
Porous materials as carriers of gasotransmitters towards gas biology and therapeutic applications
Arnau
Carné-Sánchez
 ab,
Francisco J.
Carmona
a,
Chiwon
Kim
ac and
Shuhei
Furukawa
*ac
ab,
Francisco J.
Carmona
a,
Chiwon
Kim
ac and
Shuhei
Furukawa
*ac
aInstitute for Integrated Cell-Material Sciences (WPI-iCeMS), Kyoto University, Yoshida, Sakyo-ku, Kyoto, 606-8501, Japan. E-mail: shuhei.furukawa@icems.kyoto-u.ac.jp
bCatalan Institute of Nanoscience and Nanotechnology (ICN2), CSIC and Barcelona Institute of Science and Technology, Campus UAB, Bellaterra 08193, Barcelona, Spain
cDepartment of Synthetic Chemistry and Biological Chemistry, Graduate School of Engineering, Kyoto University, Katsura, Nishikyo-ku, Kyoto, 615-8510, Japan
First published on 15th July 2020
Abstract
The discovery of NO, CO, and H2S as gasotransmitters and their beneficial role in multiple physiological functions opened an era of research devoted to exogenously delivering them as therapeutic agents. However, the gaseous nature of these molecules demands new forms of administration that enable one to control the location, dosage and timing of their delivery. Porous materials are among the most suitable scaffolds to store, deliver and release gasotransmitters due to their high surface area, tunable composition and reactivity. This review highlights the strategies employed to load and release gasotransmitters from different kinds of porous materials, including zeolites, mesoporous silica, metal–organic frameworks and protein assemblies.
 Arnau Carné-Sánchez | Arnau Carné-Sánchez was born in Manresa, Catalonia, Spain, in 1985. He received his BSc degree in Chemistry from the Universitat de Barcelona (UAB) in 2008. In 2014 he got his PhD in Chemistry from the UAB under the supervision of Prof. Daniel Maspoch and Dr Inhar Imaz. In 2014 he joined Prof. Susumu Kitagawa and Prof. Shuhei Furukawa's group in iCeMS at Kyoto University (Japan) under a JSPS postdoctoral fellowship. In 2016 he joined ICN2 as a postdoctoral researcher under a Juan de la Cierva fellowship. He is currently researching the synthesis of nano-sized molecular porous materials. |
 Francisco J. Carmona | Francisco J. Carmona was born in Granada, Spain, in 1987. He received his BSc degree in Chemical Engineering from the University of Granada (UGR) in 2011. In 2017 he got his PhD in Chemistry from the UGR under the supervision of Dr Elisa Barea and Dr Carmen R. Maldonado. In 2017 he joined the research group of Prof. Norberto Masciocchi at Insubria University (Italy). In 2019 he joined Prof. Shuhei Furukawa's group in iCeMS at Kyoto University (Japan). His current research is focused on the development of metal–organic porous frameworks as gas-releasing materials. |
 Shuhei Furukawa | Shuhei Furukawa was born in Nagasaki, Japan, in 1978. He received his PhD in chemistry from Kyoto University in 2005 under the supervision of Prof. Susumu Kitagawa. After two years postdoctoral research in the Prof. Steven De Feyter's group at Katholieke Universiteit Leuven, he returned to Kyoto as assistant professor. In 2010 he joined iCeMS, Kyoto University, as associate professor and since 2020 he has been a full professor at iCeMS. His main interest is particularly synthesis and properties of porous materials, controlling their structures over multiple length scales, and their applications in electronics and cell biology. |
1. Introduction
Signaling molecules behave as messengers between cells, which trigger subsequent physiological and chemical transformations that are vital for the correct function of organisms. Among these, gaseous signaling molecules, also termed as gasotransmitters, represent a unique class of signaling molecules because their gaseous nature allows them to diffuse through the cell membrane and to trigger a broad range of catalytic, metabolic processes within a short time after their release. To date, three main gasotransmitters have been identified: nitric oxide (NO), carbon monoxide (CO) and hydrogen sulphide (H2S).1–3 Chronologically, NO was the first gaseous molecule identified as a signaling molecule. In 1987 NO was identified as the endothelium-derived relaxing factor (EDRF) responsible for vein and artery dilation.4 Since then, interest in the biological relevance of NO has increased and its biological synthesis from L-arginine by a family of enzymes named as NO synthases (NOS) has been discovered.5 Unlike NO, the biosynthesis of CO from haem degradation catalysed by haem oxygenase (HO) was known long before and for a long time CO was considered as a toxic by-product of metabolism.6 The discovery of the biological function of NO triggered a renovated research effort focused on the biological role of CO. These efforts yielded the identification of CO as the second gasotransmitter with an important physiological impact in a variety of processes such as vasodilation, anti-inflammation, anti-proliferation and anti-apoptosis.7,8 H2S was included in the family of gasotransmitters in 2002 and is the last molecule added to this kind of signaling molecules thus far. The presence of H2S in the organism was already described and, as in the case of CO, its role was supposed to be as a merely noxious toxin.2 Further research demonstrated that H2S is involved in many intracellular signaling pathways with important functions in cytoprotection, anti-oxidation, vasodilation and angiogenesis.9,10The beneficial effects of gasotransmitters in various physiological functions open the possibility to deliver them exogenously as therapeutic agents. However, all three gasotransmitters identified so far behave as a double-edged sword. They possess acute toxicity at high concentration while they present therapeutic effects at low concentration. In addition, their activity is also dictated by where they are located. For instance, high accumulation of CO or NO in the lungs might result in oxygen replacement in heme and severe hypoxia. On the other hand, vasodilation is induced when these gases are released into blood vessels, which is an effective treatment against coronary diseases. Another example is provided by the anti-inflammatory effect of CO when delivered in the organs or tissue, which postulates CO as a promising non-steroidal anti-inflammatory drug.11
In addition, their gaseous nature implies that challenges related with their handling, storage and appropriate delivery have to be overcome before considering the use of gasotransmitters as drugs. In this context, the use of scaffolds able to immobilize bioactive gas molecules in solid state (or in solution) and liberate them on demand is highly desirable. Molecules that release bioactive gases gradually or upon external stimuli have been extensively studied and excellent reviews are available.12–16 However, the fast diffusion of molecules once administered limits the scope of this strategy as they may cause toxicity to untargeted healthy tissues either by the action of the liberated gasotransmitters or by the release of by-products after gas release. Thus, structuring of gasotransmitters into functional scaffolds in the solid state is a promising strategy to achieve higher payloads of the bioactive gas in targeted areas because they are easier to localize. To date, many kinds of solid-state materials are utilized to release bioactive gases such as inorganic and silica nanoparticles, liposomes and organic polymers.17–19 However, in most cases these materials suffer from low payloads, which limits their release capacity. This is due to the fact that only the surface of the materials is available for drug loading (through surface functionalization). A promising strategy to overcome this issue is to use porous materials as functional scaffolds. The high surface area that these materials possess enables incorporation of gasotransmitters not only on their material surfaces but also within the pores, which guarantees a higher drug loading.
The progress in nanomaterials releasing bioactive gases (including gasotransmitters) with specific applications in cancer therapy/bioimaging has been recently revised.20 Here, we analyze the current advances in loading/release of gasotransmitters inside porous materials in the nano-mesoscale (i.e. zeolites, mesoporous silica, metal–organic frameworks and protein assemblies) toward a broader variety of bio-applications. The existing gas-releasing materials are classified according to the porous component, as well as to the specific strategy used to load the gas inside them.
2. Strategies to load gases into porous materials
To incorporate gasotransmitters or gas-donor molecules into porous materials one can choose among several strategies. However, the loading methodology will influence not only the loading efficiency but also the rate of release and stimuli that will trigger the release. We categorized the existing methodologies to incorporate bioactive gases into porous materials as follows (Fig. 1): | ||
| Fig. 1 Scheme of the different strategies to prepare gasotransmitter-releasing materials based on porous frameworks. The existing combinations between strategy and porous framework are indicated. The nitric oxide molecule is used as a gas model. | ||
(1) Use of open metal sites (OMSs) as binding moieties. All porous materials listed here can incorporate metal ions into scaffolds and expose an unsaturated coordination site on metal ions as an OMS. The three gasotransmitters, NO, CO and H2S, have a strong coordination capability to metal ions, and thus, can be anchored on the pore walls of materials containing OMSs.
(2) Use of the catalytic properties of the OMSs to trigger chemical reactions that produce gasotransmitters as products. OMSs can be used not only as an anchoring site, but also as a catalytic site. In this case, the porous material behaves similarly to an enzyme, which generates a gasotransmitter from a bio-available substrate or an exogenous pro-drug.
(3) Immobilization of donor molecules of gasotransmitters on the pores by post-synthetic modification. In this strategy, coordination or covalent bonds are often used to fix donor molecules on the pore walls after forming porous materials.
(4) Physical encapsulation of donor molecules of gasotransmitters in porous materials. By taking advantage of porous materials, pro-drugs can be incorporated into the pores. However, due to the low host–guest interaction, the molecules can be leaked out of pores. Several strategies are available to limit the extent of the leakage and will be discussed in this review.
3. Zeolites
Zeolites are a family of crystalline porous aluminosilicates with the general formula Mx/nn+[(AlO2)x(SiO2)y]x−. With their well-organized and regular system of channels and pores, zeolites have been applied as ion exchangers, catalysts and molecular sieves.21 The non-toxic behavior of zeolites combined with their molecular storage property based on microporosity provides an opportunity to use them as drug carriers.22 The presence of OMSs in zeolites, upon guest removal, is advantageous towards trapping and releasing gasotransmitters. Likewise, the inherent OMSs together with the feasibility of generating catalytic sites in their frameworks (e.g. acidic sites) could enable the production of gasotransmitters from bioavailable substrates. Even though one can find reports on the adsorption of H2S and CO in zeolites,23,24 they are solely focused on their adsorption for removal applications and not on their release for therapeutic applications. Therefore, in this section the loading and release of NO will be exclusively discussed.3.1 Coordination on open metal sites
The inherent or post-synthetically generated OMSs in zeolites can act as anchoring points, where gasotransmitters can be coordinated. This induces a slower and more controlled release than when gasotransmitters are merely physisorbed.Morris and co-workers demonstrated and quantified the high affinity of NO towards zeolites by means of gas sorption experiments. In these experiments, the Co-exchanged zeolite A (Co-LTA) showed an uptake of 1.7 mmol g−1 at 800 mmHg. More interesting is the fact that 76% of NO was not released when the pressure was reduced, which demonstrated the strong affinity of NO towards the OMSs present in the structure (Fig. 2a).25 However, the release of NO could be triggered by water molecules, which have stronger affinity towards open metal sites than NO. Indeed, 1.0 mmol g−1 of NO was released from NO-loaded Co-LTA by flowing wet gas (11% relative humidity). In addition, the release kinetics could be tuned by varying the relative humidity of the wet gas. This degree of control of the NO delivery from the zeolite was exploited to probe the biological effect of exogenously delivered NO in cells by testing its role in the inhibition of platelet aggregation. A key step towards the use of NO releasing materials in real world applications is their integration into shaped, easy-handling macroscopic materials. In this context, Balkus et al. hybridized NO-loaded Co-LTA crystals with polymeric fibers of polylactic acid (PLA) with a bandage shape (Fig. 2b).26 Interestingly, the hybridization induced a more prolonged release by preventing water from coming into contact with the dispersed crystals. Such hybrid nature offers the opportunity to gain additional control over the NO release rate.
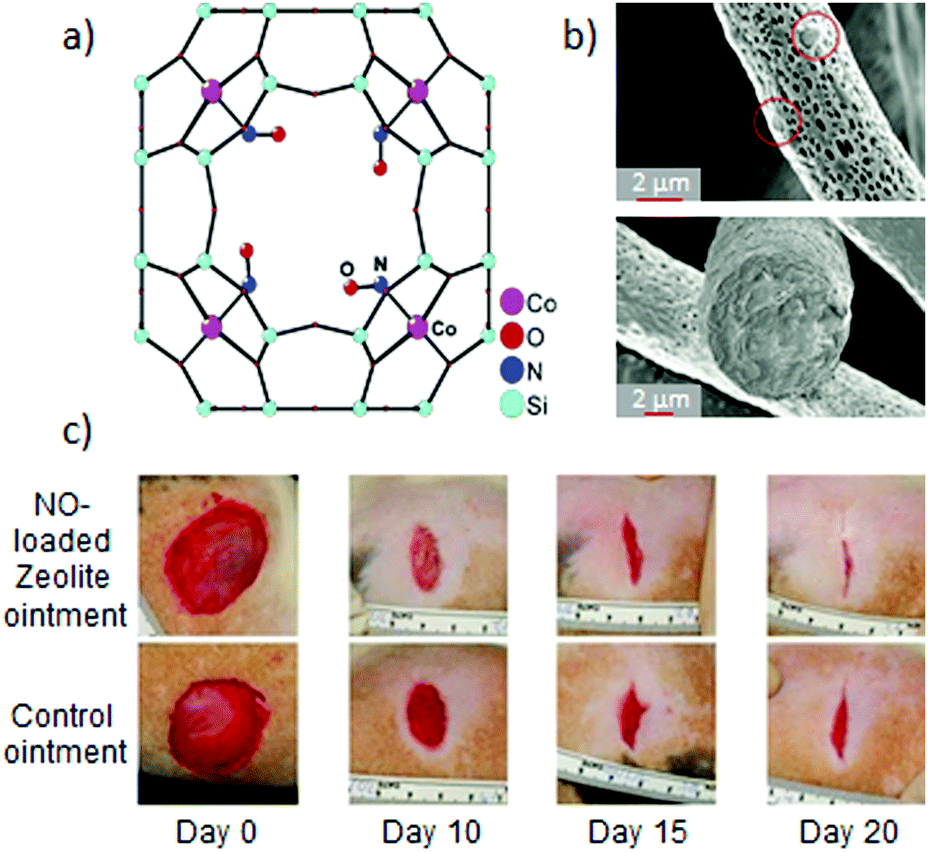 | ||
| Fig. 2 NO-releasing systems prepared by the chemisorption of NO to open metal sites in zeolites and their beneficial effects: (a) structure of the cobalt–NO complex in zeolite-A as elucidated by single-crystal X-ray diffraction. Reprinted with permission from ref. 25. Copyright 2006 American Chemical Society. (b) SEM images of electrospun polylactic acid fibers containing 13% of cobalt–Zeolite A. The hybridization of the zeolite into a macroscale material allows one to prepare humidity-triggered NO-releasing bandages with gradual gas release. Reprinted with permission from ref. 26. Copyright 2009 American Chemical Society. (c) Enhanced wound-healing due to the topical application of an ointment containing 33% of NO-loaded Zn–zeolite-A in comparison with the application of the pristine ointment. Reprinted with permission from ref. 28. Copyright 2014 Microbiology Society. | ||
Zeolites can be easily functionalized by exchanging with transition metal ions in their structure. Thus, in addition to Co-LTA, other transition metal ion-exchanged LTA zeolites were evaluated as NO delivery vehicles, such as pristine LTA, nickel, copper, manganese25 or zinc zeolites.27 This tunability of the zeolite composition has an impact on the maximum loading capacity, release kinetics and also the appearance of synergistic effects. For example, the burst antibacterial effect of the NO-loaded Zn-LTA against Gram-negative and Gram-positive bacteria was coupled with the long lasting antibacterial effect of the slowly leached Zn(II) ions from the zeolite. Likewise, its hybridization with macroscopic materials could not only modulate the properties of the resulting materials, but also favor its development toward real applications. In this regard, Joshi et al. developed an ointment containing 33% of NO-loaded Zn-LTA.28 The hybrid system showed a slower NO release than the pristine NO-zeolite, when suspended in phosphate buffered saline (PBS). The higher hydrophobic character of the ointment hindered NO-exchange with water molecules at the OMSs. This more sustained NO-release resulted in a significant microbicidal activity, and under longer times, against common wound pathogens. Likewise, the topical application of the hybrid ointment was demonstrated to enhance the healing of cutaneous wounds in rats (Fig. 2c). These examples bring to light the feasibility of modulating the release of bioactive agents from zeolites (NO, Zn(II) ions), which determines the biological response. They show the potential of NO-zeolite systems to be incorporated in wound dressing towards the treatment of infected wounds.
Besides the naturally occurring LTA-type of zeolites, titanosilicates have also shown their potential in storing and releasing NO. These materials possess stoichiometric amounts of OMSs in their structures, which largely affect the adsorption and release of NO. Rocha and co-workers focused on the OMSs of the titanosilicate ETS-4 ([Na9Si12Ti5O38(OH)·xH2O]), which is based on pentacoordinated Ti(IV).29 In order to systematically study the impact of the OMSs on the performance of titanosilicates, ETS-4 was compared with ETS-10 ([(Na,K)2Si5TiO13·xH2O]), which possesses only fully coordinated (hexacoordinated) Ti(IV). As expected, ETS-4 largely outperformed ETS-10 with respect to uptake (11 wt% and 5.5%, respectively). The further partial replacement of extraframework Na(I) cations by Cu(II) increased slightly the NO loading. However, the post-functionalization led to an increase in the toxicity of the material, probably because of the leaching of the extraframework copper cations.30 Accordingly, they explored the effects of the isomorphic substitution of framework silicium groups in ETS-10 by the less toxic Al(III) and Ga(III) cations.31 The exchanged materials offered higher NO payloads than the unexchanged ETS-10, due to the chemisorption of the gas to the exchanged cations, being also higher than the previously reported Cu(II)-exchanged ETS-4. Furthermore, the Al- and Ga-exchanged materials exhibited a significant lower toxicity than the Cu-ETS-4 analogs and no metal leaching was observed when incubated in cell culture medium.32 Although the properties of titanosilicates as NO-storing solids are promising, the assessment of their ability to regulate biological functions is essential toward their evolution to real therapeutic applications. With this aim in mind, Pinto and co-workers studied the biocompatibility, chemical stability and NO-release in biological medium of the above-mentioned materials. They selected the unexchanged ETS-4 matrix as the most promising material in terms of amount and sustained NO-release, toxicity and stability. NO-loaded ETS-4 proved to elicit biological responses due to NO-release by reversibly inhibiting mitochondrial oxygen consumption in HeLa cells and promote cell migration, highlighting its potential application in wound healing.32
3.2 Catalysis
As explained in the previous section, great advances have been made in the development of zeolites for storing and releasing NO towards therapeutic applications. However, this strategy has two main drawbacks: (i) the stored gas is finite and only suitable for short-term applications and (ii) the materials usually need to be conserved under special conditions (e.g. away from moisture). One alternative is the chemical production of the gas from a bioavailable substrate.It is known that nitrite reductase enzymes in bacteria transform NO2− to NO mediated by Cu(I) metal sites.33 Based on this, Morris and co-workers prepared Cu(II)-Zeolite-X and Cu(II)-ZSM-5 by extraframework cation exchange.34 The thermal activation of the materials led to generation of Cu(I) sites by self-reduction reaction.35 Although the materials generated a significant amount of NO when suspended in nitrite aqueous solution, the NO production lasted only a few hours. The concurrent action of both water and dioxygen molecules led to the re-oxidation of the Cu(I) centers to Cu(II), inhibiting the further transformation of nitrites to NO. More interesting is the fact that the pre-adsorption of NO resulted in a constant production of NO during 10 h. The exposition of the materials to gaseous nitric oxide results in the chemisorption of NO to the Cu(I) centers. In the presence of water, only a small fraction of the loaded NO is released, while a significant amount of NO is maintained in the material. The irreversibility of chemisorbed NO hindered the interaction of both water and dioxygen with the Cu(I) sites and inhibited their re-oxidation, and therefore, favored the sustained conversion of nitrite to nitric oxide. This demonstrated the cooperative effect of NO adsorption on the NO-production process.
The acidic transformation of nitrites is an alternative route to produce NO. Morris and co-workers studied the effect of the pore size of acidic zeolites on the production of NO from nitrites by testing four different zeolites: Al-UTL, Al-IPC-2, Al-IPC-4 and Al-IPC-6. To this end, they employed the ADOR (Assembly-Disassembly-Organization-Reassembly) method,36 which allows one to selectively control the pore size of the zeolite by altering the linkers between silicate layers. They observed that the higher the pore size, the larger the amount of NO generated and the faster the kinetics, demonstrating the feasibility of tuning the NO-generation according to the targeted application.37 In another work, Morris et al. prepared several acidic and copper-exchanged zeolites based on mordenite, ferrierite, ZSM-5 and SSZ-13.38 Although all materials were able to produce NO from nitrite sources, the copper-loaded materials showed a higher activity. In addition, given the known presence of cysteine in the body, the ability of the materials to generate NO in a mixture of nitrite/cysteine at biologically relevant concentration was assessed. As the acidic sites were consumed during NO-production, the continuous NO-generation after subsequent addition of the substrate was inhibited. Conversely, the Cu(I) sites were regenerated by the oxidation of the cysteine present in the media, allowing the continuous conversion of nitrite (Fig. 3). Among the different Cu-zeolites, Cu-SSZ-13 showed the best biocompatibility, making it a promising zeolite to use in the body.
 | ||
| Fig. 3 Scheme of the generation of NO from nitrites mediated by Cu(I) ions and acidic sites in zeolites. Bioavailable compounds like cysteine (RS-H) are able to regenerate the Cu(I) sites, allowing a continuous release after the addition of the substrate (e.g. NaNO2). In contrast, the acidic groups in zeolites are not regenerated. Therefore, the repeated addition of the substrate does not result in a continuous release of NO when the active sites are consumed. Adapted from ref. 38 with permission from The Royal Society of Chemistry. | ||
4. Mesoporous silica
Mesoporous silica is a porous inorganic solid formed by amorphous silicon dioxide containing periodic arrays of channels and cavities. It has been widely used as a drug delivery platform due to its 2 to 50 nm pore size and biocompatibility.39,40 Compared to zeolites (described in Section 3) that store gasotransmitters within their small cavities, mesoporous silica offers new strategies to accommodate gasotransmitters. For example, one can encapsulate gas-donor molecules, much bigger than the gas itself, which can trigger the release of gasotransmitters by different stimuli such as light, heat or ultrasound. This strategy can be enriched by the fact that the pore surface can be covalently modified by silanol chemistry.41 In the following sub-sections, representative works of these two strategies are summarized.4.1 Encapsulation of gas-donor molecules
The encapsulation of molecules into the pores of mesoporous silica is basically driven by physisorption: the physical interaction between guest molecules and the pore surface of host materials. Consequently, encapsulated guest molecules can escape from the pores by diffusion due to the lack of strong chemical bonding. Therefore, the extent of leaching is the most important issue in this strategy.To prevent the escape of guest molecules, Rickus and co-workers used liposome vesicles as capsules for NO donor molecules.42 Liposomes, phospholipid bilayer vesicles, are also widely used as drug nanocarriers because they prevent the leaching of the entrapped molecule.43,44S-Nitroso-N-acetylpenicillamine (SNAP), which is known to be a light-induced NO donor molecule, was encapsulated into liposomes, followed by doping them into the synthetic solution of the mesoporous silica. The sol–gel reaction of this mixture containing SNAP, liposomes and tetraethyl orthosilicate (TEOS) provided the liposome/mesoporous silica composite materials. Therefore, the NO release from the functional substrate was measured by using a self-referencing amperometric NO microsensor. It was found that the evolution of NO from the mesoporous silica surface was dependent on light irradiation and was within the range of biological relevance (14 pmol cm−2 s−1).45 In addition, the mesoporous silica composite could be used as a functional growth surface for mammalian cells.46,47
Mascharak and co-workers attempted to limit the leaching of NO donor molecules by using host–guest electrostatic interactions.48 Thus, they compared the uptake amount of the positively charged metal-nitrosyl NO donor complex ([Mn(PaPy3)(NO)]ClO4, denoted as Mn–NO) into a negatively charged aluminosilicate mesoporous material (denoted as Al-MCM-41) and a neutral mesoporous silica material (Si-MCM-41). Al-MCM-41 showed 20% higher uptake when compared to Si-MCM-41 (Al-MCM-41: 2.39 wt%, Si-MCM-41: 2.04 wt%,). In addition, Al-MCM-41 showed less leaching of Mn–NO than Si-MCM-41 in saline solution after 24 h incubation (2.92% and 3.44% of adsorbed Mn-NO was leached from Al-MCM-41 and Si-MCM-41, respectively). The mechanism of Mn–NO loading into Al-MCM-41 was supposed to entail cation exchange between [Mn(PaPy3)(NO)]+ and Na(I) ions inside the host and passive diffusion. On the other hand, passive diffusion was the only driving force for Mn–NO loading into Si-MCM-41. Congruent with the higher loading capacity of Al-MCM-41, its release amount of NO by light irradiation was also higher than Si-MCM-41 (0.43 mmol g−1 for Al-MCM-41 and 0.30 mmol g−1 for Si-MCM-41). The control on NO release from Al-MCM-41 allowed studying its antibiotic effect on A. baumannii, a skin and soft-tissue infection (SSTI) model showing effective clearance upon light-triggered NO release (Fig. 4a).
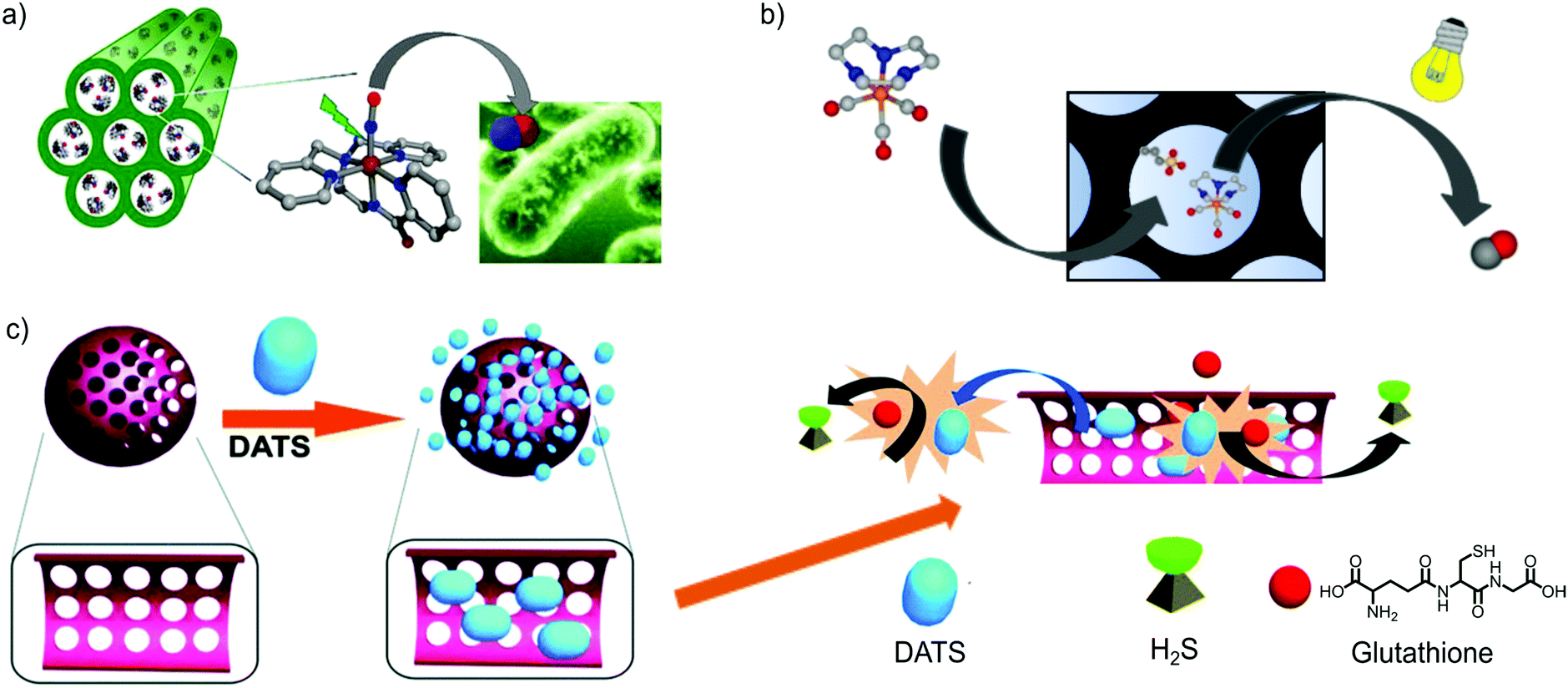 | ||
| Fig. 4 Gas-releasing frameworks prepared by the encapsulation of gas donors into the cavities of mesoporous silica. There exist examples of gas-donor@Mesoporous silica systems for all gasotransmitters (e.g. CO, NO and H2S), demonstrating the high versatility of mesoporous silica to prepare gas-releasing materials by means of the gas-donor encapsulation strategy. (a) Representation of Al-MCM-41 mesoporous silica loading the cationic [Mn(PaPy3)(NO)]+ complex. The irradiation with visible light elicits the NO-release response, exerting an antibiotic effect against a skin pathogen (e.g. A. baumannii). Reprinted with permission from ref. 48. Copyright 2012 American Chemical Society. (b) Encapsulation of the cationic photoCORM [Mn(tacn)(CO)3]+ into mesoporous silica functionalized with anionic sulfonic groups. The photoresponsive properties of the pristine CORM are maintained and the hybrid material releases CO under photoactivation with visible light. Adapted with permission from ref. 51. Copyright 2016 American Chemical Society. (c) Schematic representation of the encapsulation of Diallyl trisulfide (DATS) in mesoporous silica. The interaction of DATS with the bioavailable glutathione results in the release of H2S. The slower diffusion of glutathione inside the pores and DATS outside the pores enables a more controlled release of H2S. Reproduced from ref. 56 with permission from The Royal Society of Chemistry. | ||
Zhao and co-workers took advantage of the porosity of mesoporous silica to accommodate a photolytic NO-donor ([NH4][Fe4S3(NO)7], Roussin's Black Salt) in a core–shell upconversion nanoparticle@mesoporous silica nanocomposite.49 Irradiation of the nanoparticles with low-damage near-infrared irradiation (λ = 980 nm, within the therapeutic window) activated the NO-release, being only possible for the bare NO-donor under higher energetic irradiation. By modulating the laser output power, the amount of gas generated was controlled, resulting in a dose-controlled NO-release system. This feature was later demonstrated by in vitro cell studies, where different biological responses with NO-dose dependence were achieved by simply tuning the irradiation power of the NIR light used as a trigger.
Mesoporous silica can also be employed as a vehicle to transport guest drugs to the targeted tissue. In addition, the extent of the leaching of the encapsulated NO-prodrug could enable a more sustained NO-release. This strategy was employed by Deng et al. toward the efficient treatment of Primary Open-Angle Glaucoma (POAG).50 Nitric oxide has proved to reduce elevated intraocular pressure (IOP), a strong risk factor for developing POAG. Likewise, sodium nitroprusside (SNP), a N-donor, has been proposed as a potential therapeutic agent against this disease. However, its low uptake through the cornea together with its poor stability limits its practical use. The encapsulation of SNP inside mesoporous silica resulted in a slower release of the N-donor in PBS, extended during 50 hours. In vivo studies on Cav1 gene knockout mice demonstrated that SNP@mesoporous silica nanoparticles were able to pass through the cornea and reach the target tissues. In addition, the topical eye drop application of SNP@mesoporous silica (0.004 mg of SNP) resulted in IOP reduction of 22% after 3.5 h, followed by a sustained IOP reduction during 48 h. In contrast, the dose of the same amount of SNP solution did not affect IOP, demonstrating the beneficial effect of NO-donor encapsulation.
CO-prodrugs, known as CO-releasing molecules (CORMs), have also been incorporated into the cavities of mesoporous silica to generate CO-releasing materials. For instance, Barea and co-workers functionalized the pore surface of MCM-41 and SBA-15 mesoporous silica with alkanesulfonic groups to confer an anionic character to the frameworks. By cation exchange, they successfully encapsulated the cationic photoactive CO releasing molecule (photoCORM) [Mn(tacn)(CO)3]+ (where tacn is 1,4,7-triazacyclononane), which could not be encapsulated into the neutral matrices (Fig. 4b). Both hybrid materials kept the photoactivity of the pristine CORM, but offering a more sustained CO-delivery kinetics in physiological media when irradiated with white light. Unfortunately, the host–guest interactions were not strong enough and led to partial leaching of the cationic CORM when incubated in saline buffer.51
As was done with the NO-donor ([Mn(PaPy3)(NO)]ClO4, Mascharak et al. also used the cation exchange strategy to incorporate a cationic photoCORM into the negatively charged Al-MCM-41 nanoparticles (approx. 60 nm).52 For this, the manganese carbonyl complex (fac-[Mn(pqa)(CO)3]+, pqa = (2-pyridylmethyl)(2-quinolylmethyl)amine), denoted as Mn–CO, was exchanged with the Na(I) ions presenting in the initial Al-MCM-41 structure. In this case, the host–guest electrostatic interactions ensured a small percentage of leaching when the composite was incubated in PBS (i.e. 2% and 12% of Mn(I) leaching after incubating for 24 h and 60 h, respectively). The released CO from the [Mn-CO]@Al-MCM-41 nanoparticles upon light irradiation was used to induce vasorelaxation of rat aorta muscle rings. A similar strategy was employed to incorporate the luminescent photoCORM [Re(CO)3(pbt) (PPh3)], pbt = 2-(2-pyridyl)-benzothiazole) into the pores of Al-MCM-41.53 In this case, not only the intracellular delivery of biologically relevant amounts of CO was demonstrated, but also the nanoparticles could be easily tracked inside the cells, owing to their inherent luminescence. The good encapsulating features of the anionic Al-MCM-41, its stability in physiological media and its large pores were exploited by Barea and co-workers to encapsulate both photoCORM [Mn(tacn)(CO)3]+ and cisplatin within the same carrier, suggesting the possibility of producing CO releasing solid platforms with multiple therapeutic effects.54 Diallyl trisulfide (DATS) is known as a glutathione (GSH)-triggered H2S donor; however, its fast release kinetics hinders its therapeutic use as a rapid increase of H2S concentration may have adverse effects on the metabolism.55 Wang and co-workers encapsulated DATS inside mesoporous silica nanoparticles in order to control the release kinetics of H2S (Fig. 4c).56 DATS was loaded into mesoporous silica nanoparticles (MSNs) by stirring a mixture of DATS, MSNs and distilled water for 12 h. Surprisingly, 99% of DATS molecules were encapsulated into MSNs (which accounts for a loading of 2.5 mmol g−1). This very high loading indicates that there is a strong interaction between DATS and Si–OH of the MSN surface. The low leaching (less than 10%) observed upon 24 h incubation in PBS further supported this hypothesis. The H2S releasing experiments performed in the presence of GSH showed slower release kinetics from the composite nanoparticles in comparison with free DATS. The biomedical applications of the DATS-MSN nanoparticles were demonstrated by injecting them into mouse's veins. The nanoparticles effectively released H2S into the vein without causing any of the adverse effects caused by high concentration of H2S (such as an increase in blood pressure and heart beat rate). Later, the same group demonstrated that the hybrid system DATS@MSNs promoted the proliferation and differentiation of endothelial cells in vitro and alleviated inflammatory response caused by hypoxia–reoxygenation. Likewise, in vivo studies in mouse models confirmed these benefits, showing that DATS@MSN was able to protect the endothelium of aortic allografts from ischemia–reperfusion injury.57
4.2 Chemical attachment of gas donors
The diversity of chemical functionalities that one can use in the sol–gel process (amine, hydroxyl and carboxylate groups among others) enables functionalization of mesoporous silica through post-synthetic reactions. One can use this chemistry to covalently attach donor molecules of gasotransmitters on the surface of these materials. This approach has the disadvantage that it generally involves a multi-step chemical reaction. On the other hand, it reduces the extent of leaching due to the covalent interaction between the donor and the surface of the mesoporous silica.Surface modification of mesoporous silica with coordination complexes has been researched by a number of groups in the late 1990s and the early 2000s.58,59 Since then, surface modified fumed silica, silica gel and silica nanoparticles have been extensively developed and applied in the field of gas biology.60–62 Longhinotti et al. immobilized cis-[Ru(bpy)2(L)NO]3+ and [Fe(CN)4(L)NO]− complexes on surface functionalized mesoporous silica.63 The procedure entailed four sequential surface functionalization reactions, including amine functionalized mesoporous silica (Si-APTS), amine terminal converted to isonicotinamide (ISN) and Ru(II) and Fe(II) metal complex immobilization on the surface of Si-APTS-ISN by reacting the functionalized silica with cis-[Ru(bpy)2Cl2] and Na2[Fe(CN)4(dmso)2], respectively. Finally, metal-nitrosyl functionalities were introduced by bubbling NO gas into the prepared silica materials in acetone. Light-induced release of NO in the PBS from both the prepared materials was estimated to be 1.8 mmol g−1.
An alternative moiety that is able to store and deliver NO is the amine group, which can store NO in the form of nitrite and release it under acidic conditions. To immobilize the amine functionality on the surface of mesoporous silica, γ-aminopropyltriethoxysilane (APTES) was attached on the surface of MCM-41 and mesoporous MFI zeolite,64 which was synthesized from MCM-41 by dry-gel conversion.65 Before the modification of APTES, NO was loaded on MCM-41 and MFI zeolite, which released 0.77 and 0.67 μmol g−1 of NO in the acidic aqueous solution, respectively. In contrast, APTES modified MCM-41 and MFI zeolite showed a release of 27.43 and 17.99 μmol g−1, respectively. The higher release observed in the case of MCM-41 is attributed to its higher surface area, which implies a higher degree of surface functionalization. This high release amount in acidic conditions postulates these functionalized materials as efficient NO delivery vehicles for the gastrointestinal organs. The functionalization of the porous framework of mesoporous silica with aminosilanes was also employed by Schoenfisch et al. to generate novel NO-releasing materials.66 They prepared mesoporous silica with different particle size (ranging from 30 to 1100 nm) and functionalized covalently the pores with terminal secondary amines. By reacting with NO, they were transformed into N-diazeniumdiolates able to release NO in physiological media. The materials showed different NO-payloads and release kinetics in PBS. However, these differences could not be correlated with the distinct particle size of the carriers. A further analysis by powder small-angle X-ray scattering (SAXS) suggested that the different pore structures found in the functionalized systems determined these features. While a more ordered pore system enhanced the diffusion of water through the cavities, resulting in faster NO-release kinetics, more heterogeneous pore structures hindered the solvent diffusion and resulted in more sustained NO-release profiles. In another work,67 the amine groups in the outer surface of NH2-functionalized mesoporous silica were used to covalently attach erythrosine, a singlet oxygen photosensitizer (PS), while the internal surfaces were used to conjugate a nitro aniline-derivative as a photoactive NO-donor. The antitumoral drug doxorubicin (Dox) was further encapsulated into the porous matrix, generating a multidrug carrier. In vitro studies on the melanoma A375 cell line showed that, under light irradiation, the triple-drug nanomaterial exhibited a higher cytotoxicity than the dual system Dox-PS@mesoporous silica, suggesting an increased activity due to the nitric oxide release. A similar strategy was used to confer NO-releasing functionalities to a fluorescent carbon dot@mesoporous silica nanocomposite.68 The conjugation of a nitro aniline-derivative as a photoactive NO-donor to the pore surface quenched the emission from carbon dots by means of the Förster resonance energy transfer (FRET) process. More interestingly, visible light irradiation triggered the NO-release, with the concomitant fluorescence restoration of the carbon dots. Therefore, the nanocomposites showed a dual-function, because they not only implemented a controlled NO-release under light irradiation, but also incorporated a fluorescence reporter that allows the tracking of the gas delivery process by fluorescence microscopy and spectroscopy.
S-Nitrosothiols (RSNO), a kind of heat-sensitive NO-donor, have also been incorporated in mesoporous silica by surface functionalization. The feasibility of functionalizing mesoporous silica with thiol-terminated organosilanes was used by Malone-Povolny and Schoenfisch for, subsequently, generating the S-nitrosothiol sites.69 The same procedure was performed on non-porous silica nanoparticles to assess the influence of porosity on the NO-release kinetics. While the non-porous materials showed a half-life (t1/2) of NO release of 2.92 h in PBS at 37 °C, mesoporous silica particles exhibited a much more sustained release (t1/2 of 26.6 h). These differences were explained by the “cage effect” shown by the porous material. The confined microenvironment in the nanometer-scale pores favored the recombination of the thiyl and NO radical pair after the homolytic cleavage of the S–N bond, extending the duration of NO delivery. They also dispersed the mesoporous nanoparticles in several polyurethane membranes towards their potential applications as coating of medical devices. Note that the nanocomposites exhibited a much longer prolonged NO-release (until 30 days), with negligible particle leaching after suspension in PBS for 21 days.
In another work, the terminate-thiol groups were used for both generating S-nitrosothiols and conjugating S-transferrin (a cancer-cell targeting group) on a core–shell Fe3O4@mesoporous silica nanocomposite (Fig. 5a). The antitumoral drug doxorubicin was encapsulated into the cavities of the mesoporous shell, generating a multidrug carrier.70 The nanocomposite was able to convert near-infrared irradiation into heat, enabling to trigger the NO-release by photoactivation. Remarkably, the concomitant release of doxorubicin and NO, triggered by NIR photoactivation for 5 min, showed a synergistic effect toward the reduction of the cell-viability of a multidrug resistant breast cancer cell-line (MDR-MCF-7) (Fig. 5b and c). In vivo studies on MCF-7/ADR tumor-bearing BALB/c nude mice confirmed these results. The treatment with the multifunctional nanocomposite and NIR showed a significant tumor inhibition, superior to the parallel treatments with the pristine drugs.
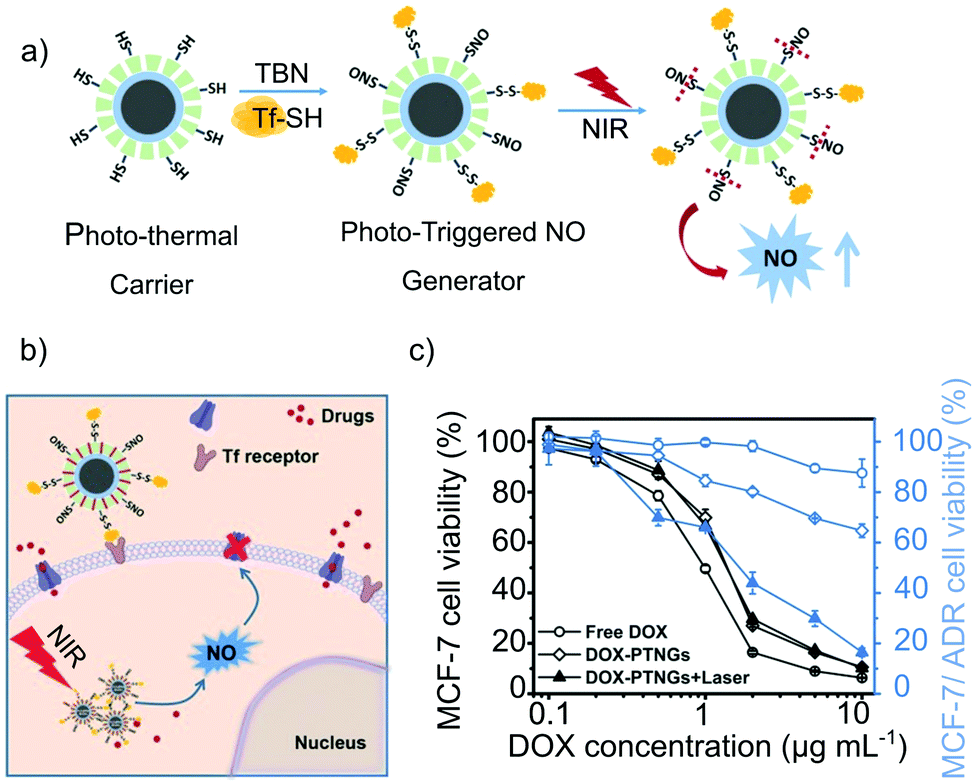 | ||
| Fig. 5 Core–shell Fe3O4@Mesoporous Silica nanocomposite with photoresponsive antitumoral properties: (a) scheme of the generation of S-nitrosothiols and S-transferrin in the mesoporous silica shell and of the photo-thermal responsive release of nitric oxide under near-infrared irradiation. (b) and (c) Antitumoral mechanism of the nanocomposite. The presence of transferring increases the specificity of the system toward tumoral cells while the NIR irradiation activates the NO-release. Likewise, the NO-release exerts a signaling function, favoring the accumulation of doxorubicin in the cells, resulting in a high antitumoral activity against Multidrug Resistant cancer. Reprinted with permission from ref. 70. Copyright 2017 John Wiley & Sons, Inc. | ||
5. Metal–organic frameworks
Metal–organic frameworks (MOFs), also called porous coordination polymers (PCPs), which are constructed via the assembly of metal ions with organic linkers, are receiving considerable attention due to their structural characteristics, highly ordered microporosity, large surface area and the wide range of potential applications in gas storage, gas separation, catalysis and sensors.71–73 Recently, by taking advantage of MOFs’ porosity, researchers have used these materials in biomedical applications, in particular to store and release drug molecules.74–76 Compared to other porous materials introduced above, the most characteristic feature of MOFs is their inherent inorganic–organic hybrid nature, which enables one to design and synthesize a virtually unlimited number of structures by appropriate combination of metal ions and organic linkers. Furthermore, the inherent hybrid nature of MOFs provides an opportunity to expand the possible strategies towards gasotransmitter delivery by taking advantage of either the inorganic part (i.e. OMSs, catalysis) or the organic part (i.e. post-synthetic modifications, prodrug ligands) or both parts acting synergistically. Their inherent porosity can be also used to host gas-donor molecules. Thus, the release of gasotransmitters from MOFs has been achieved by four characteristic strategies: (1) the release of coordinating gas molecules at open metal sites by exchanging with water molecules, (2) the catalytic decomposition of gas precursors at open metal sites, (3) encapsulation of gas-donor molecules inside their cavities and (4) incorporation of organic ligands within the MOF structure with the ability to adsorb and release gasotransmitters, or that are themselves gasotransmitter donor moieties that can be activated upon external stimuli.5.1 Coordination on open metal sites
The presence of OMSs decorating the pores of MOFs has shown great potential for selective adsorption of molecules with lone pair of electrons and, as in the case of zeolites, OMSs can be used to store and release gasotransmitters.77Morris and co-workers were pioneers in exploiting the OMSs in MOFs to incorporate NO.78 In the first study, [Cu3(btc)2]n (btc = 1,3,5-benzentricarboxylic acid, also known as HKUST-1), which has Cu(II) OMSs, was used. The first evidence of Cu–NO coordination was provided by the NO sorption isotherms. These isotherms were characterized by a hysteresis in the desorption branch. The number of NO molecules that were not desorbed corresponded to the predicted number of OMSs in the HKUST-1 structure (i.e. 2.21 mmol g−1). Further evidence was provided by infrared spectroscopy performed in the NO-loaded HKUST-1, which showed the characteristic peak at 1887 cm−1 corresponding to the stretching vibration mode of NO coordinating to Cu(II) ions. The release of NO was triggered by treating the sample with water vapor, similar to the case of zeolites described in Section 3. However, a moderate release of 2 μmol g−1 corresponding to 0.1% of the initially adsorbed NO was found. The low NO release is rationalized by the strong coordination of the Cu–NO bond. Despite the low release efficiency, the release amount was enough to inhibit platelet aggregation confirming the potential of MOFs in the field of gasotransmitters release.
Suitable coordination strength between the metal and NO is a key factor to obtain a material that performs well over the whole adsorption-storage-delivery cycle. In order to study the influence of the transition metal ions on the NO release, the same research group employed isostructural MOFs containing different metal ions for the adsorption/release of NO. Namely, they used [Ni2(dobdc)]n (denoted as CPO-27-Ni, dobdc = 2,5-dioxido-1,4-benzenedicarboxylate) and [Co2(dobdc)]n (denoted as CPO-27-Co) for NO storage and delivery (Fig. 6a).79 CPO-27-Ni and CPO-27-Co showed a total NO uptake of 7 mmol g−1 and 6 mmol g−1, respectively, at 25 °C and 6 mmol g−1 and 5 mmol g−1, respectively, of hysteresis in the desorption branch, which correspond to the number of OMSs in each structure. These results indicate that most of NO was adsorbed through coordination to the OMSs and approximately 1 mmol g−1 of NO was physically adsorbed to the frameworks. Crystal structures of both frameworks80 clearly show that NO is coordinated to metal sites with a bent geometry, which corresponds to NO acting as a one-electron donor. NO release from CPO-27-Ni and CPO-27-Co was triggered by wet gas (11% relative humidity). Unlike HKUST-1, these two MOFs released practically the same amount of NO as previously adsorbed. It is noteworthy that a NO release capacity of 7 mmol g−1 is the highest reported value for porous materials thus far. In order to test the biomedical applications of these materials, the vasodilation of pre-contracted pig coronary arteries induced by NO-loaded CPO-27-Ni was tested. The results showed a rapid relaxation of pig coronary arteries immediately after exposing them to an aqueous suspension containing pellets of CPO-27-Ni.
 | ||
| Fig. 6 Examples of NO-releasing metal–organic frameworks showing open metal sites. (a) Crystal structure of the [Co(C8H2O6)(NO)2] MOF prepared by the chemisorption of nitric oxide to the unsaturated-cobalt sites. In detail, the view of NO bound to the cobalt atoms in the MOF structure. Reprinted with permission from ref. 79. Copyright 2008 American Chemical Society. (b) Schematic representation of the formation of chemisorbed nitrates on the titanium-based MOF Ti12O15(mdip)3(OH)6(H2O)6. Reprinted with permission from ref. 89. Copyright 2020 John Wiley & Sons, Inc. | ||
The possibility to synthesize CPO-27 analogue structures with different divalent metal ions enabled one to systematically study the effect of the nature of the OMSs on the adsorption and release of NO.81 Long and co-workers reported the NO loading and releasing properties of CPO-27-Fe (based on Fe(II)).82 Interestingly, while the loading capacity was similar to the examples shown above (i.e. 6 mmol g−1), the releasing profile was different, showing an initial burst release followed by a slow and sustained release of NO that occurs over the course of 10 days. These results further prove the key role of the OMSs in the loading and releasing profiles of NO.
A unique feature of MOFs is that they combine OMSs with tunable microporosity. Therefore, one can take advantage of these properties to design multifunctional carriers, delivering more than one substance. Barea and co-workers explored this strategy using CPO-27-Ni as the host structure.83 First, the metallodrug with antitumor activity RAPTA-C ([Ru(p-cymene)Cl2(pta)], pta = 1,3,5-triaza-7-phosphaadamantane) was introduced into the pores of the framework by physisorption. Subsequently, NO was loaded into the composite material. Interestingly, the NO loading capacity (6 mmol g−1) remained almost unaltered after the incorporation of RAPTA-c, which indicates that the coordination of NO at the OMSs of the framework was not influenced by incorporated RAPTA-C molecules. Importantly, the release of both bioactive molecules was very similar as in the case in which only one of the molecules was encapsulated, indicating the potential use of MOFs as multi drug delivery vehicles.
One of the key aspects when considering MOFs for biomedical applications is their toxicity both in vivo and in vitro.84 Therefore, the synthesis of MOFs with gasotransmitter releasing properties with non-toxic building blocks is a promising strategy to make these materials more biocompatible. The MIL-88 series of MOFs composed by iron(III) carboxylates combine a non-toxic behaviour and the presence of OMSs, which can be used to capture NO.85 Therefore, they are suitable candidates as bio-friendly NO delivery vehicles. Serre and co-workers systematically studied the NO loading/release properties of these materials by synthesizing them with different functionalities ([Fe3OX(–O2C–R–CO2–)3]n, R = terephthalic acid, fumaric acid, 2-nitroterephthalic acid and dobdc; X = F, OH, Cl or –O2C–CH3).86 The uptake in all cases fell between 1 and 2.5 mmol g−1. However, the release amount was limited in the range of 0.1 mmol g−1 due to the narrow pores in the structure and the strong binding of Fe–NO. In order to improve the NO releasing capacity of the Fe(III) based frameworks, the performance of Fe-MIL-100 (Fe3OX[btc]2·xH2O), which contains big mesoporous cavities within its structure, was evaluated. As expected, the release efficiency was improved when compared to the MIL-88 structure releasing up to 15% of the initially chemisorbed NO (0.35 mmol g−1) over the course of 7 h.87 In this regard, the MOF bioMIL-3 [Ca2(azbz-TC)(H2O)(DMF)], (azbc-TC = 3,3′,5,5′-azobenzenetetracarboxylate), which possesses OMS in its structure, was also assessed as a NO-releasing material due to the high biocompatibility of calcium cations.88 It showed a NO payload of 0.8 mmol g−1 and a sustained release during 10 hours under wet nitrogen gas. Recently, Serre and co-workers have proved the feasibility of coordinating NO to the OMSs of a titanium-based MOF, namely Ti12O15(mdip)3(OH)6(H2O)6 (mdip: 3,3′,5,5′-tetracarboxydiphenylmethane) by the in situ formation of complexed nitrites (Fig. 6b).89 After a thermal treatment, bridged Ti–OH–Ti groups were generated in the secondary-building units, which after reacting with NO formed chemisorbed nitrites (NO payload of 1.3 mmol g−1). The material showed a slow NO release when suspended in PBS, not previously observed in NO-releasing MOFs due to the lability of the metal–NO bonds and/or the low stability of the frameworks in physiological media. In this case, given the high stability of the MOF, the sustained release was associated to the water-exchange of the chemisorbed nitrites. The material inhibited mitochondrial respiration in a concentration-dependent manner, demonstrating the control of the NO-releasing amount. The treatment of HeLa cells with the NO-releasing material favored the migration of the cells compared to the unloaded one, postulating its employment in wound-healing applications.
As mentioned above, all three gasotransmitters have lone pair electrons. Therefore, it is possible to use OMSs in MOFs to store and deliver not only NO but also CO and H2S. N. Metzler-Nolte and co-workers used the MIL-88 (Fe) analogues for CO adsorption and release.90In situ ultrahigh vacuum-FTIR and Mössbauer spectroscopy studies of MIL-88B and MIL-88B-NH2 confirmed the presence of Fe(II)–CO and Fe(III)–CO coordinations after the desorption cycle. In addition, the effect of the ligand functionality was also studied. It was found that the binding strength decreased in the case of MIL-88B-NH2 due to the electron donating effect of the amino group. Quantification of CO release was carried out through the myoglobin assay under physiological conditions in phosphate buffer at pH = 7.4.91 The CO release for bare MIL-88B and the amino functionalized version was 0.36 mmol g−1 and 0.69 mmol g−1 respectively. These values are consistent with the higher coordination strength in the Fe–CO bond in the case of the unfunctionalized MIL-88B.
Morris and co-workers further widened the scope of MOFs in the delivery of gasotransmitters by incorporating H2S in the structure of CPO-27-M (M = Ni, Zn).92 In close analogy to the case of NO, H2S was coordinated to the OMSs of both frameworks showing a maximum uptake close to 6.2 mmol g−1. The differences between the Ni and Zn analogues appeared in their releasing performance. While the H2S releasing capacity of CPO-27-Ni was 1.8 mmol g−1, in the case of CPO-27-Zn it was 0.5 mmol g−1. This difference is attributed to the difference in the coordination strength between the OMSs and H2S. The capacity of CPO-27-Zn to store H2S and release it under physiological conditions enabled testing its vasodilation-induced properties in pre-contracted pig arteries. The immersion of the pre-contracted arteries in an organ bath containing H2S-loaded CPO-27-Zn resulted in an artery relaxation of 39%.
5.2 Catalysis
OMSs located inside MOFs can be used as catalytic sites, which are easily accessible by the substrates owing to the intrinsic porosity of MOFs. Owing to these complementary properties, MOFs have shown outstanding potential as catalysts.93 On the other hand, several catalytic mechanisms of NO release by decomposition of RSNO are widely known including the copper-mediated pathway.94Reynolds and co-workers used the Cu(II) OMSs in the HKUST-1 structure to catalyze the decomposition reaction of S-nitrosothiols into NO. Thus, HKUST-1 and S-nitrosocysteine (CysNO) were suspended in an ethanolic solution and the NO generation was monitored. It was found that when the MOF was present the NO generation was sustained over the course of 12 h. In contrast, when only Cu(II) salts such as CuCl2 and Cu(CH3COO)2 were employed only a burst release was found. These results suggest that the combination of OMSs and narrow pore architecture is responsible for the sustained NO release.95
A further step towards the integration of the catalytic NO releasing property into functional materials was provided by the same authors. To this end, HKUST-1 was immobilized in cotton fabrics to produce NO on the surfaces.96 The composite material was shown to be active in the catalytic decomposition of S-nitrosocysteamine (CysamNO) to produce NO. In fact, a NO release of 7.1 mmol g−1 was found over a period of 4 h in ethanol. However, as in earlier cases, the poor hydrolytic stability of HKUST-1 hindered its application in the biomedical field. In order to overcome this shortcoming, the water stable MOF [(Cu4Cl)3(BTTri)8(H2O)12]·72H2O (where BTTri is 1,3,5-tris(1H-1,2,3-triazol-5-yl)benzene) was synthesized and evaluated. The high chemical stability of the MOF allowed measuring its catalytic performance and NO release in PBS buffered solution. Thus, CuBTTri was able to release 2.7 mmol g−1 of NO in the course of 4 hours. The biomedical applicability of this MOF was further optimized by hybridizing it with biocompatible polymers such as polyurethane, poly(vinyl alcohol) and chitosan. Remarkably, the performance of the MOF was not diminished by its incorporation into polymeric supports, further suggesting its practical implementation.97–99
5.3 Encapsulation of gas-donor molecules
The almost infinite possible combinations of metal ions/clusters and organic linkers to prepare metal–organic frameworks have resulted in a huge library of structures, some of them showing mesoporosity. Owing to the larger size of the cavities, the framework can host bigger molecules than the gas itself (i.e. gas-donor molecules). The resulting gas-donor@MOF systems can modify the gas-release kinetics of the bare gas-donor (i.e. slowing down the gas release) and/or reduce the toxicity risks associated to the guest molecule by trapping it inside the pores and, therefore, limiting its leaching to the physiological media.Barea et al. were pioneers in using the guest-encapsulation strategy to prepare a novel CO-releasing material based on a MOF.51 The cationic photoCORM [Mn(tacn)(CO)3]Br (tacn = 1,4,7-triazacyclononane) was incorporated in the anionic metal–organic framework bio-MOF-1 (NH2(CH3)2)2[Zn8(adeninate)4-(BPDC)6]·8DMF·11H2O (BPDC = 4,4′-biphenyldicarboxylate) by means of a cationic exchange approach (Fig. 7a). Although the material showed a high payload (0.25 mmol of CORM per gram of material), the scarce stability of the framework in physiological media led to almost full leaching of the CORM after 6 hours of incubation in PBS.
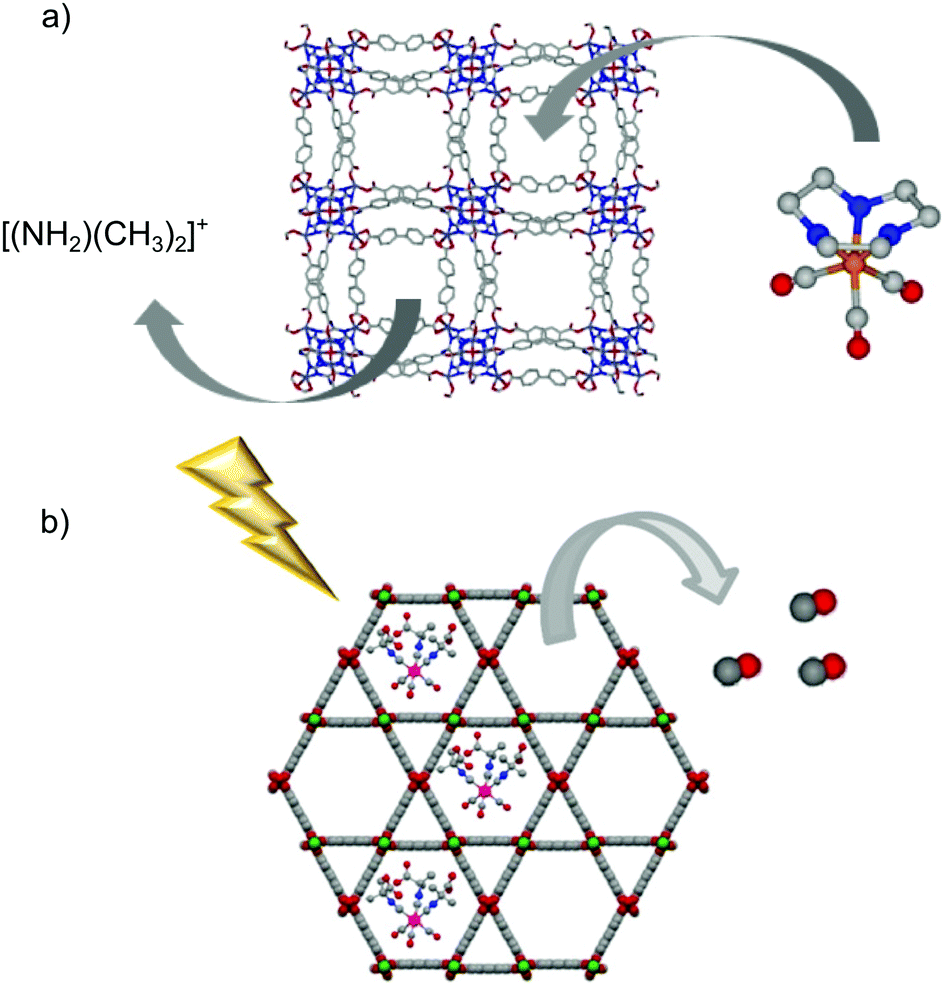 | ||
| Fig. 7 Schematic representations of the encapsulation of gas-donors into the cavities of metal–organic frameworks. (a) Encapsulation of the photoCORM [Mn(tacn)(CO)3]Br into the porous framework of bio-MOF-1 ((NH2(CH3)2)2[Zn8(adeninate)4(BPDC)6]·8DMF·11H2O) by cationic exchange. (b) Representation of the light triggered CO-release from the hybrid system photoCORM@MOF [Mo(CNCMe2CO2H)3(CO)3]@[Al(OH)(stilbenedicarboxylate)]n. The MOF-based system releases CO under light irradiation while it keeps trapped most of the metal-fragments encapsulated when suspended in physiological buffer. Adapted with permission from ref. 101. Copyright 2018 American Chemical Society. | ||
A CO-releasing material was prepared by the encapsulation of the photoCORM [Mo(CNCMe2CO2H)3(CO)3] in a hierarchical structure of [Zn2(dobdc)]n.100 The microporous nature of the Zn-MOF with a pore aperture of 1.0 × 1.4 nm may hamper the diffusion of the bulky Mo-CORM (ca. 1.3 nm). To overcome these problems, the authors used a hierarchical structure of [Zn2(dobdc)]n showing textural mesopores. The CORM was successfully encapsulated by one-pot synthesis-encapsulation (0.3 mmol of CORM per gram of material). The hybrid system exhibited a more sustained CO-release than the pristine CORM when irradiated with UV light. In addition, the material was able to keep trapped 87% of the Mo-fragment incorporated after incubation in physiological media, limiting its potential toxicity. The same CORM was also encapsulated in the cavities of the mesoporous MOF CYCU-3 [Al(OH)(SDC)]n (H2SDC: 4,4-stilbenedicarboxylic acid) (Fig. 7b).101 The coordination-modulation method102 was used to prepare several materials with different particle features, ranging from pseudospherical nanoparticles to micrometric needles. Noteworthily, the use of higher concentration of the modulator (50 times the linker concentration) led to a denser framework (CYCU-3′), whose structure was elucidated by continuous rotation electron diffraction (CRED). Although CYCU-3′ showed a lower porosity than pristine CYCU-3, the channels were big enough to accommodate the CORM molecules, with both systems showing similar payloads. Remarkably, the lower porosity in CYCU-3′ resulted in higher stability, enabling it to retain 75% of Mo-fragments encapsulated after being suspended in PBS during 72 hours. In addition, the hybrid CYCU-3′ system enabled a more sustained release of CO, both in solution and in solid state, showing increased potential toward different applications.
5.4 Chemical attachment of gas-donors
The versatility of MOFs enables the use of functional ligands with the ability to store and deliver gasotransmitters in their synthesis or their incorporation by post-synthetic modifications. This strategy offers the advantage of ensuring a very high payload of the donor moiety because it is incorporated in the structure. It also limits the extent of leaching, as in most of the cases the gas molecules are strongly attached to the organic moiety.Diazeniumdiolate (NONOate) is probably the most famous NO-releasing functionality. It is easily synthesized from secondary amines under the high pressure of NO gas and releases two molecules of NO by acid-catalyzed spontaneous decomposition with the first-order kinetics, and thus the releasing rate of NO can be precisely predicted. Moreover, its decomposition rate can be tuned by subtle changes in its structure.103 Therefore, many kinds of NONOate derivatives, which have various half-lives ranging from few seconds to several hours, have been tested in experimental models of cardiovascular diseases.
Rosseinsky and co-workers were the first to synthesize the NONOate moiety inside a framework structure.104 In this study, a two-step post synthetic modification of HKUST-1 was implemented. First, 4-(methylamino)-pyridine (denoted as 4-map) was coordinated to the OMSs. Subsequently, the secondary amines were converted to the NONOate functionality by reacting them with NO gas at high pressure. This second step was conditioned by the first step. The reaction did not occur when the loading of 4-map was close to 100% due to the pore blocking effect. On the other hand, the reaction proceeded when the loading was lower than 60%. As a proof of concept, the NO delivery was confirmed in deionized water. However, the NO release was accompanied by significant leaching of 4-map. Cohen and co-workers simplified the strategy by implementing it in two different MOFs, which contain the 2-aminoterephthalate moiety in their structure.105 Therefore, the NONOate functionality was introduced into [Zn4O(NH2-bdc)]n (denoted as IRMOF-3)106 and [Zn4O(NH2-bdc)(btb)4/3]n (denoted as UMCM-1-NH2, btb = benzene-1,3,5-tribenzoate).107 The yield of the reaction (NONOate formation) for IRMOF-3 and UMCM-NH2 was 44% and 83% respectively, which is consistent with the higher porosity of the latter. The release amount of NO in water was moderate in both cases (less than 10%) due to the very rapid degradation of both structures.
The poor hydrolytic stability of the NONOate functionalized MOFs limits their scope as NO releasing vehicles in water (or physiological media). However, they can be employed in the sustained release of NO in solid state upon exposure to humid gas. To this end, a ligand bearing three amine functionalities was employed in the synthesis of the MOF [Cu3(TDPAT)(H2O)3]n framework (denoted as Cu-TDPAT, TDPAT = 2,4,6-tris(3,5-dicarboxylphenylamino)-1,3,5-triazine).108 The amine functionalities were converted to NONOate functionalities by reaction with NO gas in a similar fashion to previously described for IRMOF-3 and UMCM-1-NH2. The NO releasing experiments in the solid state revealed that the Cu-TDPAT NONOate released NO when exposed to humid gas (RH = 85%) over the course of 7 days and with a maximum release of 0.175 mmol g−1.
Very recently, Xu et al. combined the 4-map@HKUST-1 system, showing NONOate functionalities, with a co-axial core–shell polycaprolactene-gelatin fiber to generate a NO-releasing micropatterned scaffold.109 While the gelatin shell improves the biocompatibility of the nanofibers, the MOF particles are embedded in the hydrophobic polycaprolactene core, which protects them from water. As a result, the hybrid system shows a more gradual NO-release under incubation in PBS than the pristine NO-loaded 4-map@HKUST-1 system. The copper leaching, due to the degradation of the MOF, is also more sustained in the case of the hybridized system. Given that both NO and copper ions elicit positive response toward angiogenesis, they tested the micropatterned scaffold as a bandage for wound healing in mice. The NO-loaded scaffold showed a notable reduction of the wound, much higher than the untreated control after 7 days (85.23% and 34.89%, respectively). Significantly, the micropatterned scaffold not loaded with NO shows a better performance than the control (71.9% of reduction vs. 34.89%), confirming the activity of the leached copper ions towards wound healing. This example brings to light the potentiality of hybridizing MOFs with other materials to generate macroscale scaffolds with improved properties towards their real use in gas therapy.
Most of the NO-releasing MOFs discussed so far used humidity (water molecule) to trigger the release. However, humidity is a not an easy to control parameter in real physiological conditions, which implies that the degree of control in the delivery of the bio gas is limited. On the other hand, light is a more suitable stimulus because it is non-invasive and can be manipulated in terms of intensity, wavelength and location. Therefore, the use of photodonor ligands as part of the framework is a promising strategy to develop new photoresponsive gasotransmitter releasing materials. Furukawa, Kamei and co-workers first implemented this strategy by using imidazole ligands bearing the nitro functionality in the synthesis of zeolitic imidazole frameworks (ZIF) (Fig. 8a). Aromatic molecules with nitro groups are well known for their NO photogeneration.110,111 Thus, the reaction of 2-nitroimidazole (2-nIm) or 5-methyl-4-nitroimidazole (mnIm) with Zn(II) ions yielded [Zn(2nIm)2]n (NOF-1) and [Zn(mnIm)2]n (NOF-2).112 Interestingly, both frameworks showed light triggered NO release in the solid-state with a maximum release of 3 mmol g−1. The light used for these experiments was set at 365 nm. Interestingly, the NO release duration and rate could be controlled by light on/off and intensity. It is noteworthy that the light triggered NO release efficiency of the ligands themselves was significantly lower (less than 0.5 mmol g−1) than the respective frameworks. The difference in photoreactivity between the frameworks and the ligands is attributed to the structuring effect of the frameworks. The spatial segregation of the photoreactive moieties within the framework avoids the aggregation induced quenching observed in the ligands in the solid-state, which paves the way to new solid-state NO releasing materials. The solid-state delivery of NO of NOF-1 crystals enabled their integration into substrates that can then be used as functional extracellular scaffolds. Using this cell culture platform, the spatiotemporal stimulation of cells by NO was demonstrated, which subsequently caused an increase of intracellular Ca2+ ion concentration in the genetically modified HEK293 cells with TRPC5 channels.
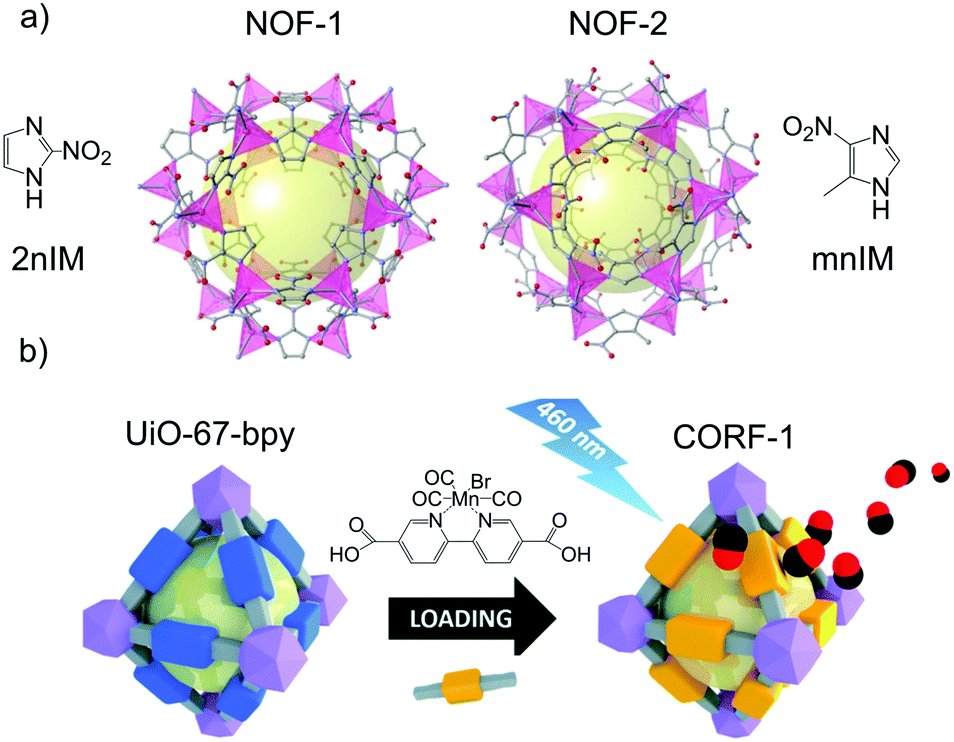 | ||
| Fig. 8 Examples of photoresponsive gas-releasing metal–organic frameworks prepared by the chemical attachment of gas donors. (a) Chemical structures of [Zn(2nIm)2]n (NOF-1) and [Zn(mnIm)2]n (NOF-2). Reprinted with permission from ref. 112. (b) Post-functionalization of the linker of the MOF [Zr6O4(OH)4(bpydc)6] with a manganese-carbonyl group to generate a photoresponsive CO-releasing material. Reprinted from ref. 117 with permission from the Royal Society of Chemistry. | ||
Besides the nitro aromatic groups, the bis-N-nitroso (BNN) moiety is also known to release NO upon light irradiation. However, its low thermal stability prevents its use in the direct synthesis of the MOF. Thus, it must be incorporated in the framework structure by post-synthetic modification through nitrosation of secondary amines. Therefore, two MOFs with bis-amino moieties in their structure were synthesized: [Ti8O8(OH)4(MeNH-bdc)6]n (preNOF-11) and [Al8(OMe)8(OH)4(MeNH-bdc)6]n (preNOF-12), where MeNH-bdc is 2,5-bis(methylamino)-1,mend4-benzenedicarboxylic acid. Subsequently, the BNN moiety was introduced by post-synthetic nitrosation.113 NOF-11 and NOF-12 released 1.60 mmol g−1 and 2.78 mmol g−1, respectively, upon white light (300–600 nm) irradiation in the solid-state. In contrast to the above-mentioned examples, both frameworks showed remarkable stability not only in water but also in phosphate buffered solutions. This high stability stems from two facts: the strong coordination of high valence metal ions, such as Ti(IV) and Al(III), with carboxylates,114 and the BNN moiety decorating the pores, which hinders water and phosphate inclusion preventing hydrolytic degradation.
Even though the most common strategy consists in introducing in the MOF synthesis ligands with the potential to release gasotransmitter molecules under certain stimuli, behaving as pro-drugs, it is also possible to exploit the affinity of these gases towards certain organic groups. This was recently demonstrated by Alkordi and co-workers. They showed that the NH2 functionality decorating the pores of UiO-66-NH2 can trap up to 6.2 mmol g−1 by chemisorption. The loaded compound released up to 1.1 mmol g−1 in acidified PBS media.115
The presence of free moieties with coordination capabilities decorating the pores of the MOF can be exploited to incorporate metal complexes to the structure through post synthetic modifications. A paradigmatic example of this approach is provided by the reactivity of ligands bearing free 2,2-bipyridine moieties towards transition metal ions.116 This reactivity was exploited to incorporate the CO photoreleasing moiety MnBr(bpydc)(CO)3 (bpydc is 5,5-dicarboxylate-2,2-bipyridine) within the porous structure of the Zr(IV) based UiO-67 [Zr6O4(OH)4(bpydc)6] through post synthetic metalation of the free 2,2-bipyridine centers with MnBr(CO)5 (Fig. 8b).117 MnBr(bpydc)(CO)3@UiO-67 (named as CORF-1) was able to efficiently release CO in the solid-state upon visible light irradiation (465 nm) with a maximum release of 4.7 mmol g−1, which was easily controlled by turning on and off the light source. These features enabled the preparation of photoresponsive CO releasing cell-growth substrates by immobilizing CORF-1 crystals within the biocompatible PDMS polymer. These functional cellular scaffolds were used to observe the cellular uptake of the on-demand generated CO from the CORF-1 crystals.
6. Protein assemblies
Protein crystals, which are the solid-state assembly of proteins, have attracted interest as porous materials. Typical protein crystals show from 30 to 65% of void volume that are filled by solvent molecules.118 The pore surface is composed of residual amino acid chains, which have potential as metal coordination sites and, therefore, can be used for anchoring gas-donors to the biomaterial.119,120 The low yield in the synthesis of protein crystals and their low stability are the main disadvantages of this strategy. On the other hand, their inherent lack of toxicity is a great advantage. In fact, protein assemblies are the more biocompatible gasotransmitter carriers discussed in this review.6.1 Chemical functionalization with gas-donors
Romao and co-workers studied, for the first time, the interaction of CO-releasing molecules, namely [fac-Ru(CO)3Cl(glycinate)], (CORM-3), and different proteins. They observed the coordination of the carbonyl-metal to side chains of amino acids, forming Ru(CO)2-protein adducts with the concomitant loss of one CO and glycinate ligands. However, they did not study the potential CO-releasing properties of these new materials.121,122 Later, Ueno and co-workers proved the feasibility of using ferritin as a CO-donor carrier.123 This protein includes 24 subunits with 8 nm inner diameter (Fig. 9). By reacting with [Ru(CO)3Cl2]2, protein-Ru(CO)2 was formed. With the aim to modulate the CO-releasing properties of the system, they used three different ferritin mutants. Each mutant contained a different amount of aminoamides side chains in their structures, resulting in three novel CO-releasing materials. All composites slowed down the CO-release rate in comparison to the pristine CORM when suspended in physiological conditions. In addition, the cellular uptake of the composites was superior to that of the bare CORM, proving the utility of protein cages as a carrier of CO-releasing molecules. In another work, they extended this approach to attach ruthenium carbonyls to a cross-linked hen egg with lysozyme crystals. As in the case of ferritin, the novel nanocomposites showed a slower CO-release than the pristine CORM when suspended in physiological media. Furthermore, they elicited biological response when incubated in living cells.124 The same authors further gained control in the delivery of CO from protein assemblies by introducing a Mn(I) based photoCORM.125 Thus, a mutant version of ferritin rich in cysteines was used to coordinate Mn(I) carbonyl species. Interestingly the composite protein assembly released CO only upon light irradiation at 456 nm. Owing to the biocompatible nature of the CO carrier, it could be used as an intracellular CO delivery vehicle. The high degree of control achieved in the CO delivery together with the biocompatibility of the carrier makes this material highly promising to further understand the role of CO at the cellular level.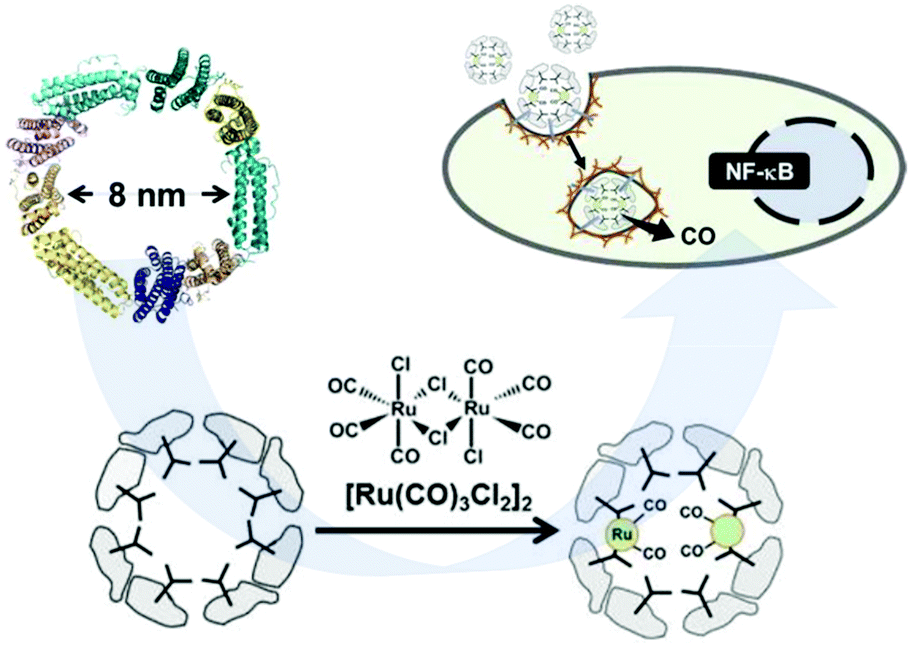 | ||
| Fig. 9 Schematic representation of the subunit of ferritin with an inner diameter of 8 nm, formation of the Ru(CO)2–protein adduct and cellular uptake of the Ferritin–Ru(CO)2 composite and intracellular CO-release. Adapted with permission from ref. 123. Copyright 2014 American Chemical Society. | ||
Very recently, Cai et al. designed a novel CO-releasing material based on a self-assembly of fluorinated amphiphilic dendritic peptide (FADP) with a hydrophobic core, co-encapsulating the photosensitizer chlorin e6 (Ce6) and the CO-releasing molecule [Mn(CO)4(S2CNMe)(CH2CO2H)] (CORM-401).126 As CORM-401 is a H2O2-responsive CORM, the presence of Ce6 enabled indirect control of CO-release by light. Under irradiation at 665 nm in water, Ce6 generates relevant concentrations of 1O2 and H2O2. The reaction of generated H2O2 with the CORM-401 triggers the CO-release. While the fluorinated surface enhances the binding affinity towards bacterial membranes, the co-delivery of CO and 1O2 exerts a significant synergistic bactericidal effect. In vitro and in vivo studies proved that the hybrid system showed a high antibacterial activity under NIR irradiation, together with good biocompatibility. Likewise, the hybridized system had a significant activity towards the ablation of biofilms. These features make CORM-401-Ce6@FADP a promising formulation toward its real application as a bactericidal agent.
7. Conclusions and outlook
After nearly fifteen years of multidisciplinary research that combined gas biology and porous materials, the resulting knowledge has led to a versatile toolbox of materials and strategies to load and release gasotransmitters from porous materials. Thus, researchers can tune the stimuli to deliver gases from porous materials as well as to control their release kinetics to meet the criteria for selected biomedical applications, such as wound healing, vasodilation and anti-inflammation among others.As we have summarized in this review, zeolites offer a robust and non-toxic framework with well-ordered OMSs being able to store NO that can be spontaneously released through water exchange or produce it in situ through catalytic reactions. Therefore, while zeolites excel in the prolonged and spontaneous release of NO, it is challenging to modulate their performance toward NO release on demand because humidity and concentration of reactants cannot be easily modulated in vivo. Alternatively, the large pores of mesoporous silica allow encapsulating gas releasing molecules that are sensitive to different stimuli such as light and heat. This strategy offers the advantage of easily incorporating the advances achieved in the field of gasotransmitter-releasing molecules into the porous material field. Thus, mesoporous silica has proven to be a versatile material for releasing all known gasotransmitters through different stimuli, which made it possible to attain important goals in the field of gas biology such as on-demand gasotransmitter release. MOFs provided their versatility in terms of structures, composition and reactivity to the field of gas biology, which enabled the development of novel routes toward the controlled release of gasotransmitters. For example, the incorporation of gas releasing moieties in the organic linker of the MOF not only achieved high payloads of gases but also their light triggered release in the solid-state paving the way for its implementation in novel devices. All these materials share the fact that they are exogenous, which might rise some concerns on their biocompatibility. In this regard, protein assemblies offer inherently biocompatible scaffolds in which to localize gasotransmitters at the cost of providing low payloads and stability.
However, as highlighted in several examples across this review, the successful implementation of porous materials for the therapeutic release of gasotransmitters in the real world application requires an appropriate strategy for their materialization/formulation and administration. Thus, processing of gasotransmitter releasing porous materials into stable colloids, gels, or ointments or shape them into macroscopic objects through hybridization with other materials such as polymers is a promising strategy toward their practical use. All these strategies entail specific challenges that still need to be overcome. For instance, the intravenous administration of the porous carriers implies the use of nanoscopic carriers with narrow size distribution and structural and colloidal stability in physiological media. In this regard, coating the porous carriers with biocompatible polymers and lipids has been shown to be a promising strategy to prevent degradation and increase colloidal stability. Alternatively, the use of non-toxic building blocks held by strong interactions in the synthesis of nanoscopic porous carriers has been shown to increase the stability/biocompatibility of the porous carrier as exemplified by titanosilicates and MOFs made with high valence metal ions (i.e. Fe(III), Zr(IV) and Ti(IV)). The synthesis of stable nano-sized porous carriers for gasotransmitters together with further studies on their biodistribution will pave the way for their efficient intravenous administration. On the other hand, the use of macroscopic devices to exogenously deliver gasotransmitters to targeted organs must ensure a robust and reproducible performance over several cycles. In this regard, the possibility to shape porous materials into gels,127 liquids,128 melted liquids129 or monoliths130 seems to be a promising strategy to process these materials into macroscopic objects without the need of an external matrix that dilutes them.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge iCeMS for supporting the gas biology materials project. This research was supported by AMED under Grant Number 20lm0203013j0002.Notes and references
- A. K. Mustafa, M. M. Gadalla, N. Sen, S. Kim, W. Mu, S. K. Gazi, R. K. Barrow, G. Yang, R. Wang and S. H. Snyder, Sci. Signaling, 2009, 2, ra72 Search PubMed.
- R. U. I. Wang, FASEB J., 2002, 16, 1792–1798 CrossRef CAS.
- R. Wang, Trends Biochem. Sci., 2014, 39, 227–232 CrossRef CAS.
- R. M. J. Palmer, A. G. Ferrige and S. Moncada, Nature, 1987, 327, 524 CrossRef CAS.
- D. J. Stuehr, Biochim. Biophys. Acta, Bioenerg., 1999, 1411, 217–230 CrossRef CAS.
- R. Tenhunen, H. S. Marver and R. Schmid, Proc. Natl. Acad. Sci. U. S. A., 1968, 61, 748–755 CrossRef CAS.
- L. Wu and R. Wang, Pharmacol. Rev., 2005, 57, 585–630 CrossRef CAS.
- R. Motterlini and L. E. Otterbein, Nat. Rev. Drug Discovery, 2010, 9, 728–743 CrossRef CAS.
- M. S. Vandiver and S. H. Snyder, J. Mol. Med., 2012, 90, 255–263 CrossRef CAS.
- R. Wang, Physiol. Rev., 2012, 92, 791–896 CrossRef CAS.
- D. Babu, R. Motterlini and R. A. Lefebvre, Br. J. Pharmacol., 2015, 172, 1557–1573 CrossRef CAS.
- C. C. Romão, W. A. Blättler, J. D. Seixas and G. J. L. Bernardes, Chem. Soc. Rev., 2012, 41, 3571–3583 RSC.
- U. Schatzschneider, Br. J. Pharmacol., 2015, 172, 1638–1650 CrossRef CAS.
- P. G. Wang, M. Xian, X. Tang, X. Wu, Z. Wen, T. Cai and A. J. Janczuk, Chem. Rev., 2002, 102, 1091–1134 CrossRef CAS.
- N. Naghavi, A. de Mel, O. S. Alavijeh, B. G. Cousins and A. M. Seifalian, Small, 2013, 9, 22–35 CrossRef CAS.
- Y. Zhao, T. D. Biggs and M. Xian, Chem. Commun., 2014, 50, 11788–11805 RSC.
- S. M. Fix, M. A. Borden and P. A. Dayton, J. Controlled Release, 2015, 209, 139–149 CrossRef CAS.
- T. Yang, A. N. Zelikin and R. Chandrawati, Adv. Sci., 2018, 5, 1701043 CrossRef.
- K. Ling, F. Men, W.-C. Wang, Y.-Q. Zhou, H.-W. Zhang and D.-W. Ye, J. Med. Chem., 2018, 61, 2611–2635 CrossRef CAS.
- L. Chen, S.-F. Zhou, L. Su and J. Song, ACS Nano, 2019, 13, 10887–10917 CrossRef CAS.
- M. E. Davis, Nature, 2002, 417, 813 CrossRef CAS.
- S. Wang, Microporous Mesoporous Mater., 2009, 117, 1–9 CrossRef CAS.
- K. Hadjiivanov, E. Ivanova and H. Knözinger, Microporous Mesoporous Mater., 2003, 58, 225–236 CrossRef CAS.
- M. Ozekmekci, G. Salkic and M. F. Fellah, Fuel Process. Technol., 2015, 139, 49–60 CrossRef CAS.
- P. S. Wheatley, A. R. Butler, M. S. Crane, S. Fox, B. Xiao, A. G. Rossi, I. L. Megson and R. E. Morris, J. Am. Chem. Soc., 2006, 128, 502–509 CrossRef CAS.
- H. A. Liu and K. J. Balkus, Chem. Mater., 2009, 21, 5032–5041 CrossRef CAS.
- S. Fox, T. S. Wilkinson, P. S. Wheatley, B. Xiao, R. E. Morris, A. Sutherland, A. J. Simpson, P. G. Barlow, A. R. Butler, I. L. Megson and A. G. Rossi, Acta Biomater., 2010, 6, 1515–1521 CrossRef CAS.
- M. Neidrauer, U. K. Ercan, A. Bhattacharyya, J. Samuels, J. Sedlak, R. Trikha, K. A. Barbee, M. S. Weingarten and S. G. Joshi, J. Med. Microbiol., 2014, 63, 203–209 CrossRef CAS.
- M. L. Pinto, J. Rocha, J. R. B. Gomes and J. Pires, J. Am. Chem. Soc., 2011, 133, 6396–6402 CrossRef CAS.
- M. L. Pinto, A. C. Fernandes, J. Rocha, A. Ferreira, F. Antunes and J. Pires, J. Mater. Chem. B, 2014, 2, 224–230 RSC.
- M. L. Pinto, A. C. Fernandes, F. Antunes, J. Pires and J. Rocha, Microporous Mesoporous Mater., 2016, 229, 83–89 CrossRef CAS.
- R. V. Pinto, A. C. Fernandes, F. Antunes, Z. Lin, J. Rocha, J. Pires and M. L. Pinto, Nitric oxide, 2019, 90, 29–36 CrossRef CAS.
- E. I. Tocheva, F. I. Rosell, A. G. Mauk and M. E. P. Murphy, Science, 2004, 304, 867 CrossRef CAS.
- A.-K. Boes, P. S. Wheatley, B. Xiao, I. L. Megson and R. E. Morris, Chem. Commun., 2008, 6146–6148 RSC.
- G. Turnes Palomino, P. Fisicaro, S. Bordiga, A. Zecchina, E. Giamello and C. Lamberti, J. Phys. Chem. B, 2000, 104, 4064–4073 CrossRef CAS.
- W. J. Roth, P. Nachtigall, R. E. Morris, P. S. Wheatley, V. R. Seymour, S. E. Ashbrook, P. Chlubná, L. Grajciar, M. Položij, A. Zukal, O. Shvets and J. Čejka, Nat. Chem., 2013, 5, 628–633 CrossRef CAS.
- R. A. Doyle, S. E. Russell and R. E. Morris, Microporous Mesoporous Mater., 2019, 280, 367–371 CrossRef CAS.
- S. E. Russell, J. M. González Carballo, C. Orellana-Tavra, D. Fairen-Jimenez and R. E. Morris, Dalton Trans., 2017, 46, 3915–3920 RSC.
- J. L. Vivero-Escoto, I. I. Slowing, B. G. Trewyn and V. S. Y. Lin, Small, 2010, 6, 1952–1967 CrossRef CAS.
- Z. Li, J. C. Barnes, A. Bosoy, J. F. Stoddart and J. I. Zink, Chem. Soc. Rev., 2012, 41, 2590–2605 RSC.
- F. Hoffmann, M. Cornelius, J. Morell and M. Fröba, Angew. Chem., Int. Ed., 2006, 45, 3216–3251 CrossRef CAS.
- J. J. Koehler, J. Zhao, S. S. Jedlicka, D. M. Porterfield and J. L. Rickus, J. Phys. Chem. B, 2008, 112, 15086–15093 CrossRef CAS.
- V. P. Torchilin, Adv. Drug Delivery Rev., 2006, 58, 1532–1555 CrossRef CAS.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751 CrossRef CAS.
- D. M. Porterfield, J. D. Laskin, S.-K. Jung, R. P. Malchow, B. Billack, P. J. S. Smith and D. E. Heck, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2001, 281, L904–L912 CrossRef CAS.
- C. Zolkov, D. Avnir and R. Armon, J. Mater. Chem., 2004, 14, 2200–2205 RSC.
- S. S. Jedlicka, J. L. McKenzie, S. J. Leavesley, K. M. Little, T. J. Webster, J. P. Robinson, D. E. Nivens and J. L. Rickus, J. Mater. Chem., 2006, 16, 3221–3230 RSC.
- B. J. Heilman, J. St. John, S. R. J. Oliver and P. K. Mascharak, J. Am. Chem. Soc., 2012, 134, 11573–11582 CrossRef CAS.
- X. Zhang, G. Tian, W. Yin, L. Wang, X. Zheng, L. Yan, J. Li, H. Su, C. Chen, Z. Gu and Y. Zhao, Adv. Funct. Mater., 2015, 25, 3049–3056 CrossRef CAS.
- C. Hu, J. Sun, Y. Zhang, J. Chen, Y. Lei, X. Sun and Y. Deng, Adv. Healthcare Mater., 2018, 7, 1801047 CrossRef.
- F. J. Carmona, S. Rojas, P. Sánchez, H. Jeremias, A. R. Marques, C. C. Romão, D. Choquesillo-Lazarte, J. A. R. Navarro, C. R. Maldonado and E. Barea, Inorg. Chem., 2016, 55, 6525–6531 CrossRef CAS.
- M. A. Gonzales, H. Han, A. Moyes, A. Radinos, A. J. Hobbs, N. Coombs, S. R. J. Oliver and P. K. Mascharak, J. Mater. Chem. B, 2014, 2, 2107–2113 RSC.
- I. Chakraborty, S. J. Carrington, J. Hauser, S. R. J. Oliver and P. K. Mascharak, Chem. Mater., 2015, 27, 8387–8397 CrossRef CAS.
- F. J. Carmona, I. Jiménez-Amezcua, S. Rojas, C. C. Romão, J. A. R. Navarro, C. R. Maldonado and E. Barea, Inorg. Chem., 2017, 56, 10474–10480 CrossRef CAS.
- G. Caliendo, G. Cirino, V. Santagada and J. L. Wallace, J. Med. Chem., 2010, 53, 6275–6286 CrossRef CAS.
- X. Sun, B. Kong, W. Wang, P. Chandran, C. Selomulya, H. Zhang, K. Zhu, Y. Liu, W. Yang, C. Guo, D. Zhao and C. Wang, J. Mater. Chem. B, 2015, 3, 4451–4457 RSC.
- W. Wang, X. Sun, H. Zhang, C. Yang, Y. Liu, W. Yang, C. Guo and C. Wang, Int. J. Nanomed., 2016, 11, 3255–3263 CrossRef CAS.
- S. M. C. Neiva, J. A. V. Santos, J. C. Moreira, Y. Gushikem, H. Vargas and D. W. Franco, Langmuir, 1993, 9, 2982–2985 CAS.
- B. J. Nablo, T.-Y. Chen and M. H. Schoenfisch, J. Am. Chem. Soc., 2001, 123, 9712–9713 CrossRef CAS.
- P. G. Zanichelli, R. L. Sernaglia and D. W. Franco, Langmuir, 2006, 22, 203–208 CrossRef CAS.
- F. G. Doro, U. P. Rodrigues-Filho and E. Tfouni, J. Colloid Interface Sci., 2007, 307, 405–417 CrossRef CAS.
- J. H. Shin, S. K. Metzger and M. H. Schoenfisch, J. Am. Chem. Soc., 2007, 129, 4612–4619 CrossRef CAS.
- F. O. N. da Silva, E. C. C. Gomes, T. D. S. Francisco, A. K. M. Holanda, I. C. N. Diógenes, E. H. S. de Sousa, L. G. F. Lopes and E. Longhinotti, Polyhedron, 2010, 29, 3349–3354 CrossRef CAS.
- F. Wei, Q. Hou, J. Y. Yang and J. H. Zhu, J. Colloid Interface Sci., 2011, 356, 526–535 CrossRef CAS.
- M. B. Yue, L. B. Sun, T. T. Zhuang, X. Dong, Y. Chun and J. H. Zhu, J. Mater. Chem., 2008, 18, 2044–2050 RSC.
- R. J. Soto, L. Yang and M. H. Schoenfisch, ACS Appl. Mater. Interfaces, 2016, 8, 2220–2231 CrossRef CAS.
- A. L. Tessaro, A. Fraix, A. C. Pedrozo da Silva, E. Gazzano, C. Riganti and S. Sortino, Nanomaterials, 2019, 9, 823 CrossRef CAS.
- D. Afonso, S. Valetti, A. Fraix, C. Bascetta, S. Petralia, S. Conoci, A. Feiler and S. Sortino, Nanoscale, 2017, 9, 13404–13408 RSC.
- M. J. Malone-Povolny and M. H. Schoenfisch, ACS Appl. Mater. Interfaces, 2019, 11, 12216–12223 CrossRef CAS.
- R. Guo, Y. Tian, Y. Wang and W. Yang, Adv. Funct. Mater., 2017, 27, 1606398 CrossRef.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705 CrossRef CAS.
- S. Kitagawa, R. Kitaura and S.-I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS.
- G. Férey, C. Mellot-Draznieks, C. Serre and F. Millange, Acc. Chem. Res., 2005, 38, 217–225 CrossRef.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS.
- J. Yang and Y.-W. Yang, Small, 2020, 16, 1906846 CrossRef CAS.
- J. Yang and Y.-W. Yang, View, 2020, 1, e20 Search PubMed.
- X. Kong, E. Scott, W. Ding, J. A. Mason, J. R. Long and J. A. Reimer, J. Am. Chem. Soc., 2012, 134, 14341–14344 CrossRef CAS.
- B. Xiao, P. S. Wheatley, X. Zhao, A. J. Fletcher, S. Fox, A. G. Rossi, I. L. Megson, S. Bordiga, L. Regli, K. M. Thomas and R. E. Morris, J. Am. Chem. Soc., 2007, 129, 1203–1209 CrossRef CAS.
- A. C. McKinlay, B. Xiao, D. S. Wragg, P. S. Wheatley, I. L. Megson and R. E. Morris, J. Am. Chem. Soc., 2008, 130, 10440–10444 CrossRef CAS.
- F. Bonino, S. Chavan, J. G. Vitillo, E. Groppo, G. Agostini, C. Lamberti, P. D. C. Dietzel, C. Prestipino and S. Bordiga, Chem. Mater., 2008, 20, 4957–4968 CrossRef CAS.
- D. Cattaneo, S. J. Warrender, M. J. Duncan, C. J. Kelsall, M. K. Doherty, P. D. Whitfield, I. L. Megson and R. E. Morris, RSC Adv., 2016, 6, 14059–14067 RSC.
- E. D. Bloch, W. L. Queen, S. Chavan, P. S. Wheatley, J. M. Zadrozny, R. Morris, C. M. Brown, C. Lamberti, S. Bordiga and J. R. Long, J. Am. Chem. Soc., 2015, 137, 3466–3469 CrossRef CAS.
- S. Rojas, P. S. Wheatley, E. Quartapelle-Procopio, B. Gil, B. Marszalek, R. E. Morris and E. Barea, CrystEngComm, 2013, 15, 9364–9367 RSC.
- À. Ruyra, A. Yazdi, J. Espín, A. Carné-Sánchez, N. Roher, J. Lorenzo, I. Imaz and D. Maspoch, Chem. – Eur. J., 2015, 21, 2508–2518 CrossRef.
- P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J.-S. Chang, Y. K. Hwang, V. Marsaud, P.-N. Bories, L. Cynober, S. Gil, G. Férey, P. Couvreur and R. Gref, Nat. Mater., 2009, 9, 172 CrossRef.
- A. C. McKinlay, J. F. Eubank, S. Wuttke, B. Xiao, P. S. Wheatley, P. Bazin, J. C. Lavalley, M. Daturi, A. Vimont, G. De Weireld, P. Horcajada, C. Serre and R. E. Morris, Chem. Mater., 2013, 25, 1592–1599 CrossRef CAS.
- J. F. Eubank, P. S. Wheatley, G. Lebars, A. C. McKinlay, H. Leclerc, P. Horcajada, M. Daturi, A. Vimont, R. E. Morris and C. Serre, APL Mater., 2014, 2, 124112 CrossRef.
- S. R. Miller, E. Alvarez, L. Fradcourt, T. Devic, S. Wuttke, P. S. Wheatley, N. Steunou, C. Bonhomme, C. Gervais, D. Laurencin, R. E. Morris, A. Vimont, M. Daturi, P. Horcajada and C. Serre, Chem. Commun., 2013, 49, 7773–7775 RSC.
- R. V. Pinto, S. Wang, S. R. Tavares, J. Pires, F. Antunes, A. Vimont, G. Clet, M. Daturi, G. Maurin, C. Serre and M. L. Pinto, Angew. Chem., Int. Ed., 2020, 59, 5135–5143 CrossRef CAS.
- M. Ma, H. Noei, B. Mienert, J. Niesel, E. Bill, M. Muhler, R. A. Fischer, Y. Wang, U. Schatzschneider and N. Metzler-Nolte, Chem. – Eur. J., 2013, 19, 6785–6790 CrossRef CAS.
- A. J. Atkin, J. M. Lynam, B. E. Moulton, P. Sawle, R. Motterlini, N. M. Boyle, M. T. Pryce and I. J. S. Fairlamb, Dalton Trans., 2011, 40, 5755–5761 RSC.
- P. K. Allan, P. S. Wheatley, D. Aldous, M. I. Mohideen, C. Tang, J. A. Hriljac, I. L. Megson, K. W. Chapman, G. De Weireld, S. Vaesen and R. E. Morris, Dalton Trans., 2012, 41, 4060–4066 RSC.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- D. L. H. Williams, Acc. Chem. Res., 1999, 32, 869–876 CrossRef CAS.
- J. L. Harding and M. M. Reynolds, J. Am. Chem. Soc., 2012, 134, 3330–3333 CrossRef CAS.
- M. J. Neufeld, J. L. Harding and M. M. Reynolds, ACS Appl. Mater. Interfaces, 2015, 7, 26742–26750 CrossRef CAS.
- J. L. Harding, J. M. Metz and M. M. Reynolds, Adv. Funct. Mater., 2014, 24, 7503–7509 CrossRef CAS.
- M. J. Neufeld, A. Lutzke, J. B. Tapia and M. M. Reynolds, ACS Appl. Mater. Interfaces, 2017, 9, 5139–5148 CrossRef.
- M. J. Neufeld, A. Lutzke, W. M. Jones and M. M. Reynolds, ACS Appl. Mater. Interfaces, 2017, 9, 35628–35641 CrossRef CAS.
- F. J. Carmona, S. Rojas, C. C. Romão, J. A. R. Navarro, E. Barea and C. R. Maldonado, Chem. Commun., 2017, 53, 6581–6584 RSC.
- F. J. Carmona, C. R. Maldonado, S. Ikemura, C. C. Romão, Z. Huang, H. Xu, X. Zou, S. Kitagawa, S. Furukawa and E. Barea, ACS Appl. Mater. Interfaces, 2018, 10, 31158–31167 CrossRef CAS.
- T. Tsuruoka, S. Furukawa, Y. Takashima, K. Yoshida, S. Isoda and S. Kitagawa, Angew. Chem., Int. Ed., 2009, 48, 4739–4743 CrossRef CAS.
- J. A. Hrabie, J. R. Klose, D. A. Wink and L. K. Keefer, J. Org. Chem., 1993, 58, 1472–1476 CrossRef CAS.
- M. J. Ingleson, R. Heck, J. A. Gould and M. J. Rosseinsky, Inorg. Chem., 2009, 48, 9986–9988 CrossRef CAS.
- J. G. Nguyen, K. K. Tanabe and S. M. Cohen, CrystEngComm, 2010, 12, 2335–2338 RSC.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS.
- Z. Wang, K. K. Tanabe and S. M. Cohen, Inorg. Chem., 2009, 48, 296–306 CrossRef CAS.
- A. Lowe, P. Chittajallu, Q. Gong, J. Li and K. J. Balkus, Microporous Mesoporous Mater., 2013, 181, 17–22 CrossRef CAS.
- P. Zhang, Y. Li, Y. Tang, H. Shen, J. Li, Z. Yi, Q. Ke and H. Xu, ACS Appl. Mater. Interfaces, 2020, 12, 18319–18331 CrossRef CAS.
- K. Hishikawa, H. Nakagawa, T. Furuta, K. Fukuhara, H. Tsumoto, T. Suzuki and N. Miyata, J. Am. Chem. Soc., 2009, 131, 7488–7489 CrossRef CAS.
- S. Sortino, Chem. Soc. Rev., 2010, 39, 2903–2913 RSC.
- S. Diring, D. O. Wang, C. Kim, M. Kondo, Y. Chen, S. Kitagawa, K.-I. Kamei and S. Furukawa, Nat. Commun., 2013, 4, 2684 CrossRef.
- C. Kim, S. Diring, S. Furukawa and S. Kitagawa, Dalton Trans., 2015, 44, 15324–15333 RSC.
- T. Devic and C. Serre, Chem. Soc. Rev., 2014, 43, 6097–6115 RSC.
- R. R. Haikal, C. Hua, J. J. Perry, D. O’Nolan, I. Syed, A. Kumar, A. H. Chester, M. J. Zaworotko, M. H. Yacoub and M. H. Alkordi, ACS Appl. Mater. Interfaces, 2017, 9, 43520–43528 CrossRef CAS.
- E. D. Bloch, D. Britt, C. Lee, C. J. Doonan, F. J. Uribe-Romo, H. Furukawa, J. R. Long and O. M. Yaghi, J. Am. Chem. Soc., 2010, 132, 14382–14384 CrossRef CAS.
- S. Diring, A. Carne-Sanchez, J. Zhang, S. Ikemura, C. Kim, H. Inaba, S. Kitagawa and S. Furukawa, Chem. Sci., 2017, 8, 2381–2386 RSC.
- T. Ueno, Chem. – Eur. J., 2013, 19, 9096–9102 CrossRef CAS.
- T. O. Yeates and J. E. Padilla, Curr. Opin. Struct. Biol., 2002, 12, 464–470 CrossRef CAS.
- Y.-T. Lai, E. Reading, G. L. Hura, K.-L. Tsai, A. Laganowsky, F. J. Asturias, J. A. Tainer, C. V. Robinson and T. O. Yeates, Nat. Chem., 2014, 6, 1065 CrossRef CAS.
- T. Santos-Silva, A. Mukhopadhyay, J. D. Seixas, G. J. L. Bernardes, C. C. Romão and M. J. Romão, J. Am. Chem. Soc., 2011, 133, 1192–1195 CrossRef CAS.
- M. F. A. Santos, J. D. Seixas, A. C. Coelho, A. Mukhopadhyay, P. M. Reis, M. J. Romão, C. C. Romão and T. Santos-Silva, J. Inorg. Biochem., 2012, 117, 285–291 CrossRef CAS.
- K. Fujita, Y. Tanaka, T. Sho, S. Ozeki, S. Abe, T. Hikage, T. Kuchimaru, S. Kizaka-Kondoh and T. Ueno, J. Am. Chem. Soc., 2014, 136, 16902–16908 CrossRef CAS.
- H. Tabe, K. Fujita, S. Abe, M. Tsujimoto, T. Kuchimaru, S. Kizaka-Kondoh, M. Takano, S. Kitagawa and T. Ueno, Inorg. Chem., 2015, 54, 215–220 CrossRef CAS.
- K. Fujita, Y. Tanaka, S. Abe and T. Ueno, Angew. Chem., Int. Ed., 2016, 55, 1056–1060 CrossRef CAS.
- W. Ma, X. Chen, L. Fu, J. Zhu, M. Fan, J. Chen, C. Yang, G. Yang, L. Wu, G. Mao, X. Yang, X. Mou, Z. Gu and X. Cai, ACS Appl. Mater. Interfaces, 2020, 12, 22479–22491 CrossRef CAS.
- A. Carné-Sánchez, G. A. Craig, P. Larpent, T. Hirose, M. Higuchi, S. Kitagawa, K. Matsuda, K. Urayama and S. Furukawa, Nat. Commun., 2018, 9, 2506 CrossRef.
- N. Giri, M. G. Del Pópolo, G. Melaugh, R. L. Greenaway, K. Rätzke, T. Koschine, L. Pison, M. F. C. Gomes, A. I. Cooper and S. L. James, Nature, 2015, 527, 216–220 CrossRef CAS.
- R. Gaillac, P. Pullumbi, K. A. Beyer, K. W. Chapman, D. A. Keen, T. D. Bennett and F.-X. Coudert, Nat. Mater., 2017, 16, 1149–1154 CrossRef CAS.
- Y. Hara, K. Kanamori and K. Nakanishi, Angew. Chem., Int. Ed., 2019, 58, 19047–19053 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |