-resolution expansion microscopy (ExM) to precisely localize the viral RNA in the model infected cells in a three-dimensional (3D) matrix. ExM is a powerful tool for imaging preserved cells and tissues, providing nanoscale resolution using conventional diffraction-limited microscopes.16 ExM involves embedding fixed cells and tissues inside a swellable acrylic-based hydrogel.17 Following tissue softening and solvent exchange, the hydrogel–specimen composite typically expands 4.5-fold the original size of the hydrogel in pure water by isotropic swelling. As a result, subcellular structures and biomolecules can be separated in space and imaged by fluorescent probes in super-resolution mode on a confocal microscope. When the expansion is uniform in the three dimensions and the fluorescent probes are preserved, the final imaging resolution is improved by a factor of 4, that is, ∼70 nm resolution using a ∼300 nm diffraction-limited objective lens. ExM protocols for imaging of subcellular structures,18 proteins,19 nucleic acids,20 and lipids21 have been recently developed and are continuously being improved.22 Previous studies have reported that biomolecules, such as RNA, proteins and lipids, tend to be washed away or structurally altered during sample mechanical homogenization and expansion, and thus it is necessary to chemically anchor the biomolecules to the polymer network. For instance, RNA molecules were quantified post-expansion in cell culture and intact mouse brain tissues using a custom reagent, LabelX, containing an alkylating group, which reacts with the nucleobase guanine, and a polymerizable acrylamide moiety.23 Unlike previous studies, herein, we exploited the size exclusion properties of the acrylic cross-linked hydrogel upon expansion in hypotonic conditions to detect and image model HIV-1 RNA. By physically entrapping the RNA molecules in the highly cross-linked intracellular hydrogel, we probed and localized the HIV-1 RNA target at nanoscale resolution post-expansion without resorting to synthetic chemical linkers or additional preparation steps.
Experimental
Materials
HPLC-purified DNA sequences were purchased from Biosearch Technologies (Risskov, Denmark). PAGE-purified DNA–LNA sequence was purchased from Exiqon (Vedbaek, Denmark). DNAse I, DNase I buffer, RIPA lysis and extraction buffer, and SYBR Safe DNA gel stain were obtained from Thermo Fisher Scientific (Waltham, MA, USA). RNeasy mini kit was purchased from Qiagen. LysoTracker® Green DND-26 was purchased from Cell Signaling Technology (MA, USA). EZ Load 20 bp Molecular Ruler, EZ Load 100 bp Molecular Ruler were purchased from Bio-Rad. HeLa cells were purchased from the American Type Culture Collection (Manassas, VA, USA). TZM-bl cells were obtained through the National Institutes of Health (NIH) AIDS Reagent Program, Division of AIDS, NIAID, NIH (Dr John C. Kappes, Dr Xiaoyun Wu and Tranzyme Inc.).24–28 Dulbecco's phosphate-buffered saline, RIPA buffer, poly(ethylene glycol) (PEG) (Mw 3350), and phosphate-buffered saline (PBS) were purchased from Sigma-Aldrich (St Louis, MO, USA). Trypsin and Dulbecco's modified Eagle's medium (DMEM) were purchased from Lonza (Allendale, USA). Fetal bovine serum (FBS) was purchased from Bovogen (Keilor East, Australia). All chemicals were used without further purification.Oligonucleotide sequences
The following DNA and DNA–LNA oligonucleotides modified and non-modified were used. All oligonucleotides were suspended to a final concentration of 100 µM in UltraPure™ DNase/RNase-free distilled water and stored at −20 °C.H1: 5′-CTTTCCGCTGGGGACT ACAAAC AGTCCCCAGCGGAAAG TCCCTT-3′
H1LNA: 5′-CTTTCCGCTGGGGACT ![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif)
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) AGTCCCCAGCGGAAAG TCCCTT-3′
AGTCCCCAGCGGAAAG TCCCTT-3′
H2570: 5′-GTTTGT AGT(BHQ-2)CCCCAGCGGAAAG ![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif) CTTTCCGCTG GGGA CT(Quasar 570)-3′
CTTTCCGCTG GGGA CT(Quasar 570)-3′
H2670: 5-GTTTGT AGT(BHQ-2)CCCCAGCGGAAAG CTTTCCGCTG GGGA CT(Quasar 670)-3′
I22: 5′-AAGGGACTTTCCGCTGGGGACT-3′
I60: 5′-CTGACATCGAGCTTGCTACAAGGGACTTTCCGCTGGGGACTTTCCAGGGAGGCGTGGCCT-3′
I*60: 5′-AGGCCACGCCTCCCTGGAAAGTCCCCAGCGGAAAGTCCCTTGTAGCAAGCTCGATGTCAG-3′
I NF-κB binding domain: 5′-GGGACTTTCCTAGAAATTAT-3′
In the hairpin sequences noted above, bases in bold represent the LNA bases, the underlined bases represent the hairpin loops, and those in italic represent the sticky end portions. The * symbol denotes complementary strand. For all experiments, the hairpins (H1LNA, H1DNA, H2570, and H2670) were heated at 95 °C for 5 min and allowed to cool to room temperature (25 °C) for 1 h.
Fluorescence measurements
Fluorescence measurements were performed on a Tecan Infinite M200 Pro microplate reader (Männedorf, Switzerland) with the following settings: λex 520 nm and emission wavelength, λem, 550–600 nm (for H2570) and λex 620 nm and λem 650–700 nm (for H2670) and the following protocols were employed.In Fig. 1d and e, the concentration of the hairpins (H1LNA, H1DNA, H2570, and H2670) was 100 nM. The fluorescence signal was monitored for 2 h at 37 °C in PBS pH 7.4 or PBS + 30% PEG3350 w/v.
Fig. S2:† The concentration of the hairpins (H2570 and H2670) was 100 nM. The fluorescence signal was monitored for 2 h at 37 °C in PBS pH 7.4 or PBS + 30% PEG3350 w/v. Different concentrations of I22 or I60, (5, 10, 50, 100, and 500 nM) were added to the mixture reactions containing H2570 or H2670.
Fig. 2a–h and S4, S6, S8:† The concentration of the hairpins (H1DNA, H1LNA, H2570, and H2670) was 100 nM. Different concentrations of I22 (Fig. 2a, b, e, f and Fig. S6†), I60 (Fig. 2c, d, g, h, and Fig. S8†) or I*I60 (Fig. S4†) (5, 10, 50, 100, and 500 nM) were added to the reaction mixtures containing H1DNA + H2570, H1DNA + H2670, H1LNA + H2570, or H1LNA + H2670. The reaction mixtures were incubated at 37 °C for 2 h in PBS pH 7.4 (Fig. 2a–h and S4, S8†) or PBS + 30% w/v PEG3350 (Fig. S6 and S8†).
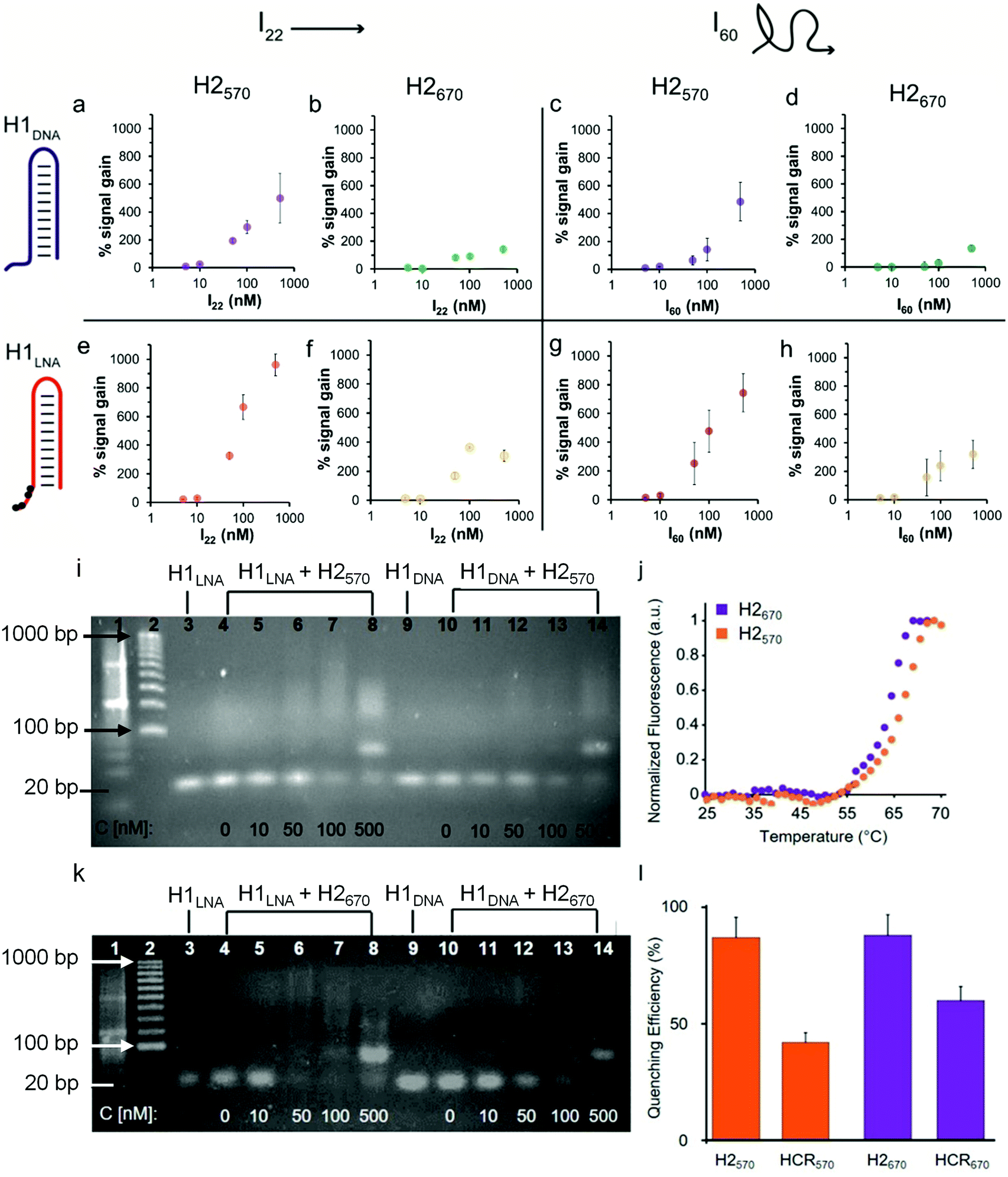 | ||
| Fig. 2 Efficiency of HCR using initiators I22 and I60. Percentage signal increase as a function of (a, b, e and f) I22 or (c, d, g and h) I60 concentration using H2570 and H1DNA (a and c) or H1LNA (e and g) as the detection element or H2670 and H1DNA (b and d) or H1LNA (f and h) as the detection element. The concentration of the hairpins (H1DNA, H1LNA, H1570, and H1670) in the reaction mixture was constant (100 nM). The reactions were performed in PBS, pH 7.4, and incubated for 2 h at 37 °C. (i) Agarose gel electrophoresis of HCR products using H1LNA or H1DNA as detection element and increasing concentrations of I60. Lane 1, ladder 20 bp; lane 2, ladder 100 bp; lane 3, H1LNA (100 nM); lanes 4–8, H1LNA (100 nM) + H2570 (100 nM) with increasing concentrations of I60 (0 nM (lane 4), 10 nM (lane 5), 50 nM (lane 6), 100 nM (lane 7), and 500 nM (lane 8)); lane 9, H1DNA (100 nM); lanes 10–14, H1DNA (100 nM) + H2570 (100 nM) with increasing concentrations of I60 (0 nM (lane 10), 10 nM (lane 11), 50 nM (lane 12), 100 nM (lane 13), and 500 nM (lane 14)). (j) Melting curves of H2570 and H2670. (k) Agarose gel electrophoresis of HCR products using H1LNA or H1DNA as detection element and increasing concentrations of I60. Lane 1, ladder 20 bp; lane 2, ladder 100 bp; lane 3: H1LNA (100 nM); lanes 4–8, H1LNA (100 nM) + H2670 (100 nM) with increasing concentrations of I60 (0 nM (lane 4), 10 nM (lane 5), 50 nM (lane 6), 100 nM (lane 7), and 500 nM (lane 8)); lane 9, H1DNA (100 nM); lane 10–14, H1DNA (100 nM) + H2670 (100 nM) with increasing concentrations of I60 (0 nM (lane 10), 10 nM (lane 11), 50 nM (lane 12), 100 nM (lane 13), and 500 nM (lane 14)). (l) Quenching efficiency (%) after treatment with DNAse I of H2570, the product of HCR using H1LNA + H2570, H2670, or the product of HCR using H1LNA + H2670 (values are shown as mean ± SD, averaged over at least three independent measurements). | ||
Fig. S3:† The concentration of the hairpins (H1DNA, H1LNA, H2570, and H2670) was 100 nM. I60 at a given concentration (from 5 to 500 nM) was added to the reaction mixtures containing H1DNA + H2570, H1DNA + H2670, H1LNA + H2570, or H1LNA + H2670 after 10 min to allow a stable baseline. The fluorescence signal was monitored for 2 h at 37 °C in PBS pH 7.4. Signal increase of H2570 or H2670 was monitored for about 2 h after I60 addition.
Fig. 2j: Fluorescence melting curves were obtained on a Horiba FL-322 Fluorolog-3 spectrometer equipped with a 450 W xenon lamp as the excitation source. All measurements were performed in quartz cuvettes with a volume of 200 µL. For H2570, λex was 520 nm (slit = 5 nm) and emission was acquired in the range from 550 to 600 nm (slit = 5 nm). Melting curves were recorded for H2570 (at 20 nM), diluted in PBS 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 at pH 7.4. A heating range of 24–84 °C with increments of 1.5 °C was applied. For H2670, λex was 620 nm (slit = 5 nm) and emission was acquired in the range from 650 to 700 nm (slit = 5 nm). Melting curves were recorded for H2670 (at 20 nM), diluted in PBS 1:100 at pH 7.4, within a heating range of 24–84 °C with increments of 1.5 °C. The obtained melting curves were normalized using the interpolation model.29Tm's were obtained using the same model from the intersection of the calculated median and the experimental melting curve.
:100 at pH 7.4. A heating range of 24–84 °C with increments of 1.5 °C was applied. For H2670, λex was 620 nm (slit = 5 nm) and emission was acquired in the range from 650 to 700 nm (slit = 5 nm). Melting curves were recorded for H2670 (at 20 nM), diluted in PBS 1:100 at pH 7.4, within a heating range of 24–84 °C with increments of 1.5 °C. The obtained melting curves were normalized using the interpolation model.29Tm's were obtained using the same model from the intersection of the calculated median and the experimental melting curve.
Native gel electrophoresis
The gel experiments were performed 24 h after the addition of I (see above description). The 1% agarose gels were prepared using 1× sodium borate buffer (from Sigma-Aldrich, St Louis, MO, USA), run at 150 V for 1 h, and stained in 1× SYRB Safe for 30 min. The gels were visualized using a Gel-Doc XR+ system (Bio-Rad).Energy transfer properties
Fluorescence measurements of the signaling hairpins (H2570 and H2670) were performed on a Horiba FL-322 Fluorolog-3 spectrometer equipped with a 450 W xenon lamp as the excitation source. All measurements were performed at 37 °C in quartz cuvettes with a volume of 200 µL, λex was 520 nm (slit = 5 nm), and emission was acquired in the range from 550 to 600 nm (slit = 5 nm). Fluorescence emission spectra were recorded for four samples: H2570 (100 nM), H2670 (100 nM), HCR reaction products obtained by incubating H2570 (100 nM) + H1LNA (100 nM) + I60 (500 nM) for 2 h at 37 °C, and HCR reaction products obtained by incubating H2670 (100 nM) + H1LNA (100 nM) + I60 (500 nM) for 2 h at 37 °C. Maximal fluorescence signal was measured after incubation of each sample with DNase I for 60 min at 37 °C. The Förster resonance energy transfer (FRET) efficiency (%) in solution was calculated using eqn (1):| FRET (%) = (Imax − IDA)/Imax × 100 | (1) |
Cell lysate
The cell lysates were prepared according to the RIPA lysis buffer protocol. Briefly, approximately 1 × 107 cells (HeLa or TZM-bl cell line) were pelleted by centrifugation at 1000g for 5 min and the supernatant was discarded. The cells were then washed twice in cold PBS and pelleted further by centrifugation at 1000g for 5 min. The RIPA lysis buffer was then added (2 mL) to the washed pellet, together with 25 μL of RiboLock RNase. The reaction mixture was then gently mixed for 15 min and centrifuged at 14000g for 15 min to pellet the cell debris. The supernatant was then transferred to a new tube for the reaction. In the control, H2570 only at a concentration of 100 nM was added to the supernatant. In the other sample, both H2570 and H1LNA were added at a concentration of 100 nM. The reaction mixtures were incubated at 37 °C for 2 h. The fluorescence was then measured as per the protocols described prior.
RNA extraction
RNA extraction from HeLa and TZM-bl cells was performed using the RNeasy mini kit according to the RNeasy mini kit protocol. The samples were first digested with DNase I and then incubated with H2570 (for the control) or H2570 + H1LNA at a concentration of 100 nM. The reaction mixtures were incubated at 37 °C for 2 h. The fluorescence was then measured as per the protocols described prior.Confocal laser scanning microscopy (CLSM) analysis
A Nikon A1 laser scanning confocal microscope was used for the confocal analysis. All samples were imaged ensuring that pixel saturation is avoided and using identical acquisition settings for comparison of fluorescence intensities. The images were analyzed with Fiji software (http://fiji.sc/).HCR studies in fixed cells
For the HCR studies in fixed cells, 40000 TZM-bl or HeLa cells per well were plated in 12 mm coverslip in 24-multiwell plates and allowed to adhere for 24 h. The cells were then washed once with PBS (1× PBS, 10 mM) and fixed with 4% (w/v) paraformaldehyde (PFA) at room temperature for 10 min, followed by three washes with 1× PBS. The cells were then permeabilized with Triton 0.1% for 5 min and washed three times. Afterwards, the following solutions H2570 or H2570 + H1LNA in 0.5× PBS were added to the cells and incubated for 1 h. The fixed cells were incubated with 0.5× PBS for 1 h for the HCR reaction to proceed. Finally, the cells were washed with a solution of 0.5× PBS and collected for the subsequent step. The samples were then mounted using ProLong Gold Antifade Mountant (Thermo Fisher Scientific, USA) and analyzed by CLSM at λex 546 nm.
HCR studies in live cells
For the HCR studies in live cells, 40000 TZM-bl cells per well were plated on Nunc™ Lab-Tek™ II chambered coverglass (Life Technologies, Scoresby, Australia) in DMEM medium supplemented with 10% FBS and allowed to adhere for 24 h. The cells were washed with 1× PBS prior to the replacement of DMEM with OPTI-MEM medium. The samples were treated with the following solutions prepared in 0.5× PBS and lipofectamine RNAiMAX (used as transfecting agent according to the supplier protocol): H2570 (final concentration of 50 nM); H2570 + H1LNA (1:1) (final concentration of 50 nM); or I*60 (final concentration 500 nM) 1 h prior to transfection with solution H2570 + H1LNA. After 2 h of incubation, the cells were washed three times with 1× PBS and the OPTI-MEM medium was replaced with DMEM. After additional 2 and 4 h, DMEM medium was swapped with Leibovitz medium supplemented with 10% FBS, and the samples were analyzed by CLSM at λex 546 nm. For each time point, LysoTracker® Green DND-26 was added to cell culture media to stain the endo–lysosome compartments and the colocalization of HCR products with endo–lysosome compartments was studied. The PCC values were calculated using Fiji ImageJ software.
ExM
Solution A (polymerization buffer) consisted of the following: 100 mM PBS, 2 M NaCl, 9% sodium acrylate, 2.5% acrylamide, and 0.15% methylenebis(acrylamide). Prior to use, 0.2% tetramethylethylenediamine and 0.2% ammonium persulfate were added to the buffer. Solution B (digestion buffer) consisted of the following: 0.8 M guanidine chloride, 1× TAE (40 mM Tris, 20 mM acetic acid, and 1 mM ethylenediaminetetraacetic acid), and 0.5% Triton. Prior to use, 8 U mL−1 of proteinase K was added to the buffer. The hold chamber was prepared by laying three stripes of parafilm onto a microscope slide and cutting holes of 10 mm in diameter, which can hold 70 µL of solution A and an 11 mm round cover glass.For the ExM experiments, control HeLa cells and target TZM-bl cells were grown on a 12 mm round cover glass in a 24 multiwell plate. When the cells reached 80% of confluency, they were fixed with 4% PFA for 10 min. After three washes with 1× PBS, the cells were treated with 0.1% Triton for 5 min and washed three times with 1× PBS. Subsequently, the sample was reacted for 1 h with 25 mM methacrylic acid N-hydroxysuccinimide ester (MA-NHS) at room temperature. Upon reaction of MA-NHS monomer with the primary amine groups on the proteins, MA-NHS monomer will anchor native proteins to the hydrogel during polymerization and cross-linking. After three washes with 1× PBS, freshly prepared solution A was poured onto the cover glass, which was then placed in the hold chamber for gelation to proceed for 1 h at 37 °C. Then, the cover glass with the polymerized hydrogel was collected, submerged in solution B and kept overnight (12 h) at 37 °C. The sample was then washed with 0.5× PBS three times for gel expansion to proceed. After the third wash, the hydrogel was cut and used for the different experiments in which the following solutions prepared in 0.5× PBS were added to the hydrogel: H2570; H2570 + H1LNA; or H2570 + H1LNA + I*60. Incubation of the hydrogel in these solutions proceeded for 1 h to allow permeation of the reagents in the hydrogel pores. The gel was then incubated with 0.5× PBS for 1 h, allowing the HCR reaction to occur. Finally, the hydrogel was washed with 0.5× PBS and collected for the subsequent step. The gel was placed onto a 1.5 mm thick microscope slide and analyzed by CLSM at λex 546 nm.
Cell incubation with dextran-fluorescein isothiocyanate (Dx-FITC)
TZM-bl or HeLa cells (40000 per well) were plated in 12 mm coverslip in a 24 multiwell plate and allowed to adhere for 24 h. The cells were then washed once with 1× PBS and fixed with 4% (w/v) PFA at room temperature for 10 min, followed by three washes with 1× PBS. The cells were then permeabilized with 0.1% Triton for 5 min and washed three times. Subsequently, the sample was either treated as per the same procedure employed for ExM studies and the formed hydrogel was incubated with 500 ng mL−1 Dx-FITC (10 kDa) for 2 h or directly incubated with 500 ng mL−1 Dx-FITC for 2 h. In the latter case, after the incubation, the sample was treated accordingly to the ExM protocol. After incubation in digestion buffer, the hydrogel was washed three times either with Milli-Q water or 0.5× PBS for 20 min each wash.
Statistics
Data are presented as means ± standard deviations (SDs) and analyzed using the software package GraphPad Prism v.6.0 (USA, CA). In all cases, p < 0.05 was considered statistically significant.Results and discussion
Rational design of chimeric LNA–DNA sensor to detect HIV-1 mRNA
We designed two 16 bp-stem detection hairpins, H1DNA and H1LNA, partially complementary to the tandem repeat of NF-κB binding sites (PromA target sequence GGGACTTTCGTGGGGACTT) found in the HIV-1 3′LTR region, hereafter referred to as mRNAPromA (Fig. 1a, b and Fig. S1†). The signaling hairpin, H2, was also designed (Fig. 1b) to provide the fluorescence signal upon hybridization with H1DNA and H1LNA. The target sequence, mRNAPromA, acts as the initiator strand (I) to trigger a chain reaction, resulting in the formation of a fluorescent nicked double strand (Fig. 1c). The complementary detection and signaling hairpins (H1DNA or H1LNA and H2, respectively) were engineered in silico using mfold web server with a 16 bp-stem, 6-base loop, and 6-base sticky end to reside in a metastable equilibrium.30 The sticky end of the H1DNA element was entirely composed of nucleotides, whereas four LNA bases (black circles in Fig. 1c) were introduced in the sticky end of H1LNA.LNA are nucleotides bearing a ribose ring “locked” by a 2′-O-CH2-4′ linkage. In the rigid bicycle, the number of conformational degrees of freedom, including rotations around internal bonds, is reduced. As a result, the methylene bridge locks and preorganizes the sugar into an RNA-like C3′-endo configuration.31 The preorganization of the sugar moiety reduces the hybridization entropy penalty upon base pairing. In addition to the net reduction in the entropy change, a favorable hybridization enthalpic contribution can be observed due to the enhancement of hydrophobic interactions and π–π-stacking between the bases.31 Overall, the incorporation of LNA nucleotides into a strand of DNA or RNA improves the thermal stability and selectivity of oligonucleotides when hybridized to a complementary DNA or RNA strand.32–34 We postulate that introducing LNA in the sticky end may affect the kinetics and thermodynamics of the HCR process, thus leading to an increase in HCR yield (i.e., high fluorescence output), a faster response, and a decrease in the effective amount of probe necessary for hybridization. To provide a fluorescent signal, H2 was modified with a fluorophore–quencher pair QUASAR®-Black Hole Quencher®-2 (BHQ-2). In addition, to explore the effect of the fluorophore–quencher pair on the HCR efficiency, H2 modified with QUASAR® 570 (H2570) was compared with H2 modified with QUASAR® 670 (H2670) (Fig. S1†).
The stability of the engineered hairpins was first examined in PBS at 37 °C (Fig. 1d and e). The mixture containing both hairpins, i.e., H1DNA or H1LNA and H2, remained stable in the absence of strand initiator over at least 2 h (Fig. 1d and e) and the signal elements, i.e., H2570 and H2670, also remained stable in the presence of the initiator strand (Fig. S2†). These results indicate that the engineered sets of hairpins possess the required features to be used as building blocks of the HCR system.
To demonstrate that the addition of IN, where N denotes the number of bases, triggers HCR in the presence of H1 and H2, we used a 22-base sequence (thereafter referred to as I22) fully complementary to the sticky end and the stem of H1. I22 has a sequence similar to that of PromA (Fig. 1a and Fig. S1†). To determine the HCR efficiency of the different systems, different combinations of H1 and H2 were examined: (i) H1DNA + H2570; (ii) H1DNA + H2670; (iii) H1LNA + H2570; and (iv) H1LNA + H2670 (Fig. 2). An increasing amount of I22 was added to the reaction mixture to assess the efficiency of HCR as a function of I22 concentration. The reaction mixtures were incubated under physiological conditions at 37 °C for 2 h. As expected, increasing concentrations of I22 increased the fluorescence signal in all four combinations (Fig. 2a, b, e and f). However, the results suggest that HCR efficiency is affected by the different formats of both H1 and H2. Specifically, the percentage signal gain for the different combinations decreased in the order of H1LNA + H2570 > H1DNA + H2570 > H1LNA + H2670 > H1DNA + H2670. A 60-base initiator (I60) was also examined, comprising the 22 nt target sequence, 3′LTRPromA with additional 19 upstream and downstream nucleotides of the LTR sequence (see Experimental). Although the target binding site embedded in I60 is likely less exposed when compared with I22, similar fluorescence signals were obtained with both I22 and I60 (Fig. 2c, d, g and h). The kinetics of the HCR process for the different systems was also investigated (Fig. S3†). The fastest kinetics was obtained using H1LNA as the detection element and H2570 as the signal element (Fig. S3c†). It is noted that a double strand DNA containing the 3′LTRPromA sequence (I*I60) is unable to act as initiator to trigger a chain reaction as shown in Fig. S4.† This observation suggests that in live cells, only single strand viral RNA transcripts can be detected by HCR signal transduction.
The specificity of H1LNA for PromA sequence was confirmed by using a control sequence 5′-GGGACTTTCCTAGAAATTAT-3′ (Icontr) consisting of 50% of the bases matching the target sequence PromA and 50% scrambled bases. A negligible fluorescence signal gain (10%) was detected when the Icontr sequence was employed as the initiator to trigger the chain reaction (Fig. S5†). To mimic the intracellular crowded milieu in vitro, a “crowding agent”, a concentrated solution of PEG, was used.35 The HCR efficiency in the presence of PEG is shown in Fig. S6 and S7.† The crowding agent improved the efficiency of the HCR process at low concentrations of I by up to 150%, possibly owing to excluded volume effects of the system.36
Collectively, a comparison of signal gains as a function of I22 and I60 concentrations, including statistical data analysis (Fig. S8†), indicates that the presence of the LNA bases in the H1LNA detection element improves the overall HCR efficiency by 1.3–3.3-fold compared with the conventional HCR process that uses DNA bases. As shown in Table S2,† the chimeric LNA–DNA sensor enabled the detection of the target sequence with higher sensitivity compared to the DNA–DNA sensor over the range of concentrations of 5 to 500 nM with a calculated limit of detection of 8–15 nM for the H1LNA/H2570 systems. Interestingly, we found that the fluorophore–quencher pair of H2 can influence the HCR process.
To investigate the structure of the concatemers (Fig. 1c) obtained by HCR using all H1–H2 combinations (i)–(iv), agarose gel electrophoresis experiments were conducted on the reaction mixtures obtained by adding different concentrations of I60 (Fig. 2). Fig. 2i shows the formation of smeared bands at higher molecular weights when H1LNA was used as the detection element at both low and high and concentrations of I60 (Fig. 2i, lanes 4–8). In contrast, faint bands were observed for the same concentrations when H1DNA was used as the detection element (Fig. 2i, lanes 10–14). These results confirm that in the presence of H2570, H1LNA as the detection element significantly improves the efficiency of the HCR process when compared with H1DNA. When H2670 was incubated with either H1DNA or H1LNA, a distinct intense band (Fig. 2k, lanes 8 and 14) was observed at the highest concentration of I60 examined, corresponding to the 80 bp concatemer I·H1·H2·H1·H2. This indicates that H2670 promotes the formation of low molecular weight concatemers and the interaction between the fluorophore–quencher pair can dictate the structure and length of the concatemers. To further study this effect, the melting curves of both hairpins (Fig. 2j) were examined, showing that H2570 with a melting temperature Tm of 66.6 °C is more stable than H2670 (Tm = 63.8 °C). Thus, relative to H2570, H2670 is more prone to nucleate with the complex I·H1, leading to a larger number of concatemers with lower molecular weights.
The lower percentage signal gain (Fig. 2b, f, d, h and Fig. S8†) resulting from these short and rigid concatemers can be ascribed to inter-molecular collisional quenching effects. Conversely, longer concatemers can assume a random coil conformation that prevents inter-molecular quenching of the fluorophores. The fluorescence emission of fluorophores in the presence of quencher molecules, such as BHQ, is typically affected by collisional quenching and is dependent on environmental factors. The conformational properties of the concatemers modulate the environment around the fluorophore and the probability of collisional quenching events to occur. To confirm this hypothesis, the efficiency of FRET- and contact-mediated quenching in the hairpins and the HCR product after treatment with deoxyribonuclease (DNase) I was measured. As expected, the FRET efficiency (85%) in the hairpins H2570 and H2670 was similar (Fig. 2l). However, 1.5-fold higher quenching in the concatemers obtained with the H2670 system than with the H2570 system was observed (Fig. 2l). The results suggest that the interactions between the fluorophore and quencher can contribute to the stability of the hairpin and thus affect the efficiency of the HCR process. Both QUASAR® 570 and QUASAR® 670 are polymethine fluorescent dyes, however the additional conjugated double bond in QUASAR® 670 may weaken the π–π interactions between the fluorophore and quencher. Overall, the engineered hairpins show potential toward the detection of mRNAPromAin vitro. The use of the chimeric LNA–DNA sensor as the recognition element and H2570 as the signal element results in high HCR efficiency.
Detection of HIV-1 in infected live cells using a chimeric LNA–DNA sensor
Following validation of the in vitro performance of the engineered chimeric LNA–DNA sensor for mRNAPromA by HCR-mediated signal amplification, the activity of the sensor was examined in live cells. To demonstrate its efficacy and selectivity in cellular extracts, H1LNA + H2570 was incubated with RNA isolated from TZM-bl (Fig. S9†) and HeLa (control cell lacking 3′LTRPromA expression) cell lines or with the whole cell lysate of both cell lines (Fig. S9†). TZM-bl cells were used as a model HIV-1 reporter cell line that contains the HIV-1 5′ and 3′LTR region sequence and thus PromA target sequence in its genome.24 In both systems, the fluorescence signal gain obtained in TZM-bl cell extracts was significantly higher than that obtained in HeLa cells, indicating that qualitative detection of mRNAPromA in TZM-bl cells can be achieved using the chimeric LNA–DNA sensor.The intracellular activity and selectivity of the chimeric LNA–DNA sensor were then examined on TZM-bl live cells (Fig. 3). TZM-bl cells were transfected with H2570 or H1LNA + H2570 using lipofectamine to enable cell internalization and endosomal escape (Table S3†).
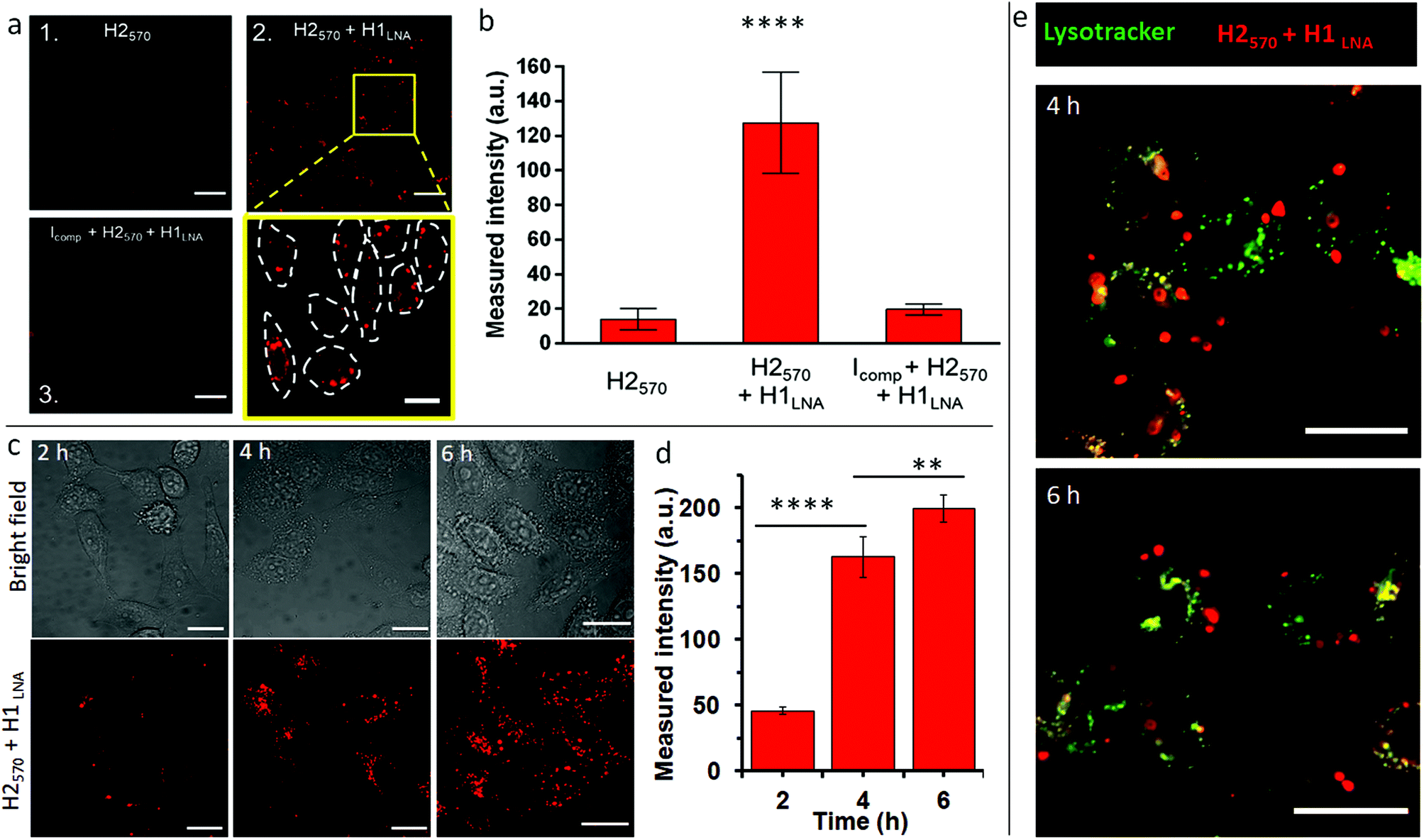 | ||
| Fig. 3 The intracellular activity and selectivity of the chimeric LNA–DNA in live cells. (a) Live CLSM imaging of TZM-bl cells incubated with H2570 only (panel 1), H2570 + H1LNA (panel 2), or H2570 + H1LNA + inhibitor Icomp (panel 3). Excitation wavelength, λex, is 546 nm. Scale bars are 50 µm in panels 1–3. A magnified area of the inset in panel 2 of the HCR signal from the cells incubated with both detection and initiator elements is also shown; scale bar is 20 µm. (b) Bar plot of the HCR signal intensity of the different systems in panels 1–3 (independent t-test, N = 30 cells. **** indicates p < 0.0001). (c) CLSM imaging of live TZM-bl cells after transfection with H2570 + H1LNA for 2, 4, and 6 h. Excitation wavelength, λex, is 546 nm. Scale bars are 20 µm. (d) Bar plot of the HCR signal intensity of the H2570 + H1LNA within the cells after incubation for 2, 4, and 6 h (independent t-test, N ≥ 200 cells. **** indicates p < 0.0001, ** indicates p < 0.01). (e) CLSM imaging of live TZM-bl cells after transfection with H2570 + H1LNA (red) for 4 and 6 h, showing limited colocalization with endo–lysosome compartments (green, stained with lysotracker green). Scale bars are 20 µm. | ||
The stability of H1LNA + H2570 was assessed upon complexation with lipofectamine. As observed from Fig. S10,† complexation of the hairpins with lipofectamine did not affect the stability of the hairpins or their ability to undergo HCR in the presence of an initiator. The TZM-bl cells were transfected with H2570 or H1LNA + H2570 for 2 h and the fluorescence signal was analyzed after 5 h incubation by CLSM (Fig. 3a). A standard and validated transfection protocol using lipofectamine was used to avoid significant toxicity to the cells. After transfection with lipofectamine-H2570 only or H2570 + H1LNA complexes, the TZM-bl cells did not show any significant reduction in cell viability (Fig. S11†). Previous studies on the intracellular delivery of oligonucleotide sensors mediated by lipofectamine have shown that at least 2 h incubation is sufficient to achieve cell internalization and binding that result in a fluorescence signal that can be detected by CLSM.37 It is noted that major interferences and false positive signals in the HCR fluorescence readout in live cells arise from the intracellular degradation of the H2570 hairpin, possibly mediated by lysosomal nucleases. Therefore, the transfection of TZM-bl cells with the H2570 hairpin only is a control experiment that allows us to rule out possible false positive fluorescence signals. When the cells were incubated with the H2570 hairpin only, a faint signal was detected (Fig. 3a, panel 1, Fig. S12†). This indicates that the lipofectamine-H2570 complex is able to avoid entrapment within the endo–lysosomal compartments and degradation by nucleases. Conversely, transfection of both HCR elements H1LNA and H2570 resulted in a strong signal (Fig. 3a, magnified area of panel 2, Fig. S12†).
Furthermore, to demonstrate that the HCR reaction was specifically triggered by mRNAPromA, TZM-bl cells were first transfected with a DNA strand that can hybridize with mRNAPromA, Icomp, therefore preventing HCR between the transfected signal (H2570) and detection (H1LNA) elements (Table S3†). Under this condition, only a faint signal was observed in cells pretreated with Icomp (Fig. 3a, panel 3, Fig. S12†), confirming the specificity of the developed chimeric LNA–DNA sensor toward mRNAPromA and the limited intracellular degradation of H2570. A quantitative evaluation of the fluorescence signal acquired under the three experimental conditions is shown in Fig. 3b.
In addition, a time course experiment, up to 6 h incubation, was performed to demonstrate that the fluorescence signal provided by live cells transfected by H2570 + H1LNA was enhanced as a function of time, as confirmed by confocal microscopy images (Fig. 3c) and quantitative evaluation of the fluorescence signal (Fig. 3d).
These results suggest that the endosomal escape and cytosolic delivery of both H1LNA and H2570 hairpins enabled the occurrence of HCR triggered by mRNAPromA. Confocal microscopy images (Fig. 3a, c, and S12†) revealed a cytosolic distribution of punctate signals in approximately 100% live cells, highlighting that the engineered chimeric LNA–DNA sensor enables the detection of mRNAPromA by HCR-mediated signal amplification with a high detection rate.
To further investigate the localization of the punctuate signals in live cells, we performed colocalization studies after 4 and 6 h incubation by staining the late endosomes/lysosomes with Lysotracker (Fig. 3e). The images were analyzed by Pearson's correlation coefficients (PCC). The images show that the red punctuate signals poorly colocalize with the late endosomes and lysosomes (PCC = 0.1). These data suggest that the spot signals localized in the cytosol do not arise from the H2570 hairpin degraded and entrapped in the endo–lysosomal vesicles. The limited colocalization between the widespread red signal and green signal (yellow signal) may be attributed to the negligible degradation of H2570 hairpin or to the limited resolution of confocal microscopy. We note that the spatial and temporal resolution of confocal microscopy does not allow precise and accurate detection and localization of nanosized structures in the cells.
Overall, these results indicate that the engineered chimeric LNA–DNA sensor enables live cell imaging and qualitative evaluation of mRNA expression. We have shown that cellular transcripts of the HIV-1 3′LTR promoter region are valid specific biomarkers that can be used to identify infected cells. The HIV-1 genome integrated in TZM-bl cells contains two identical LTR regions, i.e., 5′LTR and 3′LTR (Fig. 1a). Although these LTR regions share identical nucleotide sequences, 5′LTR serves as a virus promoter and initiates transcription, whereas 3′LTR functions as the terminator for transcription via 3′ end cleavage and polyadenylation of viral mRNA transcripts. In the absence of 5′LTR, 3′LTR has also been reported to function as a promoter. The 3′ end processing and poly(A) tail addition to pre-mRNA is derived from 3′LTR, resulting in the incorporation of the 3′LTR sequence in the 3′ end of viral mRNA transcripts.38 The HIV-1 LTR consists of two unique regions, U3 and U5, that flank a central repeat element region, R, which is the transcription start site (+1) in 5′LTR.38,39 HIV-1 mRNA transcripts incorporate a 5′ cap, including the R and U5 region.40 The HIV-1 sequence targeted by the chimeric LNA–DNA sensor is located in the U3 region of LTR and is thus present in both 5′ and 3′LTRs of the integrated provirus. The observed detection of the PromA sequence in the cytoplasm by the chimeric LNA–DNA probe is likely due to the presence of the U3 region in the 3′LTR sequence at the 3′ end of all HIV-1 mRNA transcripts, which are exported to the cytoplasm upon maturation.
Nanoscale imaging and localization of HIV-1 LTR transcript by ExM
mRNAPromA in TZM-bl cells was visualized by super-resolution ExM using the protocol detailed in the Experimental section. The expansion factors of the hydrogels and embedded cells were determined by measuring the diameter of the specimens and via nuclei staining with 4′,6-diamidino-2-phenylindole (DAPI) (Fig. S13 and S14†). Under the experimental conditions used, the hydrogels (with the embedded cells) expanded by 4.5-fold in water (Fig. S13†), with the cell nuclei expanding approximately 3.8-fold (Fig. S14†). Notably, the expansion of the samples was achieved without resorting to a linker to chemically immobilize RNA inside the hydrogels. MA-NHS was used as a monomer, which is known to anchor native proteins to the hydrogel during polymerization and cross-linking.41 We postulate that the use of MA-NHS could also enhance the cross-linking degree of the intracellular hydrogel and enable RNA retention inside the network by physical entrapment. In fact, the reaction of MA-NHS with primary amine groups on proteins generates protein-based macromers, which likely contribute to the polymerization and cross-linking process, ultimately affecting the structure of the 3D network after digestion and expansion. Furthermore, the mesh size of the formed intracellular hydrogel was tuned by optimizing the ionic strength of the buffer used during the expansion process with the aim to prevent the diffusion of RNA molecules through the pores of the swollen hydrogel. During washing of the expanded samples, charged RNA chains embedded in the hydrogel diffuse through the pores via a reptation mechanism under the influence of osmotic pressure.42 The mobility of mRNA chains is dictated by many factors including folded or unfolded RNA conformations, hydrogel pore size, and electrostatic repulsive forces between RNA and the polyacrylate network. Therefore, ionic strength, i.e., salt concentration, plays an essential role in controlling the diffusive properties of mRNA and mesh size of the hydrogel. We hypothesize that expansion of specimens prepared with MA-NHS at low ionic strength (i.e., 0.5× PBS) could limit the diffusional movements of mRNA by both reducing the hydrogel mesh size and the unfolding of the nucleic acid chains.To test our hypothesis, the cell sample obtained after hydrogel expansion in 0.5× PBS (Fig. 4a) was incubated with three different solutions containing H2570, H2570 + H1LNA, and inhibitor Icomp + H2570 + H1LNA, and the cells were analyzed with a confocal microscope (Fig. 4b and S15†). As indicated in Fig. 4b, cells incubated with H2570 only did not show any fluorescence. In contrast, red fluorescence was visible in the TZM-bl cell sample incubated with H2570 + H1LNA owing to the occurrence of HCR triggered by mRNAPromA. This was also confirmed by the lack of signal when the expanded cell samples were first incubated with the inhibitor Icomp and then with H2570 + H1LNA, indicating that the inhibitor prevents HCR signaling by hybridizing with the mRNAPromA. In addition, no signal was detected in the HeLa cell control when incubated with either H2570 or H2570 + H1LNA (Fig. 4c and S15†). The CLSM image of the (non-ExM) TZM-b1 cells prior to expansion, shown in Fig. 4d, indicates a high density of HCR-related spots, whereas decrowding of individual spots localized in the cytosol and perinuclear region was observed for the post-expansion (ExM) cells (Fig. 4d, e and S16†). Furthermore, imaging by ExM provided information about the size and the number of HCR products (Fig. 5 and S17†).
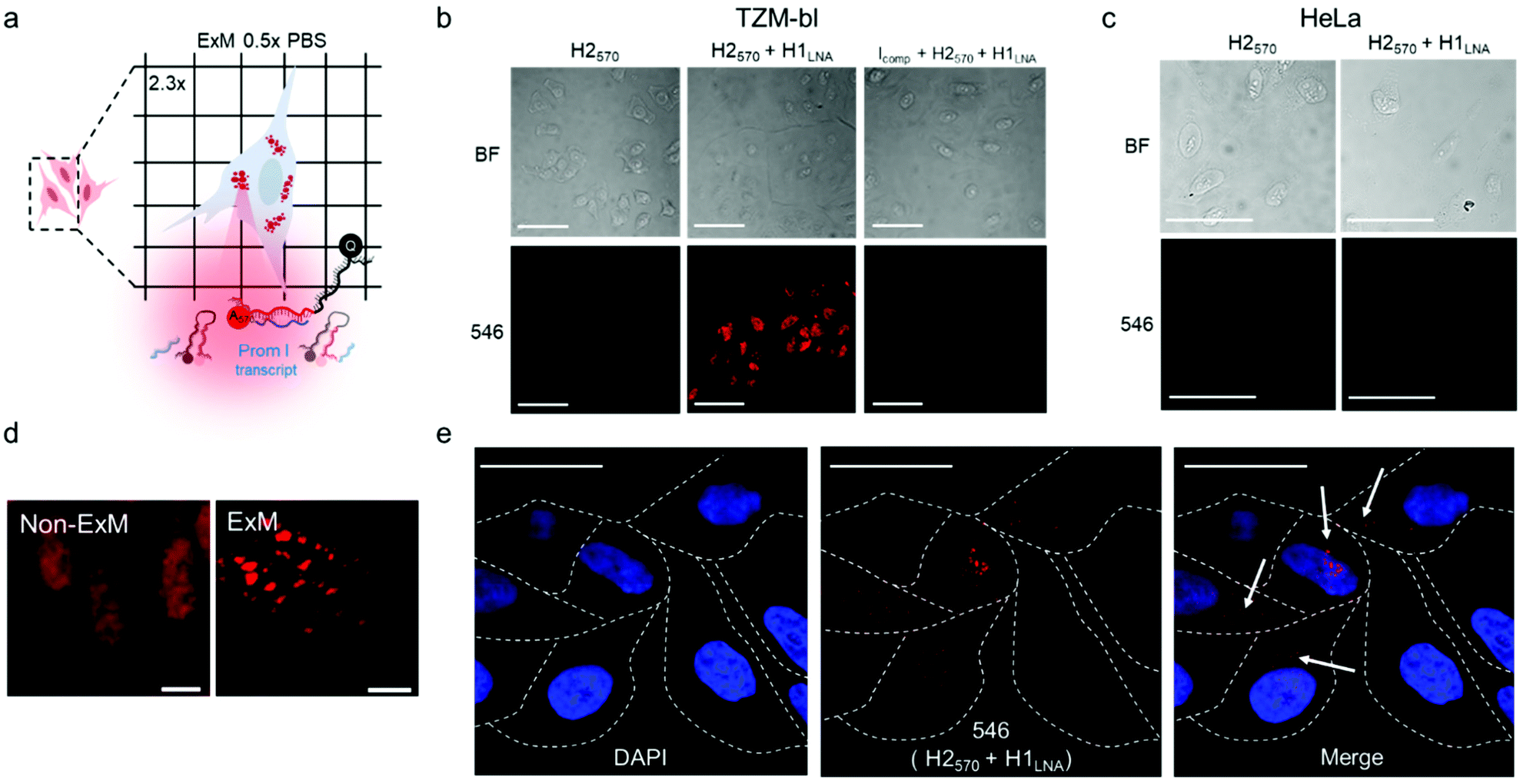 | ||
| Fig. 4 Nanoscale imaging of HIV-1 RNA with a chimeric LNA–DNA sensor and expansion microscopy. (a) Depiction of ExM experiment. (b) CLSM images of TZM-bl cells subjected to ExM procedure involving incubation with H2570, H2570 + H1LNA, or Icomp + H2570 + H1LNA: bright-field (BF) images and images showing cells excited with λex = 546 nm (546). (c) CLSM images of the expanded hydrogels containing HeLa cells incubated with H2570 alone or H2570 + H1LNA: BF images and images showing cells excited with λex = 546 nm (546). (d) Magnification of fixed non-ExM and ExM TZM-bl cells incubated with H2570 + H1LNA. (e) CLSM images of the expanded hydrogels containing TZM-bl cells incubated with H2570 + H1LNA. Nuclei are stained with DAPI. Dashed lines indicate the perimeter of the cells. White arrows indicate the localization of HCR loci inside the cells. λex = 405 and 546 nm. Scale bars are 100 μm (b and c), 10 μm (d) and 50 μm (e) (expansion factor 2.3×). | ||
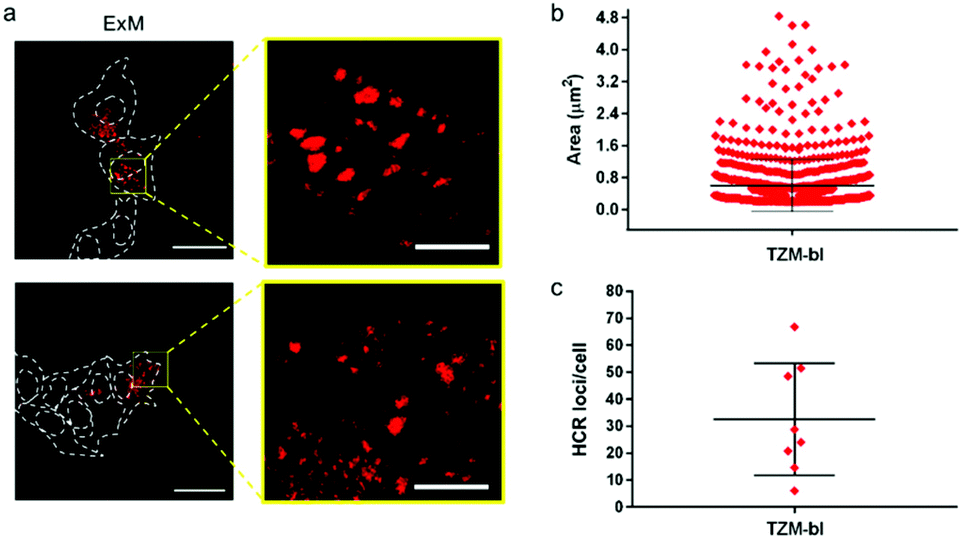 | ||
| Fig. 5 HCR loci analysis. (a) Representative CLSM images of TZM-bl cells post-ExM expansion showing HCR spots at low and high magnification; the ExM samples were incubated with H2570 + H1LNA. Dashed lines indicate the cell and nuclei perimeters. λex = 546 nm. Scale bars are 50 μm and 10 µm in the low- and high-magnification images respectively (expansion factor 2.3×). (b) Quantification of the area (µm2) of HCR loci found in TZM-bl cells. Values are shown as mean ± SD, N > 1000 loci. (c) Quantification of the HCR loci number found in TZM-bl cells (values are shown as mean ± SD, N > 50 cells). | ||
The size of the individual puncta (Fig. 5a and S17†), which likely corresponds to the amplification products of the HCR process triggered by the mRNA molecules inside the cell, was analyzed. The average area of the detected spots spanned between 100 nm2 and 2.4 µm2, taking into account the expansion factor, and an average number of ∼30 spots were detected per cell (Fig. 5b, c and S17†). Although the physical entrapment of RNA in the intracellular polymer network may result in a loss of some RNA molecules (35% of infected cells appeared positive) these results show that the engineered chimeric LNA–DNA sensor enables the nanoscale detection of HCR products in HIV-1-infected TZM-bl cells after expansion in a 3D hydrogel.
To obtain insight into the network mesh size in the expanded sample, we performed an in situ size exclusion experiment using Dx-FITC as a probe (10 kDa, hydrodynamic radius ∼2.3 nm), as shown in Fig. 6. This method allowed us to characterize the mesh size of intracellular and extracellular hydrogels. The pore size of the hydrogels/network prepared for ExM, ∼1–2 nm, was previously indirectly estimated by comparison with gels produced in test tube at the same salt concentration and acrylamide/N,N′-methylenebis(acrylamide) ratio.43 However, to our knowledge, the direct measurement of pore sizes in expanded hydrogel samples has not been reported to date.
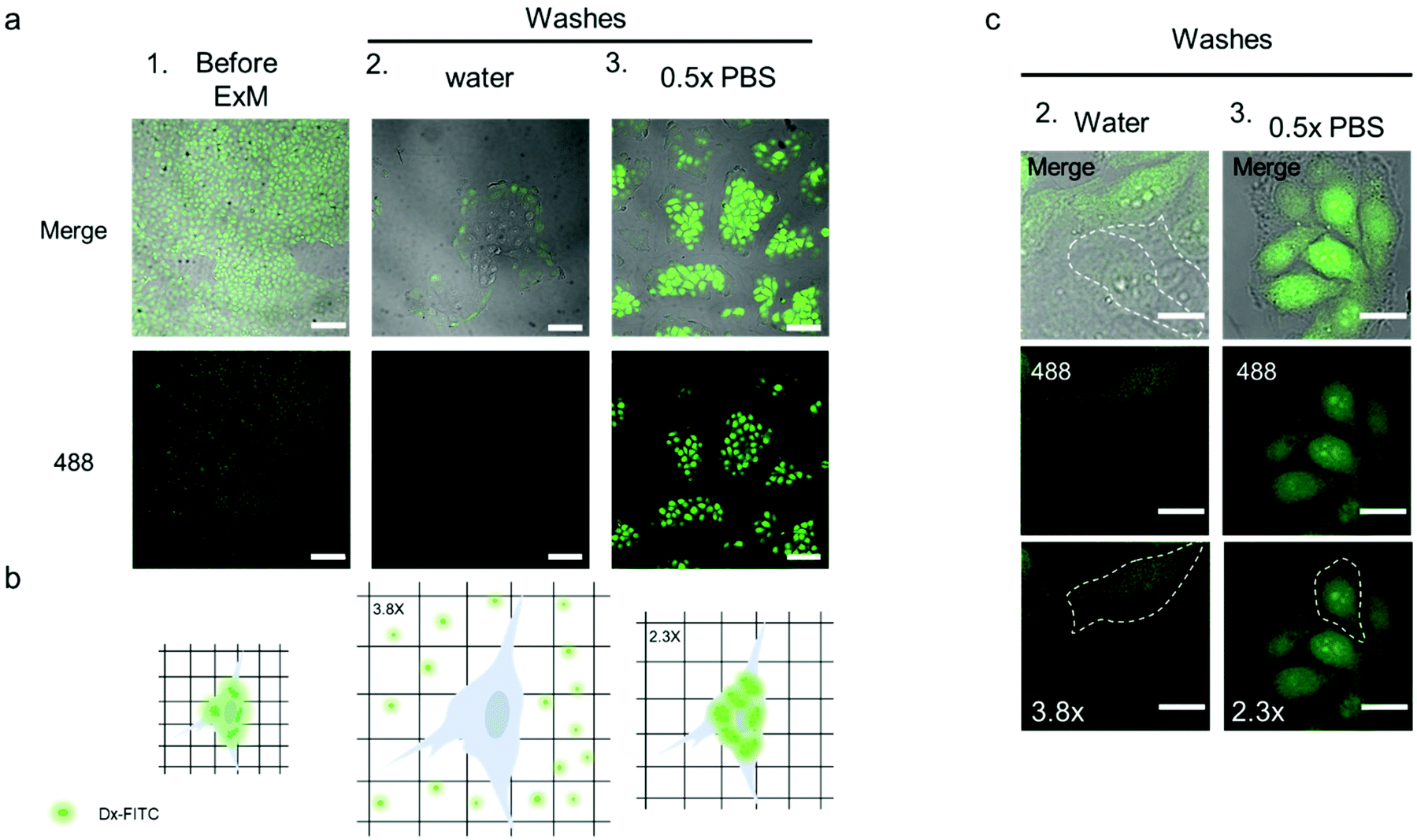 | ||
| Fig. 6 Intracellular hydrogel mesh size evaluation with standard dextran-FITC. (a) CLSM images of TZM-bl cell sample incubated with Dx-FITC and collected as is (1.), treated as per the ExM protocol and washed with water (2.) or 0.5× PBS (3.). λex = 488 nm. Scale bars are 100 μm (expansion factors are 3.8× (a 2.) and 2.3× (a 3.)). (b) Schematic of the cells containing Dx-FITC before washes (left), after hydrogel expansion following washes in Milli-Q water and diffusion of Dx-FITC from the cells (middle), and after hydrogel expansion following washes in 0.5× PBS (right). (c) Magnification of the cells embedded into the hydrogel and washed with Milli-Q water (2., left) or 0.5× PBS (3., right). The limited fluorescence in the cells washed with Milli-Q water and the difference in size of the cells treated in the two different conditions is indicated by the dashed white line, which defines the perimeter of the cell. λex = 488 nm. Scale bars are 25 μm (expansion factors are 3.8× (c 2.) and 2.3× (c 3.)). | ||
Before processing the sample for ExM, fixed TZM-bl cells were incubated with 1 mL Dx-FITC (500 ng mL−1) solution for 2 h to allow the biomacromolecule to enter the cells. The diffusion of Dx-FITC into the fixed cells was confirmed by the green fluorescence signal displayed by cells in CLSM images (Fig. 6a and b column 1). Following incubation, the sample was fixed and treated according to the ExM protocol, as described in the Experimental section. After incubation in the digestion buffer, the sample was expanded by washing in Milli-Q water or 0.5× PBS three times and imaged via confocal microscopy. Noteworthy, Dx-FITC was washed out from the hydrogel matrix after repeated washing with Milli-Q water, as indicated by the marked decrease in green fluorescence signal inside the cells (Fig. 6a, column 2; Fig. 6c, column 2; and Fig. S18†). In contrast, the fluorescence signal remained visible inside the cells when the sample was washed with 0.5× PBS (Fig. 6a, column 3; Fig. 6c, column 3; and Fig. S18†), suggesting that Dx-FITC was retained when expansion was performed in a solution of given ionic strength. These results indicate that the mesh size of the intracellular network is smaller than 4.6 nm when the composite specimen is expanded in 0.5× PBS but is larger than 4.6 nm when Milli-Q water is used. To obtain further insight into the mesh size of the hydrogel, the specimen was first expanded in 0.5× PBS and subsequently incubated with a Dx-FITC (500 ng mL−1) solution for 2 h to allow the biomacromolecule to diffuse inside the hydrogel (Fig. S19a†). In agreement with previous results, the green fluorescence signal was mainly detected in the extracellular region but not in the intracellular space (Fig. S19b†). This difference in diffusion behavior indicates the different degree of cross-linking in the intracellular and extracellular hydrogels. This was further investigated by comparing the expansion factor of the macroscopic hydrogels and nuclei following expansion of the samples under different conditions. Although the specimen incubated with 0.5× PBS shows significant macroscopic shrinking (∼2.4-fold), primarily ascribed to shrinking of the extracellular hydrogel, the nuclei shrunk only 1.6-fold when compared with the sample incubated in Milli-Q water (Fig. S20†). These data suggest that the use of MA-NHS monomer during the polymerization and cross-linking process confers a high cross-linking degree to the intracellular compartment and that the sieving properties of the intracellular hydrogel can be tuned at low ionic strength to inhibit the diffusion of biomacromolecules such as RNA.
Conclusions
In the present study, we developed chimeric LNA–DNA probes as HCR building blocks to detect HIV-1 LTR RNA in a model infected cell line. The engineered hairpins display high efficiency and specificity and are suitable for biosensing applications in live cells. We employed ExM and validated a new approach to detect HIV-1 RNA in model infected cells embedded and expanded in a 3D matrix, mimicking a tissue. The developed LNA–DNA amplification sensor combined with ExM enabled the selective imaging of individual viral transcripts with nanoscale precision in a 3D specimen in super-resolution mode. The sieving properties of the composite expanded sample were characterized, and the results revealed that the pore size of the intracellular hydrogel could be tuned by using MA-NHS monomer during the gelation and low ionic strength during the expansion. The RNA transcripts entrapped inside the expanded cell were detected without requiring additional chemicals for cross-linking of mRNA to the hydrogel matrix. The proposed combination of DNA nanotechnology and ExM provides a valuable tool for the detection of mRNAs inside cells and may find application in the biosensing of HIV-1-infected cells in complex tissues.Author contributions
AA carried out the design and characterization of the chimeric LNA–DNA sensor and wrote a draft of manuscript. MC carried out the expansion microscopy experiments on fixed cells and wrote a draft of manuscript. LM carried out confocal microscopy experiment on live cells. CC-J carried out gel electrophoresis, viability experiments and wrote a draft of the manuscript. SKB carried out confocal microscopy on live cells and statistical analysis of data. JS prepared cell extracts. CLA contributed to the experimental design of LNA–DNA sensor. GF contributed to the expansion microscopy experiments on fixed cells. FR contributed to the experimental design of LNA–DNA sensor. ADK contributed to the manuscript preparation. SRL contributed to the manuscript preparation. FCav contributed to the design and characterization of the chimeric LNA–DNA sensor in fixed and live cells and wrote the manuscript. FC contributed to experimental design and wrote the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This research was conducted and funded by the Australian Research Council Centre of Excellence in Convergent Bio-Nano Science and Technology (CE140100036) and the National Health and Medical Research Council (NHMRC) (GNT1149990). F. Caruso acknowledges the award of an NHMRC Senior Principal Research Fellowship (GNT1135806). F. Cavalieri acknowledges the award of an RMIT Senior Vice Chancellor Fellowship. This work received funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement no. 690901 (“NANOSUPREMI”). A. Amodio acknowledges funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement no. 798565 (“RE-IMMUNE”). G. Forte acknowledges the European Regional Development Fund – project MAGNET (no. CZ.02.1.01/0.0/0.0/15_003/0000492) and the European Regional Development Fund – project ENOCH (no. CZ.02.1.01/0.0/0.0/16_019/0000868). This work was performed in part at the Materials Characterization and Fabrication Platform (MCFP) at The University of Melbourne.Notes and references
- M. Arista-Romero, P. Delcanale, S. Pujals and L. Albertazzi, ACS Photonics, 2021, 9, 101 CrossRef PubMed.
- S. Pujals and L. Albertazzi, ACS Nano, 2019, 9, 9707 CrossRef PubMed.
- Q. Yan, M. Cai, L. Zhou, H. Xu, Y. Shi, J. Sun, J. Jiang, J. Gao and H. Wang, Nanoscale Adv., 2019, 1, 291 RSC.
- A. O. Pasternak and B. Berkhout, Cell Host Microbe, 2016, 20, 280 CrossRef CAS PubMed.
- M. A. Lifson, M. O. Ozen, F. Inci, S. Wang, H. Inan, M. Baday, T. J. Henrich and U. Demirci, Adv. Drug Delivery Rev., 2016, 103, 90 CrossRef CAS PubMed.
- G. M. Laird, E. E. Eisele, S. A. Rabi, J. Lai, S. Chioma, J. N. Blankson, J. D. Siliciano and R. F. Siliciano, PLoS Pathog., 2013, 9, e1003398 CrossRef CAS PubMed.
- C. Deleage, S. W. Wietgrefe, G. Del Prete, D. R. Morcock, X.-P. Hao, J. M. Piatak, J. Bess, J. L. Anderson, K. Perkey, C. Reilly, J. M. McCune, A. T. Haase, J. D. Lifson, T. W. Schacker and J. D. Estes, Pathog. Immun., 2016, 1, 68 CrossRef PubMed.
- K. M. Bruner, A. J. Murray, R. A. Pollack, M. G. Soliman, S. B. Laskey, A. A. Capoferri, J. Lai, M. C. Strain, S. M. Lada, R. Hoh, Y.-C. Ho, D. D. Richman, S. G. Deeks, J. D. Siliciano and R. F. Siliciano, Nat. Med., 2016, 22, 1043 CrossRef CAS PubMed.
- B. Hiener, B. A. Horsburgh, J.-S. Eden, K. Barton, T. E. Schlub, E. Lee, S. von Stockenstrom, L. Odevall, J. M. Milush, T. Liegler, E. Sinclair, R. Hoh, E. A. Boritz, D. Douek, R. Fromentin, N. Chomont, S. G. Deeks, F. M. Hecht and S. Palmer, Cell Rep., 2017, 21, 813 CrossRef CAS PubMed.
- C. Ahlenstiel, C. Mendez, S. T. H. Lim, K. Marks, S. Turville, D. A. Cooper, A. D. Kelleher and K. Suzuki, Mol. Ther. – Nucleic Acids, 2015, 4, e261 CrossRef CAS PubMed.
- R. M. Dirks and N. A. Pierce, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15275 CrossRef CAS PubMed.
- H. M. T. Choi, M. Schwarzkopf, M. E. Fornace, A. Acharya, G. Artavanis, J. Stegmaier, A. Cunha and N. A. Pierce, Development, 2018, 145, dev165753 CrossRef PubMed.
- J. Zheng, R. Yang, M. Shi, C. Wu, X. Fang, Y. Li, J. Li and W. Tan, Chem. Soc. Rev., 2015, 44, 3036 RSC.
- H. Wu, T.-T. Chen, X.-N. Wang, Y. Ke and J.-H. Jiang, Chem. Sci., 2020, 11, 62 RSC.
- S. Bi, S. Yue and S. Zhang, Chem. Soc. Rev., 2017, 46, 4281 RSC.
- F. Chen, P. W. Tillberg and E. S. Boyden, Science, 2015, 347, 543 CrossRef CAS PubMed.
- S. M. Asano, R. Gao, A. T. Wassie, P. W. Tillberg, F. Chen and E. S. Boyden, Curr. Protoc., 2018, 80, e56 Search PubMed.
- T. C. Kunz, R. Götz, S. Gao, M. Sauer and V. Kozjak-Pavlovic, Front. Cell Dev. Biol., 2020, 8, 617 CrossRef PubMed.
- P. W. Tillberg, F. Chen, K. D. Piatkevich, Y. Zhao, C.-C. Yu, B. P. English, L. Gao, A. Martorell, H.-J. Suk, F. Yoshida, E. M. DeGennaro, D. H. Roossien, G. Gong, U. Seneviratne, S. R. Tannenbaum, R. Desimone, D. Cai and E. S. Boyden, Nat. Biotechnol., 2016, 34, 987 CrossRef CAS PubMed.
- Y. Zhao, O. Bucur, H. Irshad, F. Chen, A. Weins, A. L. Stancu, E.-Y. Oh, M. DiStasio, V. Torous, B. Glass, I. E. Stillman, S. J. Schnitt, A. H. Beck and E. S. Boyden, Nat. Biotechnol., 2017, 35, 757 CrossRef CAS PubMed.
- E. D. Karagiannis, J. S. Kang, T. W. Shin, A. Emenari, S. Asano, L. Lin, E. K. Costa, A. H. Marblestone, N. Kasthuri and E. S. Boyden, bioRxiv, 2019 DOI:10.1101/829903 , preprint.
- A. Klimas and Y. Zhao, ACS Nano, 2020, 14, 7689 CrossRef CAS PubMed.
- F. Chen, A. T. Wassie, A. J. Cote, A. Sinha, S. Alon, S. Asano, E. R. Daugharthy, J.-B. Chang, A. Marblestone, G. M. Church, A. Raj and E. S. Boyden, Nat. Methods, 2016, 13, 679 CrossRef CAS PubMed.
- E. J. Platt, K. Wehrly, S. E. Kuhmann, B. Chesebro and D. Kabat, J. Virol., 1998, 72, 2855 CrossRef CAS PubMed.
- C. A. Derdeyn, J. M. Decker, J. N. Sfakianos, X. Wu, W. A. O'Brien, L. Ratner, J. C. Kappes, G. M. Shaw and E. Hunter, J. Virol., 2000, 74, 8358 CrossRef CAS PubMed.
- E. J. Platt, M. Bilska, S. L. Kozak, D. Kabat and D. C. Montefiori, J. Virol., 2009, 83, 8289 CrossRef CAS PubMed.
- Y. Takeuchi, M. O. McClure and M. Pizzato, J. Virol., 2008, 82, 12585 CrossRef CAS PubMed.
- X. Wei, J. M. Decker, H. Liu, Z. Zhang, R. B. Arani, J. M. Kilby, M. S. Saag, X. Wu, G. M. Shaw and J. C. Kappes, Antimicrob. Agents Chemother., 2002, 46, 1896 CrossRef CAS PubMed.
- J. L. Mergny and L. Lacroix, Oligonucleotides, 2003, 13, 515 CrossRef CAS PubMed.
- M. Zuker, Nucleic Acids Res., 2003, 31, 3406 CrossRef CAS PubMed.
- P. H. Hagedorn, R. Persson, E. D. Funder, N. Albæk, S. L. Diemer, D. J. Hansen, M. R. Møller, N. Papargyri, H. Christiansen, B. R. Hansen, H. F. Hansen, M. A. Jensen and T. Koch, Drug Discovery Today, 2018, 23, 101 CrossRef CAS PubMed.
- M. A. Campbell and J. Wengel, Chem. Soc. Rev., 2011, 40, 5680 RSC.
- L. K. McKenzie, R. El-Khoury, J. D. Thorpe, M. J. Damha and M. Hollenstein, Chem. Soc. Rev., 2021, 50, 5126 RSC.
- U. Christensen, N. Jacobsen, V. K. Rajwanshi, J. Wengel and T. Koch, Biochem. J., 2001, 354, 481 CrossRef CAS.
- S.-i. Nakano, D. Miyoshi and N. Sugimoto, Chem. Rev., 2014, 114, 2733 CrossRef CAS PubMed.
- I. M. Kuznetsova, K. K. Turoverov and V. N. Uversky, Int. J. Mol. Sci., 2014, 15, 23090 CrossRef PubMed.
- A. Glab, A. Bertucci, F. Martino, M. Wojnilowicz, A. Amodio, M. Venanzi, F. Ricci, G. Forte, F. Caruso and F. Cavalieri, Nanoscale, 2020, 12, 15402 RSC.
- B. Klaver and B. Berkhout, J. Virol., 1994, 68, 3830 CrossRef CAS PubMed.
- R. V. Guntaka, Microbiol. Rev., 1993, 57, 511 CrossRef CAS PubMed.
- J. Karn and C. M. Stoltzfus, Cold Spring Harbor Perspect. Med., 2012, 2, a006916 Search PubMed.
- T. J. Chozinski, A. R. Halpern, H. Okawa, H.-J. Kim, G. J. Tremel, R. O. L. Wong and J. C. Vaughan, Nat. Methods, 2016, 13, 485 CrossRef CAS PubMed.
- G. W. Slater, Electrophoresis, 2009, 30, S181 CrossRef PubMed.
- L. Pesce, M. Cozzolino, L. Lanzanò, A. Diaspro and P. Bianchini, J. Biophotonics, 2019, 12, e201900018 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Depiction of the HCR mechanism; stability of H2570 and H2670 in the presence of I22 and I60; in vitro HCR kinetics; efficiency of the HCR reaction using a double-stranded initiator; specificity of HCR reaction; comparison of efficiency of the HCR reaction using initiators I22 and I60; HCR in cell lysate; stability of H1 and H2 in complex with lipofectamine; viability assay; live confocal microscopy imaging of HCR reaction in live cells; representative photographs of the expanded gels; representative images of nuclei before and after ExM; detection of HCR products by confocal microscopy in non-ExM and ExM samples; quantitative analysis of size and number of the HCR products per cell; quantification of the signal intensity of Dx-FITC under different ExM hydrogel washing conditions; and extracellular hydrogel mesh size evaluation with standard Dx-FITC. See DOI: 10.1039/d1nr08418f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |