
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSequence-defined L-glutamamide oligomers with pendant supramolecular motifs via iterative synthesis and orthogonal post-functionalization†
Marcin L.
Ślęczkowski
 ab,
Ian
Segers
b,
Yiliu
Liu
*abc and
Anja R. A.
Palmans
*ab
ab,
Ian
Segers
b,
Yiliu
Liu
*abc and
Anja R. A.
Palmans
*ab
aInstitute for Complex Molecular Systems, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: liuyiliu@scut.edu.cn; a.palmans@tue.nl
bLaboratory of Macromolecular and Organic Chemistry, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands
cSouth China Advanced Institute for Soft Matter Science and Technology, South China University of Technology, 510640, Guangzhou, P. R. China
First published on 2nd November 2020
Abstract
One of the great challenges in polymer chemistry is to achieve discrete and sequence-defined synthetic polymers that fold in defined conformations and form well-defined three-dimensional structures. Here, we present our progress to arrive at functional, sequence-defined and discrete oligomers by selecting a synthetic approach based on oligo-L-glutamamides. We introduce a solution-based iterative approach and start with an orthogonally protected L-glutamic acid derivative with an alkene or alkyne side chain. Using subsequent deprotection, activation and coupling steps, discrete, sequence-defined octamers were prepared with alkene or alkyne pendants at defined positions. Benzene-1,3,5-tricarboxamide (BTA) was selected as the supramolecular motif and decyl side chains for enhancing solubility. Full functionalization of the octamer was achieved with subsequent Cu(I)-catalyzed alkyne–azide and thiol–ene “click” chemistry, affording octamers with two BTA motifs at pre-defined positions. The octamers show a strong propensity to form hydrogen bonds in bulk as evidenced by infrared spectroscopy. In dilute solution, intermolecular aggregation of the amides present in the backbone and/or BTAs is observed. Our work shows that the iterative method serves as an reliable strategy for the synthesis of functionalized sequence-defined, discrete oligomers comprising side chain supramolecular motifs, although further optimizations are required.
Introduction
Technological applications of synthetic macromolecules have driven the field of polymer chemistry to a point in which seemingly every material can be synthesized.1 Nowadays, the great variety in available polymerization methods combines a plethora of topologies with an immense choice of monomers, permitting unprecedented control over the functionalization density and molecular mass distribution of macromolecules.2–4 Yet, Nature is able to build hundreds of different types of functional proteins from only 20 different monomers.5 The secret of such diverse functionality of biomacromolecules lies in the precise control of the position of each monomer in the polymeric chain – sequence control.6 In search of developing protein-like synthetic systems,7 polymer chemists have recently been particularly interested in the development of methodologies that allow controlling both the sequence as well as the molar mass.8,9 A recent excellent review on the progress made can be found in literature.10Currently, two independent methodologies are applied to arrive at discrete, sequence-defined oligomers: solid-phase and liquid-phase. Solid-phase synthesis (SPS) was pioneered by Merrifield for peptide synthesis.11,12 This versatile approach enables the one by one build-up of oligomers by attaching monomers to the growing chain by using iterative chemical steps in a sequence-defined way. Owing to the versatility of conditions such as the used support,13 the concept of SPS evolved towards synthetic oligomers including nucleotides,14,15 peptoids,16,17 phosphodiesters,18 amides,19 triazines,20 and thiolactones.21,22 However, ultimate control of the reaction kinetics23 as well as near quantitative yields24 are required during the SPS to make the protocol applicable. As a result, most of the fixed solid-phase synthetic protocols approach their limits in terms of scalability and possibilities to incorporate more complex structures into the sequence-defined backbone chain.
The second method is based on a solution iterative (exponential or direct) growth strategy and originates from the synthesis of dendrimers.25–27 This strategy has been successfully applied to prepare a manifold of discrete, linear oligomers.10,28 While the direct growth strategy enables to attach a monomer or a short chain depending on the building blocks,![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 29–32 in the iterative exponential strategy each coupling step doubles the length of growing chain.28 The iterative exponential growth strategy, also known as the orthogonal iterative divergent/convergent approach, makes use of bifunctional monomers with orthogonally protected end-groups. The end-groups can be separately deprotected (the divergent part) and subsequently coupled with each other (the convergent part) giving an orthogonally protected dimer. The same steps can be repeated with the obtained dimer, resulting in an exponential growth of the chain. However, the required orthogonality of the protecting end-groups reduces the diversity of applicable monomers and requires chemical conversions that are compatible with each other. Nonetheless, elegant approaches show that discrete oligomers can be built up using a variety of linkages (triazoles,33–35 esters,36–41 or amides42–45) and affording high degrees of polymerizations.
29–32 in the iterative exponential strategy each coupling step doubles the length of growing chain.28 The iterative exponential growth strategy, also known as the orthogonal iterative divergent/convergent approach, makes use of bifunctional monomers with orthogonally protected end-groups. The end-groups can be separately deprotected (the divergent part) and subsequently coupled with each other (the convergent part) giving an orthogonally protected dimer. The same steps can be repeated with the obtained dimer, resulting in an exponential growth of the chain. However, the required orthogonality of the protecting end-groups reduces the diversity of applicable monomers and requires chemical conversions that are compatible with each other. Nonetheless, elegant approaches show that discrete oligomers can be built up using a variety of linkages (triazoles,33–35 esters,36–41 or amides42–45) and affording high degrees of polymerizations.
This work explores the scope and limitations of a solution-based approach to sequence-defined oligomers with pendant H-bonding motifs at predefined positions. We select an L-γ-glutamamide backbone as the scaffold holding propargylamide and trisallyloxyamide units (Scheme 1), and synthesize octamers with different sequences of monomers along the backbone with an iterative exponential growth strategy. We further show successful orthogonal post-functionalization via subsequent copper(I) catalyzed azide–alkyne cycloaddition (CuAAC) and radical thiol–ene addition. As supramolecular H-bonding motif, we select the benzene-1,3,5-tricarboxamide (BTA) unit. BTAs form one-dimensional, helical H-bonded aggregates46 and were used previously to drive the assembly of macromolecules into (single-chain) polymeric nanoparticles.47 BTAs are considered as a functional motif because of their ability to induce higher order structures, making them interesting as a test case to induce functionality into sequence defined oligomers. Ultimately, this will allow us to asses in how far properties of the oligomers are affected by the exact position of the functional side chain. The structures of all oligomers were analyzed by nuclear magnetic resonance (NMR) and mass (MS) spectroscopy, and their behavior was characterized by infrared (IR) spectroscopy in the solid state and circular dichroism (CD) spectroscopy in dilute solution.
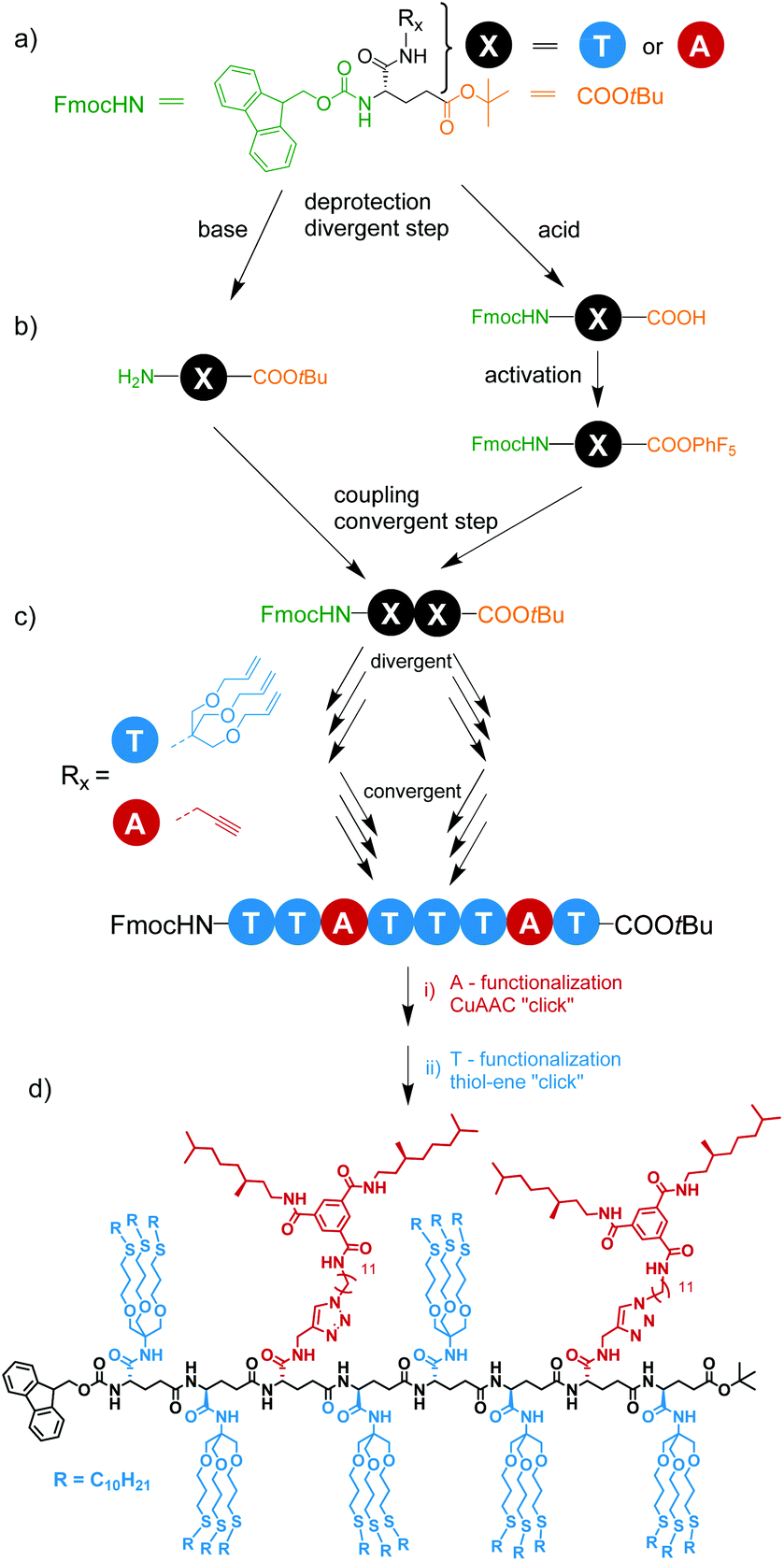 | ||
| Scheme 1 (a) Monomer design for the synthesis of L-γ-glutamamide-based octamer; (b) deprotection and amide coupling steps for the synthesis of a dimer; (c) schematic representation of the synthesis of the octamer; (d) post-functionalization of the octamer via CuAAC and thiol–ene “click” chemistry. | ||
Results and discussion
Monomer design and synthetic approach
Our selected iterative synthesis strategy of the sequence-defined octamer is shown in Scheme 1 and requires a monomer with a side chain, and two different types of end-groups. After careful evaluation, L-glutamic acid, comprising two acid functions and one amine function, was identified as a suitable monomer that fulfilled all requirements (Scheme 1a). L-Glutamic acid is commercially available with the amine group protected with fluorenylmethyloxycarbonyl (Fmoc) and the γ-carboxylic acid protected with tert-butoxy (tBuO). Whereas the tBuO is acid-labile, but base-stable, the Fmoc group is base-labile, but acid-stable (Scheme 1b). Free amine and acid groups can be readily coupled with the use of an acid activating agents to create an amide bond (Scheme 1c), and by repetitive deprotection and coupling steps the oligomers can be built up. Finally, orthogonal post-functionalization of the final oligomer backbone can be achieved via the α-carboxylic acid, modified with a functionalized group that has to react in high yields at the final steps of the synthesis (post-functionalization strategy). Thus, terminal alkyne and alkene bonds were selected as these are stable under the deprotection conditions and during amide formation but can be independently reacted using sequential CuAAC and radical thiol–ene addition (Scheme 1d). To introduce alkene and alkyne functionalities, we selected propargylamine and tris[(allyloxy)methyl]aminomethane (Scheme 2). The latter was selected to ensure sufficient solubility of the monomer and subsequent oligomers.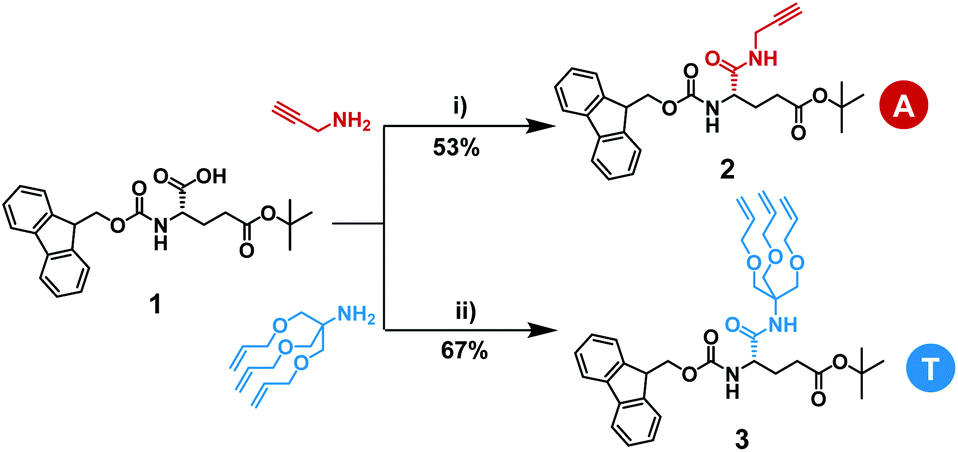 | ||
| Scheme 2 Synthesis of monomers 2 and 3. Reaction conditions: (i) propargylamine, EDC·HCl/HOBT·xH2O, DIPEA, THF, overnight at room temperature, 53%; (ii) tris[(allyloxy)methyl]aminomethane, EDC·HCl/HOBT·xH2O, DIPEA, THF, overnight at room temperature, 67%. | ||
Monomer synthesis
The synthesis of the two starting monomers with either alkene or alkyne pendants is shown in Scheme 2. The 1,2,3-tris(allyloxy) group was synthesized in 3 steps starting from 2-amino-2-(hydroxymethyl)-1,3-propanediol following a route published before.48,49 Commercially available N-protected and αCOOH-protected γ-glutamic acid 1 was modified with either commercially available propargylamine or tris[(allyloxy)methyl]aminomethane to yield monomers 2 and 3, respectively. The activation of the α-COOH in Fmoc-Glu(OtBu)-OH was achieved by N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC·HCl) and 1-hydroxybenzotriazole hydrate (HOBt·xH2O). The activated acid readily reacted with either propargylamine or tris[(allyloxy)methyl]aminomethane giving propargylamide (2) or trisallyloxy-tert-butylamide (3) as a side groups on the protected γ-glutamic acid. Partial undesired deprotection of Fmoc-protecting group by N,N-diisopropylethylamine (DIPEA) was observed, leading to a decrease in the yield. Monomers 2 and 3 were purified via column chromatography and isolated in yields of 53% and 67%, respectively. Since we did not isolate unprotected/activated intermediates, the yields of deprotection/activation steps have not been determined. Throughout this work the monomer or residue with the alkyne containing side group will be defined as an A, while the monomer or residue with the trisallyloxy-tert-butylamide side group will be defined as a T.Synthesis of L-γ-glutamamide-based octamers
In the first iterative step we synthesized dimers. Hereto, monomers 2 and 3 were selectively deprotected by removing either the Fmoc-protecting group or the tBuO protecting group. Monomers 2 and 3 were treated with TFA to remove the tBuO-protecting group, giving free carboxylic acids (4a and 6a). Next, the addition of pentafluorophenyl trifluoroacetate to these two intermediates in the presence of DIPEA activated the acids to pentafluorophenyl esters 4b and 6b. In parallel to this, monomer 3 was treated with diethylamine to remove the Fmoc-protecting group, affording monomer 5 with a terminal free amine. The isolation of free amine monomer 5 (81%) was accomplished using column chromatography. Intermediate compounds 4b and 5, and compounds 5 and 6b reacted readily with each other in DMF giving dimers FmocHN-AT-COOtBu (7) and a FmocHN-TT-COOtBu (8) (Scheme 3). Dimers 7 (80%) and 8 (74%) were purified by column chromatography and their structures were confirmed by 1H NMR, 13C NMR, and Maldi-ToF MS (Fig. S1–6, ESI†). Subsequently, dimers 7 and 8 were deprotected and activated to yield 9 and 10b (Scheme 4a). Subsequent coupling in DMF, in a similar fashion as was done to obtain the dimers, afforded tetramer FmocHN-TTAT-COOtBu (11) in a yield of 60% after column chromatography.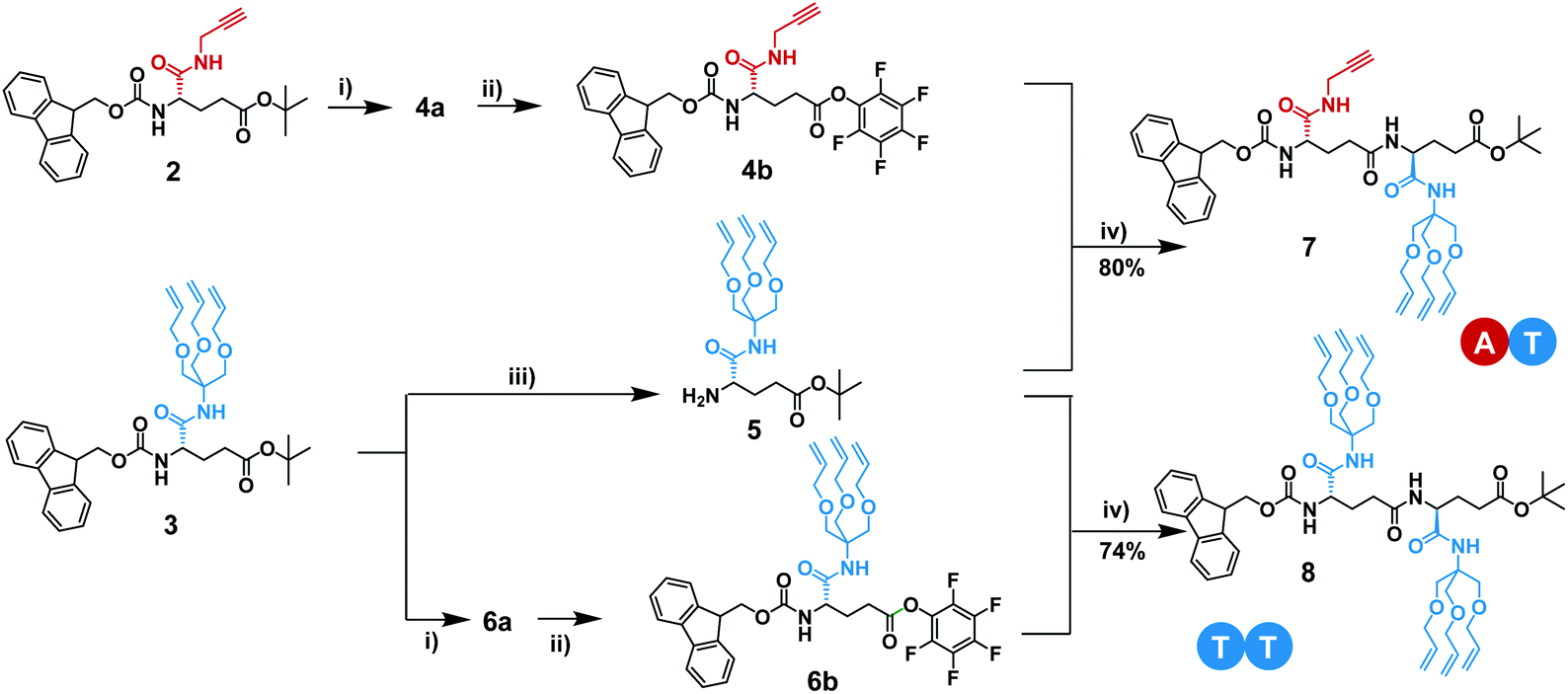 | ||
| Scheme 3 Synthesis of dimers 7 and 8. Reaction conditions: (i) TFA, DCM, 3 h at room temperature; (ii) pentafluorophenyl trifluoroacetate, DIPEA, CHCl3, 1–3 h at room temperature; (iii) diethylamine, DCM, 3 h at room temperature, 81%; (iv) DMF, 0.5–1.5 h at room temperature, 80% (for 7) and 74% (for 8). | ||
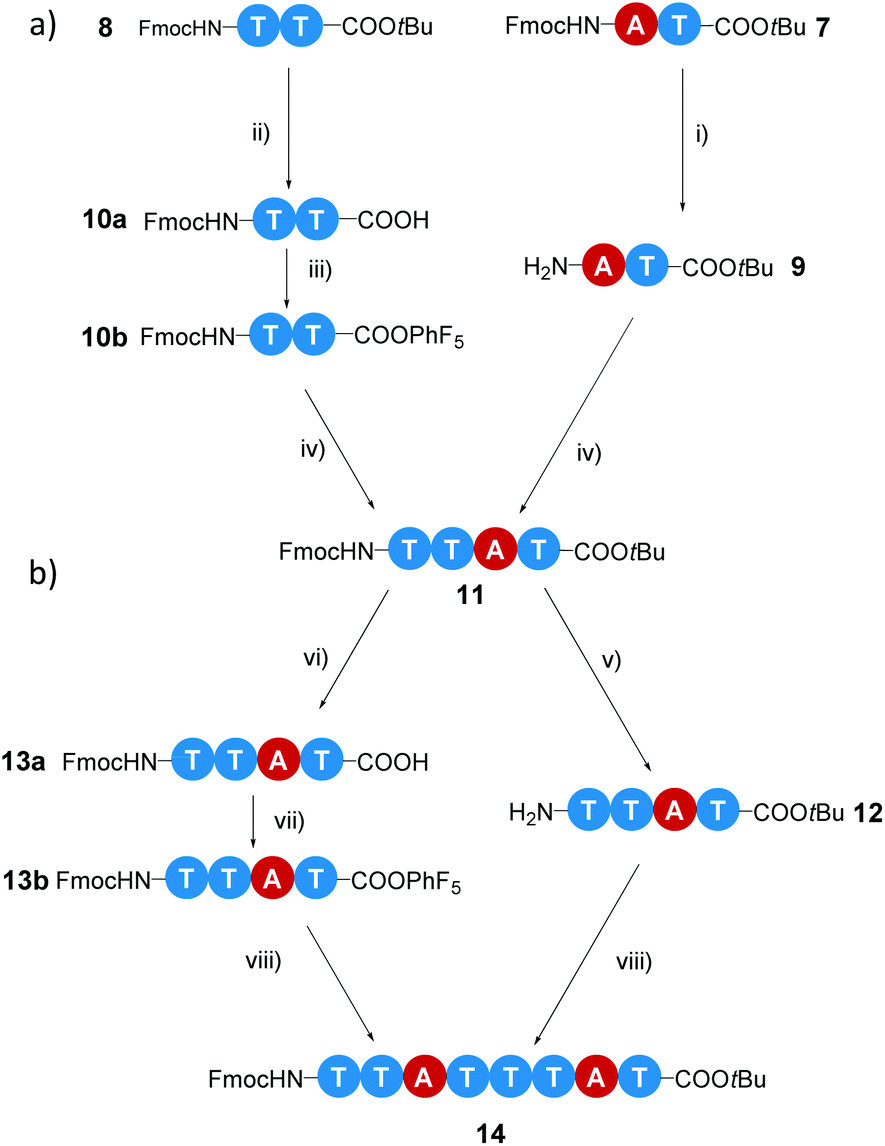 | ||
| Scheme 4 Schematic representation for the synthesis of (a) tetramer 11 and (b) octamer 14. Reaction conditions: (i) diethylamine, DCM, 3 h at room temperature; (ii) TFA, DCM, overnight at room temperature; (iii) pentafluorophenyl trifluoroacetate, DIPEA, CHCl3, 1 h at room temperature; (iv) DMF, 1 h at room temperature, 60%; (v) piperidine, CHCl3, 1 h at room temperature; (vi) TFA, CHCl3, overnight at room temperature; (vii) pentafluorophenyl trifluoroacetate, DIPEA, CHCl3, 2 h at room temperature; (viii) DMF, 1 h at room temperature, 39%. | ||
Further, deprotected tetramers 12 and 13a were obtained upon treatment of the tetramer 11 with either piperidine or TFA. Free acid 13a was activated with pentafluorophenyl trifluoroacetate to yield 13b which was subsequently coupled with 12 to yield octamer FmocHN-TTATTTAT-COOtBu (14) (Scheme 4b). The moderate yield of the final coupling (39%) was caused by the decreasing solubility of the tetramers. Octamer 14 was isolated by column chromatography and its structure was confirmed by 1H NMR, 13C NMR, and Maldi-ToF MS (Fig. 1, and Fig. S7, ESI†). The Maldi-ToF MS spectrum in Fig. 1a shows one peak at m/z 2765.5, consistent with the Na+ adduct of the octamer. Together with the observation that Maldi-ToF MS shows a similar sensitivity towards (partially) deprotected octamers (vide infra for octamer 15) and tetramers (see ESI†), this strongly points to the absence of lower oligomers and the presence of both protecting groups. The 1H NMR spectrum in DMSO-d6 (Fig. 1b) shows proton signals that are relatively sharp, which can all be assigned.
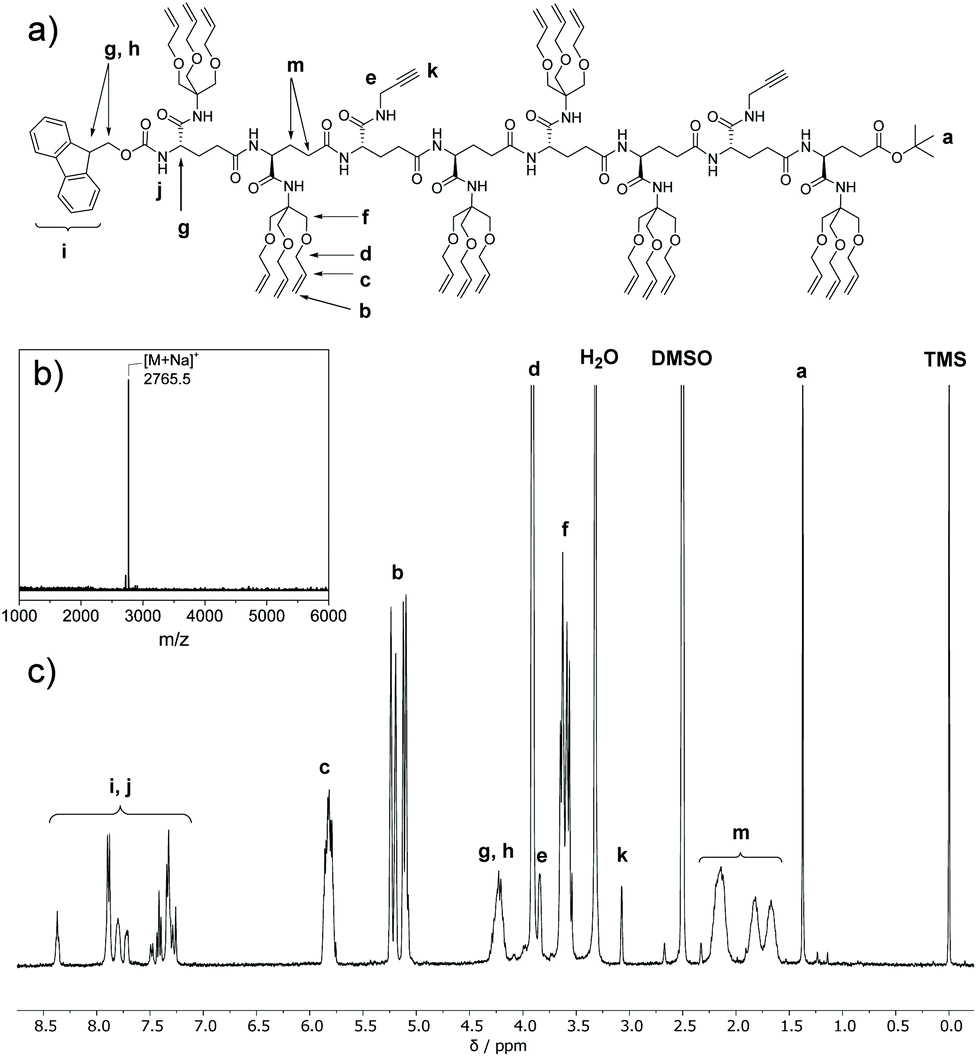 | ||
| Fig. 1 (a) Chemical structure of 14; (b) Maldi-ToF MS and; (c) 1H NMR spectrum (400 MHz, DMSO-d6) of octamer 14 (FmocHN-TTATTTAT-COOtBu). | ||
The route we selected, readily permits to make octamers of different sequence by judicious choice in deprotecting the tetramers, followed by acid activation and the amide coupling reaction. In a similar fashion as FmocHN-TTATTTAT-COOtBu (14) was prepared, two octamers having different sequences (FmocHN-TATTTATT-COOtBu (15) and Fmoc-HN-TTATTATT-COOtBu (16)) were prepared using the same synthetic procedures (Scheme S1, ESI†). This approach shows that by small changes in the coupling sequence, different topologies are easily accessible.
During the synthesis of all three octamers, we observed a significant drop in the yield during the last amide coupling reaction. The decrease in solubility already arises during the activation of the carboxylic acid group in the tetramers to the pentafluorophenyl ester. Increasing the chain length results in a further decrease in solubility, possibly as a result of the increasing number of backbone amide groups that may enhance the propensity to form hydrogen bonds and hereby increases the overall backbone rigidity. In fact, semi-crystalline polyamides such as Nylon-6,6 are hardly soluble in most of the organic solvents, also in polar aprotic solvents like DMF. The fact that the carboxylic acid group can also participate in the intermolecular hydrogen bonds potentially decreases its nucleophilic character and suppresses the activation to the pentafluorophenyl ester. Also, the solubility of the terminal free amine tetramers in chlorinated solvents and in DMF decreased. This, in combination with the use of not fully purified intermediates, resulted in incomplete conversion and the appearance of byproducts (for example FmocHN-TTAT-CO-N-piperidinyl) during the amide coupling reactions. Besides the appearance of side products, mass analysis showed the presence of unreacted substrates in the crude mixture of the amide couplings. Extensive column chromatography was required to purify the octamers. Maldi-ToF MS showed that the terminal amine group was partially deprotected in octamer 15, presumably due to the presence of residual amounts of base in the terminal free amine tetramer intermediate (12). In contrast, octamers 14 and 16 were isolated in a fully protected form.
All three octamers were obtained in moderate yields (39% and 37% for 14 and 16, respectively), but in sufficient quantities for further synthesis. The overall yield of the octamers starting from monomers A and T vary between 12% and 15%. All octamers are sparingly soluble in chlorinated solvents, such as CHCl3, and the use of polar solvents such as DMF was needed for solubilization.
Towards a sequence-defined L-γ-glutamamide hexadecamer
To assess the limits of the synthesis of oligomers based on L-γ-glutamamide, we applied the same strategy as previously, and deprotected orthogonally the octamer 14. The low solubility of the octamer hampered analysis of the reaction, making it difficult to follow the progress of the deprotection reactions. Therefore, we were not able to assess the conversion and yield of deprotected amine and acid and neither the formation of the activated ester. After stirring the activated ester and the amine, we only observed the formation of trace amounts of hexadecamer. To increase conversion, we evaluated the direct coupling of the acid and the amine using a TBTU-coupling. This method yielded a visible amount of hexadecamer by Maldi-ToF MS, but we could not find any reliable method to separate it from unreacted octamers. Thus, the formation of the hexadecamer is challenging, mainly as a result of solubility issues with the octamer precursors. Accordingly, we focused on the post-functionalization of the octamers.Post-functionalization of L-γ-glutamamide octamers
For post-functionalization, we selected an azide-functional BTA 20 and n-decanethiol for the CuAAC and thiol–ene click, respectively. To optimize the conditions for the post-functionalization of the synthesized glutamic acid-based octamers with the BTA-azide and alkyl pendant arms, we first carried out test reactions with the monomers (Scheme 5). BTA-azide 20 was synthesized from previously prepared BTA holding two (S)-3,7-dimethyloctyl and one 10-undecene arms (17),50 followed by hydroboration to the BTA-alcohol (18). The BTA alcohol was subsequently converted to the azide following a previously published protocol via tosylation to 19 and azidation (Scheme S2, ESI†).51 The coupling of the protected propargyl amide-functionalized monomer 2 with (S)-BTA-azide 20via CuAAC reaction afforded desired functionalized monomer 21 in 34% yield after purification. To achieve satisfactory yields in the post-functionalization of the octamer 14, the reaction conditions had to be further optimized (vide infra). Similarly, the tris(allyloxy) functionalized model compound and a 3-fold excess of 1-decanethiol was used in a radical thiol–ene test reaction. To validate our strategy, we monitored the conversion of the model compound 3 by 1H NMR spectroscopy (Fig. S8, ESI†). After 4 hours, no signals at 5.86 ppm and 5.18 ppm were observed, which belong to terminal olefin (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H). Concomitantly, new signals at 2.50 ppm appeared that belong to newly formed thioether (S–C–H). The functionalized tris-olefin 22 was isolated in a yield of 51%.
C–H). Concomitantly, new signals at 2.50 ppm appeared that belong to newly formed thioether (S–C–H). The functionalized tris-olefin 22 was isolated in a yield of 51%.
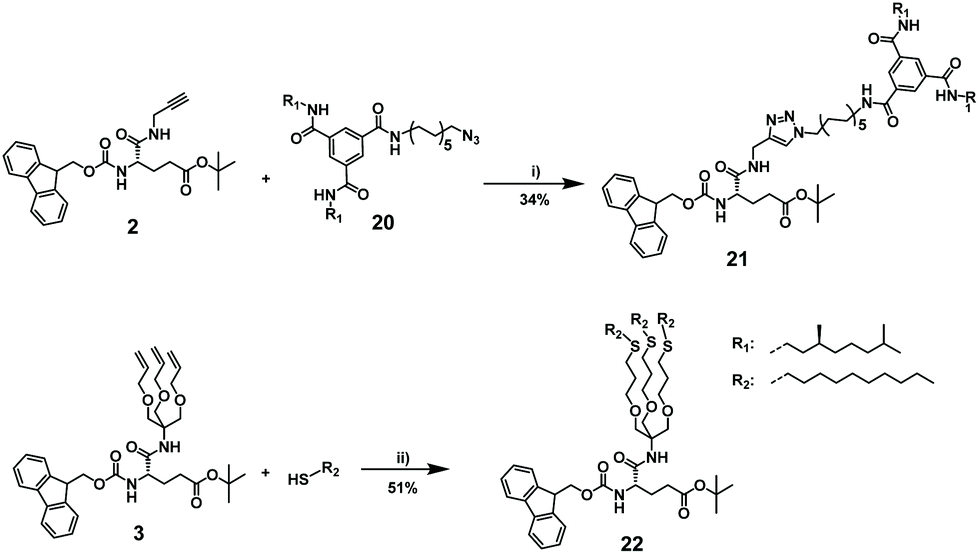 | ||
| Scheme 5 Functionalization of the monomer 2 and model tris-olefin compound via CuAAC and radical thiol–ene addition. Reagents and conditions: (i) CuSO4, sodium ascorbate, DMSO, 50 °C, argon atmosphere, 2 days at room temperature, 34% (ii) DMPA, THF, hν, argon, 4 h at room temperature, 51%. | ||
The protocol of the test reaction was then applied to the functionalization of octamer 14. In this case the reaction sequence is important, because the free alkyne is also susceptible to free radical attack. Therefore, the post-functionalization with the BTA via CuAAC click was performed first (Scheme 6a). We coupled the octamer FmocHN-TTATTTAT-COOtBu (14) with the (S)-BTA-azide (20) according to a slightly modified protocol due to low solubility of octamer 14. To avoid the problems we experienced with functionalization of model monomer, we conducted the reaction at larger scale and made sure that copper pre-catalyst was solubilized in DMSO/water, before the sodium ascorbate was added. Subsequently, the reaction was carried out for 2 days at a temperature of 50 °C, to assure good solubility. After 2 days, Maldi-ToF MS showed the presence of mono- and disubstituted octamer. Integral analysis of the 1H NMR spectrum indicated the presence of 25% unreacted terminal alkynes. Addition of another equivalent of 20 followed by stirring at 50 °C overnight afforded full functionalization. The (S)-BTA-functionalized octamer (23) was isolated via column chromatography in a 37% yield with respect to the starting octamer 14. Subsequently, 23 was functionalized with the alkyl chains via radical thiol–ene reaction with use of 2,2-dimethoxy-2-phenylacetophenone as a photoinitiator following a similar procedure as for the radical thiol–ene reaction performed on 3 (Scheme 6b). Fully functionalized octamer 24 was isolated in 31% yield with use of column chromatography and characterized by 1H NMR and Maldi-ToF MS (Fig. S9 and 10, ESI†). In this case, the Maldi-ToF MS spectrum showed a poor intensity of the molecular ion signal at m/z = 7269.65 ([M + Na+]) and 7285.39 ([M + K+]), which is likely related to the high molecular weight of 24. In addition, additional peaks were present at m/z = 7222.68 and 7301.60, which are caused by high molecular weight impurities of unknown origin.
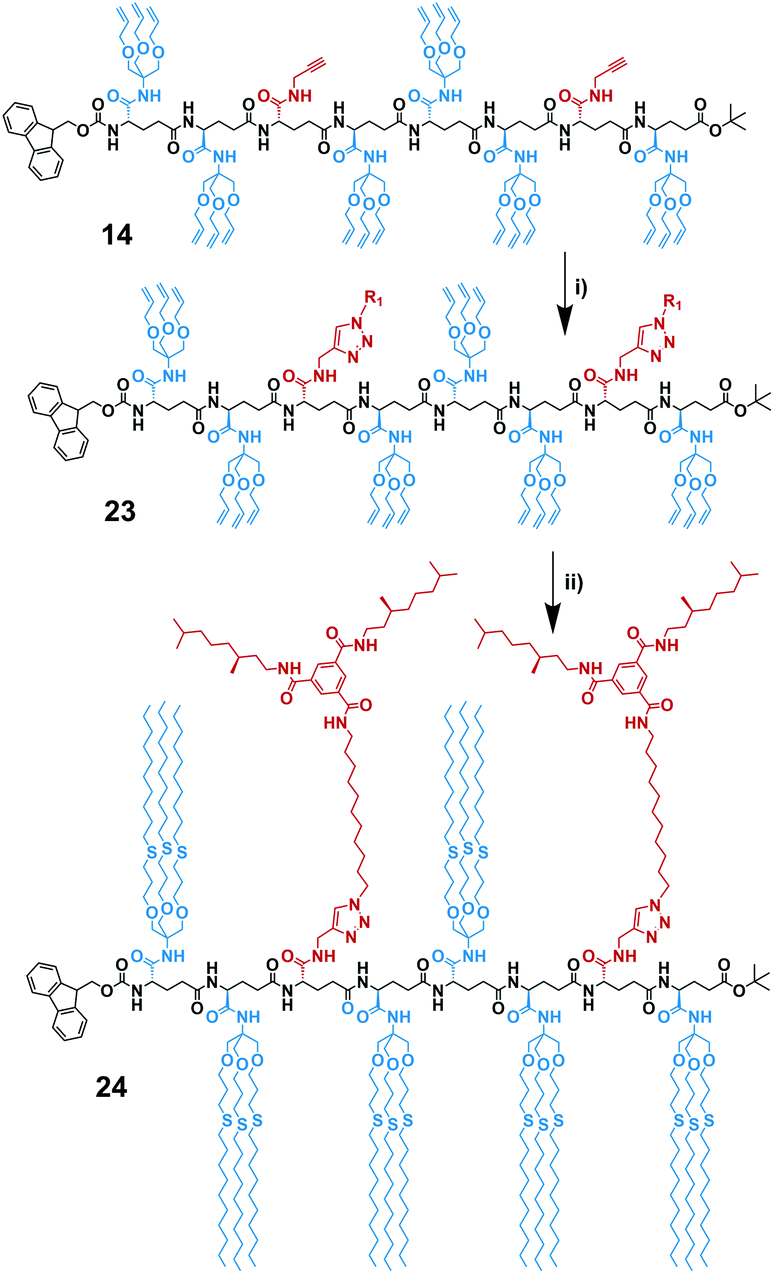 | ||
| Scheme 6 Synthesis of bis((S)-BTA)-hexa(tris(decylallyloxy))Fmoc-(TTAT2)-COOtBu (24). Reagents and conditions: (i) (1) BTA-azide 20, CuSO4, sodium ascorbate, DMSO, 50 °C → room temperature, argon atmosphere, overnight at room temperature; (2) CuSO4, sodium ascorbate, DMSO, 50 °C → RT, argon flow, 2 days at room temperature, 37%; (ii) 1-decanethiol, DMPA, DMA, hν, argon, 4 h at room temperature, 31%. | ||
Self-assembly of the L-γ-glutamamide oligomers in solid state and in solution
The poor solubility of the octamers before functionalization suggested that the growing backbone favors formation of intermolecular hydrogen bonds that cause aggregation. Infrared (IR) spectroscopy in solid state showed absorption bands at 3275 cm−1 (νN–H), 1729 cm−1, 1691 cm−1, 1661 cm−1 (amide I) and 1533 cm−1 (amide II) for tetramer 11 and octamer 14 (Fig. 2a). The position of these bands suggests the presence of hydrogen bonds with a similar pattern in both oligomers.51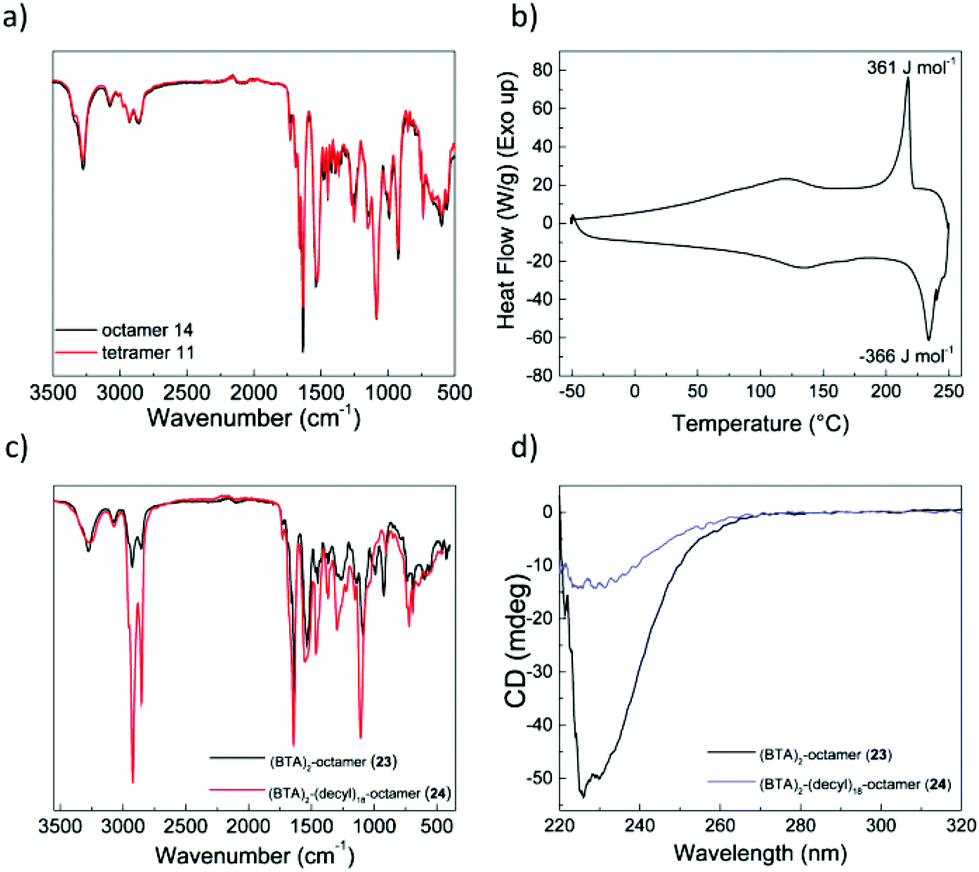 | ||
| Fig. 2 (a) IR spectra of tetramer 11 (red) and octamer 14 (black) in solid state; (b) DSC thermogram of tetramer 11 recorded at 10 °C min−1; (c) IR spectra of functionalized octamers 23 (back) and 24 (red) in the solid state; (d) CD spectra of octamers 23 (black, in DCE) and 24 (blue, in MCH), (c = 25 μM, T = 20 °C, l = 1 cm). | ||
The thermal behavior of the oligomers was studied by polarized optical microscopy (POM) and differential scanning calorimetry (DSC), and a reversible transition for tetramer 11 at 220 °C was observed with a low enthalpy of ΔH = 360 J mol−1 (Fig. 2b). This transition was accompanied by the disappearance of the texture under crossed polarizers, which suggests a melting transition (Fig. S11, ESI†). The low enthalpy of the transition suggests a low degree of crystallinity in the oligomer. None of the octamers showed any transitions in DSC nor birefringence in POM when using crossed polarizers. The combined results of IR, DSC and POM of octamer 14 suggest that very stable structures are formed in the solid state with clearing temperatures above 200 °C. However, most likely due to multiple possible conformations staying in thermodynamic equilibrium, there is no long-range order as suggested by POM.52 In conclusion, both tetramer 11 and octamer 14 showed the presence of hydrogen bonds, however the lack of long-range order suggests the presence of multiple competing supramolecular structures making a more detailed insight in the organization of the materials in the solid state challenging.
To assess, whether functionalization of the octamers with BTA and decyl chains reflects on the properties of the solid state materials, we recorded IR spectra of functionalized octamers 23 and 24. Interestingly, the pattern does not change substantially, indicating that neither of the attached units have a large influence on the supramolecular structure (Fig. 2c). H-bond stabilized BTA aggregates show characteristic absorption bands at 3226 cm−1 (νN–H), 1636 cm−1 (amide I) and 1548 cm−1 (amide II).53 Unfortunately, these absorption bands are at the same position as those arising from the backbone amides of the oligomers (Fig. 2a). Since the backbone amides have a higher concentration, the extent to which BTAs are engaged in hydrogen bonding can not be assessed. Similar to octamer 14, functionalized octamers 23 and 24 showed no birefringence and no transitions in DSC. Functionalization of the octamers did not result in a visible change of the solid state properties. We anticipated that functionalization should impact the solubility of the octamers, thus further characterization was performed in dilute solutions.
To study the behavior of the octamers in dilute solutions, we used circular dichroism (CD) spectroscopy. Due to the intrinsic low solubility in apolar solvents, CD spectra of octamer 23, functionalized with two BTAs, were recorded in 1,2-dichloroethane (DCE). On the contrary, octamer 24, functionalized with 2 BTAs and 18 decyl chains was soluble in apolar methyl cyclohexane (MCH). As a reference, we recorded also the CD spectrum of octamer 14, which shows a weak maximum around 255 nm followed by negative CD effect with an extremum around 228 nm (Fig. S12a, ESI†). The CD signal originates from presence of stereocenters with all S-configuration adjacent to the amide chromophore. The CD spectra of functionalized octamers 23 and 24 were measured after slow cooling (1 °C min−1) from 90 °C to 20 °C, and showed only one extremum at 230 nm, suggesting that the presence of BTAs leads to the formation of a different superstructure. A significantly stronger dichroic response of −50 mdeg was observed for the octamer 23 in DCE, a solvent where BTAs are normally molecularly dissolved. The aggregation of the BTAs in this case may be permitted by low solubility of the oligomeric backbone or is combined with the intermolecular aggregation of the backbone amides.54 On the other hand, octamer 24 shows hardly any CD signal (−15 mdeg) compared to the other 2 octamers, even though it was dissolved apolar MCH.
To further unravel the origin of the observed CD signal, we also measured variable-temperature CD (VT-CD) of the octamers (Fig. S12b, ESI†). We found that at high temperatures the CD signal is always present and around −10 mdeg. This signal most likely arises from the amide chromophores along the backbone and not from the BTA unit. Octamer 23 shows a clear onset of the CD increase around 50 °C, which can correspond to intermolecular BTA aggregation and/or aggregation of the amides of the oligomers. The temperature-independent signal of octamer 24 suggests that presumably no BTA aggregation is present in dilute solution. This can be a result of dense side-chain functionalization with the bulky decyl chains, which in turn effectively hampers BTA aggregation.
The results indicate that the aggregation of the BTAs in such complex systems is possible, but difficult to control when multiple binding sites compete with each other and the interactions of all the components with the solvent are not balanced. Therefore, we conclude that the combination of poorly soluble oligoamides in the backbone with a BTA motif is not an optimal design for controlled folding of sequence-defined macromolecules. Finally, the intrinsic absorption of the light of similar energy by multiple parts of the molecule impedes to unravel which chromophore contributes to which aggregation process.
Conclusions
We have demonstrated the successful synthesis of orthogonally protected L-γ-glutamamide octamers, containing alkyne- and alkene-based reactive side groups via a multi-step synthesis. Increasing the chain length resulted in decreasing solubility and reactivity, and a concomitant decrease in yield was observed. All in all, the solution-based strategy developed here allows to access sequence-defined oligomers of defined stereochemistry and with reactive handles at selected positions in overall yields of 12–15%. The reaction times of the deprotection, activation and coupling steps are relatively short. The most time-consuming step is column chromatography, and the optimization required for the purification steps. Finally, the synthetic procedures developed here are scalable to multigram scale. For example, the synthesis of the octamers afforded 1 g of end product.Orthogonal post-functionalization of one of the octamers was successfully achieved with bis((S)-3,7-dimethyloctyl)-(11-azidoundecyl)-BTA via a CuAAC reaction. Subsequently, full functionalization with solubilizing decyl chains was achieved using a radical thiol–ene addition. Furthermore, the herein synthesized oligomers formed secondary structures arising from hydrogen-bond formation in bulk as well as in dilute solutions. The initial characterization showed that despite the molecular complexity, BTA aggregation is possible in these molecules, however, improved molecular engineering is required to assess this system's particular assembly features.
Our results show that synthesis of sequence-defined oligomers holding supramolecular motifs at defined positions is possible. However, further optimization and reconsideration of the strategy and supramolecular units used is required for this approach to become applicable in the synthesis of functional, sequence-defined oligomers with high degrees of polymerization. Iterative approaches based on classical organic synthesis offer opportunities to access sequence-defined oligomers in few steps with satisfying yields and amounts. The post-functionalization strategy does not require a large excess of the reagents, in contrast to the solid-phase approach, and is therefore feasible to synthesize the oligomers in amounts permitting further modification and analysis. These advantages make that iterative solution-based approaches can offer an alternative to complement the frequently applied solid-phase strategies. While SPS method is difficult to outperform in the large scale synthesis of complex and long peptides, synthesis of non-natural oligomers in scales exceeding 1 gram remains a challenge.
The major drawback of the monomers selected here is the decreasing solubility of the backbone, which makes further elongation difficult and currently precludes isolation of the oligomers longer than the octamer in reasonable yields. To overcome the solubility issue, we will focus in the future on the pre-functionalization of the “T (olefin)” monomer with solubilizing side chains prior to the oligomer chain build-up. This should improve the solubility of the oligomers, hereby improving the reactivity. In addition, a supramolecular motif with the absorption maximum outside the absorption range of peptide bonds is also under consideration. We are currently following this strategy to synthesize longer oligomers.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors like to thank Prof. E.W. Meijer for continued support and the EUROSEQUENCES community for fruitful discussions about this project. This work was supported by the European Union's Horizon 2020 research and innovation program (Marie Skłodowska–Curie grant agreement No 642083). Y.L. acknowledges the support from the National Natural Science Foundation of China (21901077) and Guangdong Provincial Key Laboratory of Functional and Intelligent Hybrid Materials and Devices (No. 2019B121203003).References
- G. Polymeropoulos, G. Zapsas, K. Ntetsikas, P. Bilalis, Y. Gnanou and N. Hadjichristidis, Macromolecules, 2017, 50, 1253–1290 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- R. B. Grubbs and R. H. Grubbs, Macromolecules, 2017, 50, 6979–6997 CrossRef CAS.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- A. Saint-Léger, C. Bello, P. D. Dans, A. G. Torres, E. M. Novoa, N. Camacho, M. Orozco, F. A. Kondrashov and L. Ribas de Pouplana, Sci. Adv., 2016, 2, e1501860 CrossRef.
- T. N. Starr and J. W. Thornton, Nat. Biotechnol., 2017, 35, 125–126 CrossRef CAS.
- J. P. Cole, A. M. Hanlon, K. J. Rodriguez and E. B. Berda, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 191–206 CrossRef CAS.
- J.-F. Lutz, M. Ouchi, D. R. Liu and M. Sawamoto, Science, 2013, 341, 1238149–1238149 CrossRef.
- J.-F. Lutz, Macromol. Rapid Commun., 2017, 38, 1700582 CrossRef.
- S. C. Solleder, R. V. Schneider, K. S. Wetzel, A. C. Boukis and M. A. R. Meier, Macromol. Rapid Commun., 2017, 38, 1600711 CrossRef.
- B. Merrifield, Science, 1986, 232, 341–347 CrossRef CAS.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- L. Oswald, A. Al Ouahabi, C. Laure, J. Amalian, L. Charles and J. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 403–410 CrossRef CAS.
- T. G. W. Edwardson, K. M. M. Carneiro, C. J. Serpell and H. F. Sleiman, Angew. Chem., Int. Ed., 2014, 53, 4567–4571 CrossRef CAS.
- S. L. Beaucage and R. P. Iyer, Tetrahedron, 1992, 48, 2223–2311 CrossRef CAS.
- A. S. Knight, E. Y. Zhou, M. B. Francis and R. N. Zuckermann, Adv. Mater., 2015, 27, 5665–5691 CrossRef CAS.
- R. N. Zuckermann, J. M. Kerr, S. B. H. Kent and W. H. Moos, J. Am. Chem. Soc., 1992, 114, 10646–10647 CrossRef CAS.
- N. F. König, A. Al Ouahabi, S. Poyer, L. Charles and J.-F. Lutz, Angew. Chem., Int. Ed., 2017, 56, 7297–7301 CrossRef.
- Z. Wan, Y. Li, S. Bo, M. Gao, X. Wang, K. Zeng, X. Tao, X. Li, Z. Yang and Z. X. Jiang, Org. Biomol. Chem., 2016, 14, 7912–7919 RSC.
- J. W. Grate, K.-F. Mo and M. D. Daily, Angew. Chem., Int. Ed., 2016, 55, 3925–3930 CrossRef CAS.
- P. Espeel, L. L. G. Carrette, K. Bury, S. Capenberghs, J. C. Martins, F. E. Du Prez and A. Madder, Angew. Chem., Int. Ed., 2013, 52, 13261–13264 CrossRef CAS.
- S. Martens, J. Van den Begin, A. Madder, F. E. Du Prez and P. Espeel, J. Am. Chem. Soc., 2016, 138, 14182–14185 CrossRef CAS.
- P. Espeel and F. E. Du Prez, Eur. Polym. J., 2015, 62, 247–272 CrossRef CAS.
- J. M. Palomo, RSC Adv., 2014, 4, 32658–32672 RSC.
- S. M. Grayson and J. M. J. Fréchet, Chem. Rev., 2001, 101, 3819–3867 CrossRef CAS.
- C. J. Hawker and J. M. J. Frechet, J. Am. Chem. Soc., 1990, 112, 7638–7647 CrossRef CAS.
- D. A. Tomala, H. Baker, J. Dewald, M. Hall, G. Kallos, S. Martin, J. Roeck, J. Ryder and P. Smith, Polym. J., 1985, 17, 117–132 CrossRef.
- S. Binauld, D. Damiron, L. A. Connal, C. J. Hawker and E. Drockenmuller, Macromol. Rapid Commun., 2011, 32, 147–168 CrossRef CAS.
- N. Franz, G. Kreutzer and H.-A. Klok, Synlett, 2006, 1793–1815 CAS.
- S. C. Solleder, K. S. Wetzel and M. A. R. Meier, Polym. Chem., 2015, 6, 3201–3204 RSC.
- M. Porel and C. A. Alabi, J. Am. Chem. Soc., 2014, 136, 13162–13165 CrossRef CAS.
- S. C. Solleder and M. A. R. Meier, Angew. Chem., Int. Ed., 2014, 53, 711–714 CrossRef CAS.
- S. Binauld, C. J. Hawker, E. Fleury and E. Drockenmuller, Angew. Chem., Int. Ed., 2009, 48, 6654–6658 CrossRef CAS.
- J. C. Barnes, D. J. C. Ehrlich, A. X. Gao, F. A. Leibfarth, Y. Jiang, E. Zhou, T. F. Jamison and J. A. Johnson, Nat. Chem., 2015, 7, 810–815 CrossRef CAS.
- J. Li, M. Leclercq, M. Fossepré, M. Surin, K. Glinel, A. M. Jonas and A. E. Fernandes, Polym. Chem., 2020, 11, 4040–4046 RSC.
- M. B. Koo, S. W. Lee, J. M. Lee and K. T. Kim, J. Am. Chem. Soc., 2020, 142, 14028–14032 CrossRef CAS.
- N. Franz, L. Menin and H. A. Klok, Eur. J. Org. Chem., 2009, 5390–5405 CrossRef CAS.
- D. Seebach and M. G. Fritz, Int. J. Biol. Macromol., 1999, 25, 217–236 CrossRef CAS.
- K. Takizawa, C. Tang and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 1718–1726 CrossRef CAS.
- K. Takizawa, H. Nulwala, J. Hu, K. Yoshinaga and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5977–5990 CrossRef CAS.
- G. Brooke, N. Cameron, J. A. MacBride and M. Whiting, Polymer, 2002, 43, 1139–1154 CrossRef CAS.
- C. Bao, B. Kauffmann, Q. Gan, K. Srinivas, H. Jiang and I. Huc, Angew. Chem., Int. Ed., 2008, 47, 4153–4156 CrossRef CAS.
- S. S. Reddy, X. Dong, R. Murgasova, A. I. Gusev and D. M. Hercules, Macromolecules, 1999, 32, 1367–1374 CrossRef CAS.
- G. M. Brooke, S. Mohammed and M. C. Whiting, J. Chem. Soc., Perkin Trans. 1, 1997, 3371–3380 RSC.
- G. Brooke, J. A. Hugh MacBride, S. Mohammed and M. Whiting, Polymer, 2000, 41, 6457–6471 CrossRef CAS.
- S. Cantekin, T. F. A. de Greef and A. R. A. Palmans, Chem. Soc. Rev., 2012, 41, 6125 RSC.
- G. M. Huurne, A. R. A. ter Palmans and E. W. Meijer, CCS Chem., 2019, 64–82 Search PubMed.
- C. T. Seto, J. P. Mathias and G. M. Whitesides, J. Am. Chem. Soc., 1993, 115, 1321–1329 CrossRef CAS.
- M. Segura, F. Sansone, A. Casnati and R. Ungaro, Synthesis, 2001, 2105–2112 CrossRef CAS.
- M. L. Ślęczkowski, E. W. Meijer and A. R. A. Palmans, Macromol. Rapid Commun., 2017, 38, 1700566 CrossRef.
- A. Barth, Biochim. Biophys. Acta, Bioenerg., 2007, 1767, 1073–1101 CrossRef CAS.
- M. Kudo, V. Maurizot, H. Masu, A. Tanatani and I. Huc, Chem. Commun., 2014, 50, 10090–10093 RSC.
- P. J. M. Stals, M. M. J. Smulders, R. Martín-Rapún, A. R. A. Palmans and E. W. Meijer, Chem. – Eur. J., 2009, 15, 2071–2080 CrossRef CAS.
- T. Mes, R. van der Weegen, A. R. A. Palmans and E. W. Meijer, Angew. Chem., 2011, 123, 5191–5195 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, characterisation data and temperature-dependent CD measurements. See DOI: 10.1039/d0py01157f |
| This journal is © The Royal Society of Chemistry 2020 |