
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePerfluoroalkylative pyridylation of alkenes via 4-cyanopyridine-boryl radicals†
Jia
Cao
ac,
Guoqiang
Wang
a,
Liuzhou
Gao
a,
Hui
Chen
a,
Xueting
Liu
a,
Xu
Cheng
b and
Shuhua
Li
 *a
*a
aKey Laboratory of Mesoscopic Chemistry of Ministry of Education, Institute of Theoretical and Computational Chemistry, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing, 210093, P. R. China. E-mail: shuhua@nju.edu.cn
bInstitute of Chemistry and Biomedical Sciences, Jiangsu Key Laboratory of Advanced Organic Material, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing, 210093, P. R. China
cShaanxi Key Laboratory of Chemical Reaction Engineering, School of Chemistry and Chemical Engineering, Yan'an University, Yan'an 716000, P. R. China
First published on 16th January 2019
Abstract
A metal-free and photo-free method for the perfluoroalkylative pyridylation of alkenes has been developed via a combination of computational and experimental studies. Density functional theory calculations and control experiments indicate that the homolysis of Rf−X (X = Br, I) bonds by the 4-cyanopyridine-boryl radicals in situ generated from 4-cyanopyridine and B2pin2 is the key step. Sequential addition of Rf radicals to alkenes and the selective cross-coupling of the resulting alkyl radicals and 4-cyanopyridine-boryl radicals gives alkene difunctionalization products with a quaternary carbon center. This method exhibits a broad substrate scope and good functional group compatibility.
Introduction
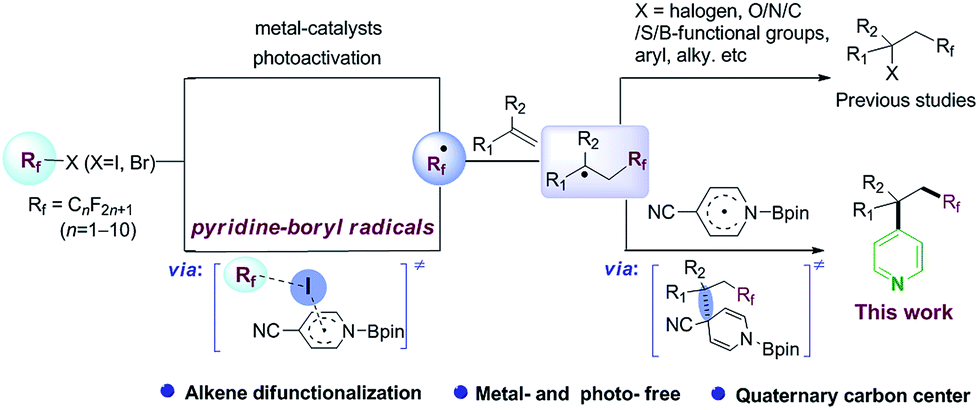
Difunctionalization of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds is a powerful strategy for the construction of complex compounds with various functional groups.1 In particular, building two C–C bonds tandemly in a single step is highly deserved in terms of structure diversity, step and atom economy. Incoporation of a perfluoroalkyl group in this tandem reaction would be attractive with potential applications in medicinal chemistry, agrochemistry, materials science.2 Along this line, radical-mediated perfluoroalkylative difunctionalization of alkenes, through transition-metal catalysis,3 photoredox catalysis,4 or visible-light activation of electron donor–acceptor (EDA) complexes,5 has played privileged roles in these transformations (Scheme 1, up). Developing a metal- and photo-free method for difunctionalization of alkenes remains an important synthetic goal.
C bonds is a powerful strategy for the construction of complex compounds with various functional groups.1 In particular, building two C–C bonds tandemly in a single step is highly deserved in terms of structure diversity, step and atom economy. Incoporation of a perfluoroalkyl group in this tandem reaction would be attractive with potential applications in medicinal chemistry, agrochemistry, materials science.2 Along this line, radical-mediated perfluoroalkylative difunctionalization of alkenes, through transition-metal catalysis,3 photoredox catalysis,4 or visible-light activation of electron donor–acceptor (EDA) complexes,5 has played privileged roles in these transformations (Scheme 1, up). Developing a metal- and photo-free method for difunctionalization of alkenes remains an important synthetic goal.
 | ||
| Scheme 1 Alkene perfluoroalkylation. | ||
Pyridine skeletons are often served as “privileged” scaffolds in drug design and discovery.6 Radical pyridylation is a useful synthetic methodology for the synthesis of value-added pyridine derivatives, due to the good functional group tolerance and broad substrate scope of these methods.7 The challenge is how to tune the reactivity and selectivity of various radicals in a system. The direct hydroarylation of alkenes with pyridines has also been investigated by transition-metal catalysts.8 However, the simultaneous introduction of the pyridine moiety and other functional groups to alkenes has been rarely reported, which might be attributed to the low reactivity and site-selectivity of the pyridine group.9 Herein, we describe a metal- and photo-free protocol for perfluoroalkylative pyridylation of alkenes, which is mediated by in situ 4-cyanopyridine-boryl radicals (Scheme 1, down).
Our investigation began with the 19F NMR observation of heating the mixture of perfluorobutyl iodide 1a, 4-cyanopyridine and B2pin2 at 80 °C (Scheme 2a, see details in ESI†), based on previous report that pyridine-stabilized boryl radical could be easily derived from 4-cyanopyridine and diboranes.10 It was found that the 19F NMR chemical shift at ∼60 ppm (which corresponds to the signal of –CF2I group) disappeared, implying that the formation of the perfluorobutyl radical might be induced by the 4-cyanopyridine-boryl radicals. The perfluorobutyl radical could be trapped by 1,1-diphenylethene in the presence of 4-cyanopyridine, perfluorooctyl iodide 1g, B2pin2, and 1,4-dihydromesitylene (as a hydrogen source) under the similar conditions (Scheme 2b). These results indicated that the 4-cyanopyridine-boryl radicals can activate the perfluoroalkyl iodides to generate perfluoroalkyl radicals. Inspired by this observation and recent progress from Studer11 and Liu12 groups that Rf–X (X = I, Br) reagents can act as inexpensive perfluoroalkyl radical precursor for difunctionalization of alkenes, we envisioned that the perfluoroalkyl radical generated from homolysis of perfluoroalkyl halides Rf–X (X = I, Br) promoted by 4-cyanopyridine radicals might be trapped by alkenes, and selective cross-coupling of the resulting alkyl radicals with the persistent 4-cyanopyridine-boryl radicals might lead to the perfluoroalkylative pyridylation of alkenes (Scheme 1, down).13
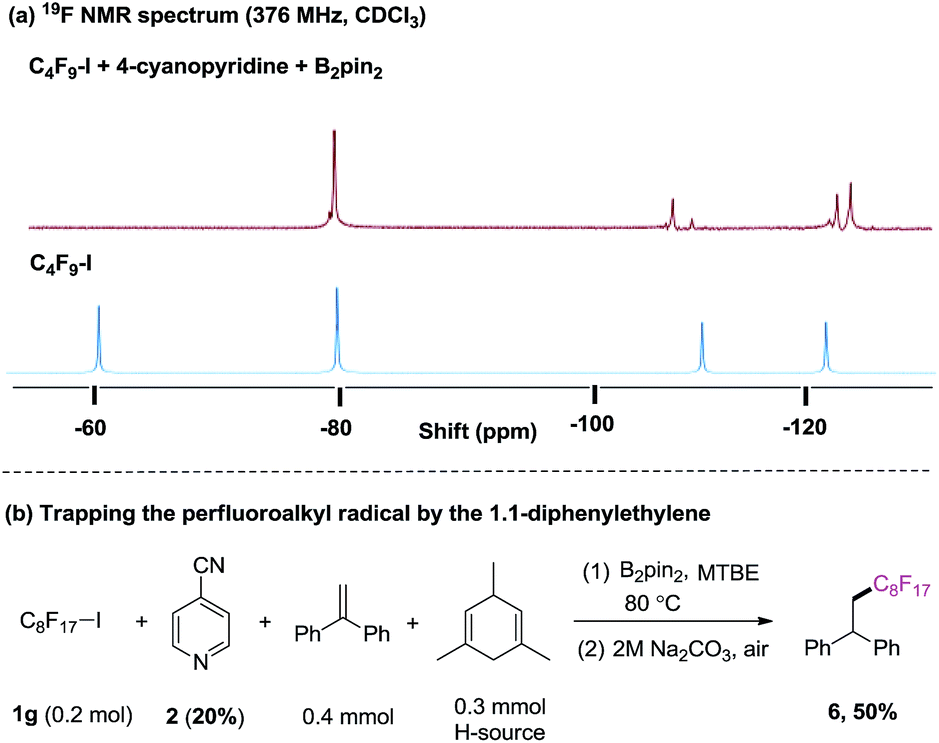 | ||
| Scheme 2 (a) 4-cyanopyridine boryl radicals-mediated C–I bond homolysis of perfluorooctyl iodide 1a monitored by 19F NMR; (b) radical trapping experiment. | ||
Results and discussion
The proposed reaction mechanism of the alkene perfluoroalkylative pyridylation was postulated in Scheme 3. The following steps may be involved: (1) 4-cyanopyridine-boryl radicals (Int1) are generated from the homolytic cleavage of the B–B bond of B2pin2 by 4-cyanopyridine; (2) Int1 activates the C–I bond of CF3I (1b) to produce the CF3 radical and Int3a, and regenerate 4-cyanopyridine; (3) CF3 radical adds to 4-methylisopropenylbenzene (3a), forming a new alkyl radical (Int4); (4) the selective cross-coupling of Int1 and Int4 by persistent radical effect,14 yields the intermediate Int5, which is hydrolysed to give the alkene perfluoroalkylation product 4b. Notably, Int1 not only catalyze the C–I bond homolysis of perfluoroalkyl halides Rf–X (X = I, Br), but also serves as the pyridine precursor.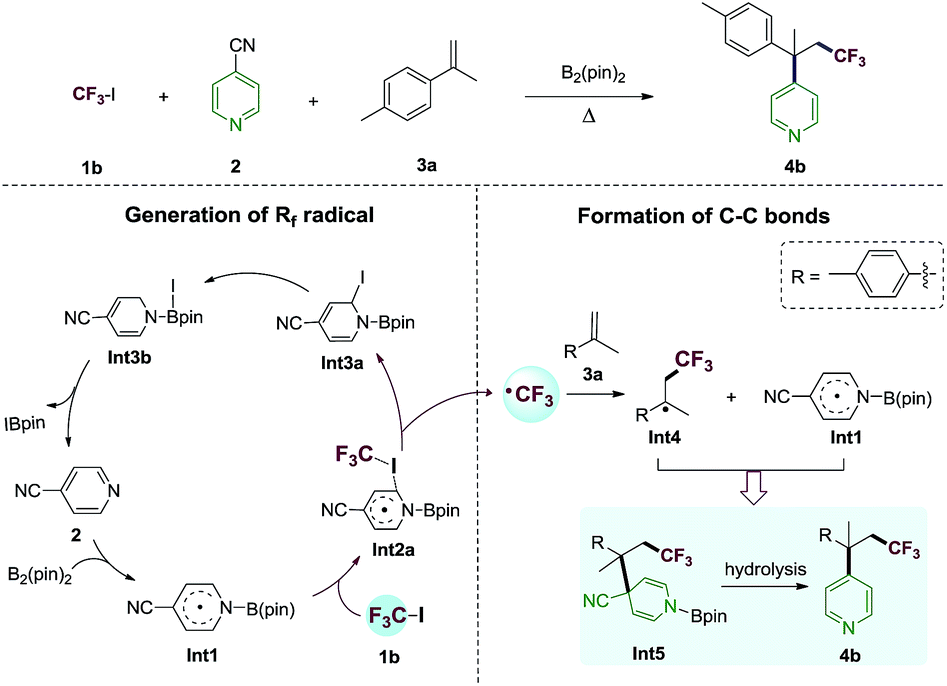 | ||
| Scheme 3 Proposed 4-cyanopyridine-boryl radicals-mediated alkene difunctionalization. | ||
To verify whether the proposed mechanism is thermodynamically or kinetically feasible, we performed DFT calculations with the M06-2X15 functional to explore the free energy profile of the proposed mechanism for the model reaction of 1b and 3a in the presence of Int1 as a reactive intermediate. The reaction mechanism of generating Int1 was reported in our previous works.10c,d,j,k The calculated free energy profile and transition state structures are listed in Fig. 1 (the optimized structures of all minimum species are shown in Fig. S1†). First, the association between the iodine atom of 1b and the carbon atom at the C2 position of the radical Int1 forms an encounter complex (Int2a), which is endergonic by 7.2 kcal mol−1. Then, the transfer of the iodine atom from 1b to Int1 to give the CF3 radical and Int3a involves a barrier of 32.6 kcal mol−1 (viaTS1) and is endergonic by 20.2 kcal mol−1 (relative to the isolated reactants Int1 and 1b). It should be mentioned that the homolytic dissociation energy of C–I bond in CF3I is 49.1 kcal mol−1. There results indicate that the homolysis of C–I bond in CF3I is indeed assisted by Int1. Subsequently, CF3 radical adds to the alkene 3a to generate a new alkyl radical Int4viaTS2, being exothermic by 11.3 kcal mol−1 with a barrier of 29.2 kcal mol−1 (with respect to the separated reactants Int1, 3a and 1b). Finally, the C–C coupling between Int1 and Int4 produces an intermediate Int5 through TS3 with a barrier of 6.8 kcal mol−1 (relative to Int4 and Int1), and the whole process is exothermic by 30.1 kcal mol−1 (with respect to the reactants Int1, 1b and 3a). In addition, the hydrolysis of the intermediate Int5 will produce the final product 4b. The results indicate that the proposed alkene perfluoroalkylation is thermodynamically favorable. Alternatively, the C–I bond homolysis by the 4-cyanopyridine-boryl radicals at the C4 position is also investigated (shown in Fig. S2†). This process is endergonic by 31.0 kcal mol−1, with a barrier of 37.7 kcal mol−1 (relative to Int1 and 1b), suggesting that the pathway is less favorable. Furthermore, we also calculate the isomerization reaction of Int3a (see Fig. S3†). Starting from Int3a, the intramolecular migration of the iodine atom from C2 atom to B atom viaTS4, could yield another isomer Int3b, which further proceeds through the breaking of the B–N bond (viaTS5) to regenerate 4-cyanopyridine. Overall, the rate-determining barrier height of this process is 10.1 kcal mol−1 and endergonic by 2.9 kcal mol−1 (relative to Int3a), indicating that the C–I bond homolysis is a catalytic process by 4-cyanopyridine. Moreover, our calculations suggest that the direct single electron transfer (SET) process between 4-cyanopyridine-boryl radicals and CF3I is highly endergonic by 60.0 kcal mol−1 (see Fig. S4†). Thus, the SET mechanism is unlikely responsible for the generation of the perfluoroalkyl radicals in the reaction.
 | ||
| Fig. 1 Computed Gibbs free energy profile of the alkene carbopyridylation via 4-cyanopyrodine boryl radicals. The optimized structures of transition states are also displayed. Interatomic distances are in Å. | ||
Based on the predicted reactivity, we first examined the proposed alkene difunctionalization using 4-cyanopyridine, perfluorobutyl iodide 1a, 4-methylisopropenylbenzene 3a as model substrates. The optimization details are given in ESI.† We found that the desired product could be obtained in 74% yield at 80 °C in the presence of B2pin2 with N,N-diisopropylethylamine (DIPEA) as additives (Scheme 4a). Control experiments suggest that this transformation occurs via a thermally induced process, as decreased yield was observed at lower temperatures. The requirement of a relatively high temperature (80 °C) are in qualitative accord with the DFT results discussed above. Moreover, the generation of intermediates Int3a (or its isomer Int3b), and the compound 7-like (from the addition of Int1 and the perfluorobutyl radical) in the presence of 1a, 4-cyanopyridine and B2pin2 under the standard conditions were confirmed by high resolution mass spectroscopy (HRMS) experiments, which provide direct evidence on the C–I homolysis mechanism of 1avia 4-cyanopyridine-boryl radicals (see Fig. S5 and S6†). In addition, the intermediacy of the cross-coupling intermediate Int5-like as well as the by-product I-Bpin could be detected by the HRMS analysis of the reaction mixture of the perfluorobutyl, 4-cyanopyridine and 4-methylisopropenylbenzene 3a under the standard conditions (Scheme 4b, Fig. S7 and S8†). Finally, the addition of the perfluoroalkyl radical to alkenes could be further confirmed by a radical clock experiment using vinyl cyclopropane 3r as the substrate (see Fig. S9† for details). In combination, the studies revealed an unique strategy for the generation of perfluoroalkyl radicals and for subsequent perfluoroalkylative pyridylation of alkenes using the inexpensive 4-cyanopyridine/B2pin2 system.
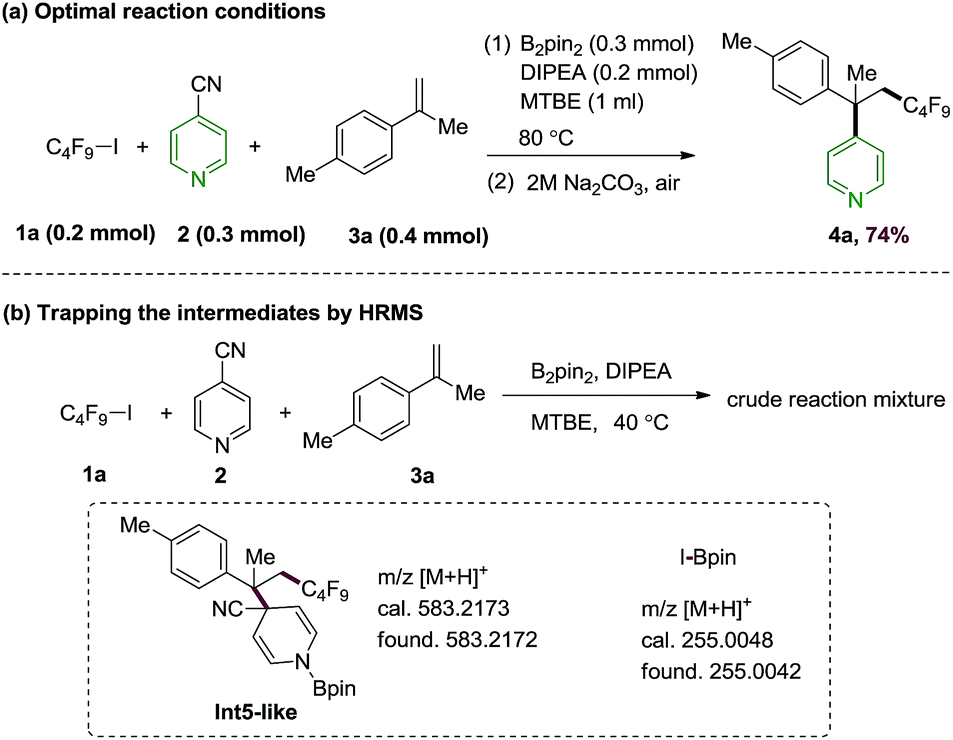 | ||
| Scheme 4 Control experiments. | ||
Then, we examined the substrate scope of the carbon radical precursor with 4-methylisopropenylbenzene 3a as the radical acceptor (see Table 1). With perfluoroalkyl iodides (CnF2n+1–I), the corresponding α-perfluoroalkyl-β-pyridylation product could be obtained in moderate to good yields (4a–4e, 4g, 4i). Notably, the sterically congested substrate, perfluoro-isopropyl iodide, reacted smoothly to afford the desired product in moderate yield (4j, 45%). 1-Chlorotetrafluoro-2-iodoethane could also be converted into the desired product 4k in good yield via the selective cleavage of C–I bond. For the perfluoroalkyl bromides, the desired products (4f, 4h, 4l, 4m) could be formed in moderate yields under the standard conditions. The reaction of C6F13I, 4-cyanopyridine and alpha-methyl styrene on a 5 mmol scale in the presence of B2pin2 readily afforded 4e in 71% yield (1.8 g).
| a Reaction conditions: 1 (0.2 mmol), B2(pin)2 (0.3 mmol), 4-cyanopyridine 2 (0.3 mmol), 4-methylisopropenylbenzene 3a (0.4 mmol), MTBE (1.0 mL), DIPEA (0.2 mmol), 24 h, 80 °C. Isolated yield. b 5 mmol scale. 1e (5 mmol), B2(pin)2 (7.5 mmol), 4-cyanopyridine (7.5 mmol), MTBE (15.0 mL), DIPEA (5.0 mmol), 24 h, 80 °C. Me = methyl, Et = ethyl. |
|---|

|
Next, the substrate scope of alkenes was evaluated. As shown in Table 2, alpha-methyl styrene bearing a variety of functional groups (such as Br, MeS, CF3O, MeSO2, CN, CF3, CO2Me etc.) on the phenyl rings, were well compatible with this protocol, offering corresponding carbopyridylation products with quaternary carbon center (5aa–5as) in moderate to good yields (43–74%). Alpha-methyl naphthalenes also reacted to provide the desired products in good yields (5ba, 73% and 5bb, 70%). Furthermore, other alpha-methyl arylethene containing fused heterocycles, such as benzofuran, phenanthrene, fluorene and carbazole, could be converted into the corresponding products 5c–5f in moderate to good yields. The reactions of more sterically congested alpha-ethyl and -propyl styrenes provided the desired products (5ga–5h) in moderate yields. In addition, 1,1-disubstituted unactivated alkenes could also smoothly transform into the corresponding products (5i–5k, 45–54% yields). However, the reactions of methacrylate and 4-methoxystyrene only afforded the carbopyridylation products 5l and 5m in lower yields. With internal alkenes as substrates, no corresponding carbopyridylation products can be detected under standard conditions. Our DFT calculations suggest that the barrier heights for the addition of trifluoromethyl radical to internal alkene or terminal monosubstituted styrene are higher than that of disubstituted styrene by 1.2–3.4 kcal mol−1 (see Table S1†). This result may be responsible for the experimental facts described above. Thus, internal alkenes or terminal monodisubstituted styrenes are not suitable for the present transformation.
| a Reaction conditions: 1a (0.2 mmol), B2(pin)2 (0.3 mmol), 4-cyanopyridine (0.3 mmol), alkene (0.4 mmol), MTBE (1.0 mL), DIPEA (0.2 mmol), 24 h, 80 °C. Isolated yield. Me = methyl, Et = ethyl, tBu = tert-butyl. |
|---|

|
Both pyridine and perfluoroalkyl groups are prevalent motifs in drugs and natural products. The simultaneous incorporation of these two groups into bioactive molecules might improve their properties, such as reactivity and metabolic stability and selectivity.2,6 As illustrated in Table 2, four complicated alkene substrates derived from abietic acid, gemfibrozil, 1-adamantaneacetic acid, and cholesterol, readily underwent the carbopyridylation to give products 5n–5q in moderate to good yields.
Conclusions
In summary, we reported a metal- and photo-free synthetic method for perfluoroalkylative pyridylation of alkenes. Density functional theory calculations and control experiments indicate the in situ prepared 4-cyanopyridine-boryl radicals from 4-cyanopyridine and B2(pin)2, which not only activates the C–I bond homolysis but also serves as a pyridine precursor, play a key role in this transformation. A high functional group tolerance and broad substrate scope were achieved. This method provides a scalable and operationally simple protocol for difunctionalization of alkenes with inexpensive 4-cyanopyridine/B2(pin)2 reagents. We anticipated that the present approach would be useful for the construction of molecules with complexity and late stage modification of drugs and natural products.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grants No. 21833002, No. 21673110, and No. 21572099), and China Postdoctoral Science Foundation (Grant No. 2017M620198). All calculations in this work have been done on the IBM Blade cluster system in the High Performance Computing Center of Nanjing University.Notes and references
- For selected examples on the difunctionalization of alkenes, see: (a) R. I. McDonald, G. Liu and S. S. Stahl, Chem. Rev., 2011, 111, 2981 CrossRef CAS PubMed; (b) R. M. Romero, T. H. Woste and K. Muniz, Chem.–Asian J., 2014, 9, 972 CrossRef CAS PubMed; (c) G. Yin, X. Mu and G. Liu, Acc. Chem. Res., 2016, 49, 2413 CrossRef CAS PubMed; (d) A. Lerchen, T. Knecht, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2016, 55, 15166 CrossRef CAS PubMed; (e) L. Pitzer, F. Sandfort, F. Strieth-Kalthoff and F. Glorius, J. Am. Chem. Soc., 2017, 139, 13652 CrossRef CAS PubMed; (f) A. Tlahuext-Aca, R. A. Garza-Sanchez and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 3708 CrossRef CAS PubMed; (g) X. W. Lan, N. X. Wang and Y. L. Xing, Eur. J. Org. Chem., 2017, 5821 CrossRef CAS; (h) T. Koike and M. Akita, Chem, 2018, 4, 409 CrossRef CAS; (i) X. Li, P. H. Chen and G. S. Liu, Beilstein J. Org. Chem., 2018, 14, 1813 CrossRef CAS PubMed; (j) J. S. Zhang, L. Liu, T. Q. Chen and L. B. Han, Chem.–Asian J., 2018, 13, 2277 CrossRef CAS PubMed; (k) A. Tlahuext-Aca, R. A. Garza-Sanchez, M. Schafer and F. Glorius, Org. Lett., 2018, 20, 1546 CrossRef CAS PubMed.
- (a) J. A. Ma and D. Cahard, Chem. Rev., 2004, 104, 6119 CrossRef CAS PubMed; (b) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS PubMed; (c) P. Jeschke, ChemBioChem, 2004, 5, 570 CrossRef CAS PubMed; (d) A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8950 CrossRef CAS PubMed; (e) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS PubMed; (f) H. Egami and M. Sodeoka, Angew. Chem., Int. Ed., 2014, 53, 8294 CrossRef CAS PubMed; (g) E. Merino and C. Nevado, Chem. Soc. Rev., 2014, 43, 6598 RSC; (h) T. Koike and M. Akita, Acc. Chem. Res., 2016, 49, 1937 CrossRef CAS PubMed; (i) S. Barata-Vallejo, M. Victoria Cooke and A. Postigo, ACS Catal., 2018, 8, 7287 CrossRef CAS; (j) D. E. Yerien, S. Barata-Vallejo and A. Postigo, Chem.–Eur. J., 2017, 23, 1 CrossRef; (k) H.-X. Song, Q.-Y. Han, C.-L. Zhao and C.-P. Zhang, Green Chem., 2018, 20, 1662 RSC; (l) X. Zhao, H. Y. Tu, L. Guo, S. Zhu, F. L. Qing and L. L. Chu, Nat. Commun., 2018, 9, 3488 CrossRef PubMed.
- (a) F. Wang, D. H. Wang, X. Mu, P. H. Chen and G. S. Liu, J. Am. Chem. Soc., 2014, 136, 10202 CrossRef CAS PubMed; (b) Y. B. Dudkina, T. V. Gryaznova, Y. N. Osin, V. V. Salnikov, N. A. Davydov, S. V. Fedorenko, A. R. Mustafina, D. A. Vicic, O. G. Sinyashina and Y. H. Budnikovaa, Dalton Trans., 2015, 44, 8833 RSC; (c) M. Y. Fu, L. Chen, Y. P. Jiang, Z. X. Jiang and Z. G. Yang, Org. Lett., 2016, 18, 348 CrossRef CAS PubMed; (d) S. Kawamura and M. Sodeoka, Angew. Chem., Int. Ed., 2016, 55, 8740 CrossRef CAS PubMed; (e) L. Li, Q. S. Gu, N. Wang, P. Song, Z. L. Li, X. H. Li, F. L. Wang and X. Y. Liu, Chem. Commun., 2017, 53, 4038 RSC; (f) W. L. Deng, Y. J. Li, Y. G. Li and H. L. Bao, Synthesis, 2018, 50, 2974 CrossRef CAS; (g) G. J. Wua and A. J. Wangelin, Chem. Sci., 2018, 9, 1795 RSC.
- (a) A. Carboni, G. Dagousset, E. Magnier and G. Masson, Chem. Commun., 2014, 50, 14197 RSC; (b) T. Koike and M. Akita, Top. Catal., 2014, 57, 967 CrossRef CAS; (c) A. J. Vázquez and N. S. Nudelman, ARKIVOC, 2015, 5, 190 Search PubMed; (d) B.-V. Sebastián, E. Y. Damian, L. Beatriz and P. Al, Curr. Org. Chem., 2016, 202, 2838 Search PubMed; (e) T. Chatterjee, N. Iqbal, Y. You and E. J. Cho, Acc. Chem. Res., 2016, 49, 2284 CrossRef CAS PubMed; (f) X. Y. Sun, W. M. Wang, Y. L. Li, J. Ma and S. Y. Yu, Org. Lett., 2016, 18, 4638 CrossRef CAS PubMed; (g) Y. X. Wang, J. H. Wang, G. X. Li, G. He and G. Chen, Org. Lett., 2017, 19, 1442 CrossRef CAS PubMed; (h) D. P. Tiwari, S. Dabral, J. Wen, J. Wiesenthal, S. Terhorst and C. Bolm, Org. Lett., 2017, 19, 4295 CrossRef CAS PubMed; (i) Y. Xu, Z. Wu, J. X. Jiang, Z. F. Ke and C. Zhu, Angew. Chem., Int. Ed., 2017, 56, 4545 CrossRef CAS PubMed; (j) X. Y. Geng, F. Lin, X. Y. Wang and N. Jiao, Org. Lett., 2017, 19, 4738 CrossRef CAS PubMed; (k) W. G. Kong, H. J. An and Q. L. Song, Chem. Commun., 2017, 53, 8968 RSC; (l) X. J. Tang and A. Studer, Chem. Sci., 2017, 8, 6888 RSC; (m) M. Alkan-Zambada and X. Hu, Organometallics, 2018, 37, 3928 CrossRef CAS; (n) R. Beniazza, L. Remisse, D. Jardel, D. Lastécouèresa and J. M. Vincent, Chem. Commun., 2018, 54, 7451 RSC; (o) H. B. Wang and N. T. Jui, J. Am. Chem. Soc., 2018, 140, 163 CrossRef CAS PubMed.
- (a) Y. Z. Cheng and S. Y. Yu, Org. Lett., 2016, 18, 2962 CrossRef CAS PubMed; (b) I. Behrends, S. Bӓhr and C. Czekelius, Chem.–Eur. J., 2016, 22, 17177 CrossRef CAS PubMed; (c) H. Jiang, Y. He, Y. Z. Cheng and S. Y. Yu, Org. Lett., 2017, 19, 1240 CrossRef CAS PubMed; (d) S. F. Zhou, T. Song, H. Chen, Z. L. Liu, H. G. Shen and C. Z. Li, Org. Lett., 2017, 19, 698 CrossRef CAS PubMed; (e) E. Valverde, S. Kawamura, D. Sekinea and M. Sodeoka, Chem. Sci., 2018, 9, 7115 RSC.
- (a) M. Schlosser, Angew. Chem., Int. Ed., 2006, 45, 5432 CrossRef CAS PubMed; (b) V. Komanduri, C. D. Grant and M. J. Krische, J. Am. Chem. Soc., 2008, 130, 12592 CrossRef CAS PubMed; (c) E. Vitaku, D. T. Smith and J. Njardarson, J. Med. Chem., 2014, 57, 10257 CrossRef CAS PubMed; (d) K. N. Lee, Z. Lei and M. Y. Ngai, J. Am. Chem. Soc., 2017, 139, 5003 CrossRef CAS PubMed.
- (a) A. McNally, C. K. Prier and D. W. C. MacMillan, Science, 2011, 334, 1114 CrossRef CAS PubMed; (b) M. T. Pirnot, D. A. Rankic, D. B. C. Martin and D. W. C. MacMillan, Science, 2013, 339, 1593 CrossRef CAS PubMed; (c) K. Qvortrup, D. A. Rankic and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 626 CrossRef CAS PubMed; (d) J. D. Cuthbertson and D. W. C. MacMillan, Nature, 2015, 519, 74 CrossRef CAS PubMed; (e) J. Streuff and A. Gansӓuer, Angew. Chem., Int. Ed., 2015, 54, 14232 CrossRef CAS PubMed; (f) J. P. Goddard, C. Ollivier and L. Fensterbank, Acc. Chem. Res., 2016, 49, 1924 CrossRef CAS PubMed; (g) F. Lima, M. A. Kabeshov, D. N. Tran, C. Battilocchio, J. Sedelmeier, G. Sedelmeier, B. Schenkel and S. V. Ley, Angew. Chem., Int. Ed., 2016, 55, 14085 CrossRef CAS PubMed; (h) M. Yan, J. L. C. Lo, J. T. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692 CrossRef CAS PubMed; (i) J. Xuan and A. Studer, Chem. Soc. Rev., 2017, 46, 4329 RSC; (j) J. M. Smith, S. J. Harwood and P. S. Baran, Acc. Chem. Res., 2018, 51, 1807 CrossRef CAS PubMed.
- (a) M. D. Hill, Chem.–Eur. J., 2010, 16, 12052 CrossRef CAS PubMed; (b) T. Andou, Y. Saga, H. Komai, S. Matsunaga and M. Kanai, Angew. Chem., Int. Ed., 2013, 52, 3213 CrossRef CAS PubMed; (c) G. Y. Song, W. W. N. O and Z. M. Hou, J. Am. Chem. Soc., 2014, 136, 12209 CrossRef CAS PubMed; (d) D. E. Stephens and O. V. Larionov, Tetrahedron, 2015, 71, 8683 CrossRef CAS PubMed; (e) X. S. Ma and S. B. Herzon, J. Am. Chem. Soc., 2016, 138, 8718 CrossRef CAS PubMed; (f) R. B. Hu, S. Sun and Y. Su, Angew. Chem., Int. Ed., 2017, 56, 10877 CrossRef CAS PubMed; (g) A. J. Boyington, M.-L. Y. Riu and N. T. Jui, J. Am. Chem. Soc., 2017, 139, 6582 CrossRef CAS PubMed; (h) R. A. Aycock, D. B. Vogt and N. T. Jui, Chem. Sci., 2017, 8, 7998 RSC; (i) A. Kundu, M. Inoue, H. Nagae, H. Tsurugi and K. Mashima, J. Am. Chem. Soc., 2018, 140, 7332 CrossRef CAS PubMed; (j) Q. Sun, P. Chen, Y. Wang, Y. Luo, D. Yuan and Y. Y. Yao, Inorg. Chem., 2018, 57, 11788 CrossRef CAS PubMed; (k) W. Zhou, T. Miura and M. Murakami, Angew. Chem., Int. Ed., 2018, 57, 5139 CrossRef CAS PubMed; (l) C. P Seath, D. B Vogt, Z. Xu, A. J. Boyington and N. T. Jui, J. Am. Chem. Soc., 2018, 140, 15525 CrossRef PubMed.
- (a) Y. Nakao, Y. Y. Yamada, N. Kashihara and T. Hiyama, J. Am. Chem. Soc., 2010, 132, 13666 CrossRef CAS PubMed; (b) Y. X. Li, G. D. Deng and X. M. Zeng, Organometallics, 2016, 35, 747 CrossRef CAS.
- (a) T. Ohmura, Y. Morimasa and M. Suginome, J. Am. Chem. Soc., 2015, 137, 2852 CrossRef CAS PubMed; (b) E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091 CrossRef CAS PubMed; (c) G. Wang, H. Zhang, J. Zhao, W. Li, J. Cao, C. Zhu and S. Li, Angew. Chem., Int. Ed., 2016, 55, 5985 CrossRef CAS PubMed; (d) G. Wang, J. Cao, L. Gao, W. Chen, W. Huang, X. Cheng and S. Li, J. Am. Chem. Soc., 2017, 139, 3904 CrossRef CAS PubMed; (e) W. M. Cheng, R. Shang, B. Zhao, W. L. Xing and Y. Fu, Org. Lett., 2017, 19, 4291 CrossRef CAS PubMed; (f) L. Zhang and L. Jiao, J. Am. Chem. Soc., 2017, 139, 607 CrossRef CAS PubMed; (g) L. Candish, M. Teders and F. Glorius, J. Am. Chem. Soc., 2017, 139, 7440 CrossRef CAS PubMed; (h) A. B. Cuenca, R. Shishido, H. Ito and E. Fernández, Chem. Soc. Rev., 2017, 46, 415 RSC; (i) G. B. Yan, D. Huang and X. Wu, Adv. Synth. Catal., 2018, 360, 1040 CrossRef CAS; (j) J. Cao, G. Wang, L. Gao, X. Cheng and S. Li, Chem. Sci., 2018, 9, 3664 RSC; (k) L. Gao, G. Wang, J. Cao, D. Yuan, W. Chen, X. Cheng, X. W. Guo and S. Li, Chem. Commun., 2018, 54, 11534 RSC.
- (a) Y. Cheng, C. Mück-Lichtenfeld and A. Studer, J. Am. Chem. Soc., 2018, 140, 6221 CrossRef CAS PubMed; (b) X. J. Tang and A. Studer, Angew. Chem., Int. Ed., 2017, 56, 1 CrossRef.
- (a) L. Fu, S. Zhou, X. L. Wan, P. H. Chen and G. S. Liu, J. Am. Chem. Soc., 2018, 140, 10965 CrossRef CAS PubMed; (b) L. Q. Wu, F. Wang, X. L. Wan, D. H. Wang, P. H. Chen and G. S. Liu, J. Am. Chem. Soc., 2017, 139, 2904 CrossRef CAS PubMed.
- During the preparation of this manuscript, Chu and Hong groups reported an intermolecular carbopyridylation of alkenes via visible-light-induced radical coupling using Togni's reagent or CF3SO2Na: (a) D. Chen, L. Xu, T. Y. Long, S. G. Zhu, J. Yang and L. L. Chu, Chem. Sci., 2018, 9, 9012 RSC; (b) Y. T. He, D. Kang, I. Kima and S. Hong, Green Chem., 2018, 20, 5209 RSC . Different from a photoinduced process in these two papers, our method represents a thermally induced perfluoroalkylative pyridylation of alkenes mediated by the in situ generated 4-cyanopyridine-boryl radicals from 4-cyanopyridine and B2pin2.
- For reviews on the persistent radical effect, see: (a) H. Fischer, Chem. Rev., 2001, 101, 3581 CrossRef CAS PubMed; (b) A. Studer, Chem.–Eur. J., 2001, 7, 1159 CrossRef CAS PubMed; (c) A. Studer, Chem. Soc. Rev., 2004, 33, 267 RSC; (d) A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58 CrossRef CAS PubMed.
- (a) Y. Zhao, N. E Schultz and D. G. Truhlar, J. Chem. Theory Comput., 2006, 2, 364 CrossRef PubMed; (b) Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed; (c) Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2006, 110, 13126 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc05237a |
| This journal is © The Royal Society of Chemistry 2019 |