
Sequence-controlled and sequence-defined polypeptoids via the Ugi reaction: synthesis and sequence-driven properties
Yinuo
Zhu
and
Youhua
Tao
 *
*
Key Laboratory of Polymer Ecomaterials, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Renmin Street 5625, Changchun 130022, People's Republic of China. E-mail: youhua.tao@ciac.ac.cn
First published on 19th July 2021
Abstract
Polypeptoids are peptidomimetic polymers, and their origin can be traced back to the early 1990s. The facile synthesis of sequence-controlled and sequence-defined polypeptoids still faces many challenges. Very recently, the Ugi reaction has been introduced into polypeptoid chemistry. In this mini review, the recent progress in the application of the Ugi reaction for the synthesis of sequence-controlled polypeptoids is summarized. The influence of the sequence structure on polypeptoid properties is presented. Moreover, the future development of the Ugi reaction for the synthesis of sequence-controlled and sequence-defined polypeptoids is discussed.
 Yinuo Zhu | Yinuo Zhu earned her B.S. degree in Chemical Engineering and Technology from Central South University in 2019. She is currently an Ph.D. candidate at the Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, in the Tao laboratory. Her present research is centered on the synthesis and application of poly(amino acid)s. |
 Youhua Tao | Prof. Youhua Tao received his Ph.D. in Polymer Chemistry in 2008 from the Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, under the direction of Professor Xianhong Wang. After postdoctoral stays (2008–2013) with Professor Masami Kamigaito (Nagoya University, Japan) and Professor Christopher N. Bowman (University of Colorado at Boulder, USA), he accepted a full professor appointment at the Changchun Institute of Applied Chemistry, Chinese Academy of Sciences. The main focus of his current research is polymerization of sustainable amino acid monomers, and organocatalyzed polymer synthesis. |
1 Introduction
In the last century, the decoding of gene and protein sequences was undoubtedly a great and far-reaching scientific research achievement.1–3 Nucleic acids and polypeptides, which are ubiquitous in organisms, maintain the normal operation of organisms, and also determine the diversity, complexity and adaptability of biological organisms. The sequence of biological macromolecules is precisely defined in nature, such as DNA, RNA and proteins.4,5 Their precise sequences drive specific functions, such as molecular recognition, biocatalysis and information storage/transport.6 After billions of years of biological evolution, nature has developed a set of sophisticated and precise sequence regulations for biomacromolecules. The synthesis of proteins through ribosomes is a well-known example.7,8 Sequence-defined polymers are synthesized from templates with different chemical structures. This is a process in which the nucleotide sequence of an mRNA template on ribosomes is translated into an amino acid sequence. Synthesizing ribosome-like catalysts is still in the infant stage of current chemical tools; however, inspired by the biological macromolecules, the synthesis of sequence-controlled and sequence-defined polymers has attracted enthusiasm, and scientists are eager to design and define precise polymers to simulate and transcend the functions of natural macromolecules.9 In recent years, a large number of sequence-controlled synthesis strategies have been developed, including the template method,10 kinetic control,11 orthogonal reaction12 and reconstitution.13 Unfortunately, the effective synthesis method still faces great challenges.14Producing polymers and realizing sequential control by step-wise synthesis are considered as a practicable way. Merrifield pioneered this strategy to synthesize peptides on solid substrates.15,16 After that, the strategy evolved into the synthesis of DNA, RNA and oligomers with diverse organic structures which can combine to various resins.17 An iterative synthesis,18–20 especially a one-by-one iterative method chiefly carried out on a solid support (such as polystyrene resin), is a typical step-wise synthesis.21,26 Scientists have developed generous current methods to improve iteration efficiency and simplify processes, and the successful examples include triazene-based polymers,22 poly(alkoxyamine amide)s,23 oligothioetheramides,24 and thiolactone aminolysis.25
Compared with homopolymers, block copolymers or random copolymers, sequence-controlled and sequence-defined polymers allow a higher level of control over structures and related properties. At present, a series of biological functions have been simulated by sequence-controlled and sequence-defined polymers, for example, the mechanical properties of biological tissues are simulated by folding and self-assembly and biocatalysis and information transmission are realized by molecular recognition; however, the heredity of organisms is mostly in the theoretical stage.27,28 As an example, sequence-controlled oligopeptides can self-assemble into various nanostructures,29 including fibers, tapes, ribbons, vesicles and tubes, and they can be further developed into biocompatible polymers, conductive polymers and other multiple applications. Artificial protein analogues with controllable sequences have attracted extensive interest. Meijer designed enzyme analogues to regulate a monomer sequence through controlled radical polymerization and single-strand folding in water to achieve special properties.30 Seeman also demonstrated that complementary DNA strands can be extended to polymers, nanoparticles and nanocrystals.31
The self-assembly of synthetic polymers not only is greatly affected by the sequence, but also changes the thermodynamic properties and other macroscopic properties of the polymers, such as the solubility,32 charge density and duration of polyelectrolytes33,34 and the mesophase and semi-crystalline phase of special polymers.35,36 Zuckermann studied the effect of a sequence-controlled fragment on the phase behavior of polypeptoid–polystyrene block copolymers; a change of the sequence structure leads to an order–disorder transition.36
Polypeptoids are a kind of pseudo-peptidic polymer, serving as a bridge between biomaterials and synthetic polymers37–39 (Fig. 1). Small changes in these skeletons have been shown to have a significant impact on overcoming the shortcomings of polypeptides, such as the stability towards enzymes40 (Fig. 2). Polypeptoids can be employed as effective siRNA transfection reagents,41 diagnostic reagents,42 pulmonary surfactant mimics,43 antibacterial agents, etc.44 Sequence-controlled polypeptoids with high biomimetic properties provide a broad range of opportunities for the development of robust artificial materials in the future. This is mainly because polypeptoids have the advantages of chemical diversity, secondary structures and biocompatibility.45–47
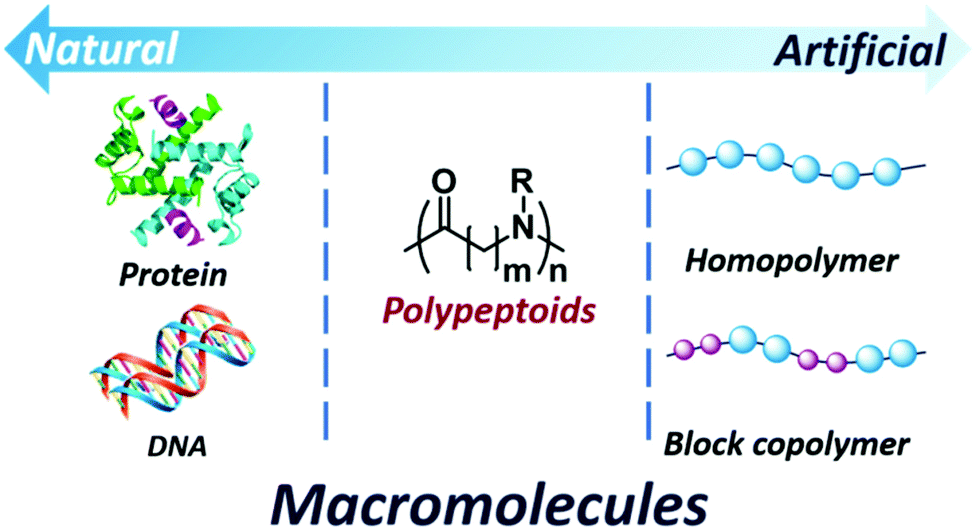 | ||
| Fig. 1 Polypeptoids build a bridge between natural and artificial macromolecules. | ||
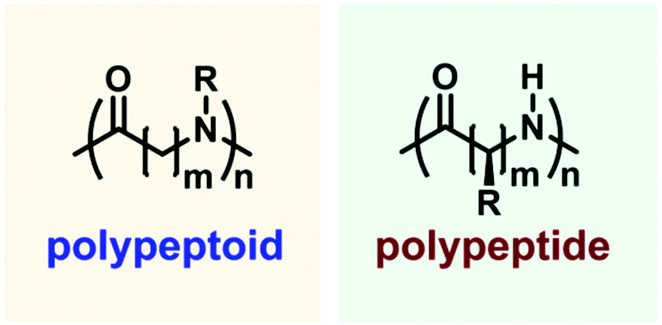 | ||
| Fig. 2 Structures of a polypeptoid and polypeptide. | ||
It is well known that there are two strategies for the synthesis of polypeptoids: solid-phase synthesis and ring-opening polymerization (ROP) of N-substituted α-amino acid-N-carboxyanhydrides (NNCAs).48 The solid-phase submonomer approach was developed by Zuckermann et al. in 1992.39 Since each monomer is individually tunable, an infinite variety of polymer sequences can be designed and synthesized.49 As for ring-opening polymerization (ROP) of N-substituted α-amino acid-N-carboxyanhydrides (NNCAs), although it overcomes the issues of low polymerization degree and molecular weight (Mw) in solid phase synthesis, it cannot synthesize polypeptoids with controllable sequences.50
Inspired by the above challenges, the Ugi reaction is considered to be a good method.51–55 The Ugi reaction was first reported by the German chemist Ivar Karl Ugi in 1959.56 In the development of a multicomponent reaction, this reaction was only used for crosslinking sodium alginate.57 Wessjohann synthesized selenocysteine containing peptoids and peptide–peptoid conjugates.58 In 2014, Meier polymerized dicarboxylic acid and diamine with isocyanides and aldehydes through the Ugi reaction to obtain polyamides.59,60 Then, sequence-defined peptoid oligomers were obtained by Meier et al. In 2016, Tao et al. introduced the Ugi reaction, which was a four-component reaction involving stoichiometric amounts of acid, amine, isocyanide, and aldehyde, into polypeptoid synthesis45 (Fig. 3A). The reaction between aldehyde and amine produces an imine, which is protonated by the carboxylic acid component. The imine is attacked by isonitriles, and carboxylic acids help to form the corresponding intermediates. Finally, the amino amide product is formed after rearrangement. They successfully obtained and poly(γ-, δ- and ε-peptoid)s by the Ugi reaction of amino acids under mild conditions45 (Fig. 3B). Subsequently, they reported the preparation of alternating polypeptoids through the Ugi reaction, and realized the synthesis of sequence-controlled polypeptoids by the Ugi reaction.61 Among the few reviews dedicated to this topic, Tao et al. documented polypeptoid preparation via the Ugi reaction.45,61,62 As a result, this mini review is not meant to offer a comprehensive review of the research activities in this field. It focuses on the recent development in the synthetic approaches to access sequence-controlled polypeptoids and highlights the sequence-driven properties that are fundamental topics in the polymer science area.
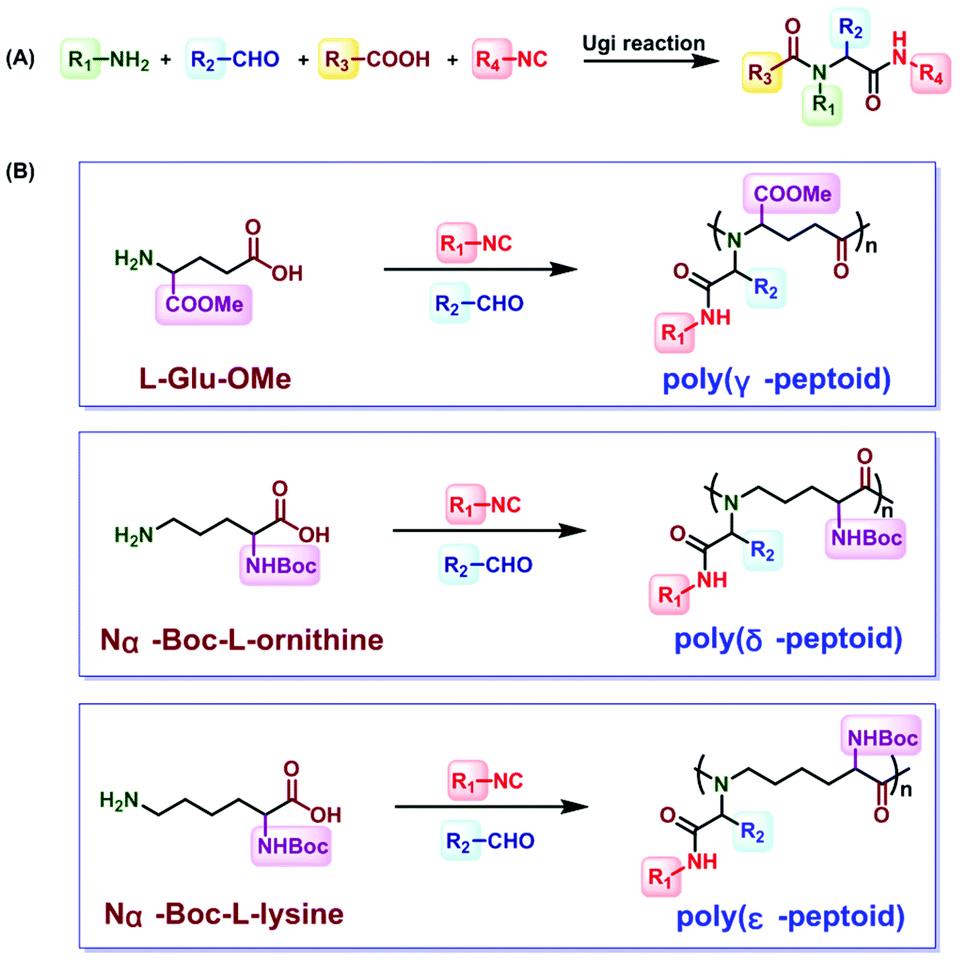 | ||
| Fig. 3 (A) General formula of the Ugi reaction. (B) Synthesis of poly(γ-, δ- and ε-peptoid)s through the Ugi reaction.45 | ||
2 Synthesis of alternating polypeptoids
Alternating polypeptoids are one of the simplest sequence-controlled and sequence-defined artificial copolymers. In recent years, the oligomeric peptoids with alternating sequences have been extensively explored. Zuckermann et al. synthesized peptoids which can self-assemble into highly ordered and free floating nanosheets with an alternating arrangement of hydrophobic and ionic monomers.63 Franzyk et al. reported the synthesis of alternating peptoids that have both stability toward enzymatic degradation and activity against multidrug-resistant bacteria.64,65In 2018, Tao et al. utilized the Ugi reaction in a one-pot procedure for the synthesis of alternating polypeptoids62 (Fig. 4A). Employing the AA′BB′-type monomers lysine methyl ester, N-Boc-glutamic acid, isobutyraldehyde, and tert-butyl isocyanide as reactants in the Ugi polymerization, the alternating polypeptoids can be easily obtained in a one-pot process, with a molecular weight of 15.1 kg mol−1 and a dispersity of 2.2. The monofunctional components should be excessive to ensure the complete consumption of the two amino acids. In addition, they found that the yield and molecular weight increased first and then decreased along the increase of the monomer concentration, which can be interpreted as the high viscosity of the system at a very high concentration. It is gratifying that when the structure of aldehydes or isocyanides is changed, the alternating polypeptoids with different side groups can be obtained in 86% yield. The structures of all the polypeptoids were confirmed by 1H, 13C, and COSY NMR spectroscopy.
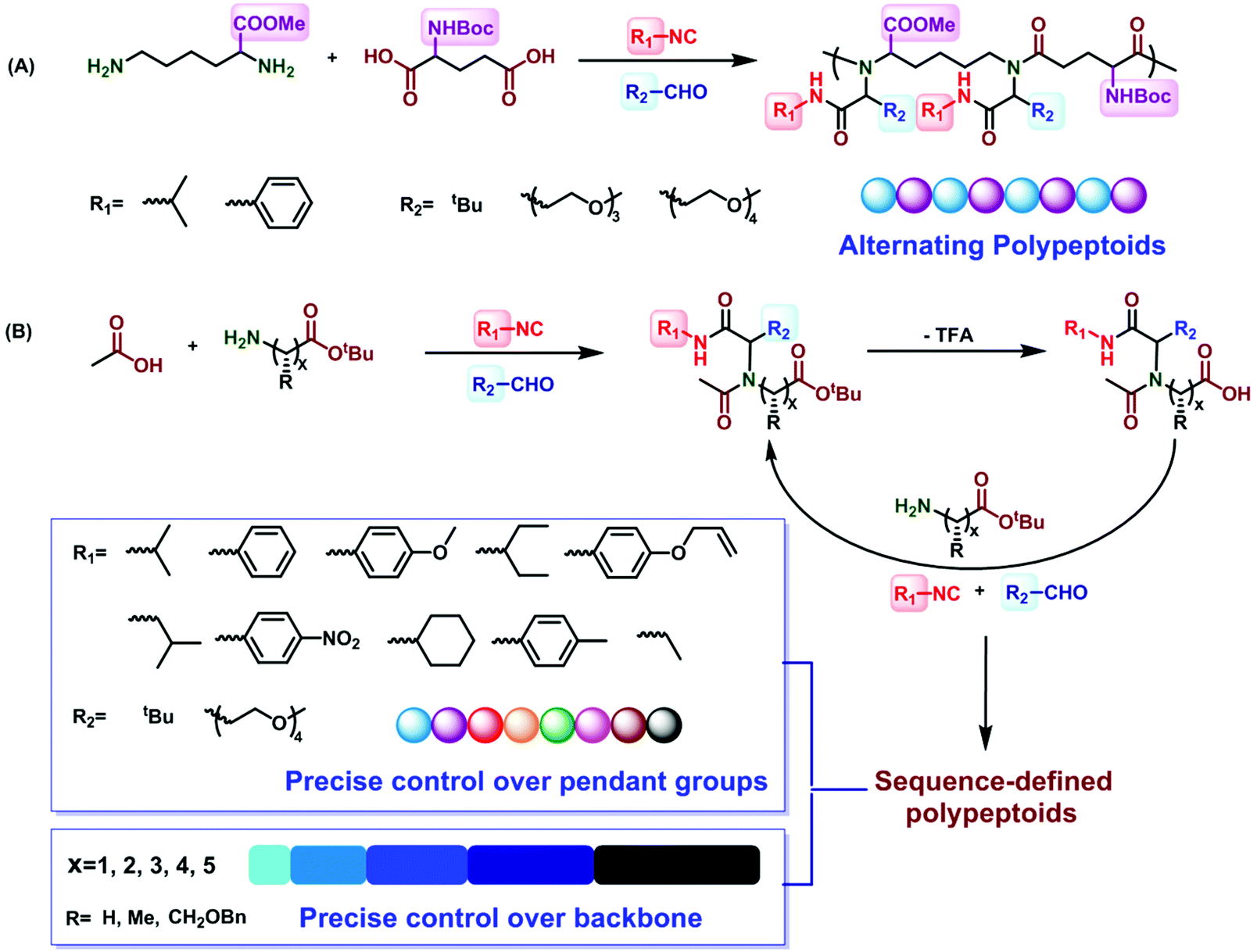 | ||
| Fig. 4 (A) Preparation of alternating polypeptoids via the Ugi reaction.62 (B) Preparation of sequence-controlled polypeptoids based on the iterative Ugi reaction.61 | ||
Debuigne et al. successfully carried out the Ugi reaction using β-alanyl-L-histidine or glycylglycine as the difunctional substrate, affording a library of structurally diverse polypeptoids featuring a high degree of peptide–peptoid alternating sequences. The principle for the choice of reactants is that the distance between amine and carboxylic acid groups is enough to prevent the cyclization of the 6-membered ring via an intramolecular side reaction. Different types of aldehydes (e.g., formaldehyde and isobutyraldehyde) and isocyanides were employed as building blocks.66
3 Synthesis of sequence-controlled polypeptoids
Sequence-controlled polymers refer to polymers with defined monomer positions along the polymer chain. The Ugi reaction can contribute to a great extent for the synthesis of sequence-controlled polypeptoids.In 2019, Tao et al. combined amino acid building blocks and iterative Ugi reactions for the synthesis of sequence-controlled polypeptoids (Fig. 4B).61 Importantly, the amino acid building block plays a crucial role in the Ugi reaction. An active amino group together with a tert-butyl ester protected carboxylic acid function that can undergo efficient deprotection consists of the building block. Subsequently, the amino acid building block reacted with other molecules (e.g., acetic acid), benzaldehyde, and tert-butyl isocyanide. The deprotection and Ugi reaction cycle was iterated, and trifluoroacetic acid (TFA) was used for deprotection. After 10 cycles, a 10-mer peptoid sequence was obtained. For purification methods, column chromatography and rotary evaporation were used. By adjusting the aldehydes or isocyanates with different side groups in each cycle, they obtained polypeptoids with diverse sequences. Using bis-COOH functionalized PEG as the starting molecule, polypeptoids with a higher Mw value (Mn ≈ 4.1 kDa) were synthesized.
Tao et al. further extended traditional sequence-controlled poly(α-peptoid)s to poly(β-, γ-, δ- and ε-peptoid)s. Thus, a 5-mer polypeptoid was constructed (Fig. 5). It is composed of five different amino acid building blocks, which greatly proves the versatility and robustness of the sequence-controlled polypeptoid by the iterative Ugi reaction.
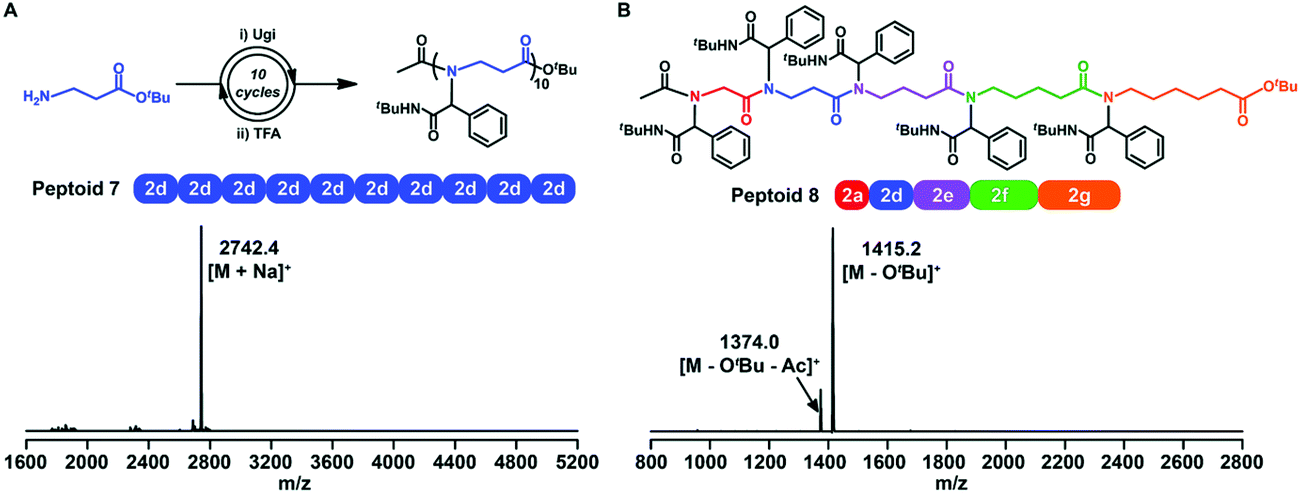 | ||
| Fig. 5 (A) Synthetic route and MALDI-TOF-MS spectrum of a 10-mer β-peptoid. (B) Chemical structure and MALDI-TOF-MS spectrum of a 5-mer peptoid consisting of all five amino acid building blocks (peptoid 8).61 | ||
Meier et al. also applied the iterative Ugi reaction to the formation of sequence-controlled peptoid oligomers. With stearic acid as the starting molecule, monodisperse four dimers (Mn = 1568.5 g mol−1) were obtained by changing the amine components in the Ugi reaction.67
In 2018, Becer and coworkers reported an iterative synthesis method combining solid-phase synthesis and the Ugi reaction to form peptides, peptoids or peptide–peptoid hybrids with well-controlled sequences.68
4 Sequence-driven properties
A monomer sequence is expected to affect polymer properties and functions. With longer range monomer sequences, diblock, triblock, and even multiblock copolymers have been prepared and are used as functional materials, which have properties quite different from those of homopolymers and random copolymers.69Tao et al. prepared a library of polypeptoids with hydrophobic and hydrophilic side chains, which have 12 sequence controllable units (Fig. 6). The arrangement of hydrophobic and hydrophilic units on the skeleton is different, and a variety of sequence controllable polypeptoids are formed by abundant arrangement and combination. The thermoresponsive behavior of these polypeptoids in water was studied at varying temperatures by measuring the light transmittance of the polypeptoid solutions. The sequence-controlled polypeptoids showed sequence-specific properties: the alternating polypeptoid was more soluble than the block polypeptoid, and thus showed higher lower critical solution temperature (LCST).
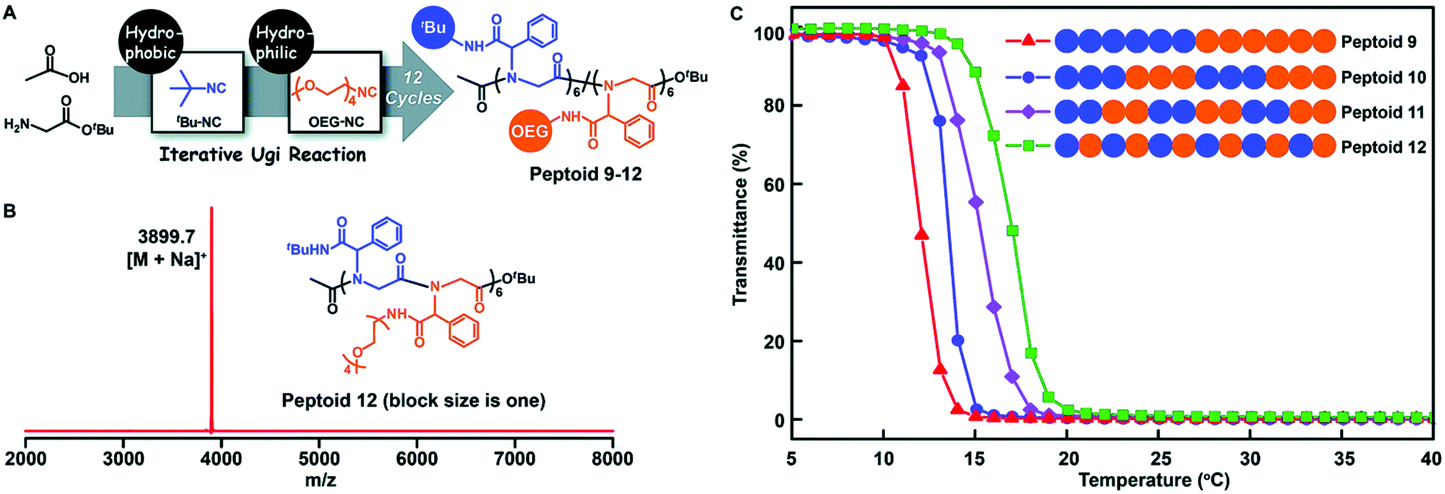 | ||
| Fig. 6 (A) Illustration of the side-chain sequence-regulated peptoids 9–12 synthesized by iterative Ugi reactions. (B) MALDI-TOF-MS spectrum of 12-mer peptoid 12. (C) Temperature dependence of the transmittance of the aqueous solutions (2 mg mL−1) of sequence-regulated peptoids.61 | ||
Tao et al. further studied the effect of the backbone structure of polypeptoids on the material properties (Fig. 7A). The structures of these sequence-controlled polypeptoids were verified by MALDI-TOF-MS and SEC tests (Fig. 7B). The solubility of these polypeptoids in mixed solvents of EtOH and H2O at different temperatures was evaluated by measuring the transmittance of the solution. The polypeptoid with an ABB alternating sequence in the backbone is more soluble than other proteins, and its cloud point is Tc = 45 °C, which is lower than that of the corresponding block copolymer (Tc = 53 °C) (Fig. 7C). These experiments indicate that not only the side chain sequence but also the backbone sequence has a great influence on the properties of polypeptoids, and the solubility can be gradually adjusted by varying the backbone and side chain monomer sequences.
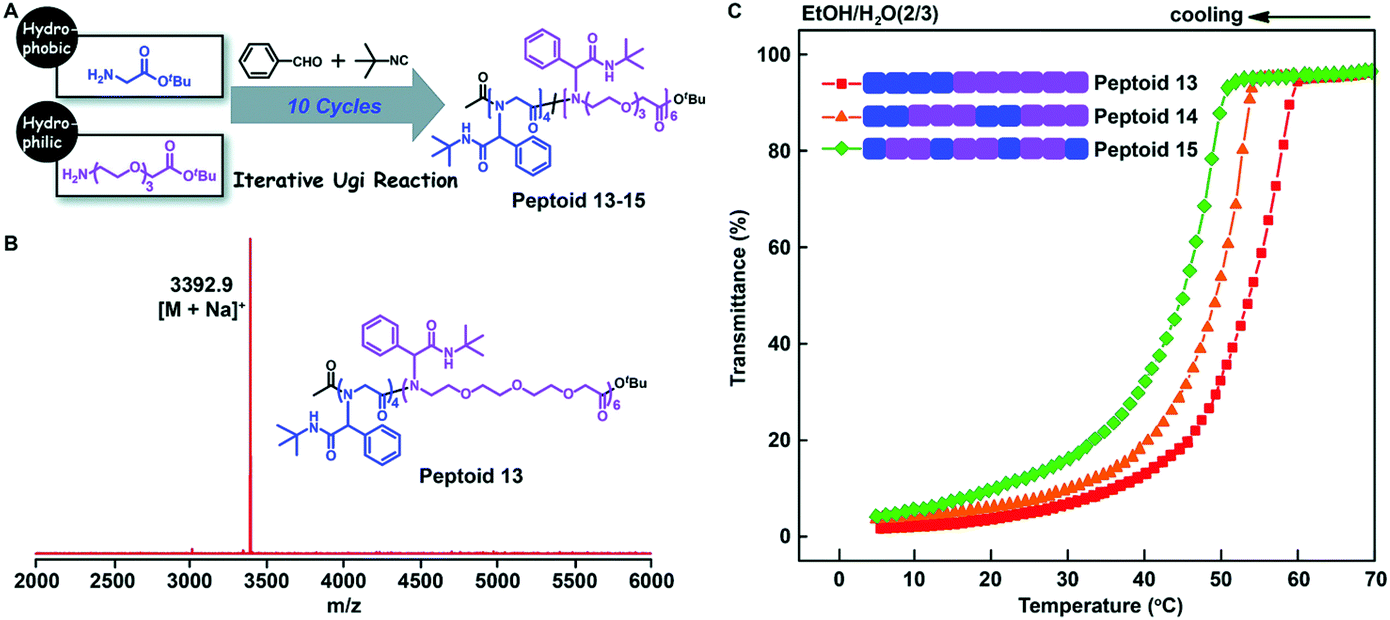 | ||
| Fig. 7 (A) Illustration of the backbone sequence-regulated peptoids 13–15 obtained by iterative Ugi reactions using glycine tert-butyl ester and oligo-ethylene-glycol based amino acid tert-butyl ester as building blocks. (B) MALDI-TOF-MS spectrum of 10-mer peptoid 13. (C) Transmittance of the backbone sequence-regulated peptoids 13–15 at varying temperatures in EtOH/H2O (2/3).61 | ||
Sequence-controlled and sequence-defined polypeptoids provide a new channel for artificial nucleic acid delivery. Nowadays, researchers are paying more and more attention to the structure of synthetic materials and biological agents to create new therapeutic methods. Therefore, Tao et al. studied whether sequence-controlled polypeptoids can bind to DNA for biomedical applications. Tao et al. added the azide function to the polypeptoids (Fig. 8A) by using the Ugi reaction and 3-azidopropan-1-amine. The azide modified polypeptoids were coupled to DNA with a dibenzocyclooctyne (DBCO) group in acetonitrile and PBS, as demonstrated by gel electrophoresis displacement (Fig. 8B). The success of conjugation was verified by MALDI-TOF-MS (Fig. 8C).
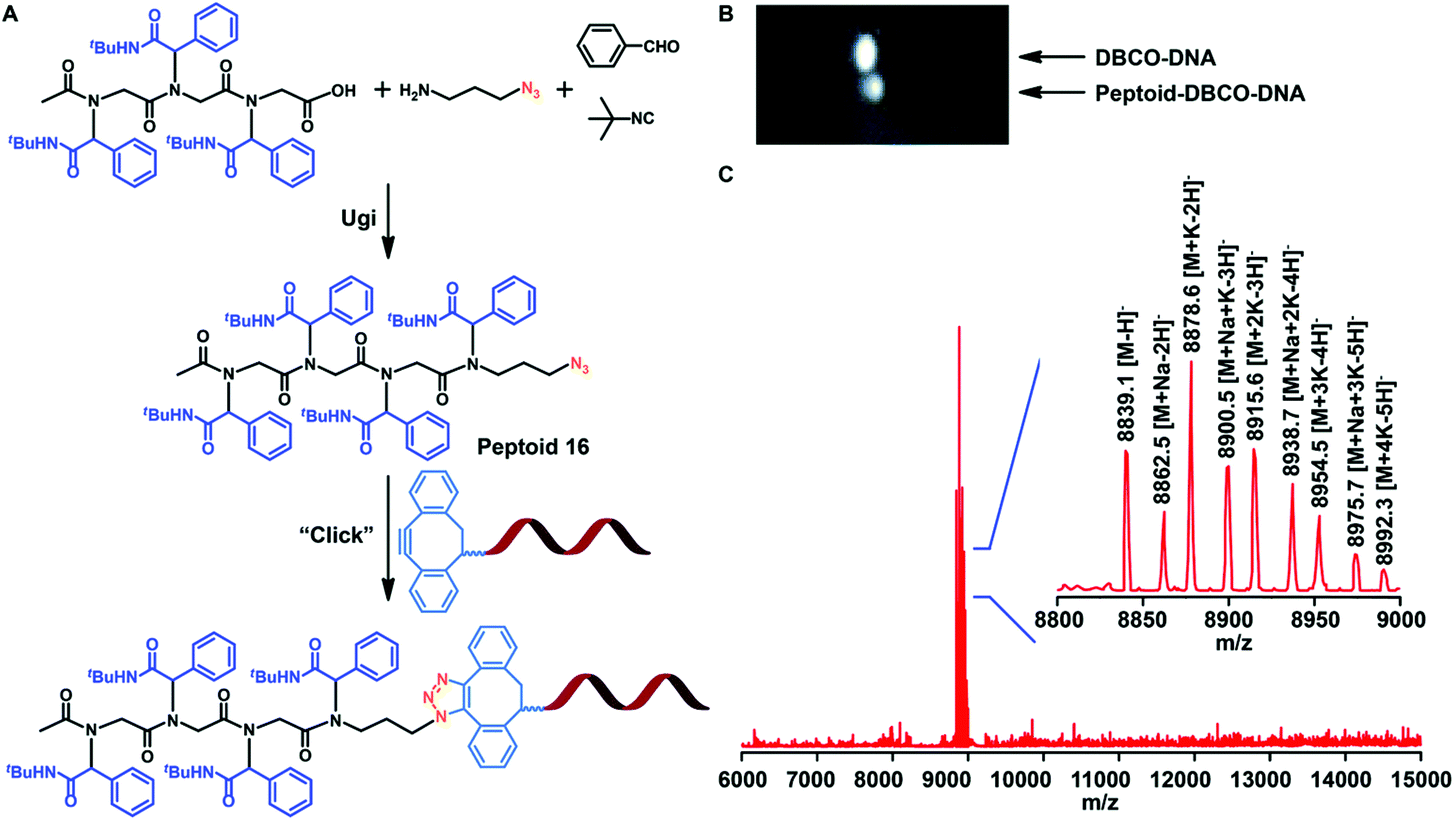 | ||
Fig. 8 (A) Conjugation of sequence-controlled peptoid 16 to DNA (DNA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :peptoid molar ratio = 1, 24 h reaction in CH3CN/PBS). (B) Gel electrophoresis result for the peptoid–DNA conjugation reaction. (C) MALDI-TOF-MS analysis of DNA conjugates. :peptoid molar ratio = 1, 24 h reaction in CH3CN/PBS). (B) Gel electrophoresis result for the peptoid–DNA conjugation reaction. (C) MALDI-TOF-MS analysis of DNA conjugates. | ||
5 Conclusions and perspectives
In summary, great progress has been made in the synthesis and sequence-driven properties of polypeptoids, including alternating polypeptoids, and sequence-controlled polypeptoids, via the Ugi reaction. Not only traditional sequence-controlled poly(α-peptoid)s, but also poly(β-, γ-, δ- and ε-peptoid)s can be obtained with high efficiency. In this review, the effect of the monomer sequence on the polypeptoid properties has also been described. It is clear that sequence-controlled polypeptoids exhibit sequence-oriented properties/functions, which could potentially lead to a ground-breaking paradigm shift for the synthesis of polymer materials.Because of the excellent performances of the Ugi reaction, it will play a more and more important role in the synthesis of sequence-controlled polypeptoids. The attraction of polypeptoids, of course, is that their designability is beyond current predictions. For instance, polypeptoids possess easily regulated thermodynamic properties. Thus, they can be coupled to biological macromolecules, which is helpful in developing new therapeutic methods.70,71 Polypeptoids can form an ideal self-assembly system, which can be assembled hierarchically to simulate proteins and other biological macromolecules. This is a complex space that has not yet been fully explored. The accurate regulation of the sequence structure of polypeptoids will bring broad applications and advanced materials.
Furthermore, how can we synthesize stereo- and sequence-controlled polypeptoids via the Ugi reaction? Although the Ugi reaction offers a good result in terms of sequence control, the synthesis of stereo- and sequence-controlled polypeptoids via the Ugi reaction remains an unmet challenge. A breakthrough work has been recently reported by Zhang and co-workers evaluating the enantioselective Ugi reaction using asymmetric phosphoric acid derivatives as catalysts.72 One can envision the application of asymmetric phosphoric acid catalysts that can enable the synthesis of stereo- and sequence-controlled polypeptoids. This will provide access to polypeptoids with different microstructures exhibiting different performances and broaden their potential applications.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work is supported by the National Natural Science Foundation of China (grants 91856113, 51873211, 22001243, and 52073274).References
- F. Sanger, Nature, 1948, 162, 491–492 CrossRef CAS.
- M. F. Perutz, Nature, 1962, 194, 914–917 CrossRef CAS.
- J. C. Venter, Science, 2001, 292, 1838–1838 CrossRef.
- J.-M. Lehn, Angew. Chem., Int. Ed., 2013, 52, 2836–2850 CrossRef CAS.
- J.-F. Lutz, J.-M. Lehn, E. W. Meijer and K. Matyjaszewski, Nat. Rev. Mater., 2016, 1, 16024 CrossRef CAS.
- Y. Brudno and D. R. Liu, Chem. Biol., 2009, 16, 265–276 CrossRef CAS PubMed.
- A. Javed, J. Christodoulou, L. D. Cabrita and E. V. Orlova, Acta Crystallogr., Sect. D: Struct. Biol., 2017, 73, 509–521 CrossRef CAS.
- D. N. Wilson and K. H. Nierhaus, Angew. Chem., Int. Ed., 2003, 42, 3464–3486 CrossRef CAS PubMed.
- C. J. Noren, S. J. Anthony-Cahill, M. C. Griffith and P. G. Schultz, Science, 1989, 244, 182 CrossRef CAS PubMed.
- X. Li and D. R. Liu, Angew. Chem., Int. Ed., 2004, 43, 4848–4870 CrossRef CAS PubMed.
- S. Pfeifer and J.-F. Lutz, J. Am. Chem. Soc., 2007, 129, 9542–9543 CrossRef CAS.
- M. Porel and C. A. Alabi, J. Am. Chem. Soc., 2014, 136, 13162–13165 CrossRef CAS PubMed.
- J.-F. Lutz, Polym. Chem., 2010, 1, 55–62 RSC.
- J. F. Lutz, M. Ouchi, D. R. Liu and M. Sawamoto, Science, 2013, 341, 628–636 CrossRef CAS.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- R. B. Merrifield, Adv. Enzymol. Relat. Areas Mol. Biol., 1969, 32, 221 CAS.
- P. Cherkupally, S. Ramesh, B. G. de la Torre, T. Govender, H. G. Kruger and F. Albericio, ACS Comb. Sci., 2014, 16, 579–601 CrossRef CAS.
- T. Soejima, K. Satoh and M. Kamigaito, J. Am. Chem. Soc., 2016, 138, 944–954 CrossRef CAS.
- F. Amir, Z. Jia and M. J. Monteiro, J. Am. Chem. Soc., 2016, 138, 16600–16603 CrossRef CAS PubMed.
- Z. Huang, J. Zhao, Z. Wang, F. Meng, K. Ding, X. Pan, N. Zhou, X. Li, Z. Zhang and X. Zhu, Angew. Chem., Int. Ed., 2017, 56, 13612–13617 CrossRef CAS PubMed.
- S. Billiet, K. De Bruycker, F. Driessen, H. Goossens, V. Van Speybroeck, J. M. Winne and F. E. Du Prez, Nat. Chem., 2014, 6, 815–821 CrossRef CAS.
- J. W. Grate, K.-F. Mo and M. D. Daily, Angew. Chem., Int. Ed., 2016, 55, 3925–3930 CrossRef CAS.
- R. K. Roy, A. Meszynska, C. Laure, L. Charles, C. Verchin and J.-F. Lutz, Nat. Commun., 2015, 6, 7237 CrossRef CAS.
- M. Porel, D. N. Thornlow, C. M. Artim and C. A. Alabi, ACS Chem. Biol., 2017, 12, 715–723 CrossRef CAS.
- P. Espeel, L. L. G. Carrette, K. Bury, S. Capenberghs, J. C. Martins, F. E. Du Prez and A. Madder, Angew. Chem., Int. Ed., 2013, 52, 13261–13264 CrossRef CAS PubMed.
- A. M. Rosales, R. A. Segalman and R. N. Zuckermann, Soft Matter, 2013, 9, 8400–8414 RSC.
- D. H. Lee, J. R. Granja, J. A. Martinez, K. Severin and M. R. Ghadiri, Nature, 1996, 382, 525–528 CrossRef CAS PubMed.
- V. B. Pinheiro, A. I. Taylor, C. Cozens, M. Abramov, M. Renders, S. Zhang, J. C. Chaput, J. Wengel, S.-Y. Peak-Chew, S. H. McLaughlin, P. Herdewijn and P. Holliger, Science, 2012, 336, 341–344 CrossRef CAS PubMed.
- H. G. Börner, Prog. Polym. Sci., 2009, 34, 811–851 CrossRef.
- T. Terashima, T. Mes, T. F. A. De Greef, M. A. J. Gillissen, P. Besenius, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2011, 133, 4742–4745 CrossRef CAS PubMed.
- N. C. Seeman, Angew. Chem., Int. Ed., 1998, 37, 3220–3238 CrossRef CAS PubMed.
- H. K. Murnen, A. R. Khokhlov, P. G. Khalatur, R. A. Segalman and R. N. Zuckermann, Macromolecules, 2012, 45, 5229–5236 CrossRef CAS.
- B. S. Aitken, C. F. Buitrago, J. D. Heffley, M. Lee, H. W. Gibson, K. I. Winey and K. B. Wagener, Macromolecules, 2012, 45, 681–687 CrossRef CAS.
- H. K. Murnen, A. M. Rosales, A. V. Dobrynin, R. N. Zuckermann and R. A. Segalman, Soft Matter, 2013, 9, 90–98 RSC.
- J. Weiss, A. Li, E. Wischerhoff and A. Laschewsky, Polym. Chem., 2012, 3, 352–361 RSC.
- A. M. Rosales, B. L. McCulloch, R. N. Zuckermann and R. A. Segalman, Macromolecules, 2012, 45, 6027–6035 CrossRef CAS.
- J. Sun and R. N. Zuckermann, ACS Nano, 2013, 7, 4715–4732 CrossRef CAS.
- C. Lavilla, G. Yilmaz, V. Uzunova, R. Napier, C. R. Becer and A. Heise, Biomacromolecules, 2017, 18, 1928–1936 CrossRef CAS PubMed.
- R. N. Zuckermann, J. M. Kerr, S. B. H. Kent and W. H. Moos, J. Am. Chem. Soc., 1992, 114, 10646–10647 CrossRef CAS.
- G. Liu and L. Jia, Angew. Chem., Int. Ed., 2006, 45, 129–131 CrossRef CAS.
- Y. Utku, E. Dehan, O. Ouerfelli, F. Piano, R. N. Zuckermann, M. Pagano and K. Kirshenbaum, Mol. BioSyst., 2006, 2, 312–317 RSC.
- M. M. Reddy, R. Wilson, J. Wilson, S. Connell, A. Gocke, L. Hynan, D. German and T. Kodadek, Cell, 2011, 144, 132–142 CrossRef CAS.
- S. L. Seurynck, J. A. Patch and A. E. Barron, Chem. Biol., 2005, 12, 77–88 CrossRef CAS.
- N. P. Chongsiriwatana, J. A. Patch, A. M. Czyzewski, M. T. Dohm, A. Ivankin, D. Gidalevitz, R. N. Zuckermann and A. E. Barron, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2794 CrossRef CAS PubMed.
- X. Zhang, S. Wang, J. Liu, Z. Xie, S. Luan, C. Xiao, Y. Tao and X. Wang, ACS Macro Lett., 2016, 5, 1049–1054 CrossRef CAS.
- A. S. Knight, E. Y. Zhou and R. N. Zuckermann, Adv. Mater, 2015, 27, 5664–5664 CrossRef.
- T. S. Burkoth, A. T. Fafarman, D. H. Charych, M. D. Connolly and R. N. Zuckermann, J. Am. Chem. Soc., 2003, 125, 8841–8845 CrossRef CAS PubMed.
- X. Tao, B. Zheng, T. Bai, M.-H. Li and J. Ling, Macromolecules, 2018, 51, 4494–4501 CrossRef CAS.
- R. Potyrailo, K. Rajan, K. Stoewe, I. Takeuchi, B. Chisholm and H. Lam, ACS Comb. Sci., 2011, 13, 579–633 CrossRef CAS.
- A. S. Culf and R. J. Ouellette, Molecules, 2010, 15, 5282–5335 CrossRef CAS PubMed.
- A. Al Samad, J. De Winter, P. Gerbaux, C. Jérôme and A. Debuigne, Chem. Commun., 2017, 53, 12240–12243 RSC.
- G. N. Kaluđerović, M. Abbas, H. C. Kautz, M. A. M. Wadaan, C. Lennicke, B. Seliger and L. A. Wessjohann, Chem. Commun., 2017, 53, 3777–3780 RSC.
- A. D. S. Barreto, V. A. dos Santos and C. K. Z. Andrade, Beilstein J. Org. Chem., 2016, 12, 2865–2872 CrossRef CAS.
- D. Previdi, S. Rodrigues, M. G. Coelho, A. Candido, L. G. Magalhaes and P. M. Donate, J. Braz. Chem. Soc., 2019, 30, 1334–1340 CAS.
- E. Ghasemi, A. S. Shahvelayati and I. Yavari, Phosphorus, Sulfur Silicon Relat. Elem., 2016, 191, 746–750 CrossRef CAS.
- I. Ugi and C. Steinbruckner, Angew. Chem., Int. Ed. Engl., 1960, 72, 267–268 CrossRef CAS.
- H. Bu, A.-L. Kjøniksen, K. D. Knudsen and B. Nyström, Biomacromolecules, 2004, 5, 1470–1479 CrossRef CAS PubMed.
- M. Abbas, J. Bethke and L. A. Wessjohann, Chem. Commun., 2006, 541–543 RSC.
- Y.-Z. Wang, X.-X. Deng, L. Li, Z.-L. Li, F.-S. Du and Z.-C. Li, Polym. Chem., 2013, 4, 444–448 RSC.
- A. Sehlinger, P.-K. Dannecker, O. Kreye and M. A. R. Meier, Macromolecules, 2014, 47, 2774–2783 CrossRef CAS.
- S. Wang, Y. Tao, J. Wang, Y. Tao and X. Wang, Chem. Sci., 2019, 10, 1531–1538 RSC.
- Y. Tao, S. Wang, X. Zhang, Z. Wang, Y. Tao and X. Wang, Biomacromolecules, 2018, 19, 936–942 CrossRef CAS.
- K. T. Nam, S. A. Shelby, P. H. Choi, A. B. Marciel, R. Chen, L. Tan, T. K. Chu, R. A. Mesch, B. C. Lee, M. D. Connolly, C. Kisielowski and R. N. Zuckermann, Nat. Mater., 2010, 9, 454–460 CrossRef CAS PubMed.
- R. D. Jahnsen, N. Frimodt-Møller and H. Franzyk, J. Med. Chem., 2012, 55, 7253–7261 CrossRef CAS PubMed.
- R. D. Jahnsen, A. Sandberg-Schaal, K. J. Vissing, H. M. Nielsen, N. Frimodt-Møller and H. Franzyk, J. Med. Chem., 2014, 57, 2864–2873 CrossRef CAS.
- P. Stiernet, B. Couturaud, V. Bertrand, G. Eppe, J. De Winter and A. Debuigne, Polym. Chem., 2021, 12, 2141–2151 RSC.
- S. C. Solleder, K. S. Wetzel and M. A. R. Meier, Polym. Chem., 2015, 6, 3201–3204 RSC.
- M. Hartweg, C. J. C. Edwards-Gayle, E. Radvar, D. Collis, M. Reza, M. Kaupp, J. Steinkoenig, J. Ruokolainen, R. Rambo, C. Barner-Kowollik, I. W. Hamley, H. S. Azevedo and C. R. Becer, Polym. Chem., 2018, 9, 482–489 RSC.
- A. M. Rosales, H. K. Murnen, R. N. Zuckermann and R. A. Segalman, Macromolecules, 2010, 43, 5627–5636 CrossRef CAS.
- C. Lambruschini, I. Demori, Z. El Rashed, L. Rovegno, E. Canessa, K. Cortese, E. Grasselli and L. Moni, Molecules, 2021, 26, 89–103 CrossRef CAS.
- A. F. de la Torre, A. Ali, F. Z. Galetto, A. L. Braga, J. A. C. Delgado and M. W. Paixão, Mol. Diversity, 2020, 24, 1–10 CrossRef CAS.
- J. Zhang, P. Yu, S. Y. Li, H. Sun, S. H. Xiang, J. J. Wang, K. N. Houk and B. Tan, Science, 2018, 361, 1087–1095 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |