
Stepwise benzylic oxygenation via uranyl-photocatalysis†
Deqing
Hu
a and
Xuefeng
Jiang
 *abc
*abc
aShanghai Key Laboratory of Green Chemistry and Chemical Process, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, China. E-mail: xfjiang@chem.ecnu.edu.cn
bState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai, 200032, China
cState Key Laboratory of Elemento-organic Chemistry, Nankai University, Tianjin 300071, China
First published on 13th December 2021
Abstract
Stepwise oxygenation at the benzylic position (1°, 2°, 3°) of aromatic molecules was comprehensively established under ambient conditions via uranyl photocatalysis to produce carboxylic acids, ketones, and alcohols, respectively. The accuracy of the stepwise oxygenation was ensured by the tunability of catalytic activity in uranyl photocatalysis, which was adjusted by solvents and additives demonstrated through Stern–Volmer analysis. Hydrogen atom transfer between the benzylic position and the uranyl catalyst facilitated oxygenation, further confirmed by kinetic studies. Considerably improved efficiency of flow operation demonstrated the potential for industrial synthetic application.
Benzylic oxygenation brings forth a good number of valuable molecules such as alcohols, ketones, and carboxylic acids. It is an indispensable process for elevating the oxidation states of fossil raw materials, widely employed in agrochemicals, pharmaceuticals and organic materials.1 Conventionally, benzylic oxygenation relied on highly active inorganic and organic oxidants, such as KMnO4, CrO3, Oxone, tBuOOH, m-CPBA, H2O2, O3, NHPI, etc.2 Oxygen, in accordance with the requirement of sustainable chemistry, plays a major role for benzylic oxygenation with the help of Fe,3 Co,4 and Mn5 catalysis.6 Pandey,7 Fukuzumi,8 Wolf,9 Cibulka,10 Shi11 and et al.12 confirmed catalytic systems for benzylic oxygenation under mild photocatalyzed conditions. However, benzylic oxygenation step by step is still an intractable challenge, owing to the slight distinction of BDEs and oxidative potentials of benzylic position, the BDE of C(sp3)–C(sp3) bond lower than C–H bonds, and the oxidative potential of alcohols being lower than that of benzylic C–H bonds (Scheme 1a).13 Based on our stepwise oxygenation of sulfides, uranyl cation (UO22+) has been proven to be a powerful photo-oxidative catalyst with tunable photoactivity supported by additives and solvents.14,15 Uranyl, activated by visible-light irradiation through ligand-to-metal charge transfer (LMCT), achieves the unique characteristic of excellent oxidizing ability (E° = + 2.6 V vs. standard hydrogen electrode), intense capability of hydrogen atom abstraction, and tunable mechanism of both hydrogen atom transfer (HAT) and single electron transfer (SET).16 Herein, stepwise oxygenation at 1°, 2°, 3° benzylic position through the process of C–H bond activation via uranyl photocatalysis was reported for the production of carboxylic acids, ketones, and alcohols comprehensively (Scheme 1b).
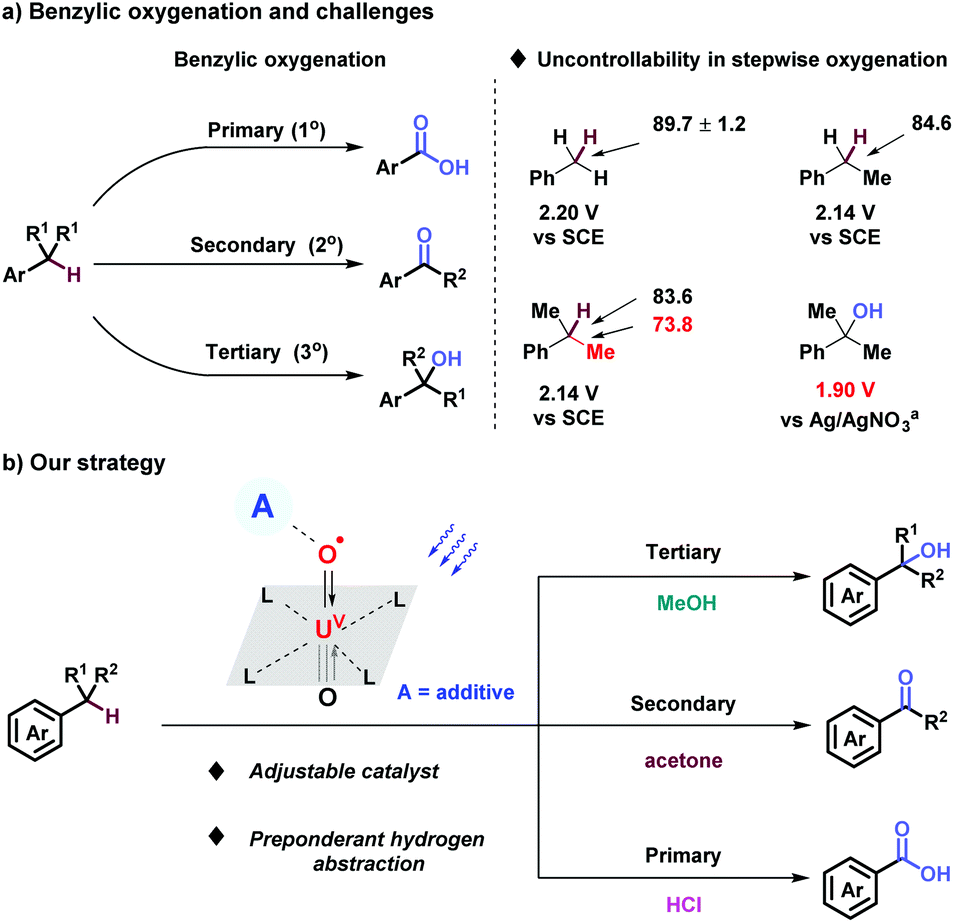 | ||
| Scheme 1 Benzylic oxygenation. BDE (kcal mol−1). Saturated calomel electrode as reference electrode. aAg/AgNO3 as reference electrode. | ||
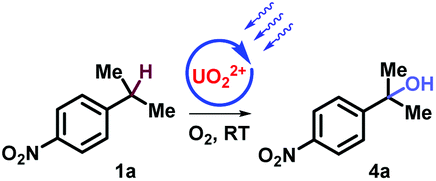
The study commenced with electron poor alkylbenzenes that bear the challenging nitro functional group (Table 1, and ESI, Part III†). Methanol was found to be crucial for the selective C–H bond activation of 4-nitro-isopropylbenzene 1a to the tertiary alcohol (Table 1, entry 1, and ESI, Part III†). In comparison, acetone as a solvent promoted the oxygenation of 4-nitro-ethylbenzene 2a to afford ketone in 81% yield (Table 1, entry 5). Furthermore, HCl as an additive was necessary for obtaining carboxylic acid in oxygenation of 4-nitro-toluene 3a (Table 1, entry 9). Noticeably, without uranyl photocatalyst, light, or O2, none of the three products could be obtained (Table 1).
| Entries | Conditions | Results | |
|---|---|---|---|
| a Substrate (1a, 2a, and 3a) (0.2 mmol), UO2(OAc)2·2H2O (2–4 mol%), additive (1 equiv.), solvent (1 mL), O2 balloon, blue light (460 nm), rt. ND means no detected. | |||
|
1 | UO2(OAc)2·2H2O (4 mol%) MeOH, 9w | 4a (60%) |
| 2 | Without [U] | ND | |
| 3 | Without light | ND | |
| 4 | Without O2 | ND | |
|
5 | UO2(OAc)2·2H2O (2 mol%) acetone, 6w | 5a (81%) |
| 6 | Without [U] | ND | |
| 7 | Without light | ND | |
| 8 | Without O2 | ND | |

|
9 | UO2(OAc)2·2H2O (2 mol%) HCI (2N) (1 equiv.), acetone, 9w | 6a (81%) |
| 10 | Without [U] | ND | |
| 11 | Without light | ND | |
| 12 | Without O2 | ND | |
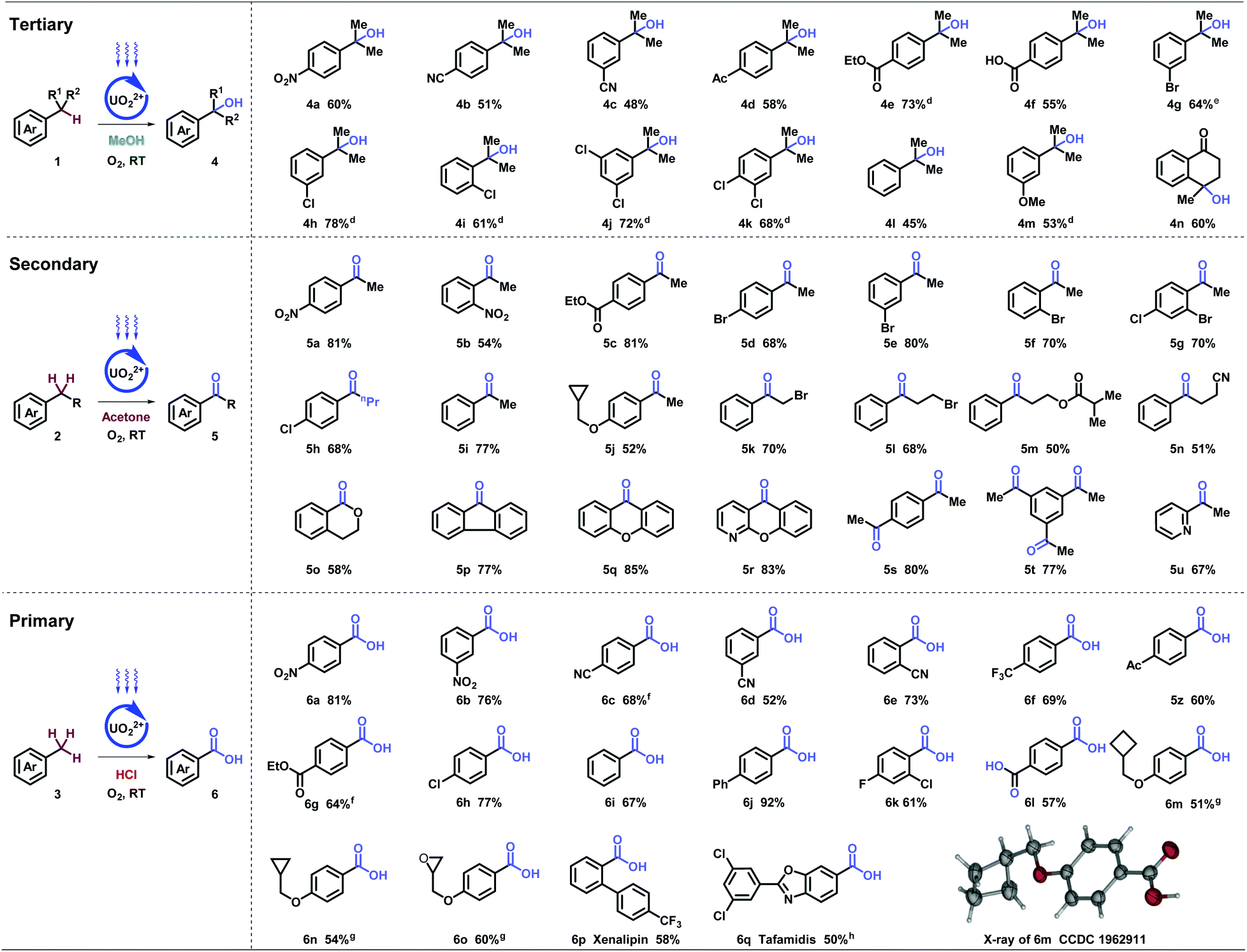
With the optimized conditions in hand, stepwise oxygenation of tertiary, secondary, and primary benzylic molecules were systematically studied (Table 2). Firstly, tough targets of diverse cumenes were tested. Besides nitro (4a), other strong electron-withdrawing groups, such as cyano- (4b and 4c), acetyl- (4d), and carboxyl (4e and 4f), were compatible in the tertiary alcohol generation. In addition, aryl halides with bromo- (4g), and choloro- (4h–4k) at different positions generated corresponding oxgynated product successfully, which provides potential for further coupling derivations. Substrates with electron-neutral (4l) and -donating (4m) groups were transformed with moderate yields. Remarkably, cyclic tertiary benzylic compound could produce desired alcohol (4n), applied in the synthesis of pharmaceuticals and natural products.17
| a General conditions for cumenes: 1 (0.2 mmol), UO2(OAc)2·2H2O (2 mol%), and MeOH (1 mL) were stirred with blue light (460 nm, 9 W), O2 balloon. b General conditions for ethylbenzenes: 2 (0.2 mmol), UO2(OAc)2·2H2O (2 mol%), and acetone (1 mL) were stirred with blue light (460 nm, 6 W), O2 balloon. c General conditions for methylbenzenes: 3 (0.2 mmol), UO2(OAc)2·2H2O (2 mol%), HCl (2 N) (1 equiv.) and acetone (1 mL) were stirred with blue light (460 nm, 9 W), O2 balloon. d 20 mol% of HBr (48% aq) was added. e TFA (1 equiv.). f Cl3CCO2H (1 equiv.) was used instead of HCl (2 N) (1 equiv.). g AcOH (1 equiv.) was used instead of HCl (2 N) (1 equiv.). h 0.1 mmol scale. |
|---|
|
Subsequently, the oxygenation of secondary benzylic was investigated. Substrates with electron-withdrawing (5a–5h) and neutral (5i) groups, delivered desired products smoothly. Electron-donating group (5j) was preserved with cyclopropylmethyl group connecting to heteroatoms. Diverse functional groups on the alkyl chain, such as halides (5k and 5l), ester (5m), and nitrile (5n), achieved desired transformations, despite other active hydrogen atoms existing. A series of benzocycloalkyls were investigated with moderate to excellent yields (5o–5r), including fluorenones (5p), xanthenone (5q), and N-xanthenone (5r), which are important building blocks in photoelectronic materials and pharmaceuticals. Remarkably, multiple-position oxygenation (5s and 5t) was afforded with excellent yields. Heteroaromatics, such as pyridine (5u), was tolerated in spite of coordinating and poisoning effects.
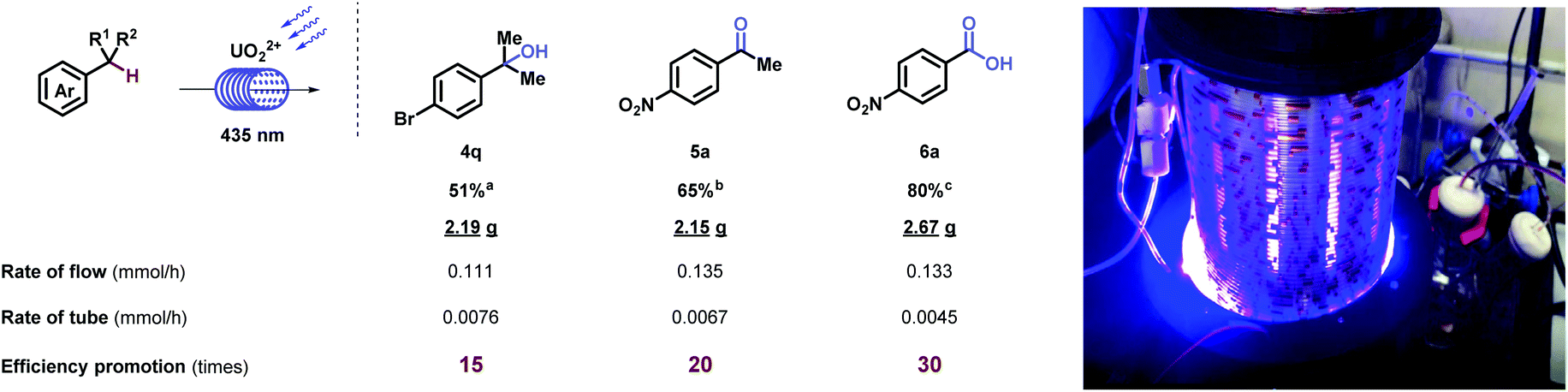
Further investigation on the oxygenation of methylbenzenes started with strong electron-withdrawing groups. Alkyl- benzenes with nitro- (6a and 6b), cyano- (6c–6e), and trifluoro- (6f) groups substituted at different positions generated desired products in good to excellent yields, which was rarely achieved under such mild conditions (room temperature, normal pressure, and oxygen as an oxidant). Other functional groups, such as acetyl- (5z), carboxyl- (6g), and halide (6h) were inspected and showed moderate to excellent yields. When molecules carrying electron-neutral (6i and 6j) groups, the transformations were efficient. Good result goes for the multi-substituted toluene (6k) as well. Terephthalic acid (6l), a significant industrial material for preparing polymers such as polyethylene terephthalate (PET) and polybutylene terephthalate (PBT), was afforded via p- xylene in moderate yields. Compatibility was further proved by strained rings (6m and 6n) and epoxy (6o). X-ray diffraction of 6m (CCDC 1962911†) further confirmed the structure. It is noteworthy that clinical pharmaceuticals were synthesized with this late-stage oxygenation straightforwardly. Xenalipin (6p), applied widely in controlling the levels of cholesterol and triglyceride in plasma,18 was afforded successfully. Tafamidis (6q) bearing two chloro- and benzoxazolyl groups that are efficient for treating TTR amyloid polyneuropathy,19 was obtained in 50% yield via direct oxygenation. Furthermore, flow apparatus was designed to pursue industrial applicability. A closed circulatory system consists of pump, polytetrafluoroethylene (PTFE) tube and round-bottom flask. The solution, filled with oxygen, was driven by the pump, and flowed through the PTFE tube irradiated by blue light. Under uranyl photocatalysis, 20 mmol-scale reactions were conducted at the rate of 0.111, 0.135, 0.133 mmol h−1, which displayed 15-, 20-, 30-fold efficiency compared with tube operation respectively (Table 3).
| a 1q (20 mmol) and UO2(OAc)2·2H2O (2 mol%) were stirred in MeOH (60 mL) in irradiation of blue light (435 nm) bubbling with O2 balloon. b 2a (20 mmol) and UO2(OAc)2·2H2O (2 mol%) were stirred in acetone (60 mL) in irradiation of blue light (435 nm) bubbling with O2 balloon. c 3a (20 mmol), UO2(OAc)2·2H2O (2 mol%), and HCl (2 N) (1 equiv.) were stirred in acetone (60 mL) in irradiation of blue light (435 nm) bubbling with O2 balloon. |
|---|
|
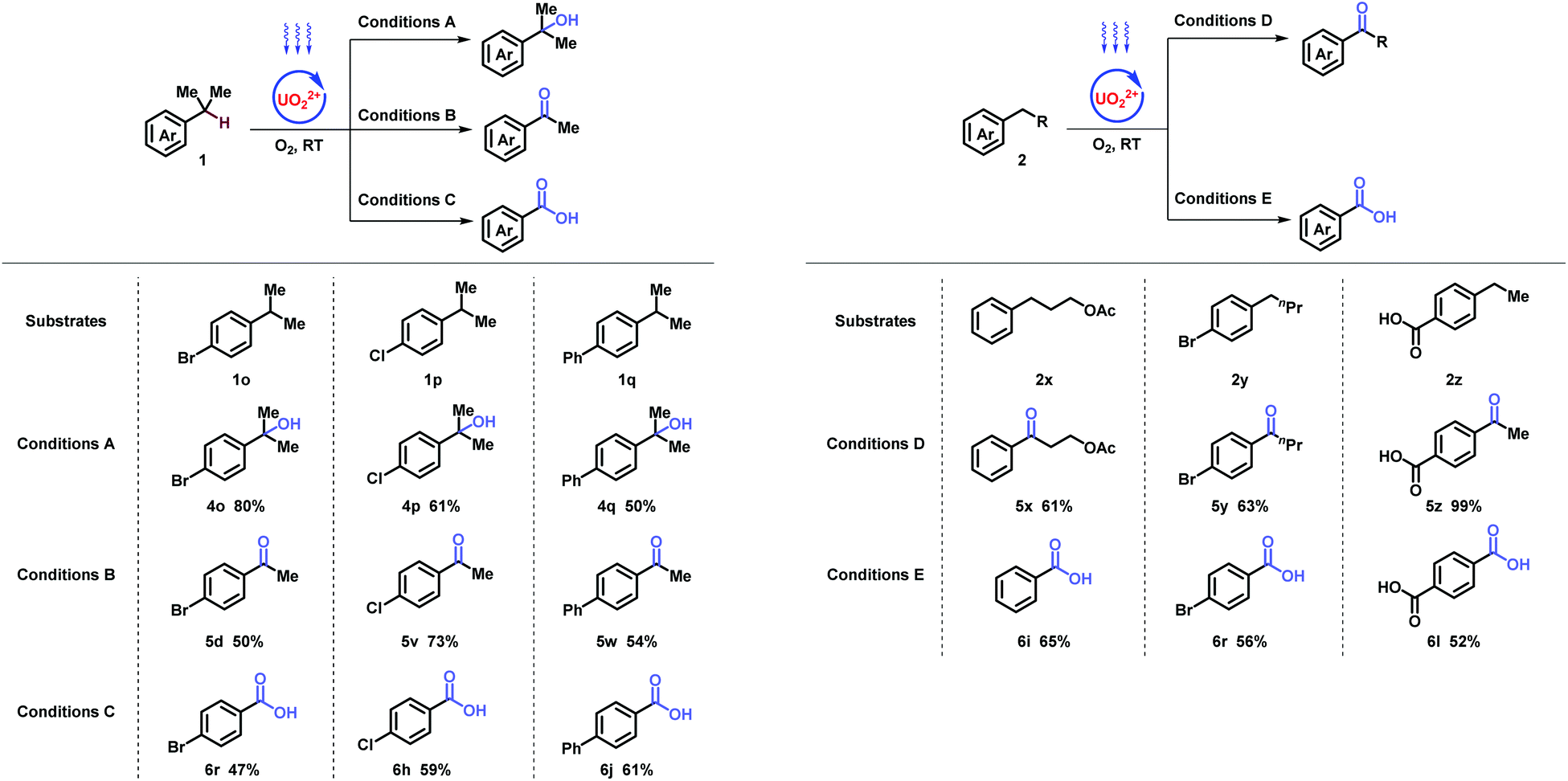
After achieving the stepwise abstraction of benzylic hydrogen atoms in 3°, 2°, 1° mode, oxygenation involving C–C activation was further studied in controllable pathway (Table 4). Firstly, cumene was investigated. Compared with methanol, acetone was inclined to single C–C bond cleavage for ketone generation. Double C–C bond cleavages were achieved in benzylic with one extra equivalent of sulfuric acid (2 N) (Table 4, left). For the deep oxygenation of ethylbenzenes, tert-Butyl alcohol assisted the corresponding cleavage of Csp2–Csp3 bonds (Table 4, right).
| General conditions: 1 or 2 (0.2 mmol) and UO2(OAc)2·2H2O (2 mol%) were stirred in solvent under the irradiation of blue light bubbling with O2 balloon. Conditions A: MeOH (1 mL), blue light (460 nm, 9 W). Conditions B: acetone (1.2 mL), blue light (460 nm, 6 W). Conditions C: H2SO4 (2 N, 1.0 equiv.), acetone (1 mL), blue light (430 nm, 12 W). Conditions D: acetone (1 mL), blue light (460 nm, 6 W). Conditions E: tBuOH (1.2 mL), blue light (430 nm, 12 W). |
|---|
|
Subsequently, the mechanism of stepwise oxygenation was further investigated. Firstly, radical quenching experiments with 2,2,6,6-tetramethyl-1-piperinedinyloxy (TEMPO) and butylated hydroxytoluene (BHT) suggested the radical property of these system (ESI, Section IV-1†). Uranyl acetate was confirmed as the photocatalyst instead of other components in the system through UV-visible light absorption experiments (ESI, Fig. S1†). Then, the interactions between substrates (e.g. 4-nitrotoluene, 4-nitro-1-ethylbenzene, and 4-nitro-1-isopropylbenzene) and active catalyst were approved via fluorescence quenching experiments (Fig. S2†). Next, the reaction ratio of alkyl benzenes with different substitutions showed the first order response to the bond dissociation energy of the benzylic C–H bonds (Scheme 2a, ESI†), which described HAT processes to be involved in these transformations.20 The following kinetic isotope effect experiments with deuterated toluene (Scheme 2b, for details, see ESI, IV5†) indicated that the HAT process was not the rate-determining step. Furthermore, the selectivity in these transformations was studied via Stern–Volmer experiments, indicating the effective quenching effect between methanol/HCl (aq) and active uranyl species. Hence, the tertiary alcohol was preserved during the oxygenation of benzylics, attributing to the lower catalytic properties of UO22+ in methanol inhibited the C–C bonds scissions, which was also proven by preliminary works.14,15c Acidic medium could increase the oxidizability of oxidant and accelerate the oxygenation of benzylics.21
 | ||
| Scheme 2 (a) Kinetic studies. (b) KIE experiments. | ||
Based on the results above, the proposed mechanism is outlined as below (Scheme 3). Blue light initiates UO22+via ligand-to-metal charge transfer (LMCT), generating oxyl radical, followed by HAT process at benzylic position. Benzyl radical A is consequently captured by oxygen molecule (3O2) to generate peroxy radical B. For tertiary benzylic molecules, methanol helps to control the activity of uranyl species via hydrogen-bond interaction, in which the comproportionation between A and B occurs and delivers intermediate C, resulting in the production of the alcohols 4via HAT. For the oxygenation of ethyl benzene, ketones 5 are prone to be obtained after interacting with OUVOH+ with water releasing. For toluenes, with the acceleration of proton acid, B reacts with OUVOH+, leading to aldehyde D and regenerating UO22+. D experiences another HAT immediately to form acyl radical E, further oxidized with dioxygen to afford acids 6.
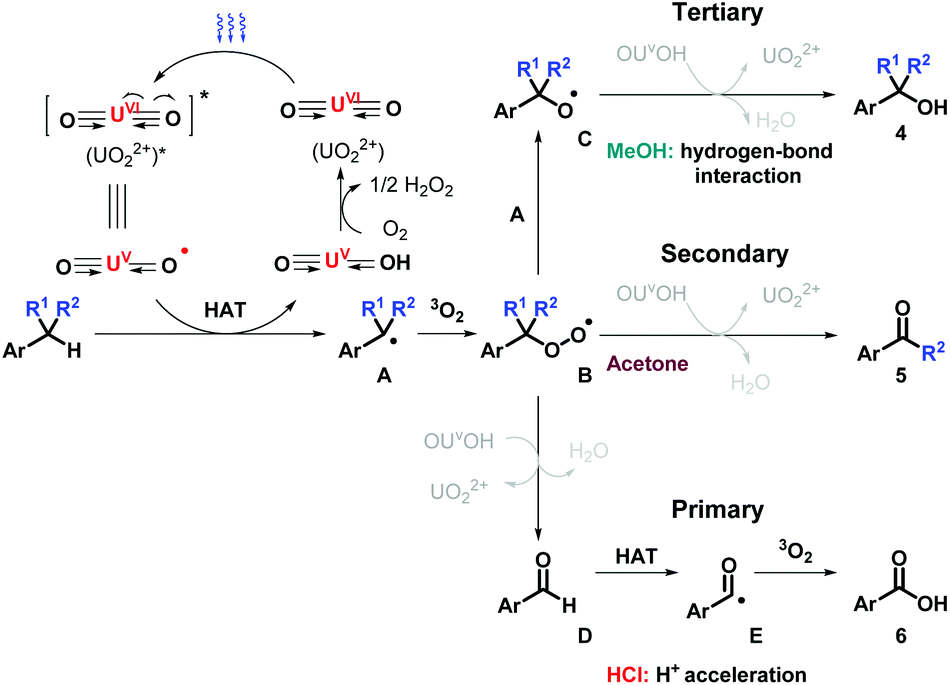 | ||
| Scheme 3 Proposed mechanism. | ||
Conclusions
In summary, a full-scale catalytic strategy for benzylic oxygenation of diverse oxidative-state molecules under ambient conditions (visible light, oxygen, normal pressure, and room temperature) was established comprehensively. Several challenges were studied: (1) the tunable stepwise oxygenation of benzylic C–H bonds; (2) the competent oxygenation of intense electron-deficient benzylics with oxygen under mild conditions; (3) the efficient catalytic oxygenation of cumenes to obtain benzylic tertiary alcohols. Flow setup design and operation pave the way for potential applications. Preliminary mechanistic studies indicated that solvent (MeOH, tBuOH) and additive (HCl, H2SO4) were proposed to be able to adjust the catalytic properties of UO22+, such as the catalytic activity be decreased in methanol, while that increased in acid, ensuring the accuracy of the stepwise oxygenation. Other strategies for selective activation of inert bonds are ongoing in our group.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for financial support provided by NSFC (22125103 and 21971065), STCSM (20XD1421500 and 20JC1416800), Innovative Research Team of High-Level Local Universities in Shanghai (SSMU-ZLCX20180501), and Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning.Notes and references
- J. M. Mayer, in Biomimetic Oxidations Catalyzed by Transition Metal Complexes, ed. B. Meunier, Imperial College Press, London, 2000 Search PubMed.
- (a) M. Costas, Coord. Chem. Rev., 2011, 255, 2912–2932 CrossRef CAS; (b) S. Caron, R. W. Dugger, S. G. Ruggeri, J. A. Ragan and D. H. B. Ripin, Chem. Rev., 2006, 106, 2943–2989 CrossRef CAS PubMed.
- (a) S. Evans and J. R. L. Smith, J. Chem. Soc., Perkin Trans. 2, 2001, 174–180 RSC; (b) S.-I. Murahashi, Y. Oda and T. Naota, J. Am. Chem. Soc., 1992, 114, 7913–7914 CrossRef CAS.
- (a) J.-Y. Qi, H.-X. Ma, X.-J. Li, Z.-Y. Zhou, M. C. K. Choi, A. S. C. Chan and Q.-Y. Yang, Chem. Commun., 2003, 1294–1295 RSC; (b) N. Hirai, N. Sawatari, N. Nakamura, S. Sakaguchi and Y. Ishii, J. Org. Chem., 2003, 68, 6587–6590 CrossRef CAS PubMed.
- (a) S. A. Chavan, D. Srinivas and P. Ratnasamy, Chem. Commun., 2001, 1124–1125 RSC; (b) A. Shibamoto, S. Sakaguchi and Y. Ishii, Org. Process Res. Dev., 2000, 4, 505–508 CrossRef CAS.
- (a) N. Jiao and S. S. Stahl, Green Oxidation in Organic Synthesis, John Wiley & Sons Ltd, 2019 CrossRef; (b) Y. Liang, J. Wei, X. Qiu and N. Jiao, Chem. Rev., 2018, 118, 4912–4945 CrossRef CAS PubMed; (c) J. H. Teles, I. Hermans, G. Franz and R. A. Sheldon, Ullmann's Encycl. Ind. Chem., 2015, 1–103 Search PubMed; (d) A. Gonzalez-de-Castro, C. M. Robertson and J. Xiao, J. Am. Chem. Soc., 2014, 136, 8350–8360 CrossRef CAS PubMed; (e) J. E. Backvall, Modern Oxidation Methods, WileyVCH, Weinhiem, 2nd edn, 2010 CrossRef; (f) T. Punniyamurthy, S. Velusamy and J. Iqbal, Chem. Rev., 2005, 105, 2329–2364 CrossRef CAS PubMed.
- G. Pandey, S. Pal and R. Laha, Angew. Chem., Int. Ed., 2013, 52, 5146–5149 CrossRef CAS PubMed.
- H. M. Neu, J. Jung, R. A. Baglia, M. A. Siegler, K. Ohkubo, S. Fukuzumi and D. P. Goldberg, J. Am. Chem. Soc., 2015, 137, 4614–4617 CrossRef CAS PubMed.
- B. Mühldorf and R. Wolf, Angew. Chem., Int. Ed., 2016, 55, 427–430 CrossRef PubMed.
- J. Zelenka, E. Svobodová, J. Tarábek, I. Hoskovcová, V. Boguschová, S. Bailly, M. Sikorski, J. Roithová and R. Cibulka, Org. Lett., 2019, 21, 114–119 CrossRef CAS PubMed.
- C. Wang, G. Zhang, Z. Zuo, R. Zeng, D. Zhai, F. Liu and Z. Shi, Sci. China: Chem., 2021, 64, 1487–1492 CrossRef CAS.
- (a) X.-Y. Yu, J.-R. Chen and W.-J. Xiao, Chem. Rev., 2021, 121, 506–561 CrossRef CAS PubMed; (b) X.-B. Li, C.-H. Tung and L.-Z. Wu, Nat. Rev. Chem., 2018, 2, 160–173 CrossRef CAS; (c) L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed; (d) D. Staveness, I. Bosque and C. R. J. Stephenson, Acc. Chem. Res., 2016, 49, 2295–2306 CrossRef CAS PubMed; (e) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed; (f) E. C. Gentry and R. R. Knowles, Acc. Chem. Res., 2016, 49, 1546–1556 CrossRef CAS PubMed; (g) D. C. Fabry and M. Rueping, Acc. Chem. Res., 2016, 49, 1969–1979 CrossRef CAS PubMed; (h) M. Majek and A. Jacobi von Wangelin, Acc. Chem. Res., 2016, 49, 2316–2327 CrossRef CAS PubMed; (i) M. N. Hopkinson, A. Tlahuext-Aca and F. Glorius, Acc. Chem. Res., 2016, 49, 2261–2272 CrossRef CAS PubMed; (j) I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725 CrossRef CAS PubMed; (k) J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 345, 433 CrossRef CAS PubMed; (l) E. Arceo, I. D. Jurberg, A. Álvarez-Fernández and P. Melchiorre, Nat. Chem., 2013, 5, 750–756 CrossRef CAS PubMed; (m) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (n) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed.
- (a) Y. R. Luo, Comprehensive Handbook of Chemical Bond Energies, Boca Raton, 2007 CrossRef; (b) S. Fukuzumi, K. Ohkubo, T. Suenobu, K. Kato, M. Fujitsuka and O. Ito, J. Am. Chem. Soc., 2001, 123, 8459–8467 CrossRef CAS PubMed; (c) E. A. Mayeda, L. L. Miller and J. F. Wolf, J. Am. Chem. Soc., 1972, 94, 6812–6816 CrossRef CAS.
- Y. Li, S. A.-e.-A. Rizvi, D. Hu, D. Sun, A. Gao, Y. Zhou, J. Li and X. Jiang, Angew. Chem., Int. Ed., 2019, 58, 13499–13506 CrossRef CAS PubMed.
- (a) D. Hu, Y. Zhou and X. Jiang, Natl. Sci. Rev., 2021, nwab156, DOI:10.1093/nsr/nwab156; (b) Y. Zhou, D. Hu and X. Jiang, JACS Au, 2021, 1, 1141–1146 CrossRef CAS PubMed; (c) J. Yu, C. Zhao, R. Zhou, W. Gao, S. Wang, K. Liu, S. Chen, K. Hu, L. Mei, L. Yuan, Z. Chai, H. Hu and W. Shi, Chem. – Eur. J., 2020, 26, 16521–16529 CrossRef CAS PubMed; (d) P. L. Arnold, J. M. Purkis, R. Rutkauskaite, D. Kovacs, J. B. Love and J. Austin, ChemCatChem, 2019, 11, 3786 CrossRef CAS; (e) L. Wu, X. Cao, X. Chen, W. Fang and M. Dolg, Angew. Chem., Int. Ed., 2018, 57, 11812–11816 CrossRef CAS PubMed; (f) J. G. West, T. Aaron Bedell and E. J. Sorensen, Angew. Chem., Int. Ed., 2016, 55, 8923–8927 CrossRef CAS PubMed; (g) Y. Mao and A. Bakac, J. Phys. Chem., 1996, 100, 4219–4223 CrossRef CAS; (h) W. Wang, A. Bakac and J. H. Espenson, Inorg. Chem., 1995, 34, 6034–6039 CrossRef CAS.
- (a) D. Hu and X. Jiang, Synlett, 2021, 32, 1330–1342 CrossRef CAS; (b) N. Behera and S. Sethi, Eur. J. Inorg. Chem., 2021, 95–111 CrossRef CAS; (c) B. E. Cowie, J. M. Purkis, J. Austin, J. B. Love and P. L. Arnold, Chem. Rev., 2019, 119, 10595–10637 CrossRef CAS PubMed; (d) Y. Li, J. Su, E. Mitchell, G. Zhang and J. Li, Sci. China: Chem., 2013, 56, 1671–1681 CrossRef CAS; (e) H. D. Burrows and S. J. Formosinho, J. Chem. Soc., Faraday Trans., 1977, 73, 201–208 RSC; (f) H. D. Burrows and T. J. Kemp, Chem. Soc. Rev., 1974, 3, 139–165 RSC.
- (a) N. A. Meanwell, J. Med. Chem., 2011, 54, 2529–2591 CrossRef CAS PubMed; (b) A. V. Kabanov, E. V. Batrakova and V. Y. Alakhov, J. Controlled Release, 2002, 82, 189–212 CrossRef CAS.
- M. C. Lewis, G. L. Hodgson, T. K. Shumaker and D. H. Namm, Atherosclerosis, 1987, 64, 27–35 CrossRef CAS PubMed.
- C. E. Bulawa, S. Connelly, M. DeVit, L. Wang, C. Weigel, J. A. Fleming, J. Packman, E. T. Powers, R. L. Wiseman, T. R. Foss, I. A. Wilson, J. W. Kelly and R. Labaudinière, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9629 CrossRef CAS PubMed.
- J. M. Mayer, Acc. Chem. Res., 2010, 44, 36–46 CrossRef PubMed.
- A.-S. Feiner and A. J. McEvoy, J. Chem. Educ., 1994, 71, 493–494 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1962911. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1gc04042a |
| This journal is © The Royal Society of Chemistry 2022 |