
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The evolution of ‘sol–gel’ chemistry as a technique for materials synthesis
A. E.
Danks
a,
S. R.
Hall
b and
Z.
Schnepp
*a
aSchool of Chemistry, University of Birmingham, Birmingham, B152TT, UK. E-mail: z.schnepp@bham.ac.uk
bComplex Functional Materials Group, School of Chemistry, University of Bristol, Bristol, BS81TS, UK
First published on 16th December 2015
Abstract
From its initial use to describe hydrolysis and condensation processes, the term ‘sol–gel’ is now used for a diverse range of chemistries. In fact, it is perhaps better defined more broadly as covering the synthesis of solid materials such as metal oxides from solution-state precursors. These can include metal alkoxides that crosslink to form metal–oxane gels, but also metal ion–chelate complexes or organic polymer gels containing metal species. What is important across all of these examples is how the choice of precursor can have a significant impact on the structure and composition of the solid product. In this review, we will attempt to classify different types of sol–gel precursor and how these can influence a sol–gel process, from self-assembly and ordering in the initial solution, to phase separation during the gelation process and finally to crystallographic transformations at high temperature.
 A. E. Danks | Ashleigh Danks is currently studying for a PhD at the University of Birmingham in the group of Zoe Schnepp. His research is focussed on the sol–gel synthesis of nanocomposites of metal carbides, oxides and carbon and he has a particular interest in materials for catalysis. During his PhD, he has spent several months at the National Institute for Materials Science in Japan as well as a 6 month project working for the Defence Science and Technology Laboratory. |
 S. R. Hall | Simon Hall joined the University of Bristol in 1997, reading for a PhD in Biomimetic Materials Chemistry in the laboratory of Professor Stephen Mann FRS. After a three year postdoctoral study, where he produced advanced functional materials for Vectura Ltd and Toyota Ltd, he was awarded a Royal Society University Research Fellowship in 2004. His research interests centre around crystal control in superconductors, semiconductors, piezoelectrics and polyaromatic hydrocarbons. He is currently a Senior Lecturer in Inorganic Chemistry at the University of Bristol, Director of the Complex Functional Materials Group and Co-I of the Bristol Centre for Functional Nanomaterials Centre for Doctoral Training. |
 Z. Schnepp | Zoe Schnepp is passionate about green chemistry, both in her research and the potential for changing negative public perceptions of chemistry. With diverse interests in nanotechnology, catalysis and materials from biomass, Zoe leads a growing group in the School of Chemistry at the University of Birmingham, UK. Prior to this, she held Postdoctoral Fellowships in the International Center for Young Scientists at the National Institute for Materials Science in Japan and the Max Planck Institute for Colloids and Interfaces in Germany. She received her PhD from the University of Bristol. |
1. Introduction
Since the discovery by early man that rocks could be modified to make tools, the world has demanded materials with increasingly complex functionality.1 Given this fact, it is not surprising that the development of new synthetic methods has been a major field of scientific endeavour. Many inorganic materials, such as metal oxides or carbides, can be prepared fairly simply by mixing powder reactants and heating to give the desired product. While reaction conditions are relatively easy to achieve (furnace technology being well established), there are some drawbacks. These centre primarily on the inhomogeneity of the starting materials. In mixtures of two or more powders, complete conversion is limited by mass transport. Initial reaction takes place at the edges of adjacent particles and if reactant diffusion is blocked there will be areas of unreacted starting material. Some of these issues can be overcome by ball-milling: reducing the particle size and increasing the sample surface area. However, extended heating or multiple treatments separated by successive sample milling steps may be required. Furthermore, it is often difficult to control particle morphology using solid-state methods.As an alternative to solid-state chemistry, a range of solution techniques have emerged, including coprecipitation, hydrothermal processing, solvothermal methods and sol–gel chemistry.2 Among these techniques, sol–gel chemistry offers some particular advantages, centred on the ability to produce a solid-state material from a chemically homogeneous precursor. By trapping the “randomness of the solution state”![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3 and thereby ensuring atomic level mixing of reagents, one should be able to produce complex inorganic materials such as ternary and quaternary oxides at lower processing temperatures and shorter synthesis times. Furthermore, sol–gel chemistry should enable greater control over particle morphology and size. In reality, producing a homogeneous precursor at room temperature does not ensure homogeneity throughout a reaction and many sol–gel routes have therefore been designed to combat or control phase segregation during synthesis. Neither is it always necessary to ensure complete ‘randomness’ in the precursor. In fact some of the most interesting advances in the sol–gel field in recent years have come from gels that have some degree of ordering and structure.
3 and thereby ensuring atomic level mixing of reagents, one should be able to produce complex inorganic materials such as ternary and quaternary oxides at lower processing temperatures and shorter synthesis times. Furthermore, sol–gel chemistry should enable greater control over particle morphology and size. In reality, producing a homogeneous precursor at room temperature does not ensure homogeneity throughout a reaction and many sol–gel routes have therefore been designed to combat or control phase segregation during synthesis. Neither is it always necessary to ensure complete ‘randomness’ in the precursor. In fact some of the most interesting advances in the sol–gel field in recent years have come from gels that have some degree of ordering and structure.
This review will give an introduction to the different types of gel and then describe the types of chemistry that may be considered under the heading ‘sol–gel’. We focus in particular on how different molecular precursors can influence composition and structure in the synthesis of materials and highlight mechanistic studies that have offered insight into the processes that occur during sol–gel synthesis.
1.1 ‘Sol’ and ‘gel’
Sol–gel chemistry is the preparation of inorganic polymers or ceramics from solution through a transformation from liquid precursors to a sol and finally to a network structure called a ‘gel’.4 Traditionally, the formation of a sol occurs through hydrolysis and condensation of metal alkoxide precursors (section 1.2), but a sol can be more generally defined as a colloidal suspension, which encompasses a wider range of systems. The International Union of Pure and Applied Chemistry (IUPAC) define a colloidal system as a dispersion of one phase in another where, “the molecules or polymolecular particles dispersed in a medium have at least in one direction a dimension roughly between 1 nm and 1 μm”. In this sense, the term ‘sol’ can be applied to most of the systems discussed in this paper, including in situ formation of inorganic polymer particles via covalent bonds such as siloxanes, as well as the solvation and subsequent ionic crosslinking of biopolymers.1.2 Types of gel
In the sol–gel process, there are many different ways that a gel can be formed. Sometimes, the same precursors can result in very different structures with only small changes in conditions. Generally, the gel state is simply defined as a non-fluid 3D network that extends through a fluid phase. Gels were grouped by Flory in 1974 into four types, including ordered, lamellar gels (e.g. clays or surfactant mesophases), covalent polymer networks, networks of physically aggregated polymers (e.g. hydrogels formed via helical junctions) and finally disordered particulate gels.5 However, for the purposes of sol–gel chemistry, which is used for the preparation of inorganic solids, a more useful classification of different gel types was given by Kakihana in 1996.3Fig. 1 outlines the five key types of gel that feature in ‘sol–gel’ chemistry. It is debatable whether metal complexes can always be classed as gels since many of these actually form viscous solutions or glassy solids rather than gels. However, the fundamental goal of forming a homogeneous metal-containing precursor is still applicable and the use of small molecules, often generalized as the ‘citrate sol–gel’ method, is frequently cited in the literature.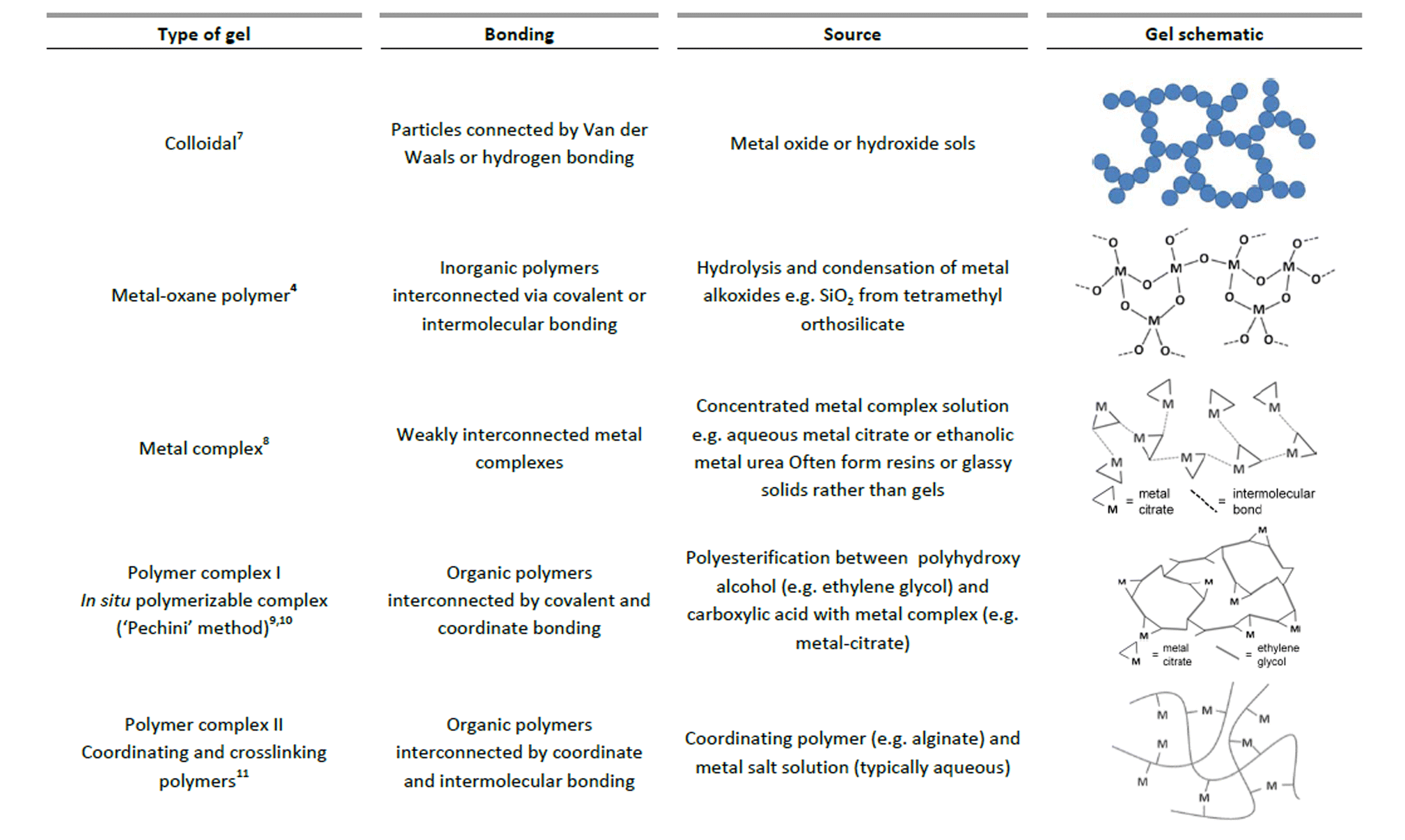 | ||
| Fig. 1 Chart classifying 5 different types of gels that are relevant in sol–gel synthesis of materials. | ||
2. Gels via hydrolysis and condensation
2.1 General introduction
The origin of sol–gel chemistry was the observation in the 19th century that an alkoxide prepared from SiCl4 started to form a gel when exposed to air.6 This was later found to be driven by atmospheric moisture causing first hydrolysis of the silicon alkoxide and then condensation. These processes have since been widely studied and can be carefully tuned, for example through acid or base catalysis, to form gels with very different structures. Hydrolysis and condensation chemistry is limited by the number of elements that readily form alkoxides and also by the high reactivity of many of these compounds. Furthermore, the very different rates of hydrolysis of different alkoxides can lead to substantial phase separation and difficulty in synthesizing ternary or quaternary systems. However, this ‘traditional’ sol–gel chemistry is still one of the most widely used and studied fields of materials chemistry.Aside from precursor preparation, the sol–gel process can be summarized in the following key steps:
(i) Synthesis of the ‘sol’ from hydrolysis and partial condensation of alkoxides.
(ii) Formation of the gel via polycondensation to form metal–oxo–metal or metal–hydroxy–metal bonds.
(iii) Syneresis or ‘aging’ where condensation continues within the gel network, often shrinking it and resulting in expulsion of solvent.
(iv) Drying the gel either to form a dense ‘xerogel’ via collapse of the porous network or an aerogel for example through supercritical drying.
(v) Removal of surface M–OH groups through calcination at high temperature up to 800 °C (if required).
2.2 Precursors
Sol–gel chemistry originated with the hydrolysis and condensation of metal alkoxides, although it can also occur between hydrated metal species. Most of the examples of alkoxide-based sol–gel chemistry involve early transition group metals (e.g. Ti, Zr) or early p-block elements (e.g. Al, Si), however there are many other examples of elemental alkoxides. Metal alkoxides can be prepared in a number of ways depending of the nature of the metal. As with the original synthesis, metal chlorides can be reacted with alcohols. Highly reducing metals, i.e. alkali metals and lanthanides, can react directly with alcohols to produce the corresponding alkoxide and hydrogen.12 A large number of alkoxides, such as Ta(OR)5 (R = Me, Et, nBu), can be produced via anodic dissolution of the metal in alcohol with an electro-conductive additive such as LiBr.13 Following the successful synthesis of many mono-metallic alkoxides many groups have also prepared bi-14 and ter-15 metallic alkoxides.The suitability of the various alkoxides for sol–gel chemistry and outcome of the reactions depends on several things. One factor is how electronegativity differences between the oxygen and metal affect the ionic character of the M–O bond, which can be predicted using the partial charge model developed by Livage et al.16 Another important effect is the electron donating/withdrawing ability of the alkyl/aryl chain on the stability of the alkoxy groups. Both of these factors ultimately direct gel structure by influencing the relative rates of hydrolysis and condensation and thus the degree of oligomerization or polymerization. Finally, physical factors such and volatility and viscosity can affect suitability of alkoxides for sol–gel chemistry.17
2.3 Hydrolysis and condensation of alkoxides
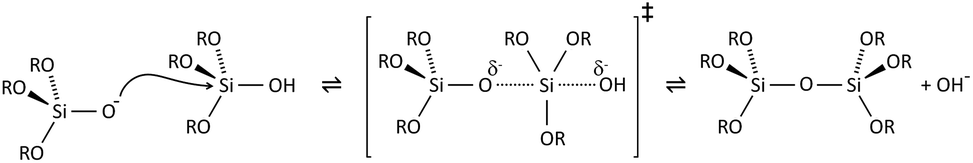
The key to mastering sol–gel chemistry of alkoxides requires understanding of the central hydrolysis and condensation reactions. These are strongly affected by process parameters such as the nature of the R-group (e.g. inductive effects), the ratio of water to alkoxide and the presence and concentration of catalysts. The sol–gel chemistry of silica is typically driven by either acid or base catalysts as the neutral reaction is very slow. The structure of the resulting gel is significantly different depending on the catalyst and this is due to the relative rates of the hydrolysis and condensation reactions. Hydrolysis results in the replacement of an alkoxy group with a hydroxyl with a pentacoordinate transition state in both the acid (Scheme 1) and base (Scheme 2) catalysed systems. Depending on the conditions and the Si/H2O ratio, more than one alkoxy group may be hydrolysed (eqn (1)). The rate of each hydrolysis step depends on the stability of the transition state which in turn depends on the relative electron withdrawing or donating power of –OH versus –OR groups. The result is that successive hydrolysis steps get progressively slower under acidic conditions and faster under basic conditions.| Si(OR)4 + nH2O → Si(OR)4−n(OH)n + nROH | (1) |
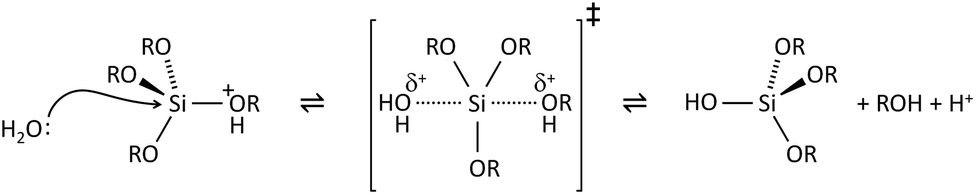 | ||
| Scheme 1 Acid catalysed hydrolysis of silicon alkoxides. | ||
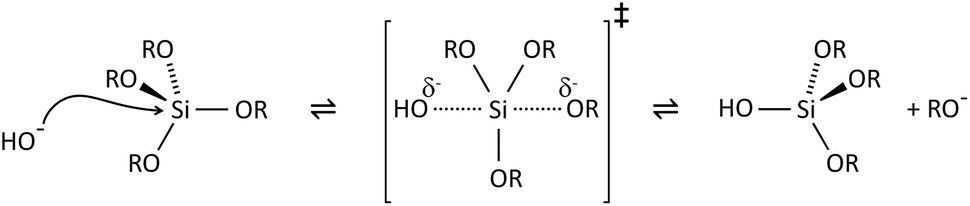 | ||
| Scheme 2 Base catalysed hydrolysis of silicon alkoxides. | ||
Condensation follows a similar pattern, being catalysed by either acid (Scheme 3) or base (Scheme 4) and resulting in the formation of siloxane bonds (or metaloxane bonds for other metals). The progression of condensation depends on the degree of hydrolysis that has already occurred as a silanol group is required on at least one silicon centre. If hydrolysis is complete before the first condensation step occurs, the resulting product (OH)3Si–O–Si(OH)3 has 6 sites for subsequent condensation steps. This is observed in basic conditions, where hydrolysis steps get progressively faster. Multiple condensation steps result in small, highly branched agglomerates in the ‘sol’ which eventually crosslink to form a colloidal gel. In acidic conditions, where the first hydrolysis step is typically the fastest, condensation begins before hydrolysis is complete. Condensation often occurs on terminal silanols, resulting in chain like structures in the sol and network-like gels. The consequences for gel morphology are represented in Fig. 2.
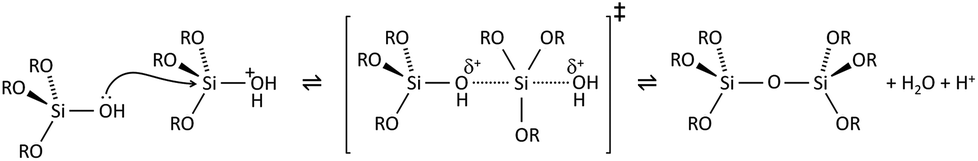 | ||
| Scheme 3 Acid catalysed condensation of silicon alkoxides. | ||
 | ||
| Scheme 4 Base catalysed condensation of silicon alkoxides. | ||
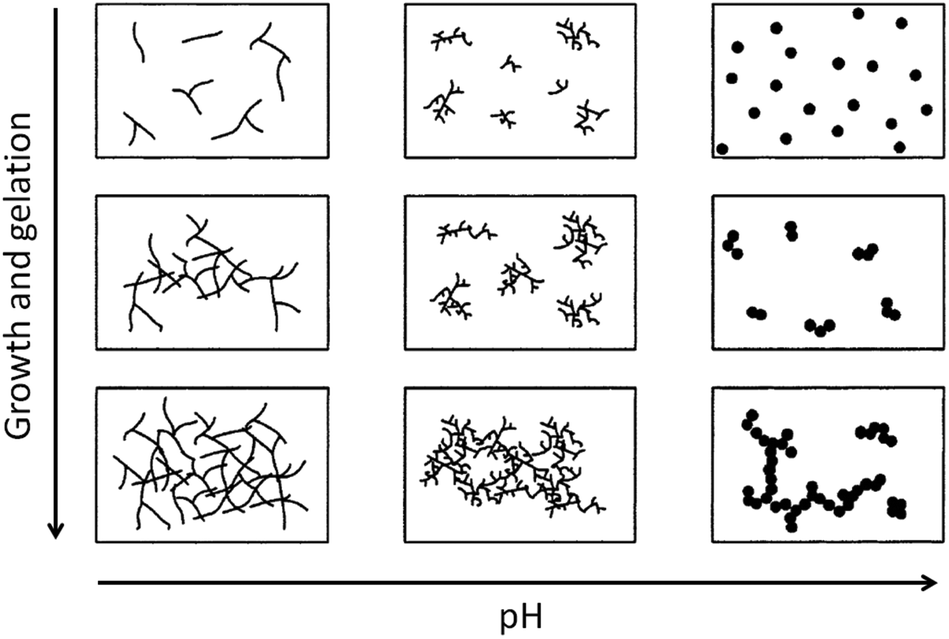 | ||
| Fig. 2 Diagram showing how pH affects the growth and structure of a gel; adapted with permission from ref. 2. Copyright (2004) American Chemical Society. | ||
In addition to acid and base catalysts, many other factors can affect the rates of hydrolysis and condensation and thus the structure of silica gels. One important influence is the presence of solvents, either to enhance mixing (many silicon alkoxides are immiscible with water) or direct interaction of solvent molecules with the silicon centre. Water itself is important and alkoxide:water ratio can be tuned to limit hydrolysis. Many different silicon alkoxides exist and the inductive and steric effects of the R group can impact on hydrolysis rates. It should also be noted that molecular silicon chemistry is far more diverse than simple tetraalkoxides and many compounds exist with the general structure SiR(OR)3, SiR2(OR)2 or SiR3OR. Finally, the presence of chelating agents such as acetylacetone can also be a method to reduce hydrolysis and condensation rates, although this becomes more important in the sol–gel chemistry of other metals.
Other metal alkoxides in sol–gel chemistry can follow similar reactions and pathways to silicon. However, most other metal alkoxides are based on elements with substantially lower electronegativity than silicon, the most important being the early transition metals such as titanium and zirconium. The partial charge model considers electronegativity differences and can be used to estimate stability and reactivity of alkoxides. For a four-coordinate tetraethoxy complex (M(OEt)4), the partial charges for Si, Ti and Zr can be calculated as +0.32, +0.63 and +0.74 respectively.2 Hydrolysis proceeds via nucleophilic attack by either water or hydroxyl groups on the central metal and the substantially higher rates of hydrolysis for Ti and Zr alkoxides are reflected in their higher partial positive charge. An important point to make here is that the variable reactivities of different metal alkoxides can lead to problems in synthesizing ternary or quaternary products due to phase separation during the condensation steps. The higher rate of hydrolysis means that many transition metal alkoxides react violently with water to the extent that most need special handling and storage. An important case where careful handling is required is for titanium alkoxides. Most of these will react vigorously with water to produce ill-defined titanium–oxo/hydroxo precipitates. Unlike silicon, where catalysts are added to enhance hydrolysis and condensation, titanium alkoxides therefore require additives to slow down the sol–gel reactions. Most of this chemistry centres on the use of bidentate or multidentate ligands such as acetylacetone to substitute for some of the alkoxide groups on the titanium, although the precise solution structure of the metal alkoxides and the formation of clusters are also important.18 The strength of binding of the chelating ligand, as well as the ligand:alkoxide ratio both impact the reactivity of the titanium precursor but also have structural implications for the resulting gel. For example if the number of OR groups is reduced, there are fewer sites for hydrolysis resulting in less crosslinking in the final gel. The chelating ligands may also have stereochemical effects by directing hydrolysis and condensation to certain sites. These effects are described in detail in an excellent review by Schubert.19 The control of metal sol–gel chemistry in this way has enabled the synthesis of a wide range of crystalline and amorphous transition metal oxide structures such as thin films20 or monodisperse particles.21
2.4 Materials from sol–gel chemistry: processing, post-processing and templating
In addition to the chemistry involved in forming a gel, there are many ways to control the sol–gel process to introduce important chemical and structural features. One important factor is how the sol or gel is physically treated because something as simple as rate of evaporation during gelation can have a substantial impact on gel structure. Heat treatment is also important for drying gels as well as removing surface hydroxyl groups, densifying the material to produce a ceramic monolith or converting to a crystalline material.There are many methods of processing a silica sol or gel and many of these can be applied to gels produced from other elements (ref. 4 and references therein). Processing sols or gels can be as simple as fast stirring during hydrolysis and condensation to produce small particles, as exemplified by the Stŏber synthesis.22 Another important feature of sol–gel processing is converting the solvent-filled gel into a dry solid. Simple evaporation of solvent from a silica gel is possible, but the movement of solvent through the gel subjects it to considerable capillary forces resulting in collapse of the network. This can be countered to some extent by aging the gel for a long time prior to drying but some densification is unavoidable due to expulsion of the sol from within the gel body (syneresis). The products of this uncontrolled drying (called xerogels) often have a high surface area due to the large number of small pores but without addition of structure-directing agents this porosity is generally disordered. If a larger pore volume is required, gels can be dried under supercritical conditions to produce aerogels with up to 98% air (or other gas) by volume.23 In fact, sol–gel synthesis can also be carried out in supercritical fluids to produce nanostructures of a wide range of materials. It is also possible to achieve high levels of porosity through freeze drying, this results in a cryogel, the porosity of which is usually between a xerogel and aerogel.24 In terms of fibres and thin films from alkoxide precursors, the most important factor is tuning the water:alkoxide:solvent ratio.25 The resulting solution can then be spun into fibres or spin/dip coated onto a surface to produce metal oxide thin films.26
In addition to physical methods to control structure in sol–gel chemistry, templates can also play an important role in introducing both ordered and disordered porosity. The most common additives have been ‘soft templates’, for example amphiphiles, block copolymers, ionic liquids, biopolymers and proteins.27 Alternatively, hard materials such as colloidal particles, bacterial filaments or cellulose nanocrystals have been employed.28 In some cases, alkoxides can be modified to enhance interaction of the sol–gel precursors with a soft or a hard template and produce ordering or porosity on multiple length scales.29 In many of these examples, the template can either be left in the oxide to produce an inorganic/organic nanocomposite or removed by dissolution or calcination. In addition, templating can be combined with functionalization of the material, for example to generate porous silica that incorporates molecular recognition sites.30
A particularly important field that uses amphiphiles for ‘templating’ sol–gel materials is evaporation-induced self-assembly (EISA). This can be used to introduce ordered mesoporosity into bulk or thin-film metal oxide materials.31 There are many excellent reviews of the field and the mechanisms of ordering in EISA.32 Briefly, the method involves a mixture of sol–gel precursors such as water, ethanol and a metal alkoxide or chloride, combined with amphiphiles such as cetyltrimethylammonium bromide (CTAB) or block copolymers. Rather than simple direction of the sol–gel condensation within solution, EISA relies on gradual evaporation of volatile species from the mixture to form a mesophase. Inorganic material accumulates around this liquid-crystal template, which results in well-ordered mesostructuring in the resulting metal oxide.
It should be noted that while this section has focussed on processing of alkoxide precursors, many of these methods can also be applied to materials and techniques discussed later in this review.
3. Small molecule ‘gels’
3.1 Hydrolysis reactions in aqueous solution
The scope of ‘traditional’ sol–gel chemistry is broad and it is only possible here to give a flavour of the diverse reactions possible with alkoxide chemistry. However, alkoxide-based sol–gel chemistry has one significant limitation, namely that many metal alkoxides either cannot be formed or are too unstable to be used. A minor additional point is that if a metal alkoxide is extremely sensitive to moisture it can often be employed in sol–gel synthesis with careful handling, but water-soluble structure directing agents or templates may not be compatible. Given the power of sol–gel chemistry, many alternative methods have been developed that can employ aqueous metal salts rather than alkoxides. The chemistry is very different but the goal is the same: the controlled formation of metal oxide or other ceramic structures from solution-phase precursors. The first main strategy involves the use of small molecules (often chelating agents) to modify the aqueous hydrolysis chemistry of metal ions. For an aqueous solution of metal ions of charge z+, water molecules coordinate to the metal via electrons in their bonding orbitals. In the case of transition metals, this density is transferred into empty d-orbitals. The result is a weakening of the O–H bonds of bound water molecules and, depending on the pH, deprotonation or hydrolysis (eqn (2)). For highly reactive metals, the high rate of hydrolysis may necessitate the control of water content and the use of nonaqueous solvents. For less reactive metals, where an aqueous solution is readily achieved, the extent of hydrolysis can be controlled easily with pH. Basic conditions will force the equilibrium to the right and favour the formation of oxo ligands, whereas acidic conditions can favour hydroxo ligands or even prevent hydrolysis. For some metals, such as Fe3+, hydrolysis can result in extended polymer-like precipitates containing oxo and hydroxo bridges. In addition to pH, highly charged metal cations tend to weaken the O–H bond in bound water and favour hydrolysis. This relationship between charge, pH and hydrolysis equilibria is shown in Fig. 3a.[M(OH2)]z+ ⇌ [M–OH](z−1)+ + H+ ⇌ [M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O](z−2)+ + 2H+ O](z−2)+ + 2H+ | (2) |
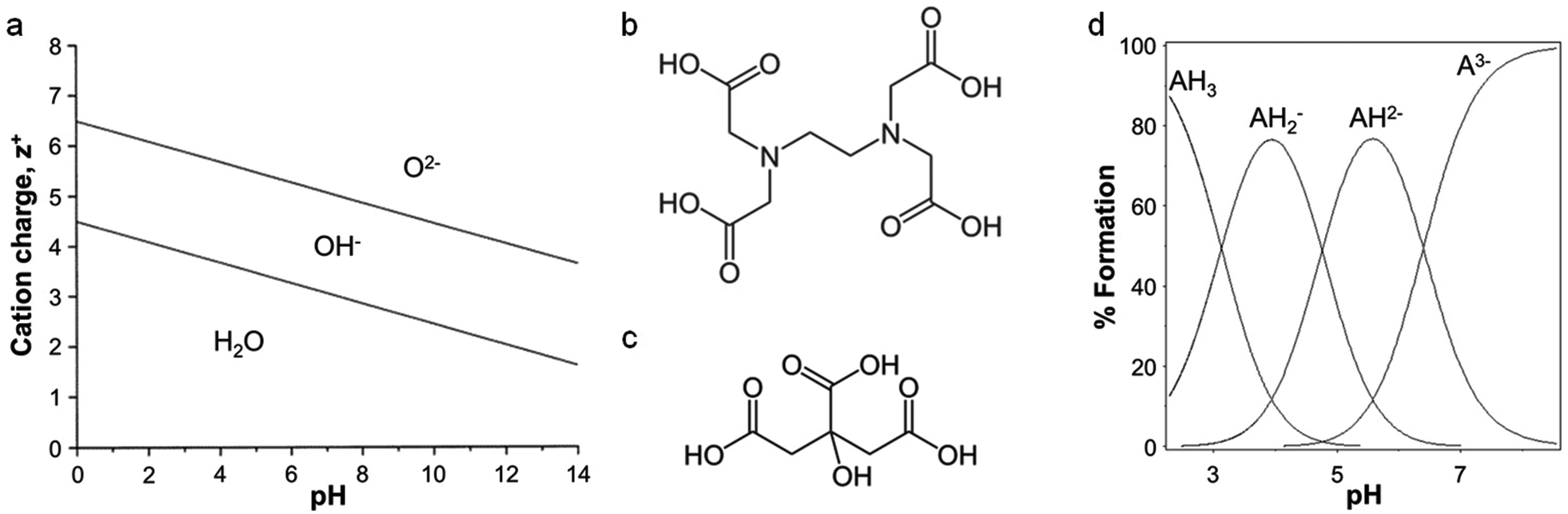 | ||
| Fig. 3 (a) Relationship between charge, pH, and hydrolysis equilibrium of cations, modified with permission from ref. 2. Structures of (b) EDTA and (c) citric acid. (d) Ion speciation graph for citric acid plotted using the program HSS. | ||
For many metal salts the aqueous solution is stable. However, while the solution is homogenous, it does not resemble the gels of sol–gel chemistry. The formation of most metal oxides (and other ceramics) requires heat treatment and simply drying a metal salt solution will result either in precipitation of the original metal salt, or amorphous oxides/hydroxides. The result is typically large crystals or aggregates, certainly not the controlled formation of porous structures or regular particles of sol–gel methods. To avoid this, many small molecules have been employed to form stable aqueous metal complexes and structures that more closely resemble ‘gels’. Many of these small molecules are chelating agents and the main function is to change the hydrolysis equilibria of dissolved metals. For example, the addition of EDTA (ethylenediaminetetraacetic acid, Fig. 3b) to aqueous iron can significantly reduce the equilibrium constant of hydrolysis from Kh = 10−3 (eqn (3))33 to Kh = 10−7.5 (eqn (4)).34 By making hydrolysis considerably less favourable, the removal of solvent from many metal–chelate solutions results in homogeneous glassy solids or resins rather than precipitates. These can be heat treated to form powders or nanostructures of a wide range of binary, ternary and ternary metal oxides as well as metal nitrides or carbides.
| [Fe(H2O)6]3+ + H2O ⇌ [Fe(OH)(H2O)5]2− + H3O+ | (3) |
| [Fe(H2O)2EDTA]− + H2O ⇌ [Fe(OH)(H2O)EDTA]2− + H3O+ | (4) |
In this section, we will consider some of the most important examples of small molecule sol–gel chemistry in detail. This field has a huge scope, with multiple factors that can be tuned such as pH, concentration and nature of complexing ligand, temperature of gelation, rate and final temperature of calcination.35
3.2 Citrate
One of the most common small organic molecules used in sol–gel chemistry is citric acid. Citric acid is a weak triprotic acid (Fig. 3c) with three carboxylic acid moieties that are able to dissociate (eqn (5)–(7)).36 While being readily available and cheap it is also an effective chelating agent. In a typical synthesis, aqueous metal salts (e.g. nitrates) are mixed with citric acid and the resulting solution heated to form a viscous solution or gel. Some reports describe the addition of bases such as ammonia or ethylene diamine to modify the pH and enhance cation binding to the citrate. The homogeneity and stability of metal citrate solutions can thus depend strongly on pH (Fig. 3d).37 Tuning of pH appears to be particularly important in systems with several different metals, in order to optimize the formation of stable metal citrate species and prevent precipitation of individual hydroxides.38,39| H3Cit ⇌ H2Cit− + H+ Ka1 = 7.10 × 10−4 | (5) |
| H2Cit− ⇌ HCit2− + H+ Ka2 = 1.68 × 10−5 | (6) |
| HCit2− ⇌ Cit3− + H+ Ka1 = 6.40 × 10−6 | (7) |
The citric acid sol–gel method (also referred to as the citrate sol–gel method) is normally used for the synthesis of metal oxide powders. Conversion of the ‘gel’ to a metal oxide is simply achieved by pyrolysis in air, with the maximum temperature depending on the specific system. The method has been used to synthesize binary, ternary and quaternary metal oxides in both crystalline and amorphous forms. The key advantage of this method, as with more traditional sol–gel chemistry, is the homogeneity of the starting material. As the metal–citrate ‘gels’ are heated, the organic component undergoes combustion at ∼300–400 °C, depending on the metal counterion and presence of additives. The presence of the organic matrix during the first stages of synthesis can ensure that when nucleation occurs, the sites are evenly dispersed and numerous, ensuring a small crystallite size. In the case of ternary or quaternary systems, the other purpose of the matrix is to ensure that the different metals remain mixed on an atomic scale. Even in systems where the desired compound may not crystallise until >700 °C, long after the organic component has been combusted, the homogeneity of the precursor gel can ensure that the system remains amorphous until the final product begins to nucleate. This is important in ternary and quaternary metal oxides such as doped yttrium aluminium garnets (e.g. Y3Al5O12, YAG), where the nucleation and growth of intermediate phases can disrupt the homogeneity of the system and result in impurities or particle size irregularity in the final product.40 Another impact of the homogeneity of citrate sol–gel precursors is on reaction temperature since the final crystalline metal oxide may be formed at considerably lower temperatures than powder solid-state methods where mass transport between grains limits the reaction.41
Although most reports describe an air atmosphere, it should be noted that inert atmospheres can also be used to produce ceramic/carbon composites where the citrate provides the carbon source. One example is the synthesis of carbon/LiFePO4, where the reducing conditions also have the effect of preserving the ferrous oxidation state of the iron precursor. In this case, the resulting material is a composite of small particles of the LiFePO4 (an important cathode material for lithium ion batteries) dispersed in a carbon matrix that enhances electronic conductivity.42 In addition to oxide/carbon composites, inert atmospheres can be used to transform citrate precursors into reduced materials such as metal carbides43 or metal borides.44
The majority of work reported on the citrate sol–gel method uses metal nitrate precursors. Rather than simply being a convenient source of aqueous metal ions, the nitrate counterion plays an important role in citrate sol–gel chemistry. Thermogravimetric analysis of various metal nitrate/citrate combinations reveal a very sharp mass loss step associated with an exothermic peak in the differential thermal analysis (DTA) trace (Fig. 4a).45 The mass loss typically occurs around 200 °C and is associated with a rapid, self-propagating combustion where the nitrate acts as the oxidant and the citrate as the organic fuel (Fig. 4b–e).46 In some systems, the combustion can be triggered by ignition of the sample at room temperature to form loose powder. These may require further heat treatment to achieve the desired crystalline phase but in many cases enough heat is generated during the combustion process.47 The crystallinity and morphology of powders prepared by citrate auto-combustion can depend on pH. For example, in the synthesis of NiCuZn ferrite from nitrate/citrate precursors, an increase of pH in the precursor solution resulted in oxide products with a more open and porous network structure (Fig. 4f).48 In fact, many citrate combustion syntheses result in ‘sponge-like’ products due to the large volume of gases evolved during the reaction of nitrate with the organic component.49 In this particular example, the pH was modified via addition of ammonia, resulting in a build-up of NH4NO3. The exothermic peak for high pH (>6) was sharp and the product more porous due to the decomposition of NH4NO3 into NOx and O2 which accelerates combustion.
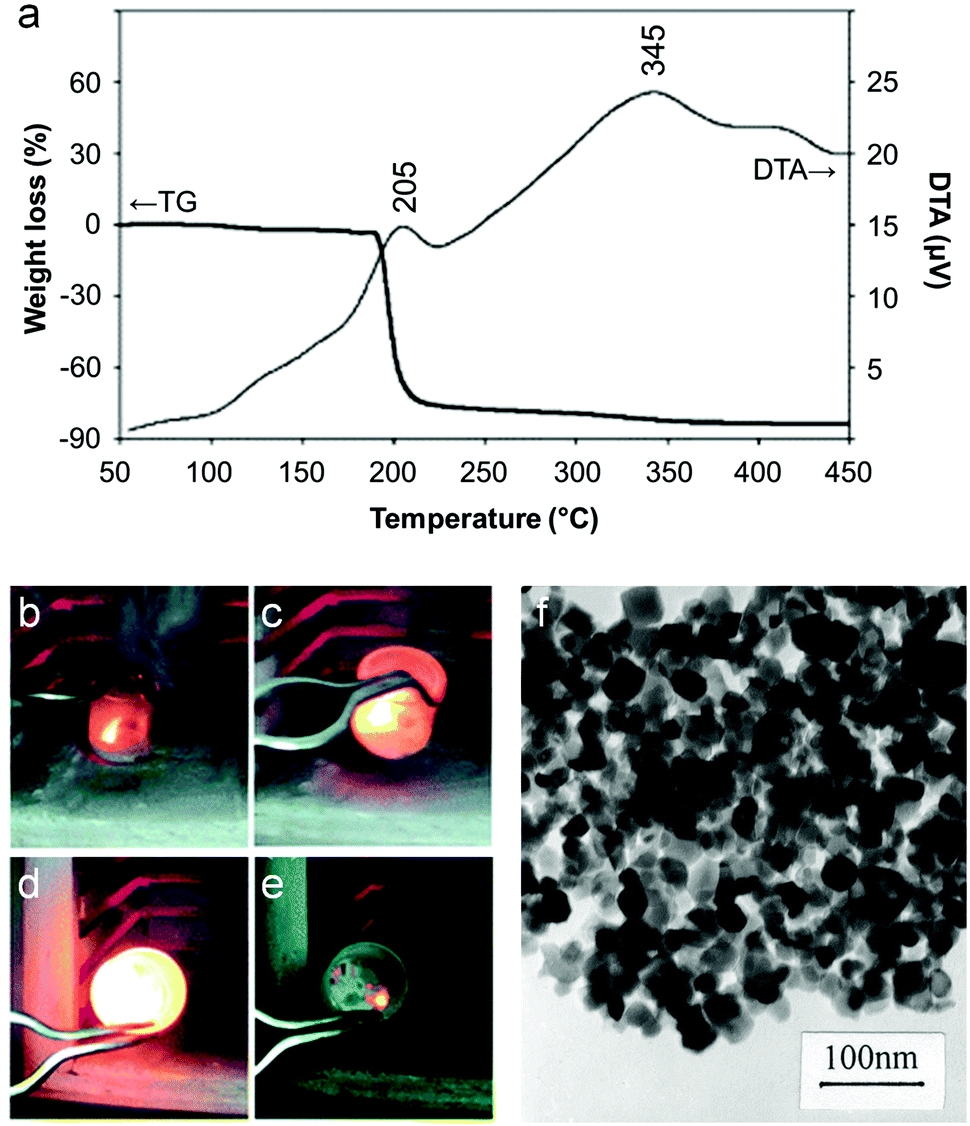 | ||
| Fig. 4 (a) TGA/DTA trace for a mixture of citrate with barium and iron nitrates showing a sharp combustion at ∼200 °C. (b–e) Images from a video of a combustion synthesis showing rapid progression of the reaction. (f) TEM image of a sample of a NiCuZn ferrite synthesized from a citrate/nitrate combustion method. Figures modified with permission from ref. 45, 46 and 48 respectively. | ||
3.3 Other aqueous small molecule gelators
Given the success and wide application of citric acid in sol–gel processing, it is perhaps not surprising that other mono- and di-carboxylic acids have also been employed. These include tartaric acid,50 glycolic acid51 and oxalic acid.52 As with citric acid, the pH of the system is important to optimize binding of the carboxylate to the metal and many of these examples use ammonia or other bases as an additive.53 Interestingly, the large range of dicarboxylic acids offers a way to control the decomposition profile of a nitrate/carboxylate gel in combustion synthesis. Linear aliphatic dicarboxylic acids from oxalic (HO2CCO2H) to sebacic (HO2C(CH2)8CO2H) acid were used to synthesize LiNi0.8Co0.2O2. The gels showed a linear increase in decomposition temperature with increasing length of the dicarboxylic acid.54 Given that persistence of the organic matrix may have some influence on particle size in sol–gel synthesis, this offers a useful way to study aqueous sol–gel processes across a wide range of systems.Many other small molecules have been combined with metal nitrates as a fuel/oxidant combination for oxide powder synthesis.55 Glycine is a common chelator and ‘fuel’ and has a lower ignition temperature of combustion than citrate.56 This can be advantageous. Glycine–nitrate mixtures burn quickly, releasing gases and forming ‘fluffy’ powders.57 However, the reaction is highly exothermic and in some cases explosive, which may preclude large scale-up of the process.58 A modification to the glycine–nitrate method that helps to mitigate the violent reaction and gas evolution is to soak the precursors into cellulose fibres.59 The cellulose fibres act as a micro-reactor for the sol–gel process and also help to ensure a very small particle size (15–20 nm) of the resulting La0.6Sr0.4Co0.2Fe0.8O3 by maintaining spatial separation of the nucleation points and growing crystallites. Other notable small molecule gelators to be used in sol–gel synthesis are ethylenediaminetetraacetic acid (EDTA),60 glucose61 and amino acids such as glutamine and histidine.62
3.4 Epoxide sol–gel
The chemistry involved in the epoxide ‘sol–gel’ method is somewhat different from the other examples in this section. However, the overall strategy still involves the addition of a small molecule to influence hydrolysis of a dissolved metal salt and so it is useful to discuss it here. In general, the method involves dissolving hydrated metal salts in ethanol (or other polar protic solvents). Propylene oxide is then added to drive formation of a gel. The nature of the metal counterion (e.g. Cl−, NO3−) the solvent and the amount of water and propylene oxide are all critical to the appearance and also rate of formation of the gel.63 Unlike the chelating agents in the previous examples, the role of propylene oxide in this approach is as a proton scavenger, driving the formation of metal–oxo bonds. For example, in the synthesis of iron oxide using propylene oxide, the epoxide additive is believed to drive firstly the hydrolysis of hydrated iron species (eqn (8)), which then leads to condensation to form iron oxo (Fe–O–Fe) bonds (eqn (9)). In this sense, the epoxide sol–gel method bears some resemblance to silica sol–gel chemistry.| [Fe(H2O)6]3+ + H2O ⇌ [Fe(OH)(H2O)5]2+ + H3O+ | (8) |
| 2Fe(OH)(H2O)5]2+ ⇌ [(H2O)5FeOFe(H2O)5]4+ + H2O | (9) |
3.5 Urea
Like the epoxide method, the use of urea in sol–gel chemistry is somewhat different to many examples of ‘small molecule gelators’ in that water is generally not used as the solvent. There are examples of urea being used in a standard fuel/nitrate sol–gel combustion synthesis to form metal oxides.64 However, the most recent urea chemistry has involved the synthesis of metal nitrides and carbides. The earliest examples of using urea to make nitrides involved heating metals (e.g. gallium or indium) with urea and so cannot be considered as sol–gel chemistry.65 However, the author performed a detailed analysis of the decomposition products during synthesis which is instructive when considering later ‘gel’ examples. It should also be noted that gallium and indium have very low melting points and most of the reactions in this paper would occur above this, therefore it is certainly not a ‘normal’ solid-state process. Urea undergoes endothermic decomposition to form numerous compounds, for example biuret and triuret via initial condensation of two and three urea molecules respectively. Subsequent condensation and dehydration reactions result in release of NH3 and H2O and formation of compounds including dicyandiamide, cyanuric acid, melamine and melem (a tri-s-triazine). In the reaction of gallium with urea,66 the author concludes that gallium reacts with some of the urea decomposition products to generate polymeric intermediates. These undergo further decomposition to produce GaN although pure GaN could only be isolated if the process was carried out in an ammonia atmosphere.In later work, it was demonstrated that phase-pure metal nitrides could be synthesized under an inert atmosphere (e.g. N2 or Ar) directly from ‘gel-like’ precursors.67 In general, metal chlorides such as MoCl5 or WCl4 are dissolved in ethanol, releasing HCl gas and producing and ethanolic solution of the metal alkoxides. Solid urea is then added and the resulting solution dried to give a glassy solid. Heating these solids to 800 °C under nitrogen results in molybdenum or tungsten nitrides or carbides depending on the metal:urea ratio. The products are comprised of nanoparticles (diameter 4–20 nm) embedded in amorphous carbon. The method was later extended to synthesize nanoparticles of nitrides and carbides of Ga, Ti, Nb and V. The formation of nanoparticles is a significant step, given the importance of metal carbides and nitrides in catalysis.68 In the case of iron carbide (Fe3C), the ability to form small nanoparticles (5–10 nm) using urea meant that the product was superparamagnetic (each nanoparticle is a single magnetic domain).69
Calcium can assist the urea sol–gel synthesis. This has proven effective in isolating TaON and Ta3N5, both important semiconductor photocatalysts with a smaller bandgap than the corresponding oxide. Previous methods result in the formation of complex mixtures of oxynitrides and nitrides but the use of calcium/urea precursors enabled the tunable formation of single phases by simply changing the Ta/urea ratio.70 The authors use infrared spectroscopy and thermogravimetric analysis to propose a mechanism that involves Ca2+ binding to urea via the carbonyl oxygen, weakening the CO bond and strengthening the C–N bonds (Fig. 5). The result is that the urea decomposition is delayed, slowing the release of NH3 and creating a more homogeneous and persistent atmosphere within the evolving system. Importantly, the use of calcium also produces a much more homogeneous product with smaller crystallites and higher surface area.
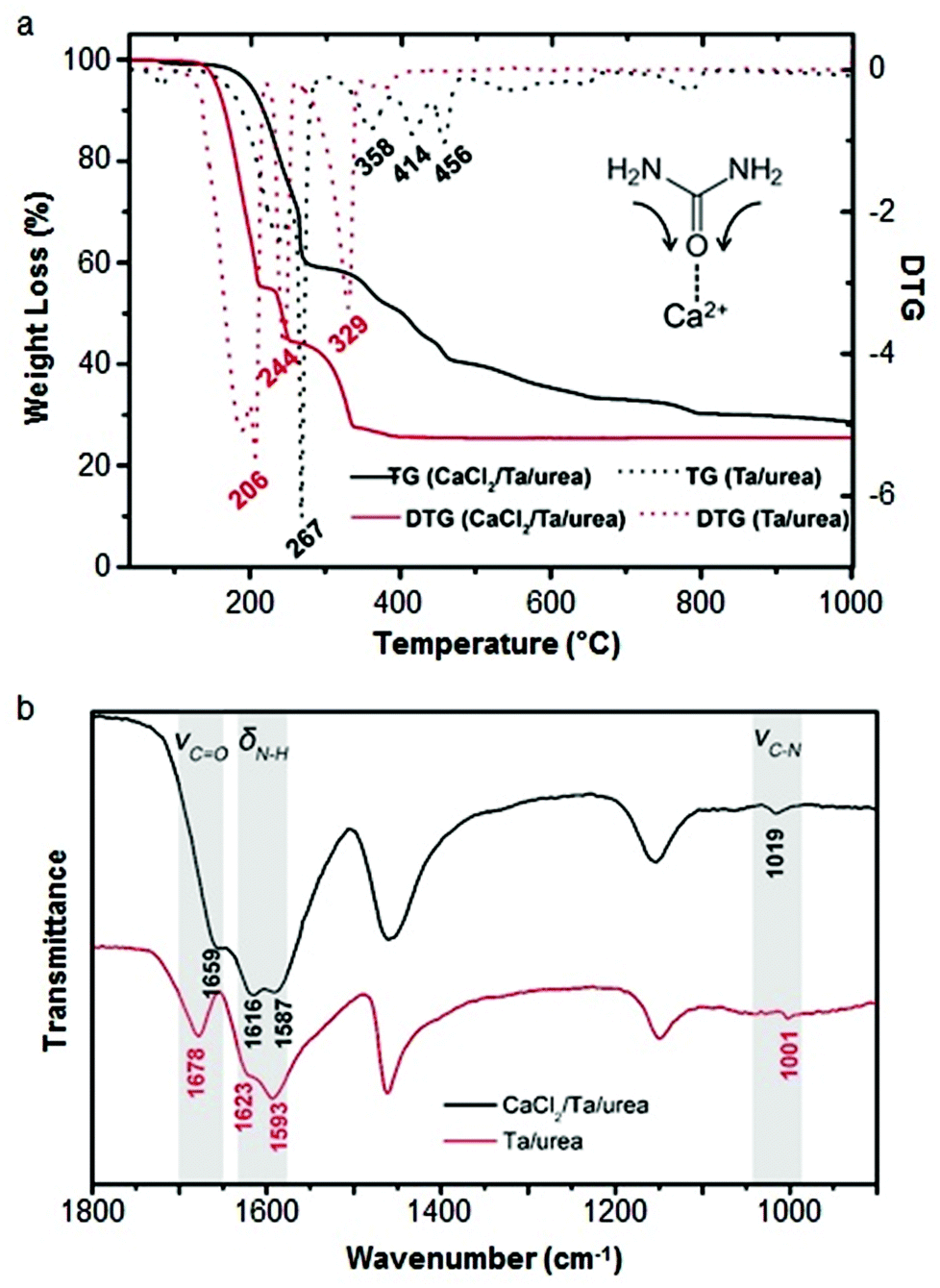 | ||
| Fig. 5 (a) TGA/DTG of tantalum/urea mixtures both with (black) and without (red) calcium showing the later onset of decomposition with calcium. (b) IR spectra of the same samples showing a shift and reduction in intensity of the CO stretch of urea when calcium is added. Modified with permission from ref. 70. | ||
4. Pechini method
4.1 Chemistry of the Pechini method
The Pechini method, named after the author of the original patent,9 builds on the principles of sol–gel chemistry involving small molecule chelating ligands in that the initial step is to form a homogeneous solution of metal/citrate complexes. However, the Pechini method takes this further to convert the mixture into a covalent polymer network to entrap the metal ions. The reasoning for the method was to delay the thermal decomposition of the organic matrix in order to afford more control over the growing ceramic product. The key reaction used in Pechini synthesis is transesterification between citrate and ethylene glycol (Scheme 5).71 In a typical synthesis, a metal salt is dissolved in water with citric acid and ethylene glycol to form a homogeneous precursor solution containing metal–citrate chelate complexes. This solution is heated to initiate polyesterification between the citrate and ethylene glycol, forming an extended covalent network. Fig. 6 shows a proposed schematic of this process. Following formation of the polymer network, the material is heated in a furnace to combust the organic matrix and form the ceramic product. One of the most significant advantages of the Pechini method is the ability to form a polymeric precursor where two or more metals may be dispersed homogeneously throughout the network.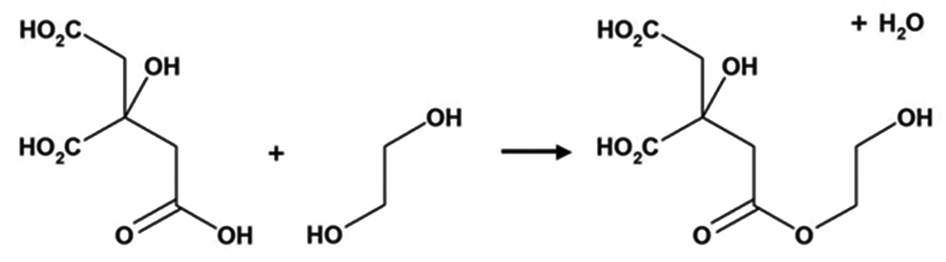 | ||
| Scheme 5 The transesterification reaction that occurs between citric acid and ethylene glycol in the Pechini process. | ||
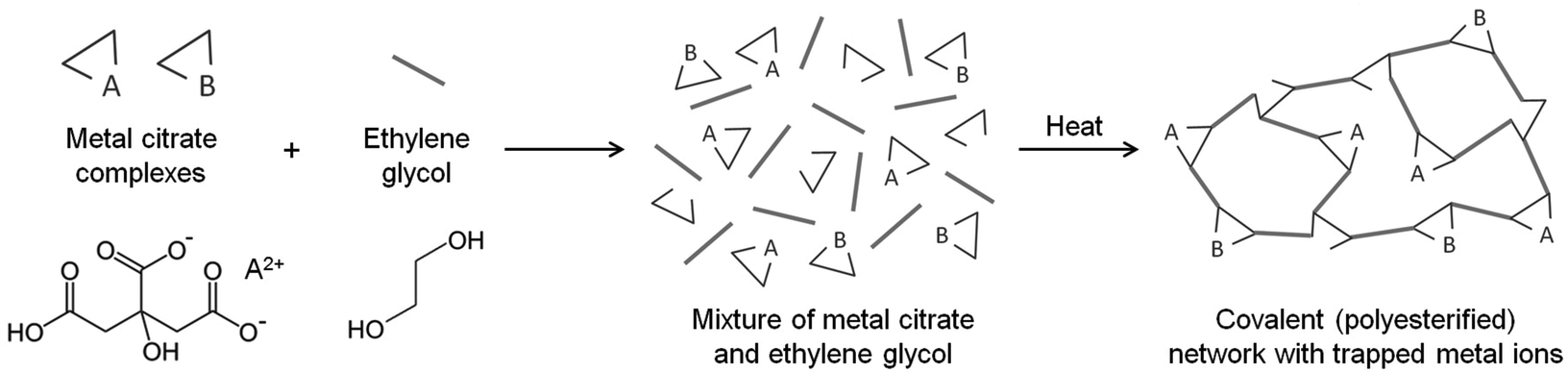 | ||
| Fig. 6 Schematic of the Pechini method of making metal/organic gels. | ||
4.2 Materials from the Pechini method
Many authors have employed the Pechini method to synthesize metal oxides by combining metal salts with ethylene glycol and citric acid and treating the resulting resin in a furnace in air. Multiple variations have been reported in order to optimize the method to a particular system. As described in the citrate sol–gel section above, the binding of citrate to metal ions depends on the pH of the solution, with low pH resulting in protonation of the citrate and high pH risking precipitation of metal hydroxides. The control of pH in the citrate sol–gel method is thus very important in controlling homogeneity in the gel and particle size in the final product. Similarly, many examples of the Pechini method report optimizing the pH in the initial metal–citrate solution using ammonia, ammonium hydroxide or other bases.72 One method of adjusting citrate binding in the Pechini method was proposed by Abreu Jr. et al. Rather than using a base such as ammonium hydroxide, the authors use urea, which gradually decomposes in the system to release ammonia and result in a more controlled rise in pH.73 The effect on the product is striking. The urea-modified method produces much smaller crystallites of Pb(Zr0.52Ti0.48)O3, although it is unclear whether this is exclusively due to the pH control or also the increased spatial separation of the nuclei by urea condensation products such as melamine.In addition to metal oxides, the Pechini method has been used to synthesize transition metal carbides. These have been employed for many years for their mechanical properties but more recently have attracted interest as catalysts. The method is essentially the same as for oxides, but the precursor gel or resin must be heated in an inert or reducing atmosphere instead of air. For example, Ni6Mo6C can be synthesized by heating metal acetates, citric acid and ethylene glycol under hydrogen to 600–900 °C The product structure depends strongly on the maximum temperature, being mesoporous with a surface area of 96 m2 g−1 when heated to 800 °C but sintering to a surface area of only 5 m2 g−1 after heating to 900 °C.74
Most examples of Pechini synthesis of ceramics result in powders of agglomerated crystallites. However, if metal nitrates are used in the initial mixture, it is possible to generate foams through release of nitrous oxides during the reaction, analogous to many examples of combustion synthesis.75 Alternatively, it is possible to fill a porous template with the mixture of metal salt, ethylene glycol (EG) and citric acid (CA) and heat to polymerise the network inside the template. This has been used in several cases to produce polycrystalline wires using anodised alumina templates.76 Pechini precursors can also be used to create thin films. This is possible with alkoxide-based sol–gel precursors. However, the aqueous nature of the Pechini method enables the production of films from a wider range of elements as water-soluble salts are more readily available and easier to work with than metal alkoxides. For example, Eu-doped Lu2O3 films can be prepared by mixing LuCl3, water, ethanol, citric acid, polyethylene glycol and Eu(NO3)3 and dip coating the resulting solution onto silicon wafers.77 The ability to create films of ceramics is important in various applications, particularly those involving light absorption and emission such as displays.
Mesoporous oxides have also been produced in a similar way by templating with colloidal crystals and this latter example has given some remarkable insights into the mechanism of how the Pechini precursors decompose to form a ceramic. By changing various experimental parameters, Rudisill et al. showed that the structure of CeO2 and Mg/Ca/Sr-doped ceria could be tuned to form either mesoporous microspheres or a bicontinuous mesoporous network.78 The authors showed that the structure was dictated by phase separation in the early stages of the synthesis i.e. during the polyesterification process, analogous to the polymerization-induced phase separation that can be achieved in sol–gel synthesis of silica from alkoxides.79 This resulted in either a nucleation mechanism or the development of a bicontinuous structure through spinodal decomposition (Fig. 7). A detailed investigation showed that many factors could affect the final structure, including the nature of the alkaline earth metal in the ternary systems (e.g. Ce0.5Mg0.5O1.5vs. Ce0.5Ca0.5O1.5), the metal:CA ratio and the amount of EG. The confinement effect of the template was critical for the formation of the unusual structures. This is due to both the physical effect of confining the precursors as well as electrostatic interactions of the soluble precursors with the charged surface of the PMMA (polymethylmethacrylate) spheres of the colloidal crystal.
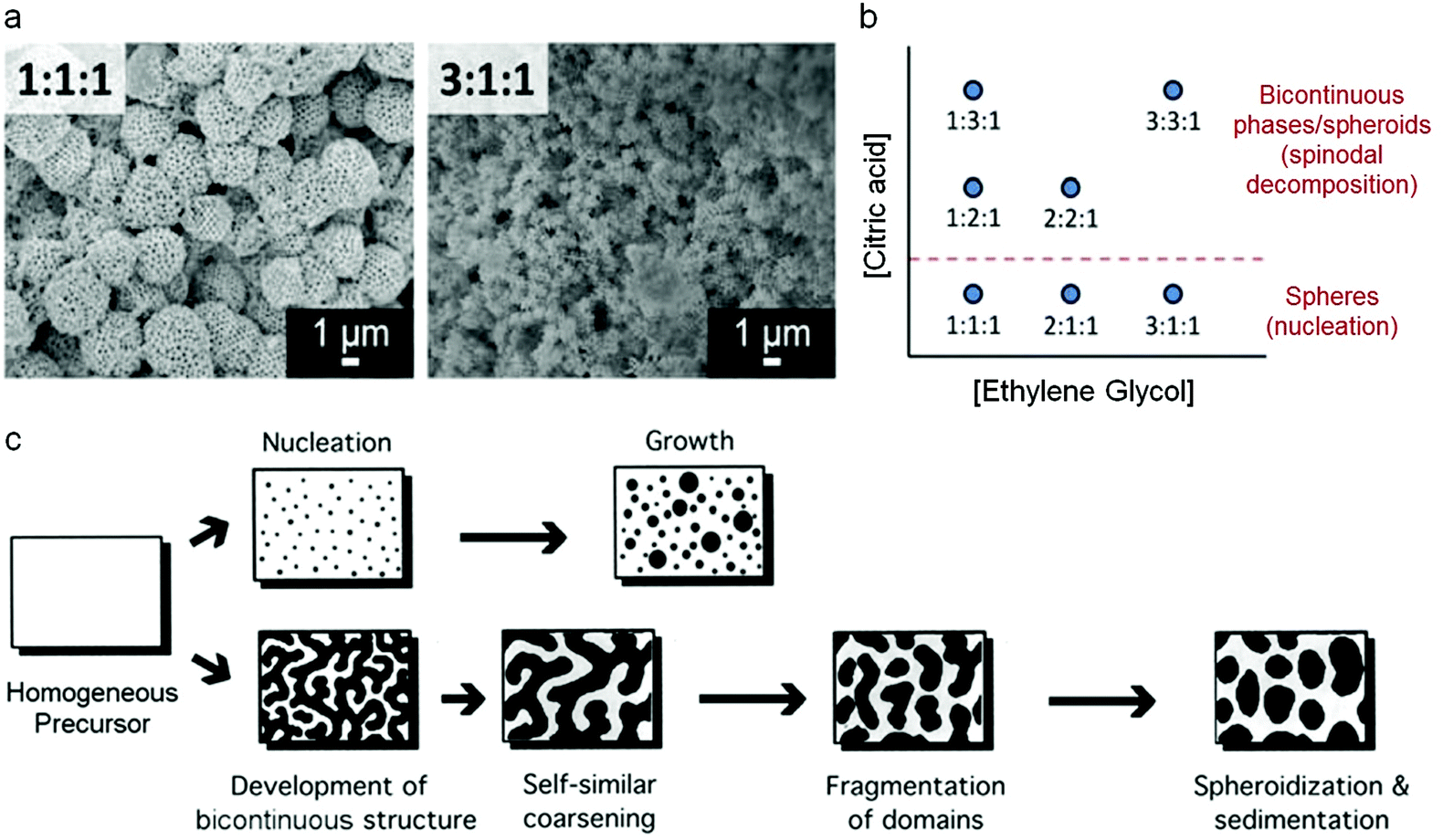 | ||
| Fig. 7 (a) SEM images of samples prepared at two EG:CA:TMI (ethylene glycol to citric acid to total metal ion) ratios, (b) schematic showing how EG:CA:TMI ratio affects structure and (c) schematic of nucleation and spinodal decomposition mechanisms. Modified with permission from ref. 78. Copyright (2012) ACS. | ||
4.3 Modifications to the Pechini method
Since the early examples of Pechini synthesis, there have been many developments to enhance the range of materials and structures that can be achieved via this process. Most of these have centred on replacing the citric acid with other di-, tri- or tetra-carboxylic acids and/or the ethylene glycol with other polyols. Early examples focussed on the substitution of citric acid with chelating agents that have a higher decomposition temperature, such as EDTA (ethylenediaminetetraacetic acid). In the synthesis of the YBCO superconductor YBa2Cu3O7−x, this was shown to aid the formation of a phase pure product. The synthesis of quaternary oxides like YBCO superconductors is often hampered by the uncontrolled nucleation and growth of intermediate phases that impede the reaction and lead to impurities in the final product. Replacement of citric acid with tartaric acid or EDTA was shown to limit the formation of unwanted barium carbonate. This was proposed to be due to the later onset of thermal decomposition of the EDTA/ethylene glycol polymer i.e. a longer period of homogeneity at the start of the heating process. Interestingly, the EDTA system produced larger particles than citric or tartaric acids, which could indicate that the structure and degree of branching of the polyester influences particle size.80Further insight into the specific effect of different precursors in Pechini synthesis came from some work by Rudisill et al.81 These authors previously discussed the synthesis of mesoporous microspheres or a bicontinuous mesoporous network of doped cerium oxides from Pechini precursors in a PMMA (polymethylmethacrylate) opal template.78 In a more recent publication, the authors investigated the substitution of citric acid (tricarboxylic acid) and ethylene glycol (diol) with malic acid (dicarboxylic acid) and glycerin (triol) respectively (Fig. 8a and b). Phase separation in the PMMA-templated systems is dependent on the polyesterification process and, importantly, the degree of polymerization. Changing the ratio of carboxylic acid:hydroxyl moieties therefore enables a study of functional group balance without changing the overall metal:organic ratio in the system. In other words, the number of reactive groups available for polyesterification can be varied in a controlled way. The result was a dramatic change in sample morphology (microspheres vs. bicontinuous network) with a simple change in the carboxyl:hydroxyl ratio i.e. extent of polyesterification (Fig. 8c–f) The authors postulate that a low level of polyesterification would lead to small and relatively soluble oligomers that can evenly fill the space in the PMMA template, minimizing polymerization-induced phase separation and leading to a continuous structure. A high level of polymerization and a correspondingly high molecular weight of the polyester would lead to polymer-rich regions in the aqueous solution. The difference in polarity between the solvent-rich and polymer-rich regions drives the formation of microspheres as the system attempts to minimize interfacial energy.
 | ||
| Fig. 8 Structures of (a) malic acid and (b) glycerine. SEM images of samples of Ce0.5Mg0.5O1.5 prepared at a 2:1:1 molar ratio of EG:CA:TMI (ethylene glycol to citric acid to total metal ions) in a PMMA opal template showing (c) no reagent substitutions, (d) substitution of glycerine for EG, (e) substitution of malic acid for CA, and (f) substitution of both glycerine and malic acid for EG and CA, respectively (c–f modified with permission from ref. 81). Copyright (2015) American Chemical Society. | ||
5. Polymers
The purpose of the Pechini synthesis is to synthesize a polymer in situ around metal ions and thus ensure the metals are mixed and stabilized homogeneously within a covalent matrix or ‘gel’. Therefore, it seems logical that the next step in the evolution of sol–gel chemistry involved the direct combination of polymers with metal salts to form sol–gel precursors. Polymer chemistry is diverse and many polymers form well-defined structures in solution, some of these with quite long-range order. Many polymers also exhibit strong interactions with metal ions. Despite the superficial similarity between polymer sol–gel methods and Pechini synthesis, there are specific advantages to using polymers. In particular, the ability of some polymers to form ordered superstructures can be used to control morphology in ceramic synthesis. However, polymers can bring some drawbacks, as will be discussed in this section.5.1 Synthetic polymers
Many reports using synthetic polymers in sol–gel chemistry focus on polyvinyl alcohol (PVA). The method is simple: aqueous metal salts (e.g. metal nitrates) are mixed with polyvinyl alcohol to form a homogeneous precursor that is heated at moderate temperatures (∼80 °C) to form a gel.82 This gel is typically dried and then heated in a furnace to obtain the required metal oxide ceramic. In some cases, the dried gel is powdered before the final heat treatment. The presence of the polymer ensures that particle size is kept very small (∼25 nm), even in the case of complex quaternary oxides such as YBCO (YBa2Cu3O7−x).83 This control over ceramic crystallite growth is believed to be due to complexation of the metal ions by hydroxyl substituents on the PVA. This was postulated by Liu et al. who showed that particle size homogeneity and phase purity of BiFeO3 was dependent on Mx+/–OH ratio in the PVA precursor.84 These authors also demonstrated the presence of carboxylate moieties, thought to be caused by oxidation of the PVA by nitrates in the system and this could conceivably aid metal binding. As with small-molecule gels, the metal nitrate precursors in a PVA sol–gel method can be exploited to initiate self-propagating combustion. This can be enhanced by the addition of supplementary ‘fuels’ to the PVA/metal nitrate mixture, such as urea, hexamine or citric acid.85In addition to PVA, several other synthetic polymers have been employed in sol–gel synthesis of ceramics such as polyethylene glycol (PEG)86 or polyvinylpyrrolidone (PVP).87 As with PVA, most of the methods involve dissolving metal salts such as nitrates or acetates with the polymer in a solvent such as water and heating to form a gel. The main benefit of the polymer is the same in all cases. Functional groups on the polymer (e.g. imide on PVP) bind to the dissolved metal ions to form a homogeneous gel, which constrains particle nucleation and growth, generating a nanoparticulate ceramic product. The benefit of different polymers comes primarily from their physical properties. For example, PVA and PVP have both been employed in sol–gel precursors that can be spin-coated onto a substrate to form ceramic thin films such as PLZT (Pb0.92La0.08Zr0.52Ti0.48O3).88 A sol–gel precursor of polyacrylonitrile (PAN) and poly(urea-co-formaldehyde) methylated resin (PUF) with chromium chloride in N,N-dimethylformamide (DMF) can be electrospun to give thin fibres. Heating these to 1000 °C in a nitrogen atmosphere resulted in carbon fibres that contain nanoparticles of chromium nitride/carbide.89
5.2 Biopolymers
As highlighted in the section on synthetic polymers, the main requirements for polymers in sol–gel chemistry is that they are soluble in at least one solvent (preferably water) and also contain functional groups that can bind to metal ions. In this sense, the polymers that are available in nature are the ideal resource as many of them dissolve readily in hot water. Although this chemistry is quite different from the examples of silicon alkoxides, biopolymers in solution still fall within standard definitions of a ‘sol’ in that their chains extend for 1 nm–1 μm. The sol–gel transition can then be triggered by cooling the solution and/or by addition of metal ions since many biopolymers form complex, organized gels in water through binding with metal ions. As will become clear in this section, the extended, organized gel structures formed by biopolymers can be exploited in sol–gel chemistry to direct ceramic formation in a unique way.The term ‘biopolymer’ is used in several situations, but for the purposes of this review, we will consider a biopolymer to be a macromolecule that has been extracted from biomass, i.e. a polymer that has been synthesized by a living organism. With such wide range of biological sources to choose from, different biopolymers have very different properties and so some care is needed when selecting a biopolymer for sol–gel chemistry. This can be a useful handle for tuning material properties. The diverse applications of biopolymers in templating90 and nanofabrication91 have previously been reviewed. However, in this paper, we will discuss the specific use of biopolymers in sol–gel chemistry, highlighting their advantages and disadvantages.
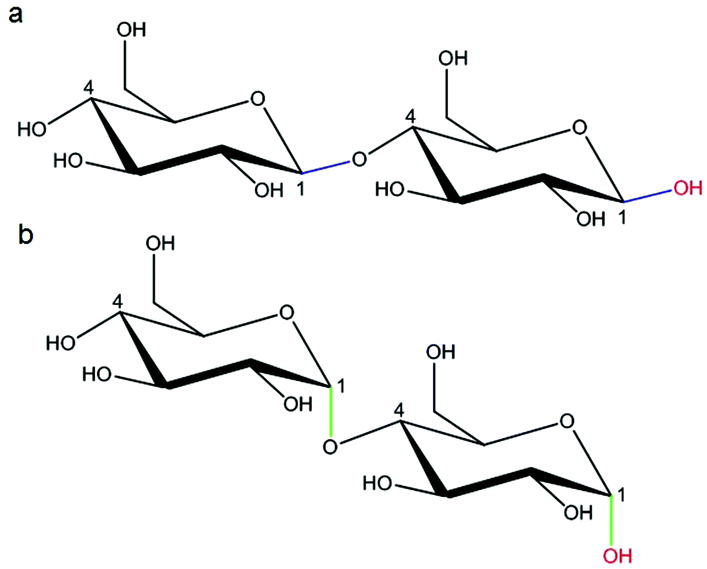 | ||
| Fig. 9 (a) Alpha and (b) beta glycosidic linkages. | ||
:4; although this can vary between species. Both amylose and amylopectin are homopolymers of α-D-glucose. Amylose contains α(1 → 4) glycosidic links and as a result is linear and can form helices. Amylopectin is a complex, highly branched polymer built from α(1 → 4) linked glucose units with non-random α(1 → 6) links approximately every 30 units, providing branch points. The solubility of starch depends mainly on the ratio of amylose to amylopectin but generally starch is insoluble in cold water. The hydroxyl substituents on starch biopolymers are readily modified, which can affect the physical properties and, importantly, metal binding.92
Starch can be used to produce nanoparticles of various oxides such as the pigment CoxZn1−xAl2O4 by simply heating a mixture of aqueous metal salts with starch to form a gel, followed by calcination in air.93 The starch behaves as a chelating agent and the long chains of the molecule restrict growth and sintering of nanoparticles. In the synthesis of many materials, for example luminescent doped YVO4, nanoparticles are important and the high level of control offered by the starch synthesis brings advantages over many other synthesis techniques.94 In addition to metal oxides, starch gels can be heated in an inert atmosphere or vacuum to produce carbons with a wide range of surface chemistries, depending on the temperature (Fig. 10), i.e. the degree of decomposition of the polysaccharide.95 These surface functionalities can be used to tune the properties of the carbon, particularly in separation or catalysis and can also be modified e.g. by post-treatment with H2SO4 to give sulfonic acid groups and a solid acid catalyst.96 The structure of these carbons depends on firstly drying the starch gel in a controlled way (e.g. supercritical drying or solvent exchange) to maintain the open gel network and produces homogeneous mesoporous solids. It is also possible to achieve metal–carbon composites, either by decorating starch-derived carbons with metal nanoparticles97 or through calcination of metal/starch gel precursors in inert atmosphere.98 A final alternative is to directly produce porous metal carbides such as SiC by using the starch as both the gel precursor and the carbon source.99
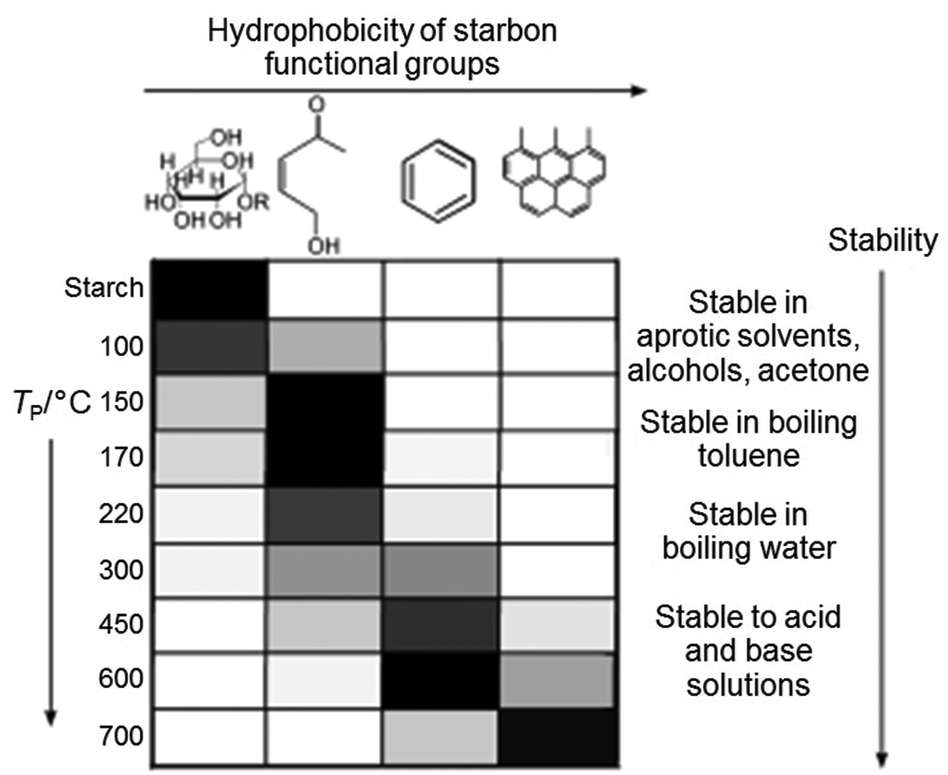 | ||
| Fig. 10 Distribution of functional groups on starch-derived carbons prepared at different temperatures (grey scale to indicate relative amounts where black is highest). Modified with permission from ref. 95. | ||
000000 Da. A typical structure of this biopolymer is a glucan formed primarily of α(1 → 6) glycosidic repeating units with side chains linked to the backbone via α(1 → 2), α(1 → 3) and α(1 → 4) glycosidic bonds (Fig. 11).100 Dextran is produced by enzymatic action of bacteria such as Leuconostoc mesenteroides on sucrose and as a result the main side groups are hydroxyls, although the polymer also contains reductive aldehyde substituents. Dextran is soluble in water over a large range of concentrations with the ability to bind to metals and potentially reduce them in solution to form metallic nano- and microstructures. Dextran is particularly useful in this type of synthesis as it is highly soluble, stable and biocompatible, which is important in biomedical applications. An advantage of dextran in sol–gel chemistry is that it is readily modified, for example to produce anionic dextran sulfate or carboxymethyl–dextran where metal binding may be enhanced.
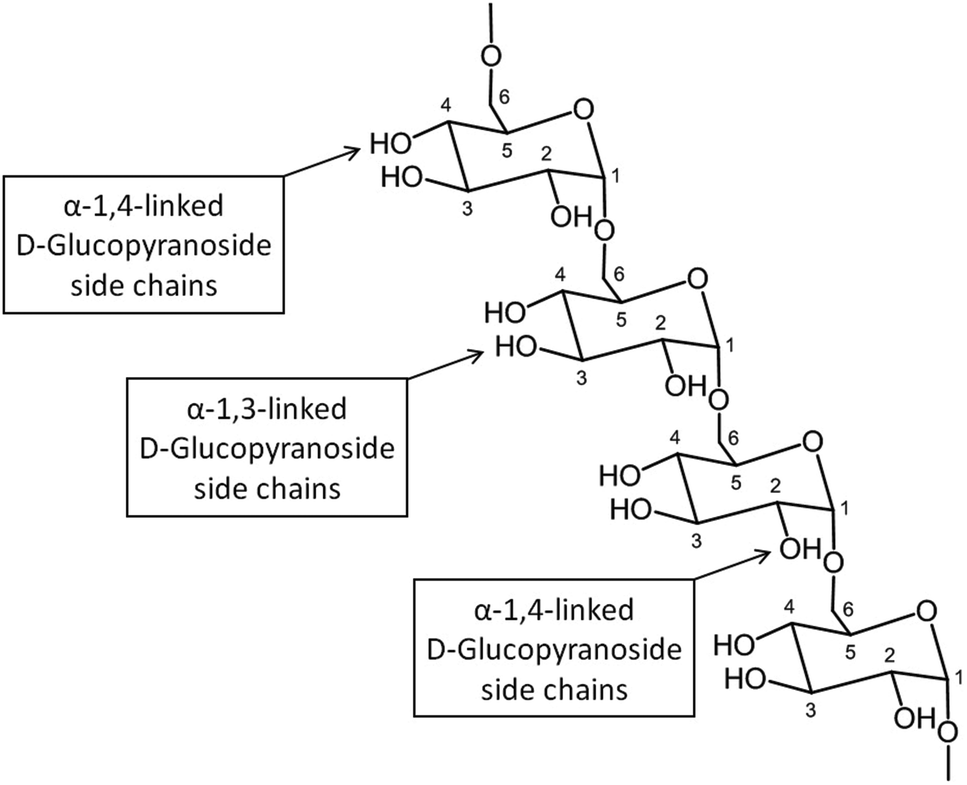 | ||
| Fig. 11 Structure of Dextran showing the different glycosidic bonds. | ||
A comprehensive study of the use of dextran in materials synthesis produced sponge-like structures of a range of compounds including metals and metal oxides.101 In general, aqueous metal salts were mixed with dextran to produce viscous liquids that could be shaped into monoliths or drawn out into macroscopic wires. Drying to remove water and then heating in air to 800 °C removed the dextran to produce reticular structures, for example gold, silver or copper oxide as well as composite materials. In the case of gold and silver, the dextran reduced the metal ions during the initial drying process although presumably some sintering occurred during the final heating process. Similarly, sponges of YBa2Cu3O7−δ can be synthesized by dissolving dextran in an aqueous solution of the relevant metal nitrates to form a paste that is dried and then heated in air to 920 °C.102 This synthesis was also extended to use carboxylated crosslinked dextran beads (CM-Sephadex®) as a precursor rather than soluble dextran. The resulting YBa2Cu3O7−δ maintains the spherical shape of the dextran precursor, with the microstructure being composed of agglomerated nanoparticles.103
 | ||
| Fig. 12 (a) Structure of chitosan. TEM images of samples of YBa2Cu4O8 synthesized using (b) acetate/tartrate and (c) chitosan. (d) Photograph and (e) SEM image of chitosan beads prepared by crosslinking with Ce3+ with (inset) SEM image of sponge-like structure formed after calcination of the beads. Images reproduced with permission from ref. 11 (b, c) and 109 (d, e). | ||
Chitosan has the ability to sequester metal cations along the length of its chain105 and can thus form homogeneous gels with various metal ions. In the synthesis of type-II superconductors, crystallite polydispersity and grain boundary misalignment can reduce the critical current density and so control over morphology is important. Producing useful shapes such as wires is also a step forward generally in the synthesis of electronic quaternary oxide ceramics. Hall showed in 2006 that the binding of Y3+, Ba2+ and Cu2+ ions by chitosan in an aqueous gel could be used to synthesize nanowires of the Y124 superconductor (YBa2Cu4O8).11 The biopolymer matrix chelates the metal precursors and ensures multiple nucleation sites. Subsequent Y124 crystal growth is along the crystallographic c-axis, controlled by the chitosan as it decomposes, to form nanowires up to 1000 nm long with an average width of 50 nm. This is in stark contrast to a small molecule sol–gel synthesis using acetate and tartrate that produces irregular, micron-sized particles (Fig. 12b and c).
It is also possible to gain control over polydispersity and size of nanoparticles by using chitosan as a chelating agent, where the biopolymer matrix slows the sintering process. In this particular case, aqueous gold and palladium salts were reduced within a chitosan film followed by heating under argon. In the absence of air, the chitosan decomposes to carbon with Au/Pd nanoparticles supported throughout the matrix, showing high and selective activity for aerobic oxidation of benzylic alcohols.106 The combustion of chitosan from a sol–gel precursor can also be used to generate pores within a material. For example, gels of chitosan with sodium silicate were prepared and calcined in air to produce silica with bimodal porosity, the macroporosity being generated by removal of chitosan.107
The insolubility of chitosan in basic solutions can be exploited to add another dimension to sol–gel synthesis in the formation of gel beads. Aqueous metal salt solutions such as Al(NO3)3108 or (NH4)2Ce(NO3)6109 can be combined with chitosan in acetic acid to produce homogeneous solutions. Dropwise addition of the chitosan/metal solutions into aqueous base such as NH4OH results in gel spheres from precipitation of the chitosan. These can be calcined in air to produce sponge-like porous spherical CeO2 or mesoporous Al2O3 (Fig. 12d and e).
:M ratio depends on the seaweed source and can vary from ∼30% G to ∼70% G. Although mannuronate and guluronate only differ in configuration at the C5 position, the conformation of the two monomers is very different leading to polymannuronate segments having a flattened ‘sheet-like’ appearance and polyguluronate forming buckled chains. Alginate is an anionic polysaccharide, each monomer containing a carboxylate moiety, and so binding to metal cations is strong. This is particularly the case for polyguluronate segments, which are crosslinked by multivalent metal cations with each cation bound to 4 guluronate monomers (known as the ‘egg-box’ model, Fig. 13a). In this way, two polyguluronate segments can be joined into a left-handed double helix complexing many metal cations. Alginate salts can be purchased in various forms (e.g. sodium or ammonium salt) that dissolve in water to form viscous solutions. Alginic acid is insoluble in water and needs to be converted to a salt by addition of base. One challenge in the use of alginate is the strength of the metal–biopolymer gelation. The strong crosslinking means that the addition of multivalent cations to alginate results in a rubbery gel that expels water if the concentration of the two solutions is high. When using alginate in sol–gel synthesis of ternary or quaternary ceramics, it is therefore important to premix the metal salts to ensure homogeneity within the alginate.
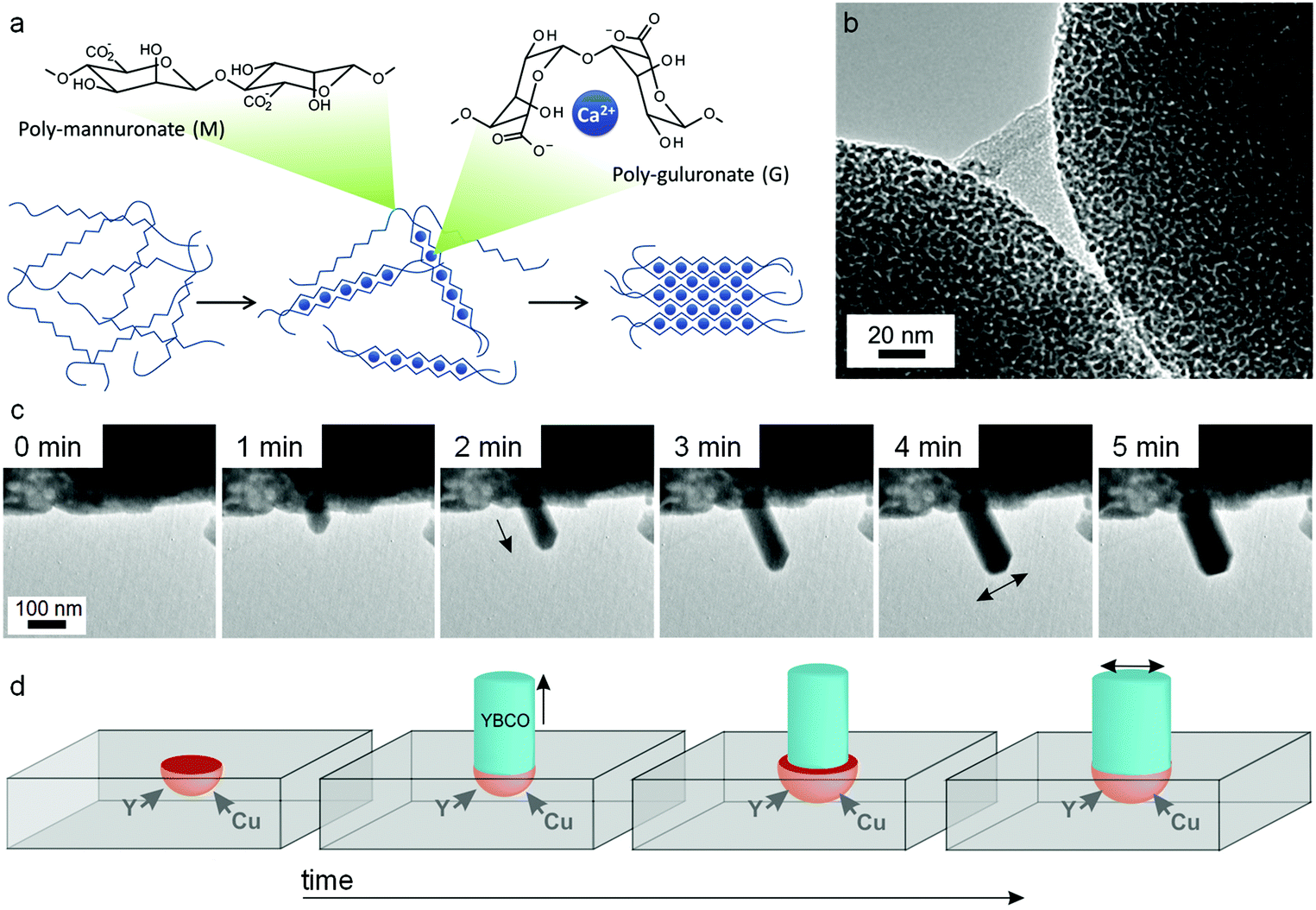 | ||
| Fig. 13 (a) Egg-box model of cation binding in alginate. TEM images of (b) YBCO nanowires synthesized from alginate with acetate/tartrate precursors with (inset) BaCO3 nanoparticles that act as sites for catalytic float outgrowth. TEM images of (c) straight nanowires growing from a precursor of alginate with metal nitrates with (d) schematic showing nanowire spontaneously broadening as a result of microcrucible creep and expansion during heating. Images modified with permission from ref. 91 and 114 (c/d). Image (b) reproduced with permission from ref. 111. Copyright (2013) American Chemical Society. | ||
A search of the literature for ‘alginate sol–gel’ will result primarily in references for synthesis of aqueous alginate gels for biomedical applications such as protein or cell encapsulation, drug delivery, or tissue engineering. Many of these exploit the sol–gel transition of alginate when it is acidified or treated with metal ions such as calcium. However, there are also many examples of alginate being used to prepare sol–gel precursors for materials synthesis. This can either involve direct addition of aqueous metal salts to sodium or ammonium alginate solutions, or preparation of a calcium alginate gel template followed by solvent exchange and infiltration with precursors such as titanium alkoxide.110 In the case of gel templates, one advantage is that the alginate can be prepared in various forms such as small beads. The resulting metal oxides retain the bead shape but with a mesoporous structure (Fig. 13b) which could be particularly useful in applications such as drug delivery111 or sorption.112
In a similar method to the chitosan synthesis of YBa2Cu4O8, alginate has been used to prepare nanowires of YBCO superconductors by mixing Y, Ba and Cu acetate/tartrate salts with alginate to form a gel and then heating to 920 °C in air. As has been mentioned above, the main limitation in the synthesis of YBCO ceramics is the uncontrolled nucleation and growth of BaCO3 crystals, which leads to inhomogeneity in the final product. In the alginate synthesis, the strong sequestration of metal cations from aqueous acetate precursors by the biopolymer leads to controlled nucleation of barium carbonate nanoparticles. A mechanistic investigation on quenched samples then showed that these barium carbonate nanoparticles act as catalysts for the outgrowth of YBCO nanowires.113 A more recent study actually observed nanowire growth in real time using transmission electron microscopy. In this case, nitrate salts were combined with alginate and resulted in similar controlled nucleation of barium carbonate but in a more porous Y/Cu matrix. The rough surface of the matrix provided sites for a microcrucible mechanism of YBCO nanowire outgrowth (Fig. 13c and d).114 It is interesting in these cases how a simple change of metal counterion can lead to such different morphologies (tapered wires from acetates and straight wires from nitrates). When the YBCO phase begins to form (>850 °C), the biopolymer has long since been combusted. However, the way that the biopolymer and the metal salts interact in the early stages of the synthesis (<400 °C) can exert remarkable control over growth of the final quaternary phase. In a subtle departure from the central theme of sol–gel chemistry, the biopolymer in this case is not ensuring homogeneity but rather controlling inhomogeneity.
As well as metal counterions, the nature of the alginate salt can also affect ceramic structure. For example, La0.67Sr0.33MnO3 (LSMO, a colossal magnetoresistant material) can be synthesized in the form of nanowires or nanoparticles from sodium alginate and ammonium alginate respectively.115 In the case of sodium alginate, a sodium carbonate phase was identified as a secondary phase and this was proposed to act as a flux, aiding the transport of other components during the formation of the LSMO nanowires. Sodium alginate is also compared to sodium ascorbate in this paper. Alginate and ascorbate have almost the same empirical formula (C6H7O6 and C6H6O6 respectively) and so enable a direct comparison of a small molecule and a polymer. The difference is stark. Ascorbate mixed with La, Sr and Mn salts results in small but irregularly-shaped particles compared to the nanowires formed from sodium alginate.
Alginate can also be used in the sol–gel synthesis of metal/metal oxide nanocomposites and in this case, the alginate has a dual function. For example, if aqueous Ce(NO3)3 and HAuCl4 are mixed with sodium alginate and dried at room temperature, the gel turns a bright fuchsia pink as it dries to a flexible film. Calcination in air at 600 °C then results in a brittle, pink/purple solid with a sponge-like network of CeO2 nanoparticles (diameter ∼20 nm) with embedded Au nanoparticles.116 In this synthesis, the alginate firstly binds to Au3+ and Ce3+ cations via the carboxylate side-groups. The Au3+ is then reduced to Au via oxidative decarboxylation of the alginate with the polymer stabilising the resulting nanoparticles. During the calcination step, the alginate then controls the nucleation and growth of CeO2 around the Au nanoparticles resulting in a composite from a single precursor.
A final important point about alginate is the possibility for tuning the material synthesis through the natural variability of the biopolymer. Different seaweed species are harvested on a large scale to extract alginates with different G:M ratios, primarily for use in the food industry and biomedical applications. In sol–gel synthesis, this has been exploited to control particle growth in Co, Ni and CoNi nanoparticles.117 In this system, the alginate with the highest G-content (i.e. providing the strongest metal binding) produced monodisperse spherical nanoparticles (∼2 nm) whereas medium and low G alginates lead to larger, less well-defined particles.
000 with a third of the amino acids being glycine, 21% proline and hydroproline, 10% alanine and the rest being amino acids in much smaller quantities (Fig. 14a).118,119 Gelatin dissolves readily in hot water, forming clear (often pale yellow) solutions with viscosity depending on the loading, source and molecular weight. On cooling, the polypeptide chains arrange themselves into left-handed helices which in turn form a right-handed super helix of 3 strands. These junction zones, which are usually rich in proline and hydroxyproline monomers, are what trigger the sol to gel transition in gelatin. Gel strength is a common factor for characterizing different gelatins and these are quoted as ‘Bloom strengths’.
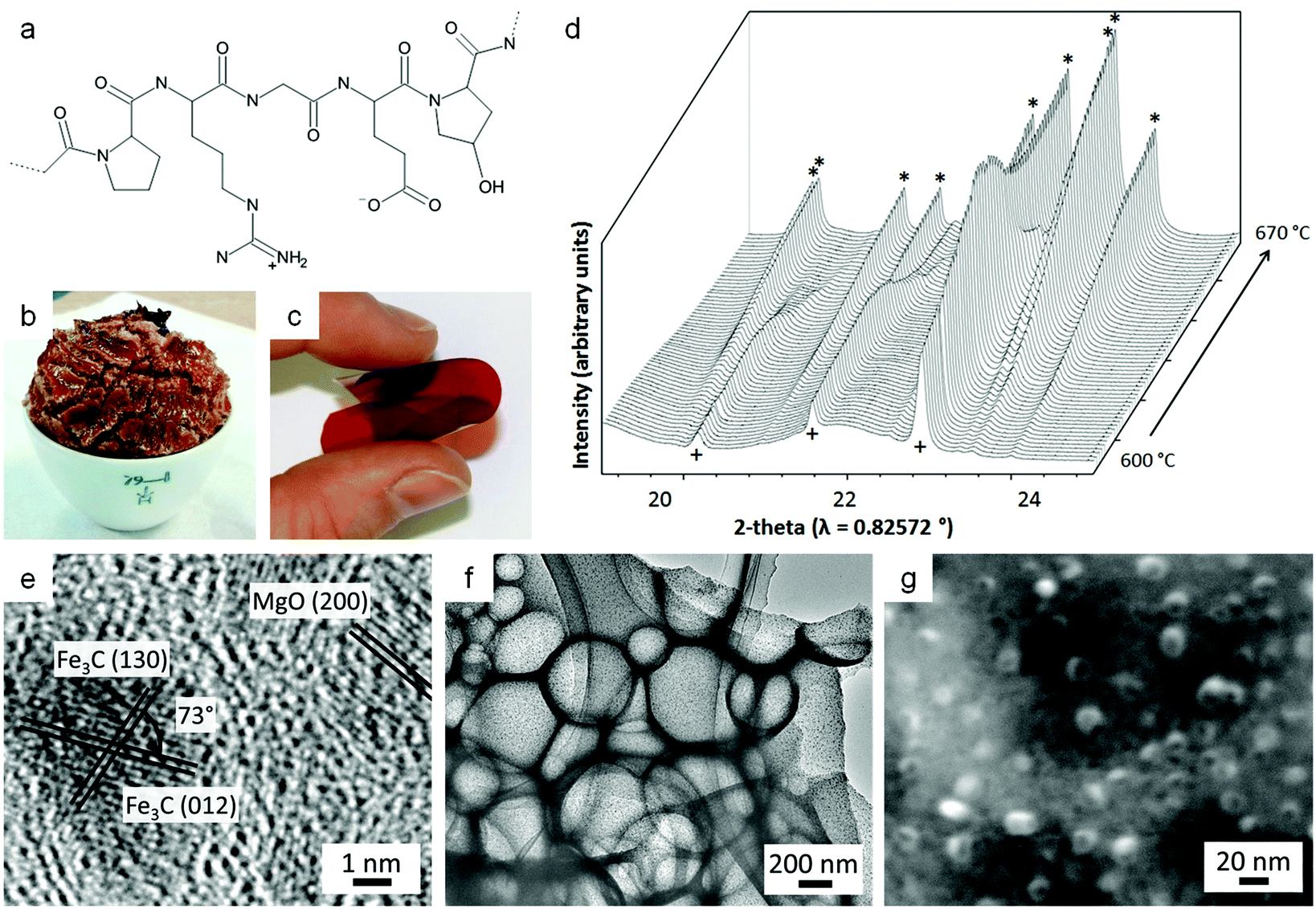 | ||
| Fig. 14 (a) Typical structure of a gelatin fragment. Images of (b) foam from gelatin and iron nitrate and (c) film from gelatin and iron acetate. (d) Synchrotron powder X-ray diffraction data showing the iron nitride (+) to carbide (*) transition during a sol–gel synthesis from gelatin. (e) TEM image of a Fe3C/MgO/C composite showing two types of nanoparticles. TEM (f) and SEM (g) images of a Fe3C/MgO/C composite after acid washing showing macropores and mesopores. Images reproduced with permission from ref. 121 (c) and 123 (d). Copyright (2010) and (2015) American Chemical society. Images (e–g) modified with permission from ref. 125. | ||
The diverse range of side-chains on a gelatin molecule make it a useful gelling-agent for sol–gel chemistry. For example, a mixture of aluminium, yttrium and terbium nitrates mixed with gelatin in hot water can be cooled to form a gel. The authors in this study then infiltrated the gelatin with ammonia to drive precipitation of amorphous hydroxide intermediates within the gel, before drying under vacuum and calcining in air to produce fine powders of YAG:Tb (Terbium-doped yttrium aluminium garnet).120 The homogeneous gelatin precursor ensures a small particle size and size range (∼40–55 nm). This offers substantial advantages over solid-state synthesis as morphology and purity are critical for the application of YAG:Tb in scintillation and CRT projection.
Gelatin has also been used to produce nanoparticles of metal nitrides and carbides, as well as oxide/carbide or oxide/nitride nanocomposites. For example, mixing iron nitrate solution with hot aqueous gelatin results in a viscous, sticky gel that forms an orange foam (Fig. 14b) on drying at 80 °C in air. In contrast, iron acetate/gelatin forms a flexible brown film (Fig. 14c) when dried from a solution. The iron acetate precursor can then be heated to ∼800 °C under nitrogen to form Fe3C nanoparticles embedded in a porous carbon matrix,121 whereas the nitrate precursor forms Fe3N or Fe3C depending on the heating conditions.122In situ synchrotron powder diffraction data showed that the mechanism proceeds via initial formation of iron oxide (FeOx) nanoparticles, <3 nm in diameter, during the initial decomposition and carbonization stages of the gelatin polymer. The iron oxide nanoparticles then react with nitrogen species in the carbon matrix to form Fe3N nanoparticles which then transform to Fe3C via an intermediate carbonitride (Fig. 14d).123 A similar approach can also be used to synthesize nanocomposites such as MIoxide/MIIcarbide (where MI and MII are different metals). Aqueous or ethanolic metal salts are mixed and combined with gelatin and the resulting gels dried to foams and calcined under nitrogen. Despite the homogeneous precursor, the different thermal stabilities of the metals can drive phase separation to form composites such as TiO2/Fe3C, TiO2/WN or MgO/Fe3C (Fig. 14e) with potential applications in catalysis.124 In the case of MgO/Fe3C, mild acid washing can be used to remove the nanoparticles to produce carbons with trimodal (macro/meso/micro) porosity (Fig. 14f and g).125
6. New advances in sol–gel chemistry
This final section highlights some of the recent or unusual developments in sol–gel chemistry, including synthetic methods and post-processing techniques. In particular, we focus on alternative physical processing techniques as well as more unconventional or non-aqueous sol–gel precursors.6.1 Alternative heating processes and lower-temperature routes
Typically, the conversion of a gel precursor to the desired inorganic material occurs during a heating step in an air or inert-gas furnace. However this can lead to a number of problems such as non-uniform heating, reaction with ceramic crucibles or container-effects influencing gas escape.126 Furthermore, the slow cooling rate of conventional furnaces can lead to sintering during cooling. As such, several authors have developed alternative heating processes, the most common being microwave heating. Microwaves have some significant advantages over conventional heating processes. The electric component of the microwave field interacts with charge carriers in the material giving rise to direct heating through dipolar polarization (more significant in the liquid phase) or conduction heating (dominant in solid materials). Due to the direct interaction of the microwaves with components of the material, heating is instantaneous and it also occurs volumetrically (uniform through the sample).127 In sol–gel chemistry, this can lead to faster reactions, lower final temperature and even improved phase purity, due to the extremely rapid temperature rise and more uniform heating.128 For example, a synthesis of barium hexaferrite (BaFe12O19) nanoparticles from metal nitrates, citric acid, EDTA and ammonia could be achieved by heating freeze dried precursors for just 15 seconds in a modified domestic microwave oven. In this case, the microwave heating triggers an autocombustion reaction that last for another 10 seconds.129 The shorter heating time and lower reaction temperature can in turn lead to smaller particle size and also a narrower particle size distribution. The authors in this study emphasize the importance of homogeneity in heating and comment that normal heating to trigger autocombustion in complex metal oxides often leads to mixed products due to inhomogeneous ignition of hydrocarbons. In addition, the turbulent flow of hot gases from autoignition often quickly breaks up samples with resulting particulates quickly losing temperature, which may also prevent formation of a desired compound. In this paper, the authors avoid this problem by containing the autocombustion reaction within a quartz vessel lined with heat-retaining barium hexaferrite strips.As previously discussed (section 2.4) hydrothermal synthesis is another alternative to standard calcination, the high auto-generated pressure and can be used to dissolve or carbonize organics as well as initiate formation of crystalline inorganics at much lower temperatures than conventional heating. In the case of sol–gel precursors, the advantage of hydrothermal processing is made clear in an article by Fang et al.130 In this paper, the authors mix iron and tin chlorides with citric acid and polyethylene glycol in water and ammonium hydroxide to form a gel. This gel is either dried and treated in a furnace in air at 350 °C, or heated in a Teflon-lined autoclave at 150 °C. The result is smaller crystallites of Fe-doped SnO2 in the hydrothermal system, although it should be noted that hydrothermal treatment requires 12 hours (typical of a hydrothermal synthesis) compared to the 1 hour at 350 °C in the furnace.
6.2 Non aqueous sol–gel chemistry
As discussed at the beginning of this paper, hydrolysis and condensation of metal alkoxides is strongly dependent on water content. Most of the other methods of sol–gel processing described in this paper have been developed as a result of this limitation of alkoxides. In these methods, the primary focus has been to form stable chelation complexes and gels from aqueous metal salts such as nitrates or acetates. However, there have also been substantial advances in developing alkoxide chemistry in water-free conditions. This can bring significant advantages, such as the ability to produce dispersible nanoparticles and also very small particle sizes.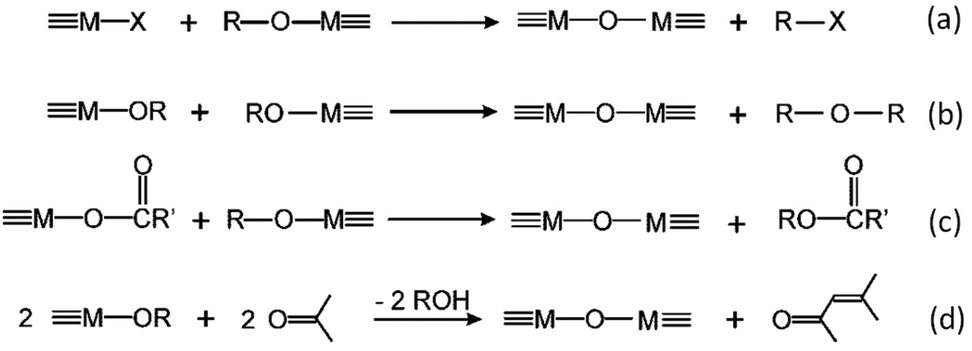 | ||
| Scheme 6 Condensation steps in non-hydrolytic sol–gel chemistry including (a) alkyl halide elimination, (b) ether elimination, (c) ester elimination and (d) aldol-like condensation. Modified with permission from ref. 133. | ||
One of the most powerful aspects of NHSG chemistry is the ability to produce dispersible nanoparticles rather than the sintered agglomerates typical of many other sol–gel processes. Non-hydrolytic conditions result in slower and more controllable kinetics than standard hydrolysis reactions, meaning it is easier to control crystallization. Crystallization of the desired phase also tends to occur at lower temperatures in NHSG chemistry. As a result, many NHSG reactions can be carried out entirely in solvent, with factors such as solvent, temperature and additives having a large influence on structure and morphology of the crystals. For example, a mixture of iron carbonyl with dioctyl ether and oleic acid can be heated under reflux to give monodisperse iron oxide nanoparticles of 6–13 nm in diameter (with 1 nm increments in diameter) depending on the molar ratio of precursors.135 Tuning conditions in NHSG chemistry (such as selective adsorption of surfactants to certain crystal faces) can also be used to introduce anisotropy into metal oxides.136
As described earlier in this paper, one of the main problems in the synthesis of ternary and quaternary metal oxides is phase separation. Even a homogeneous gel precursor may not result in a phase-pure product, if there is preferential precipitation of a binary intermediate such as the formation of BaCO3 in YBCO synthesis. Ionic liquids have been used in an ingenious way to improve homogeneity during the early stages of ceramic synthesis.143 This general method has been used to synthesize phase pure samples of a range of metal oxides, such as YBa2Cu3O7−x, Bi2Sr2CaCu2O8 (BSSCO superconductor) and yttrium-doped BiFeO3 (multiferroic material) among others. The method is simple. Aqueous metal salts are added to the ionic liquid and the mixture heated to remove water. Acetate and nitrate ionic liquids are selected to enhance metal dissolution within the ionic liquid. Microcrystalline cellulose is then added and dissolved (ionic liquids are well known to dissolve cellulose) to provide a polymeric, non-selective chelating agent to bind the metal cations within the mixture. Most aqueous sol–gel syntheses require drying before heating in a furnace (or quickly dry during the first stages of calcination). The special feature of this IL synthesis is that the system remains liquid to a much later stage of synthesis, until the IL decomposes. Future investigations of exactly how the IL decomposes and controls crystal formation should provide some fascinating insight into this system.
7. Concluding remarks
The term ‘sol–gel’ has broadened significantly from the original usage to describe hydrolysis and condensation processes and many newer methods may not involve a clear sol–gel transition. Instead, the theme that now seems to connect most sol–gel chemistries is an exploitation of the solution state in the synthesis of a solid material. By beginning a synthesis in the solution (or liquid) state, one can ensure complete homogeneity, which has considerable advantages in generating a phase-pure product and can also result in lower synthesis temperatures. If inhomogeneity is required (for example the formation of micelles or liquid crystalline phases to template a silica material), then this is readily controlled to produce well-defined morphological features in the resulting solid material.The ability to use molecular precursors to control the morphology of a ceramic product is one of the main advantages of sol–gel processing. For example, a simple change of silicon alkoxide can influence the structure of the resulting silica monolith. Alternatively, in Pechini synthesis, a simple change from a diol to a triol can alter the degree of polyesterification and drive phase separation to produce either spherical or bicontinuous mesoporous ceramics. Another powerful tool is the use of additional molecular species that interact with the sol–gel precursors or each other, often referred to as ‘soft templating’. In the case where phase separation or structural ordering happens in the solution state, this is relatively well understood (although advances in techniques such as small angle neutron scattering have added new insights into the templating process).144 What is becoming more apparent is how changes in molecular composition in the solution precursor can have a dramatic influence much later on in a ‘sol–gel’ synthesis, for example in the high-temperature synthesis of ceramics.
Combustion synthesis is perhaps the simplest example of how a choice of molecular precursor can influence a material structure even after the solution or gel has been dried and heated. In this case, a strongly exothermic reaction between an oxidant such as nitrate and fuel such as citrate or glycine produces large volumes of gas that result in an open foam-like structure in the ceramic product. The combustion does not need to be dramatic to produce outgassing and the thermal decomposition of organic precursors in many sol–gel reactions also leads to porous solids. In addition to changing the macrostructure, the choice of precursors can also influence the individual crystallite morphology. For example, in the synthesis of YBCO superconductors, a combination of a biopolymer with metal nitrate precursors results in a porous intermediate mixture of individual metal oxides and carbonates. The porous nature of this intermediate facilitates mass transport at the later stages of synthesis and results in nanowires of the complex metal oxide product.107 It is even possible to change the profile of crystallographic transformations in a ceramic synthesis through a choice of molecular precursors. For example, metastable iron nitride can be favoured over iron carbide simply through a choice of iron nitrate rather than acetate in the precursor gel.115 In this case, the higher surface area of the nitrate-derived precursor results in increases accessibility of the nitrogen atmosphere that stabilizes the iron nitride phase.
It is clear from all of these examples that there is still much to be discovered in the field of sol–gel chemistry. With the increasing power of methods to study high-temperature processes in situ, we are gaining better understanding of how molecular precursors can continue to influence a sol–gel process long after complete combustion of the starting molecules. Furthermore, there is still much to be learned about the solution state and how molecular interactions and phase separation changes during the gelation procedure or during the formation of a solid from a gel.
Acknowledgements
The authors acknowledge the EU (Marie Curie ‘SusNano’), the EPSRC, the University of Birmingham and the University of Bristol for funding.References
- J. Livage, New J. Chem., 2001, 25, 1–1 RSC.
- B. L. Cushing, V. L. Kolesnichenko and C. J. O'Connor, Chem. Rev., 2004, 104, 3893–3946 CrossRef CAS PubMed.
- M. Kakihana, J. Sol-Gel Sci. Technol., 1996, 6, 7–55 CrossRef CAS.
- C. J. Brinker and G. W. Scherer, Sol-gel Science: The physics and chemistry of sol-gel processing, Academic Press, London, 1990 Search PubMed.
- P. J. Flory, Faraday Discuss. Chem. Soc., 1974, 57, 7–18 RSC.
- E. von Ebelmen, Justus Liebigs Ann. Chem., 1846, 57, 319–355 CrossRef.
- D. Avnir, T. Coradin, O. Lev and J. Livage, J. Mater. Chem., 2006, 16, 1013–1030 RSC.
- Z. Haijun, J. Xiaolin, Y. Yongjie, L. Zhanjie, Y. Daoyuan and L. Zhenzhen, Mater. Res. Bull., 2004, 39, 839–850 CrossRef.
- M. P. Pechini, Method of preparing lead and alkaline earth titanates and niobates and coating method using the same to form a capacitor, US Patent, 3330697A, 1967 Search PubMed.
- J. Lin, M. Yu, C. Lin and X. Liu, J. Phys. Chem. C, 2007, 111, 5835–5845 CAS.
- S. R. Hall, Adv. Mater., 2006, 18, 487–490 CrossRef CAS.
- K. S. Mazdiyasni and L. M. Brown, Inorg. Chem., 1970, 9, 2783–2786 CrossRef CAS.
- N. Y. Turova, A. V. Korolev, D. E. Tchebukov, A. I. Belokon, A. I. Yanovsky and Y. T. Struchkov, Polyhedron, 1996, 15, 3869–3880 CrossRef CAS.
- R. G. Jones, E. Bindschadler, D. Blume, G. Karmas, G. A. Martin, J. R. Thirtle and H. Gilman, J. Am. Chem. Soc., 1956, 78, 6027–6030 CrossRef CAS.
- R. C. Mehrotra and M. Aggrawal, Polyhedron, 1985, 4, 1141–1142 CrossRef CAS.
- J. Livage, M. Henry and C. Sanchez, Prog. Solid State Chem., 1988, 18, 259–341 CrossRef CAS.
- M. Guglielmi and G. Carturan, J. Non-Cryst. Solids, 1988, 100, 16–30 CrossRef CAS.
- F. Babonneau, S. Doeuff, A. Leaustic, C. Sanchez, C. Cartier and M. Verdaguer, Inorg. Chem., 1988, 27, 3166–3172 CrossRef CAS.
- U. Schubert, J. Mater. Chem., 2005, 15, 3701–3715 RSC.
- T. Ohya, M. Ito, K. Yamada, T. Ban, Y. Ohya and Y. Takahashi, J. Sol-Gel Sci. Technol., 2004, 30, 71–81 CrossRef CAS.
- E. Scolan and C. Sanchez, Chem. Mater., 1998, 10, 3217–3223 CrossRef CAS.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- R. Sui and P. Charpentier, Chem. Rev., 2012, 112, 3057–3082 CrossRef CAS PubMed.
- A. Pons, L. Casas, E. Estop, E. Molins, K. D. M. Harris and M. Xu, J. Non-Cryst. Solids, 2012, 358, 461–469 CrossRef CAS.
- L. C. Klein, in Thin Film Processes II, ed. J. L. Vossen and W. Kern, Academic Press Inc., London, 1991, pp. 501–520 Search PubMed.
- R. M. Pasquarelli, D. S. Ginley and R. O'Hayre, Chem. Soc. Rev., 2011, 40, 5406–5441 RSC.
- Y. Liu, J. Goebl and Y. Yin, Chem. Soc. Rev., 2013, 42, 2610–2653 RSC.
- K. E. Shopsowitz, H. Qi, W. Y. Hamad and M. J. MacLachlan, Nature, 2010, 468, 422–425 CrossRef CAS PubMed.
- D. Brandhuber, V. Torma, C. Raab, H. Peterlik, A. Kulak and N. Hüsing, Chem. Mater., 2005, 17, 4262–4271 CrossRef CAS.
- J. E. Lofgreen and G. A. Ozin, Chem. Soc. Rev., 2014, 43, 911–933 RSC.
- T. Brezesinski, M. Groenewolt, A. Gibaud, N. Pinna, M. Antonietti and B. M. Smarsly, Adv. Mater., 2006, 18, 2260–2263 CrossRef CAS.
- D. Grosso, F. Cagnol, G. J. de, A. A. Soler-Illia, E. L. Crepaldi, H. Amenitsch, A. Brunet-Bruneau, A. Bourgeois and C. Sanchez, Adv. Funct. Mater., 2004, 14, 309–322 CrossRef CAS.
- C. M. Flynn, Chem. Rev., 1984, 84, 31–41 CrossRef CAS.
- R. C. Courtney, R. L. Gustafson, S. Chaberek and A. E. Martell, J. Am. Chem. Soc., 1958, 80, 2121–2128 CrossRef CAS.
- A. Katelnikovas, J. Barkauskas, F. Ivanauskas, A. Beganskiene and A. Kareiva, J. Sol-Gel Sci. Technol., 2007, 41, 193–201 CrossRef CAS.
- CRC Handbook of Chemistry and Physics, ed. R. C. Weast, CRC Press, Boca Raton, 53rd edn, 1972, p. D-120 Search PubMed.
- G. Xu, H. Ma, M. Zhong, J. Zhou, Y. Yue and Z. He, J. Magn. Magn. Mater., 2006, 301, 383–388 CrossRef CAS.
- P. Vaqueiro and M. A. López-Quintela, Chem. Mater., 1997, 9, 2836–2841 CrossRef CAS.
- B. J. Hwang, R. Santhanam and D. G. Liu, J. Power Sources, 2001, 101, 86–89 CrossRef CAS.
- J. Zhang, J. Ning, X. Liu, Y. Pan and L. Huang, Mater. Res. Bull., 2003, 38, 1249–1256 CrossRef CAS.
- M. Zayat and D. Levy, Chem. Mater., 2000, 12, 2763–2769 CrossRef CAS.
- K.-F. Hsu, S.-Y. Tsay and B.-J. Hwang, J. Mater. Chem., 2004, 14, 2690–2695 RSC.
- L. Zhao, K. Fang, D. Jiang, D. Li and Y. Sun, Catal. Today, 2010, 158, 490–495 CrossRef CAS.
- L. Bača and N. Stelzer, J. Eur. Ceram. Soc., 2008, 28, 907–911 CrossRef.
- A. Mali and A. Ataie, Scr. Mater., 2005, 53, 1065–1070 CrossRef CAS.
- W. Wen and J.-M. Wu, RSC Adv., 2014, 4, 58090–58100 RSC.
- L. Junliang, Z. Wei, G. Cuijing and Z. Yanwei, J. Alloys Compd., 2009, 479, 863–869 CrossRef.
- Z. Yue, J. Zhou, L. Li, H. Zhang and Z. Gui, J. Magn. Magn. Mater., 2000, 208, 55–60 CrossRef CAS.
- Y. Li, L. Xue, L. Fan and Y. Yan, J. Alloys Compd., 2009, 478, 493–497 CrossRef CAS.
- A. Kareiva, M. Jarppinen and L. Niinistö, J. Mater. Chem., 1994, 4, 1267–1270 RSC.
- S. H. Park and Y.-K. Sun, J. Power Sources, 2003, 119–121, 161–165 CrossRef CAS.
- R. Thirunakaran, K.-T. Kim, Y.-M. Kang and J. Young-Lee, Mater. Res. Bull., 2005, 40, 177–186 CrossRef CAS.
- Y.-K. Sun, Solid State Ionics, 1997, 100, 115–125 CrossRef CAS.
- G. T.-K. Fey, J.-G. Chen, Z.-F. Wang, H.-Z. Yang and T. P. Kumar, Mater. Chem. Phys., 2004, 87, 246–255 CrossRef.
- A. Sutka and G. Mezinskis, Front. Mater. Sci., 2012, 6, 128–141 CrossRef.
- Y. Y. Meng, M. H. He, Q. Zeng, D. L. Jiao, S. Shukla, R. V. Ramanujan and Z. W. Liu, J. Alloys Compd., 2014, 583, 220–225 CrossRef CAS.
- X. Guo, D. Mao, G. Lu, S. Wang and G. Wu, J. Catal., 2010, 271, 178–185 CrossRef CAS.
- L. B. Kong, Y. Huang, W. Que, T. Zhang, S. Li, J. Zhang, Z. Dong and D. Tang, in Transparent Ceramics, Springer, 2015 Search PubMed.
- W. Zhou, Z. Shao, R. Ran, H. Gu, W. Jin and N. Xu, J. Am. Ceram. Soc., 2008, 91, 1155–1162 CrossRef CAS.
- V. Rouessac, J. Wang, J. Provost and G. Desgardin, J. Mater. Sci., 1996, 31, 3387–3390 CAS.
- T. Liu and Y. Xu, Mater. Chem. Phys., 2011, 129, 1047–1050 CrossRef CAS.
- R. Norouzbeigi and M. Edrissi, J. Am. Ceram. Soc., 2011, 94, 4052–4058 CrossRef CAS.
- A. E. Gash, T. M. Tillotson, J. H. Satcher, J. F. Poco, L. W. Hrubesh and R. L. Simpson, Chem. Mater., 2001, 13, 999–1007 CrossRef CAS.
- G. Avgouropoulos and T. Ioannides, Appl. Catal., A, 2003, 244, 155–167 CrossRef CAS.
- S. Podsiadło, Thermochim. Acta, 1995, 256, 375–380 CrossRef.
- S. Podsiadło, Thermochim. Acta, 1995, 256, 367–373 CrossRef.
- C. Giordano, C. Erpen, W. Yao and M. Antonietti, Nano Lett., 2008, 8, 4659–4663 CrossRef CAS PubMed.
- R. B. Levy and M. Boudart, Science, 1973, 181, 547–549 CAS.
- C. Giordano, A. Kraupner, S. C. Wimbush and M. Antonietti, Small, 2010, 6, 1859–1862 CrossRef CAS PubMed.
- Q. Gao, C. Giordano and M. Antonietti, Small, 2011, 7, 3334–3340 CrossRef CAS PubMed.
- J. Lin, M. Yu, C. Lin and X. Liu, J. Phys. Chem. C, 2007, 111, 5835–5845 CAS.
- A. Chowdhury, S. O'Callaghan, T. A. Skidmore, C. James and S. J. Milne, J. Am. Ceram. Soc., 2009, 92, 758–761 CrossRef CAS.
- A. Abreu Jr., S. M. Zanetti, M. A. S. Oliveira and G. P. Thim, J. Eur. Ceram. Soc., 2005, 25, 743–748 CrossRef.
- A. M. Stux, C. Laberty-Robert and K. E. Swider-Lyons, J. Solid State Chem., 2009, 181, 2741–2747 CrossRef.
- C. Chen, J. K. M. Garofano, C. K. Muoto, A. L. Mercado, S. L. Suib, M. Aindow, M. Gell and E. H. Jordan, J. Am. Ceram. Soc., 2011, 94, 367–371 CrossRef CAS.
- J. Wang, A. Manivannan and N. Wu, Thin Solid Films, 2008, 517, 582–587 CrossRef CAS.
- H. Guo, M. Yin, N. Dong, M. Xu, L. Lou and W. Zhang, Appl. Surf. Sci, 2005, 243, 245–250 CrossRef CAS.
- S. G. Rudisill, N. M. Hein, D. Terzic and A. Stein, Chem. Mater., 2013, 25, 745–753 CrossRef CAS.
- K. Nakanishi and N. Soga, J. Am. Ceram. Soc., 1991, 74, 2518–2530 CrossRef CAS.
- M. Motta, C. Deimling, M. Saeki and P. Lisboa-Filho, J. Sol-Gel Sci. Technol., 2008, 46, 201–207 CrossRef CAS.
- S. G. Rudisill, S. Shaker, D. Terzic, R. Le Maire, B. Su and A. Stein, Inorg. Chem., 2015, 54, 993–1002 CrossRef CAS PubMed.
- K. Hayat, M. A. Gondal, M. M. Khaled, S. Ahmed and A. M. Shemsi, Appl. Catal., A, 2011, 393, 122–129 CrossRef CAS.
- Y.-K. Sun and I.-H. Oh, Ind. Eng. Chem. Res., 1996, 35, 4296–4300 CrossRef CAS.
- T. Liu, Y. Xu and J. Zhao, J. Am. Ceram. Soc., 2010, 93, 3637–3641 CrossRef CAS.
- A. Subramania, N. Angayarkanni and T. Vasudevan, Mater. Chem. Phys., 2007, 102, 19–23 CrossRef CAS.
- S. R. Lukić, D. M. Petrović, M. D. Dramićanin, M. Mitrić and L. Đačanin, Scr. Mater., 2008, 58, 655–658 CrossRef.
- S. Kandhasamy, A. Pandey and M. Minakshi, Electrochim. Acta, 2012, 60, 170–176 CrossRef CAS.
- S. Chao, B. Ma, S. Liu, M. Narayanan and U. Balachandran, Mater. Res. Bull., 2012, 47, 907–911 CrossRef CAS.
- A. García-Márquez, D. Portehault and C. Giordano, J. Mater. Chem., 2011, 21, 2136–2143 RSC.
- S. R. Hall, Philos. Trans. R. Soc., A, 2009, 465, 335–366 CrossRef CAS.
- Z. Schnepp, Angew. Chem., Int. Ed., 2013, 52, 1096–1108 CrossRef CAS PubMed.
- D.-K. Kweon, J.-K. Choi, E.-K. Kim and S.-T. Lim, Carbohydr. Polym., 2001, 46, 171–177 CrossRef CAS.
- D. Visinescu, C. Paraschiv, A. Ianculescu, B. Jurca, B. Vasile and O. Carp, Dyes Pigm., 2010, 87, 125–131 CrossRef CAS.
- H. Zhang, X. Fu, S. Niu and Q. Xin, J. Alloys Compd., 2008, 457, 61–65 CrossRef CAS.
- V. Budarin, J. H. Clark, J. J. E. Hardy, R. Luque, K. Milkowski, S. J. Tavener and A. J. Wilson, Angew. Chem., Int. Ed., 2006, 118, 3866–3870 CrossRef.
- V. Budarin, J. H. Clark, R. Luque and D. J. Macquarrie, Chem. Commun., 2007, 634–636 RSC.
- R. Luque, J. H. Clark, K. Yoshida and P. L. Gai, Chem. Commun., 2009, 5305–5307 RSC.
- Y. Chen, J. Cao, M. Zheng, X. Ke, H. Ji, J. Liu and G. Ji, Chem. Lett., 2006, 35, 700–701 CrossRef CAS.
- V. Raman, O. P. Bahl and U. Dhawan, J. Mater. Sci., 1995, 30, 2686–2693 CrossRef CAS.
- A. Misaki, M. Torii, T. Sawai and I. J. Goldstein, Carbohydr. Res., 1980, 84, 273–285 CrossRef CAS.
- D. Walsh, L. Arcelli, T. Ikoma, J. Tanaka and S. Mann, Nat. Mater., 2003, 2, 386–390 CrossRef CAS PubMed.
- D. Walsh, S. C. Wimbush and S. R. Hall, Chem. Mater., 2007, 19, 647–649 CrossRef CAS.
- R. Boston, A. Carrington, D. Walsh and S. R. Hall, CrystEngComm, 2013, 15, 3763–3766 RSC.
- H. Xie, S. Zhang and S. Li, Green Chem., 2006, 8, 630–633 RSC.
- A. J. Varma, S. V. Deshpande and J. F. Kennedy, Carbohydr. Polym., 2004, 55, 77–93 CrossRef CAS.
- S. R. Hall, A. M. Collins, N. J. Wood, W. Ogasawara, M. Morad, P. J. Miedziak, M. Sankar, D. W. Knight and G. J. Hutchings, RSC Adv., 2012, 2, 2217–2220 RSC.
- T. Witoon, M. Chareonpanich and J. Limtrakul, Mater. Lett., 2008, 62, 1476–1479 CrossRef CAS.
- H. V. Fajardo, A. O. Martins, R. M. de Almeida, L. K. Noda, L. F. Probst, N. L. Carreño and A. Valentini, Mater. Lett., 2005, 59, 3963–3967 CrossRef CAS.
- A. Sifontes, G. Gonzalez, J. Ochoa, L. Tovar, T. Zoltan and E. Canizales, Mater. Res. Bull., 2011, 46, 1794–1799 CrossRef CAS.
- M. C. Kimling and R. A. Caruso, J. Mater. Chem., 2012, 22, 4073–4082 RSC.
- X. Wang, D. Chen, L. Cao, Y. Li, B. J. Boyd and R. A. Caruso, ACS Appl. Mater. Interfaces, 2013, 5, 10926–10932 CAS.
- M. C. Kimling, N. Scales, T. L. Hanley and R. A. Caruso, Environ. Sci. Technol., 2012, 46, 7913–7920 CrossRef PubMed.
- Z. Schnepp, S. C. Wimbush, S. Mann and S. R. Hall, Adv. Mater., 2008, 20, 1782–1786 CrossRef CAS.
- R. Boston, Z. Schnepp, Y. Nemoto, Y. Sakka and S. R. Hall, Science, 2014, 344, 623–626 CrossRef CAS PubMed.
- Z. Schnepp, S. C. Wimbush, S. Mann and S. R. Hall, CrystEngComm, 2010, 12, 1410–1415 RSC.
- Z. Schnepp, S. R. Hall, M. J. Hollamby and S. Mann, Green Chem., 2011, 13, 272–275 RSC.
- R. Brayner, M.-J. Vaulay, F. Fiévet and T. Coradin, Chem. Mater., 2007, 19, 1190–1198 CrossRef CAS.
- M. Djabourov, J. Leblond and P. Papon, J. Phys., 1988, 49, 319–322 CAS.
- D. R. Eyre, Science, 1980, 207, 1315–1322 CrossRef CAS PubMed.
- J. Zhou, F. Zhao, X. Wang, Z. Li, Y. Zhang and L. Yang, J. Lumin., 2006, 119, 237–241 CrossRef.
- Z. Schnepp, S. C. Wimbush, C. Giordano and M. Antonietti, Chem. Mater., 2010, 22, 5340–5344 CrossRef CAS.
- Z. Schnepp, M. Thomas, S. Glatzel, K. Schlichte, R. Palkovits and C. Giordano, J. Mater. Chem., 2011, 21, 17760–17764 RSC.
- Z. Schnepp, A. E. Danks, M. J. Hollamby, B. R. Pauw, C. A. Murray and C. C. Tang, Chem. Mater., 2015, 27, 5094–5099 CrossRef CAS.
- Z. Schnepp, M. J. Hollamby, M. Tanaka, Y. Matsushita, Y. Xu and Y. Sakka, Chem. Commun., 2014, 50, 5364–5366 RSC.
- Z. Schnepp, Y. Zhang, M. J. Hollamby, B. R. Pauw, M. Tanaka, Y. Matsushita and Y. Sakka, J. Mater. Chem. A, 2013, 1, 13576–13581 CAS.
- X. Sun, Y. Shi, P. Zhang, C. Zheng, X. Zheng, F. Zhang, Y. Zhang, N. Guan, D. Zhao and G. D. Stucky, J. Am. Chem. Soc., 2011, 133, 14542–14545 CrossRef CAS PubMed.
- H. J. Kitchen, S. R. Vallance, J. L. Kennedy, N. Tapia-Ruiz, L. Carassiti, A. Harrison, A. G. Whittaker, T. D. Drysdale, S. W. Kingman and D. H. Gregory, Chem. Rev., 2014, 114, 1170–1206 CrossRef CAS PubMed.
- S. Wu, S. Zhang and J. Yang, Mater. Chem. Phys., 2007, 102, 80–85 CrossRef CAS.
- L. Junliang, Z. Yanwei, G. Cuijing, Z. Wei and Y. Xiaowei, J. Eur. Ceram. Soc., 2010, 30, 993–997 CrossRef.
- L. M. Fang, X. T. Zy, Z. J. Li, S. Zhu, C. M. Liu, W. L. Zhou and L. M. Wang, J. Alloys Compd., 2008, 454, 261–267 CrossRef CAS.
- A. Vioux, Chem. Mater., 1997, 9, 2292–2299 CrossRef.
- M. Niederberger, Acc. Chem. Res., 2007, 40, 793–800 CrossRef CAS PubMed.
- D. P. Debecker and P. H. Mutin, Chem. Soc. Rev., 2012, 41, 3624–3650 RSC.
- I. Bilecka and M. Niederberger, Electrochim. Acta, 2010, 55, 7717–7725 CrossRef CAS.
- J. Park, E. Lee, N.-M. Hwang, M. Kang, S. C. Kim, Y. Hwang, J.-G. Park, H.-J. Noh, J.-Y. Kim, J.-H. Park and T. Hyeon, Angew. Chem., Int. Ed., 2005, 44, 2873–2877 CrossRef PubMed.
- R. Buonsanti, E. Carlino, C. Giannini, D. Altamura, L. De Marco, R. Giannuzzi, M. Manca, G. Gigli and P. D. Cozzoli, J. Am. Chem. Soc., 2011, 133, 19216–19239 CrossRef CAS PubMed.
- A. Hector, Chem. Soc. Rev., 2007, 36, 1745–1753 RSC.
- I.-S. Kim and P. N. Kumta, J. Mater. Chem., 2003, 13, 2028–2035 RSC.
- Y. Shi, Y. Wan and D. Zhao, Chem. Soc. Rev., 2011, 40, 3854–3878 RSC.
- L. Viau, M. Néouze, C. Biolley, S. Volland, D. Brevet, P. Gaveau, P. Dieudonné, A. Galarneau and A. Vioux, Chem. Mater., 2012, 24, 3128–3134 CrossRef CAS.
- J. Le Bideau, L. Viau and A. Vioux, Chem. Soc. Rev., 2011, 40, 907–925 RSC.
- S. Zhang, K. Dokko and M. Watanabe, Mater. Horiz., 2015, 2, 168–197 RSC.
- D. C. Green, S. Glatzel, A. M. Collins, A. J. Patil and S. R. Hall, Adv. Mater., 2012, 24, 5767–5772 CrossRef CAS PubMed.
- M. J. Hollamby, D. Borisova, P. Brown, J. Eastoe, I. Grillo and D. Shchukin, Langmuir, 2011, 28, 4425–4433 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2016 |