
Emerging investigators series: the efficacy of chlorine photolysis as an advanced oxidation process for drinking water treatment†
C. K.
Remucal
*ab and
D.
Manley
b
aDepartment of Civil and Environmental Engineering, University of Wisconsin-Madison, 1415 Engineering Dr., Madison, WI, USA. E-mail: remucal@wisc.edu
bEnvironmental Chemistry and Technology Program, University of Wisconsin-Madison, 660 N. Park St., Madison, WI, USA
First published on 17th May 2016
Abstract
The photolysis of hypochlorous acid (HOCl) and hypochlorite (OCl−) produces a suite of reactive oxidants, including hydroxyl radical (˙OH), chlorine radical (Cl˙), and ozone (O3). Therefore, the addition of light to chlorine disinfection units could effectively convert existing drinking water treatment systems into advanced oxidation processes. This review critically examines existing studies on chlorine photolysis as a water treatment process. After describing the fundamental chemistry of chlorine photolysis, we evaluate the ability of chlorine photolysis to transform model probe compounds, target organic contaminants, and chlorine-resistant microorganisms. The efficacy of chlorine photolysis to produce reactive oxidants is dependent on solution and irradiation conditions (e.g., pH and irradiation wavelengths). For example, lower pH values result in higher steady-state concentrations of ˙OH and Cl˙, resulting in enhanced contaminant removal. We also present the current state of knowledge on the alteration of dissolved organic matter and subsequent formation of disinfection by-products (DBPs) during chlorine photolysis. Although the relative yields of DBPs during chlorine photolysis are also dependent on solution conditions (e.g., higher organic DBP yields at low pH values), there is conflicting evidence on whether chlorine photolysis increases or decreases DBP production compared to thermal reactions between chlorine and dissolved organic matter in the dark. We conclude the review by identifying knowledge gaps in the current body of literature.
 Christina Remucal | Christina Remucal is currently an assistant professor in the Department of Civil and Environmental Engineering at the University of Wisconsin-Madison where she focuses on aquatic chemistry. She is also affiliated with several interdisciplinary programs, including Environmental Chemistry and Technology, Freshwater and Marine Sciences, and Molecular and Environmental Toxicology. Dr. Remucal holds a BS (2003) in Environmental Engineering and Science from the Massachusetts Institute of Technology and an MS (2004) and PhD (2009) in Civil and Environmental Engineering from the University of California, Berkeley. She completed her postdoctoral research in the Institute of Biogeochemistry and Pollutant Dynamics at the Swiss Federal Institute of Technology in 2012. |
 Devon Manley | Devon Manley is a graduate student in the Environmental Chemistry and Technology Program at the University of Wisconsin-Madison where she is advised by Dr. Christina Remucal. Ms. Manley holds a BA (2015) in Chemistry from Grinnell College. She has previously conducted environmental chemistry research at Grinnell College, the Swiss Federal Institute of Technology, and the Center for the Environmental Implications of Nanotechnology at Duke University. |
Water impactChlorine photolysis could be used to simultaneously enhance pathogen inactivation and contaminant transformation by converting chlorine-based disinfection units into advanced oxidation processes. A complete understanding of the chemistry of chlorine photolysis is necessary to optimize conditions for a variety of applications, including municipal treatment systems, decentralized solar-based point-of-use water treatment practices, and pump-and-treat groundwater remediation operations. |
Introduction
Conventional drinking water treatment systems are primarily designed to remove particles and pathogens.1 The majority of drinking water utilities in the United States utilize chlorine-based disinfection systems, in which free chlorine is added as chlorine gas (Cl2) or sodium hypochlorite (NaOCl) to form hypochlorous acid (HOCl) and hypochlorite (OCl−).2,3 The use of free available chlorine (FAC; referred to as “chlorine” in this manuscript) has several advantages compared to other disinfectants; chlorine is inexpensive, effective against many waterborne pathogens, and provides residual disinfectant in the distribution system.1 However, concerns about the formation of disinfection by-products (DBPs) during chlorine disinfection, as well as the presence of chlorine-resistant pathogens and emerging chemical contaminants, have led utilities to consider alternative treatment approaches.2,4–6Chlorine-resistant pathogenic microorganisms include oocysts of protozoan parasites (e.g., Cryptosporidium parvum) and spores of vegetative bacteria (e.g., Bacillus subtilis).2,7–9 These organisms cannot be effectively inactivated using chlorine-based disinfectants under the conditions encountered in most treatment facilities. The use of sequential disinfectants is considered to be a viable treatment option to inactivate chlorine-resistant pathogens because it results in enhanced disinfection compared to chlorine alone.7–11 During sequential disinfection, a primary disinfectant (e.g., ozone, chlorine dioxide, or ultraviolet (UV) irradiation) is applied to achieve partial inactivation followed by the application of a chlorine-based secondary disinfectant to achieve additional inactivation and to provide residual disinfection in the water distribution system.2,7–13 Although this approach is effective against many recalcitrant pathogens, it requires additional infrastructure and can alter the yield of DBPs in the treated water.4,12–15
Emerging chemical contaminants of concern in drinking water include pharmaceuticals and personal care products (PPCPs). Many of these compounds are poorly retained in wastewater treatment systems and can be found at low levels in the environment, including in drinking water sources.6,16–19 Although some PPCPs are oxidized in chlorine-based disinfection systems through direct reaction with HOCl or Cl2,20–26 many compounds are not removed by conventional treatment processes and can be found in treated water.17,18,27–36 While the toxicological effects of constant exposure to low levels of PPCPs are unknown, the presence of these compounds is of concern.
One potential solution to remove trace organic contaminants, such as PPCPs, from drinking water is the use of advanced oxidation processes (AOPs). Conventional AOPs, such as UV/hydrogen peroxide (H2O2), UV/ozone (O3), H2O2/O3, and Fenton-based (i.e., iron/H2O2) systems, rely on the formation of hydroxyl radical (˙OH).37–42 The production of ˙OH is desirable because the non-selective radical reacts at near diffusion-controlled rates with many organic compounds, including organic contaminants and biomolecules (e.g., proteins and nucleic acids).43,44 Although the low selectivity of ˙OH means that other compounds typically present in water (e.g., dissolved organic matter (DOM) and bicarbonate) can compete with contaminants for ˙OH, its rapid reactivity facilitates its use in the remediation of numerous types of recalcitrant compounds. However, the inclusion of conventional AOPs in existing drinking water treatment facilities requires costly retrofits, increases the physical size of the plant, and can be expensive to operate due to high energy costs.
The combination of chlorine and light during water treatment could effectively turn existing chlorine-based disinfection systems into AOPs. The photolysis of HOCl and OCl− produces a suite of reactive oxidants, including ˙OH, O3, and chlorine radical (Cl˙).45–47 Chlorine photolysis could be applied to simultaneously inactivate chlorine-resistant pathogens and transform organic contaminants of concern by combining multiple mechanisms: (1) direct reaction with HOCl/OCl−, (2) direct photolysis by UV irradiation, and (3) reactive species-mediated indirect photolysis (i.e., reaction with ˙OH, O3, and/or Cl˙ produced during chlorine photolysis).39–42,48–54 In the case of municipal drinking water treatment systems, this approach would utilize existing infrastructure and require only the addition of a suitable light source (i.e., either UV-A light-emitting diodes or higher energy UV-C lamps).52,53 Additional applications include solar-based point-of-use water treatment in decentralized systems,52,55 pump-and-treat groundwater remediation,56 treatment of ballast water,57 and point-of-use treatment to remove chlorine off-flavors.58 The fundamental chemistry of this process is also relevant to chlorine photolysis in uncovered chlorine disinfection contact basins in wastewater treatment plants59,60 and swimming pools.59,61
The overall aim of this manuscript is to critically review existing studies on chlorine photolysis as a water treatment application. After describing the fundamental chemistry of chlorine photolysis, we evaluate the ability of chlorine photolysis to transform model probe compounds, target organic contaminants, and chlorine-resistant microorganisms. We also present the current state of knowledge on the alteration of DOM and the formation of DBPs during chlorine photolysis. Our review focuses on the effect of solution and irradiation conditions (e.g., pH and irradiation wavelengths) on the efficacy of chlorine photolysis because the formation of reactive oxidants is dependent on these parameters. Most studies utilized low pressure-UV (LP UV; a monochromatic light source at 254 nm) or medium pressure-UV (MP UV; a polychromatic light source with wavelengths ranging from 200–400 nm) light sources. Studies conducted with alternate light sources (e.g., light in the UV-A region or the complete solar spectrum) are included when available. We limit our discussion to combined chlorine and irradiation applications, rather than sequential treatment (i.e., UV irradiation followed by chlorine addition). Studies on chloramine photolysis, which is not as photoactive as HOCl/OCl−,39,57,58,62 are not included.
Chemistry of chlorine photolysis
The rate of chlorine photolysis is pH and wavelength (λ) dependent. Solution pH is critical because the acid dissociation constant (pKa) of hypochlorous acid is approximately 7.5 (reaction 5), which is near the pH of many natural waters. As a result, the dominant chlorine species can shift between HOCl and OCl− over the pH range expected in water treatment applications (Fig. 1). The two chlorine species have different UV-visible absorption spectra (Fig. 2). HOCl has a maximum absorption coefficient of 98–101 M−1 cm−1 at 235 nm, while OCl− has a maximum absorption coefficient of 359–365 M−1 cm−1 at 292 nm.41,63 Therefore, the effect of pH on chlorine photolysis rate depends on the light source used for irradiation. The photolysis rate of chorine is generally independent of pH using a LP UV light source because both species have similar absorptivities at 254 nm (Fig. 2).64 When UV-A, UV-B, or polychromatic (i.e., MP UV) light sources are used, the photodecomposition of chlorine is faster at higher pH values because OCl− absorbs more light at λ > 254 nm (Fig. 2).59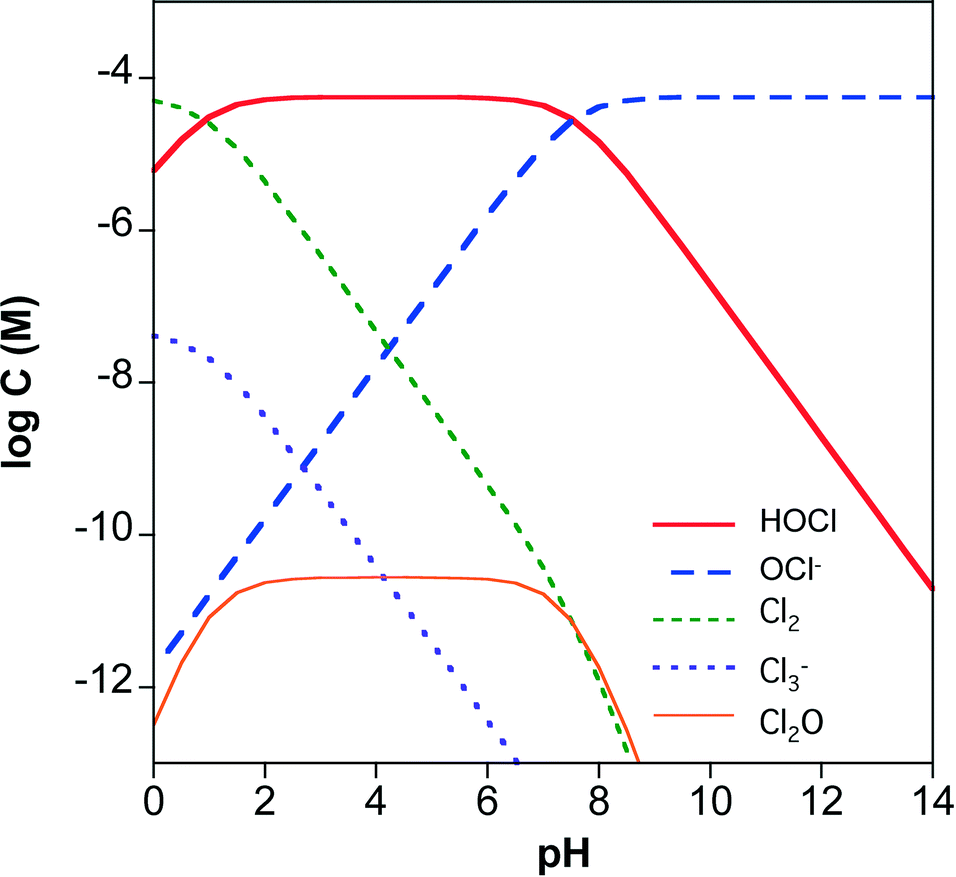 | ||
| Fig. 1 Chlorine speciation in 4 mg L−1 total chlorine and 150 mg L−1 chloride as a function of pH. Equilibrium constants are from ref. 3. | ||
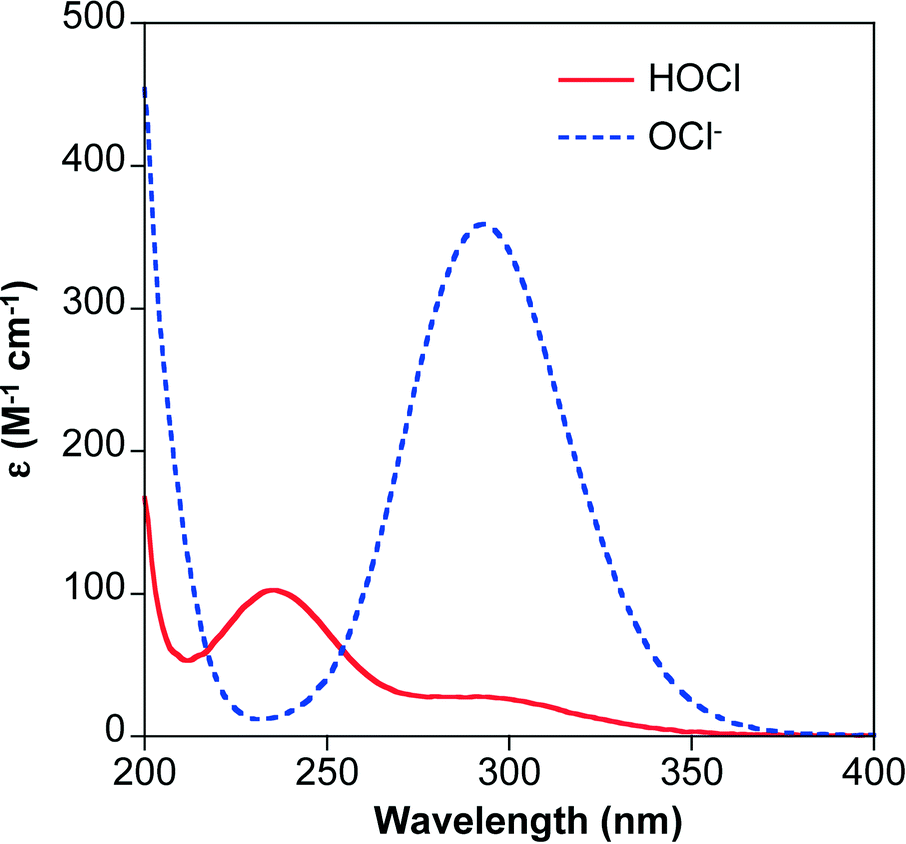 | ||
| Fig. 2 The molar absorption coefficients (ε) of HOCl and OCl− as a function of wavelength. The spectra were collected using solutions of free available chlorine in ultrapure water adjusted to pH 6 (HOCl) and pH 9 (OCl−) using HCl and NaOH, respectively. | ||
The products of chlorine photolysis are also pH and wavelength dependent. The irradiation of HOCl at λ < 400 nm produces ˙OH and Cl˙ via homolytic cleavage (reaction 1 in Table 1).47,59,65 Cl˙ can react with water to produce HOCl−˙ (reaction 16 in Table 2), which can decompose to form additional ˙OH (reaction 20). At λ < 320 nm, the irradiation of OCl− produces predominantly O−˙ (reaction 2) or excited singlet state oxygen atoms (O(1D); reaction 3). O−˙ is the conjugate base of ˙OH (reaction 6; pKa = 11.9), while O(1D) can produce ˙OH through reaction with water (reaction 7).66 At λ > 320 nm, OCl− photolysis produces ground state oxygen atoms (O(3P); reaction 4),45,69 which react with O2 to form O3 (reaction 8).46,70,71
| No. | Reaction | Parameter | 254 nm (UV-C) | 303–313 nm (UV-B) | 355–365 nm (UV-A) | >300 nm (Hg) | 200–400 nm (MP UV) |
|---|---|---|---|---|---|---|---|
| a Data collected in the gas phase. N/A = data not available. | |||||||
| 1 | HOCl + hν → ˙OH + Cl˙ | Φ HOCl | 1.0–2.839,40,53,63,64 | N/A | N/A | N/A | 1.06–3.739,41,64 |
| Φ OH | 1.439 | 1.0a,65,68 | N/A | N/A | 0.7941 | ||
| η OH | 0.46–0.8540,50 | N/A | N/A | 0.7050 | N/A | ||
| 2 | OCl− + hν → O−˙ + Cl˙ | Φ OCl− | 0.85–2.439,40,45,53,63,64 | 0.39–0.8745,51 | 0.6045 | N/A | 0.9–1.739,64 |
| Φ OH | 0.27845 | 0.12745 | 0.0845 | N/A | N/A | ||
| η OH | 050 | 0.7051 | N/A | 0.1050 | 1.1841 | ||
| 3 | OCl− + hν → Cl− + O(1D) | Φ O(1D) | 0.13345 | 0.02045 | 045 | N/A | N/A |
| 4 | OCl− + hν → Cl− + O(3P) | Φ O(3P) | 0.07445 | 0.07545 | 0.2845 | N/A | N/A |
| No. | Reaction | Rate constant | Ref. |
|---|---|---|---|
| 5 | HOCl ⇌ H+ + OCl− | pKa5 = 7.40–7.47 | 3, 74 |
| 6 | ˙OH ⇌ H+ + O−˙ | pKa6 = 11.9 ± 0.2 | 44 |
| 7 | O(1D) + H2O → 2 ˙OH | k 7 = 1.2 × 1011 M−1 s−1 | 65 |
| 8 | O(3P) + O2 → O3 | k 8 = 4 × 109 M−1 s−1 | 46, 70, 71 |
| 9 | O(3P) + OCl− → ClO2− | k 9 = 9.4 × 109 M−1 s−1 | 46, 70 |
| 10 | ˙OH + HOCl → ClO˙ + H2O | k 10 = 8.5 × 104–2.0 × 109 M−1 s−1 | 39, 75–77 |
| 11 | ˙OH + OCl− → ClO˙ + OH− | k 11 = 8.8 × 108–9.8 × 109 M−1 s−1 | 44, 50, 70, 75, 77 |
| 12 | ˙OH + Cl− → HOCl−˙ | k 12 = 4.3 × 109 M−1 s−1 | 69, 73, 78 |
| 13 | Cl˙ + HOCl → ClO˙ + H+ + Cl− | k 13 = 3 × 109 M−1 s−1 | 69 |
| 14 | Cl˙ + OCl− → ClO˙ + Cl− | k 14 = 8.2 × 109 M−1 s−1 | 69 |
| 15 | Cl˙ + Cl− → Cl2−˙ | k 15 = 6.5 × 109–2.1 × 1010 M−1 s−1 | 69, 73, 78–81 |
| 16 | Cl˙ + H2O → HOCl−˙ + H+ | k 16 = 3.0 × 102–1.8 × 104 M−1 s−1 | 69, 78, 79 |
| 17 | Cl2−˙ → Cl˙ + Cl− | k 17 = 6.0 × 104–1.1 × 105 s−1 | 73, 78–80 |
| 18 | Cl2−˙ + OH− → Cl− + HOCl−˙ | k 18 = 7.3 × 106–4.5 × 107 M−1 s−1 | 82, 83 |
| 19 | Cl2−˙ + H2O → Cl− + HOCl−˙ + H+ | k 19 = 2.4 × 101 M−1 s−1 | 73, 79 |
| 20 | HOCl−˙ → ˙OH + Cl− | k 20 = 6.1 × 109 s−1 | 69, 73, 78 |
| 21 | HOCl−˙ + Cl− → Cl2−˙ + OH− | k 21 = 1.0 × 105 M−1 s−1 | 53 |
| 22 | HOCl−˙ + H+ → Cl˙ + H2O | k 22 = 2.1 × 1010 M−1 s−1 | 69 |
| 23 | O3 + ClO2− → ClO2 + O3−˙ | k 23 = 4 × 106 M−1 s−1 | 69 |
The photochemical efficiency of chlorine photolysis can be described in three ways (Table 1). First, the quantum yield of chlorine loss (ΦHOCl and ΦOCl− in Table 1) is most commonly reported. This quantum yield is equal to the moles of free chlorine lost per mole of photons absorbed. The quantum yield of chlorine loss is dependent on solution conditions and often has a value greater than 1.0 due to radical chemistry (e.g., additional chlorine loss via reactions 10, 11, 13, and 14).39,40 Second, the quantum yield of ˙OH formation (ΦOH) represents the moles of ˙OH produced per mole of photons absorbed. This parameter represents the true quantum yield of reactions 1 and 2. Finally, the production of ˙OH may be represented by the yield factor (ηOH),50 which is defined as the moles of ˙OH produced per mole of free chlorine decomposed. Although these three parameters are sometimes compared within the literature, it is important to note that they represent different processes and cannot be used interchangeably.
The available quantum yields for reactions 1–4 are summarized in Table 1. Although OCl− can absorb more light (Fig. 2), the production of ˙OH is more efficient at low pH (i.e., when HOCl is the dominant species) and at λ < 320 nm because the quantum yield of ˙OH production by HOCl is higher than that of OCl−.39,59,63,72 To date, most of the studies focusing on contaminant transformation utilized wavelengths in the UV-C range (i.e., LP and MP UV sources) and focused on the generation of ˙OH. At higher irradiation wavelengths, HOCl photolysis becomes less important because the compound no longer absorbs light. Under these conditions, the photolysis products of OCl− shift from O−˙ and O(1D) (i.e., which subsequently generate ˙OH) to O(3P) (i.e., leading to O3 generation).46,70
The numerous reactive oxidants produced during chlorine photolysis include ˙OH, Cl˙, dichloride radical anion (Cl2−˙), and O3. The most important reactions involved in the formation of these species are summarized in Table 2, along with the range of experimentally determined rate constants. Of the oxidants produced during the AOP, ˙OH is the least selective and most reactive species, reacting at near diffusion-controlled rates with many organic and inorganic compounds.44,73 The radical reacts with organic compounds via H-atom abstraction, electron transfer, or OH addition.73 While the high reactivity of ˙OH radical makes it ideal for degrading a wide range of contaminants, it is also inefficient as it reacts quickly with many compounds present in natural waters such as DOM, carbonate/bicarbonate, and other molecules.44,59
The reactive halogen species formed during chlorine photolysis include Cl˙ and Cl2−˙. Cl˙ forms directly from HOCl and OCl− photolysis (reactions 1 and 2). Although the reactivity of Cl˙ with many compounds is similar to ˙OH, it is generally more selective and therefore less desirable as an AOP oxidant. This radical generally reacts via H-atom abstraction or Cl addition, with rate constants near the diffusion-controlled limit in some cases (i.e., 108–1010 M−1 s−1).80 Cl˙ radical can also react with Cl− to form Cl2−˙ (reaction 15) in aqueous solutions.82 Like Cl˙, Cl2−˙ can react via H-atom abstraction or Cl addition. Rate constants of Cl2−˙ are on the order of 102–106 M−1 s−1 for H-atom abstraction,82 while reaction via Cl addition is generally noted to be faster.80,82
The production of O3 in the AOP is desirable because the oxidant can lead to contaminant oxidation or pathogen inactivation through direct reaction or through the generation of ˙OH.84 Rate constants for the oxidation of organic compounds by ozone vary widely (i.e., 10−2–109 M−1 s−1)84 and are generally four orders of magnitude faster than those for HOCl.67,85
In water containing bromide (Br−), chlorine photolysis can result in the production of hypobromous acid (HOBr), hypobromate (OBr−), and a series of bromine radicals.86–88 The photolysis of HOBr and OBr− produce ˙OH and O3via similar pathways as their chlorinated analogues.46,64,69,71,89 Bromine radicals such as Br˙, ˙BrO3, Br2−˙, ˙BrO, and ˙BrO2 form via a series of electron transfer reactions when ˙OH and Cl˙ are present.46,66,69,71,90 The oxidizing strength of the bromine radicals follows the general trend ˙BrO3 > Br˙ > Br2−˙ > ˙BrO > ˙BrO2.71,91 Bromine radicals are generally more reactive than their chlorine counterparts but are found in lower concentrations and therefore are less important for organic oxidation. Note that this review focuses on chlorine photolysis of freshwater and the chemistry of bromine radicals is not discussed in detail.
Applications of chlorine photolysis
Probe compounds
A variety of model probe compounds have been used to quantify the formation of selected radicals and to assess the trends in radical formation under different experimental conditions (Table 3). An ideal probe compound should meet three criteria in order to be used during chlorine photolysis. First, the probe should react slowly with chlorine in the absence of light.49,50,53 Second, the probe should undergo minimal direct photolysis under experimental conditions.39,50,53 Rapid thermal oxidation by chlorine or rapid direct photolysis (e.g., p-chlorobenzoate photolysis at 254 nm)92 complicates interpretation of experimental results. Finally, the probe compound should react with the specific reactive species through known mechanisms at known rates. For example, nitrobenzene is an ideal ˙OH probe because the compound does not react quickly with Cl˙ or Cl2−˙.39,50 Conversely, many commonly used probes (e.g., benzoate and p-chlorobenzoate) react with both ˙OH and Cl˙ at comparable rates (Table 3).39,49,50 Due to the formation of multiple reactive species during chlorine photolysis, the use of multiple probes (i.e., an ˙OH-specific probe and one that reacts with both ˙OH and Cl˙) is a good approach to assess the yields of each species.| Probe | Reaction rate with ˙OH (M−1 s−1) | Ref. | Reaction rate with Cl˙ (M−1 s−1) | Ref. | Reaction rate with Cl2−˙ (M−1 s−1) | Ref. | Used as a probe in |
|---|---|---|---|---|---|---|---|
| a Rates were measured at pH 4 (benzoic acid pKa = 4.2). b Rates were determined in non-aqueous solvents as indicated. N/A = data not available. | |||||||
| Benzoate | 5.9 × 109 | 44 | N/A | 2 × 106 | 82 | 49, 53 | |
| Benzoic acid | 1.8 × 109 | 93 | 1.8 × 1010 (pH 4)a | 94 | 2 × 105 (pH 4)a | 94 | |
| n-Butanol | 4.2 × 109 | 44 | 4.8 × 108 (acetonitrile)b | 95 | N/A | 49 | |
| t-Butanol | 6.0 × 108 | 44 | 3 × 108 | 96 | 7 × 102 | 82 | |
| p-Chlorobenzoate | 5.0 × 109 | 97 | Does react at pH < 1 | 39 | 3 × 106 | 82 | 39, 40, 52, 64, 92, 98 |
| 1-Chlorobutane | 3.4 × 109 | 99 | Does react at pH < 1 | 50 | N/A | 50 | |
| Ethanol | 1.9 × 109 | 44 | 1.5 × 109 | 80 | 4.5 × 104 | 82 | 49 |
| Methanol | 9.7 × 108 | 44 | 5.7 × 109 (carbon tetrachloride)b | 100 | 3.5 × 103 | 82 | 40, 51 |
| Nitrobenzene | 3.9 × 109 | 44 | Negligible | 39, 50 | Negligible | 39, 50 | 39, 50, 53 |
| 2-Propanol | 1.9 × 109 | 44 | 6 × 109 | 96 | 1.2 × 105 | 82 | |
The relative importance of ˙OH and reactive halogen species (i.e., Cl˙ and Cl2−˙) as oxidants in the chlorine photolysis system has not been resolved. Multiple studies suggest that ˙OH is the dominant oxidant based on kinetic arguments. For example, experimental results obtained with both 1-chlorobutane and nitrobenzene were consistent with ˙OH as the only rate-controlling oxidant for the elimination of the probe molecules, despite the fact that 1-chlorobutane is also susceptible to reaction with Cl˙.50 Similarly, accurate prediction of p-chlorobenzoate (p-CBA) loss based on the results of nitrobenzene transformation supported the conclusion that ˙OH is the primary oxidant.39 Conversely, a comprehensive kinetic model of chlorine photolysis indicated that both ˙OH and Cl˙ contributed to the degradation of benzoate during the photolysis of chlorine at 254 nm.53 Reaction with Cl˙ was estimated to be more important because the bimolecular reaction rate between Cl˙ and benzoate is higher than that of ˙OH, assuming that benzoate reacts with Cl˙ at a similar rate to benzoic acid (Table 3). Although the limited ability of t-butanol to quench benzoate oxidation was reported as further evidence of the importance of non-˙OH oxidants,53 it is important to note that t-butanol and many other commonly used ˙OH quenchers also react with Cl˙ at near diffusion-controlled rates (Table 3). More research is needed to conclusively demonstrate the relative importance of reactive halogen species compared to ˙OH during chlorine photolysis.
Quantification of the transformation products of probe compounds could provide needed insight into the contribution of Cl˙ as an oxidant. However, the probe experiments listed in Table 3 simply quantified the loss of the probe compounds and did not identify relative yields of transformation products, with the exception of methanol oxidation to formaldehyde. Aromatic probe compounds are likely to form different products depending on the oxidant with which they react. For example, benzoate reacts with ˙OH to form hydroxybenzoate isomers101,102 and with Cl˙ to form chlorobenzoate isomers;49,94 therefore, different yields of products could be used to investigate the relative yields of ˙OH and Cl˙. However, it should be noted that the presence of halogenated products may not be due to direct reaction with Cl˙ or Cl2−˙ in the chlorine photolysis system. Phenols produced by ˙OH attack to the aromatic ring (e.g., nitrophenols produced by nitrobenzene oxidation) are more amenable to direct oxidation by free chlorine compared to the parent probe compound, leading to the production of halogenated products.49,98 The formation of multiple products, including 2-chloro-4-nitrophenol, and the absence of chloronitrobenzene isomers as products of nitrobenzene oxidation during one chlorine photolysis study39 demonstrates the relevance of this mechanism. Therefore, the role of reactive halogen species in the generation of chlorinated products from probe compounds must be interpreted carefully.
Despite the limitations in existing probe compound studies, the available data provides insight into the formation of reactive species during chlorine photolysis. Most probe studies of chlorine photolysis systems focused on the formation of ˙OH, in part because generation of the radical is critical for applications of the AOP. Reported steady-state concentrations of ˙OH ([˙OH]ss) range from 10−14 to 10−12 M, with most measured values on the order of 10−13 M.39,41,52,53,64 Details on the trends in [˙OH]ss with respect to pH and wavelength are discussed below. Although limited data is available on the steady-state concentrations of reactive halogen species, [Cl˙]ss and [Cl2−˙]ss on the order of 10−14 M and 10−14 to 10−13 M, respectively, were estimated using a kinetic model.53
Degradation rates of probe compounds and reported [˙OH]ss values are consistently higher at lower pH values.39,50,53,64,92,98 For example, the [˙OH]ss at pH 6–6.5 is generally 2–4 times the [˙OH]ss at pH 8.5–9.53,64 Although OCl− absorbs more light than HOCl (Fig. 2),59 it is less efficient at producing ˙OH (Table 1).39,50,64 Additionally, OCl− reacts more quickly with ˙OH compared to HOCl (reactions 10 and 11) and the ability of free chlorine to scavenge ˙OH increases at higher pH values.39 Production of ˙OH increases with chlorine concentration at pH values ≤6,39,53 but is independent of chlorine concentration at circumneutral pH values due to enhanced scavenging of ˙OH by OCl−.39 Collectively, these factors result in a lower apparent ˙OH yield at higher pH values. Similarly, predicted steady-state concentrations of Cl˙ and Cl2−˙ are both approximately four times higher at pH 6 compared to pH 9,53 and yields of chlorinated products from n-butanol transformation increased with decreasing pH values.49
The trends in ˙OH and Cl˙ production during chlorine photolysis due to irradiation wavelengths are less clear. At pH values >8.5, similar ˙OH yields were reported using a 254 nm lamp and a Hg lamp (λ > 300 nm)50 and consistent [˙OH]ss values were observed using both narrow-band LP UV and polychromatic MP UV irradiation sources.64 Production of ˙OH appears to be wavelength dependent at pH values <6.5 (i.e., when HOCl is dominant); both Hg lamps50 and MP UV lamps64 produced higher ˙OH yields than LP UV light sources. Limited data suggests that production of Cl˙ may also be wavelength dependent; higher yields of chlorinated n-butanol products were observed using 254 nm light compared to 365 nm light at pH 10.49
Numerous compounds typically present in natural waters can decrease loss rates of probe compounds by scavenging ˙OH and Cl˙, as is true for all ˙OH-based AOPs. Natural scavengers shown to affect probe compound degradation during chlorine photolysis include DOM, bicarbonate/carbonate, and bromide.39,53,64,98 Additionally, scavengers that trigger chain reactions (e.g., methanol)40 can lead to enhanced chlorine consumption while decreasing the oxidation rate of probe compounds. Based on relative rates of reaction, DOM preferentially scavenges ˙OH, while HCO3− preferentially scavenges Cl˙.53
The generation of O3 during chlorine photolysis (e.g., via reaction 8) has received minimal attention in the literature, likely because the generation of O3 is favored when higher wavelengths of light are used and most studies utilize light in the UV-C region (Tables 1 and 4). However, O3 can form under some conditions.52,55 For example, O3 concentrations up to 1.8 μM were quantified when 10 mg Cl2 L−1 was irradiated in a solar simulator (λ > 290 nm) at pH 8.52 O3 is a potent oxidant and can undergo further reaction to form ˙OH;84 therefore, the conditions under which chlorine photolysis results in enhanced O3 generation warrant further investigation.
| Compound | Light source | [Free chlorine] (mg Cl2 L−1) | pH | References for chlorine photolysis | |
|---|---|---|---|---|---|
| Enhanced degradation rate | Product data available | ||||
| Acids | |||||
| Acetic acid | Hg | 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 500 |
12 | 103 | 103 |
| α-Chloropropionic acid | Hg | 32500 |
12 | 103 | 103 |
| β-Chloropropionic acid | Hg | 32500 |
12 | 103 | 103 |
| α-Hydroxypropionic acid | Hg | 33250 |
12 | 103 | 103 |
| β-Hydroxypropionic acid | Hg | 32300 |
12 | 103 | 103 |
| Propionic acid | Hg | 31400 |
12 | 103 | 103 |
| Antibacterial agent | |||||
| Triclosan | LP UV | 1.4–7 | 7 | 54 | 54 |
| Aromatic sulfonic acids | |||||
| p-Cumenesulfonic acid | Hg | 710 | 12 | 104 | 104 |
| 2-Mesitylenesulfonic acid | Hg | 710 | 12 | 104 | 104 |
| 1-Napthalenesulfonic acids | Hg | 710 | 12 | 104 | 104 |
| 2-Napthalenesulfonic acids | Hg | 710 | 12 | 104 | 104 |
| Chelating agent | |||||
| Ethylenediaminetetraacetic acid (EDTA) | LP UV | 0–5.4 | 5–9 | 105 | 105 |
| Corrosion inhibitor | |||||
| Benzotriazole | LP UV | 1–6 | 7 | 42 | |
| Tolyltriazole | LP UV | 1–6 | 7 | 42 | |
| Disinfection by-product | |||||
| N-Nitrosodimethylamine (NDMA) | MP UV | 0.8–7.7 | 6.9–7.1 | 56 | |
| DOM surrogate | |||||
| 4,6-Dioxoheptanoic acid | LP UV, UV-A | 1000 | 6, 7.5, 9 | 92 | 92 |
| o-Methoxybenzoic acid | LP UV, UV-A | 1000 | 6, 7.5, 9 | 92 | 92 |
| Dye | |||||
| Methylene blue | UV-B, sunlight | 35–250 | 10 | 51 | |
| Napthenic acids and related model compounds | |||||
| Cyclohexanoic acid | UV-B, sunlight | 35–570 | 10 | 51 | |
| LP UV | 50 | 5 | 40 | ||
| Naphthenic acids in oil sands process-affected water | UV-B, sunlight | 200–300 | 8.3, 10 | 106 | |
| Pesticides and pesticide degradation products | |||||
| Chlortoluron | LP UV | 1.8–70.9 | 5–9 | 107 | 107 |
| Desethylatrazine | LP UV | 1–6 | 7 | 42 | |
| Pharmaceutical and personal care products (PPCPs) | |||||
| 17α-Ethinylestradiol (EE2) | LP UV | 1–6 | 7 | 42 | |
| Caffeine | MP UV | 2, 6, 10 | 6.5, 7.5, 8.5 | 108 | |
| Carbamazepine | LP UV | 1–6 | 7 | 42 | |
| Diclofenac | LP UV | 1–6 | 7 | 42 | |
| Metoprolol | LP UV | 1–5 | 2–9 | 109 | 109 |
| Ronidazole | LP UV | 3.5–210 | 5–9 | 110 | 110 |
| Sulfamethoxazole | LP UV | 1–6 | 7 | 42 | |
| Solvents and related transformation products | |||||
| 1,1-Dichloroethene (1,1-DCE) | MP UV | 0.8–7.7 | 6.9–7.1 | 56 | |
| 1,2-Dichloroethene (1,2-DCE) | MP UV | 0.8–7.7 | 6.9–7.1 | 56 | |
| 1,4-Dioxane | LP UV | 140–890 | 2.9–9.5 | 111 | |
| Trichloroethylene (TCE) | MP UV | 0.8–7.7 | 6.9–7.1 | 56 | |
| MP UV | 10.6 | 5, 7.5, 10 | 41 | ||
| Surfactants | |||||
| Benzenesulfonic acid | Hg | 18300 |
11 | 48 | 48 |
| Diethylene glycol | Hg | 16000 |
12 | 112 | 112 |
| Diethylene glycol dimethyl ether | Hg | 16000 |
12 | 112 | 112 |
| Diethylene glycol monomethyl ether | Hg | 16000 |
12 | 112 | 112 |
| p-Ethylbenzenesulfonic acid | Hg | 16000 |
11 | 48 | 48 |
| Ethylene glycol | Hg | 16000 |
12 | 112 | 112 |
| Ethylene glycol dimethyl ether | Hg | 16000 |
12 | 112 | 112 |
| Ethylene glycol monomethyl ether | Hg | 16000 |
12 | 112 | 112 |
| p-Toluenesulfonic acid | Hg | 2200 | 11 | 48 | 48 |
| Taste and odor compounds | |||||
| 2-Methylisoborneal | LP UV | 8 | 5–7.6 | 113 | |
| MP UV | 1–5 | 6, 7.5 | 114 | ||
| MP UV | 2, 6, 10 | 6.5, 7.5, 8.5 | 108 | ||
| Geosmin | LP UV | 8 | 5–7.6 | 113 | |
| MP UV | 2, 6, 10 | 6.5, 7.5, 8.5 | 108 | ||
| X-ray contrast media | |||||
| Iohexol | LP UV | 3.5–35 | 5–9 | 115 | 115 |
| Iopamidole | LP UV | 1–6 | 7 | 42 | |
Organic contaminants
The chlorine photolysis-based AOP is capable of transforming a wide range of target organic contaminants (Table 4). The process has been investigated in lab-, pilot-,42,108,114 and full-scale treatment reactors,56,108 with most studies utilizing LP and MP UV irradiation sources. Chlorine photolysis is able to remove recalcitrant organic compounds through three mechanisms: direct reaction with chlorine, direct photolysis, and reaction with reactive oxidants (e.g., ˙OH). Different transformation products may be produced through each mechanism. For example, ronidazole forms chlorinated products through thermal reactions with HOCl, hydroxylated products during direct photolysis, and a combination of products during chlorine photolysis.110 Therefore, contaminant transformation may result in a wide range of products (Table 4), including the formation of chlorinated products48,54,103,107,110,112,115 and mineralization to CO2 in some cases.48,92,103,105,107,110,112,115 Although degradation of many compounds is generally very efficient,42 some compounds are relatively less susceptible to oxidation in the AOP (e.g., carbamazepine,42 cyclohexanoic acid,40 desethylatrazine,42 and TCE56).Rates of contaminant removal are dependent on experimental conditions (e.g., pH, the concentration of chlorine, and irradiation intensity) and follow the trends expected based on the probe compound experiments described above. In general, the oxidation rates of target contaminants increase with decreasing pH values41,56,105–111,114,115 and with increasing chlorine concentrations.51,54,56,106,107,109–111,115 However, high concentrations of chlorine can result in decreased contaminant transformation rates due to scavenging of ˙OH, primarily at higher pH values when OCl− is the dominant species.51 Additionally, contaminant transformation product distributions can be dependent on initial chlorine concentrations, with a shift from organic products to mineralization observed at very high chlorine concentrations.48,103,107,110,112 Finally, contaminant transformation rates are also dependent on light intensity,107,109 but the effect of irradiation wavelength on organic compound removal has not been systematically investigated.
The role of reactive species in enhanced contaminant degradation rates during chlorine photolysis warrants further investigation. In most cases, ˙OH is assumed to be the dominant oxidant based on the seminal work by Hoigné discussed above.41,50,51,107,110,113 For example, a kinetic model developed for TCE oxidation assumed that the dominant loss pathways were through direct photolysis and reaction with ˙OH.41 One study provided mechanistic evidence for ˙OH by demonstrating that nitrobenzene (i.e., an ˙OH-specific scavenger) was able to quench oxidation of naphthenic acids.106 Conversely, the formation of chlorinated metoprolol products was attributed to reaction with Cl˙ and ClO˙ radicals.109 However, as discussed above, the presence of chlorinated products does not necessarily indicate direct reaction with halogen radicals; it is likely that hydroxylated products formed through ˙OH attack are more susceptible to oxidation by HOCl. Therefore, more mechanistic evidence is needed to conclusively rule out reactive halogen species as oxidants during chlorine photolysis.
Direct comparison between the UV/chlorine AOP and the commonly used UV/H2O2 AOP shows that UV/chlorine is more effective on the basis of contaminant removal, ˙OH yield, chemical costs, and energy usage under some conditions. HOCl and OCl− are more efficient at producing ˙OH because the chlorine species absorb more light and have higher quantum yields than H2O2.39,113,114 Additionally, the scavenging rate of ˙OH by HOCl (reaction 10) is lower than that of H2O2 (kH2O2 = 2.7 × 107 M−1 s−1).39,114 However, OCl− is much more reactive with ˙OH (reaction 11) than either HOCl or H2O2 and the UV/chlorine AOP becomes less competitive at higher pH values.114 In waters with low concentrations of dissolved organic carbon (DOC), UV/chlorine is more effective at producing ˙OH and at degrading target contaminants than UV/H2O2 only at slightly acidic pH values (i.e., pH ≤ 6.5).41,108,113,114 However, UV/chlorine can be as effective as UV/H2O2 at circumneutral pH values in waters with [DOC] > 2 mg C L−1.41,113 The chemical costs of chlorine are up to 50% lower than that of H2O239,56,114 and the UV/chlorine AOP could result in a 30–75% energy savings compared to UV/H2O2, depending on the target contaminant and the pH of the treated water.42,108
Chlorine-resistant microorganisms
Limited data is available on the inactivation of microorganisms during simultaneous exposure to chlorine and light. Photolysis of chlorine by simulated and real sunlight enhances the inactivation of chlorine-resistant microorganisms, including the model spore Bacillus subtilis52 and the pathogenic oocyst Cryptosporidium parvum.55 Inactivation of B. subtilis with chlorine in the dark was faster at pH 6 than at pH 8 because HOCl is a more potent disinfectant than OCl−. However, the relative enhancement of inactivation due to chlorine photolysis was greater at higher pH values; B. subtilis inactivation rates increased by a factor of 1.2 and 2.3 at pH 6 and 8, respectively.52 Increased inactivation of B. subtilis and C. parvum was observed in natural waters and in pure buffered waters, indicating that increased alkalinity and DOM did not play a major role in hindering inactivation.52,55 The results from probe and quencher experiments demonstrate that both ˙OH and O3 have complementary roles in the inactivation of B. subtilis and C. parvum, possibly by sensitizing the organisms to further oxidative attack by HOCl or O3.52,55 More research is needed on the mechanism and rates of inactivation of other chlorine-resistant microorganisms during chlorine photolysis, as well as inactivation rates under different conditions (e.g., UV-C irradiation).Formation of disinfection by-products
Alteration of bulk DOM properties
The reaction of DOM with chlorine is the major source of DBPs during conventional water disinfection.4–6 Therefore, investigating the effect of chlorine photolysis on the concentration and composition of DOM is essential to understanding the effect of the AOP on DBP production. During chlorine photolysis, DOM could react directly with HOCl/OCl−, undergo photobleaching via direct photolysis, or react with reactive oxidants (e.g., ˙OH, O3, or Cl˙). Each of these processes may change the molecular composition of DOM and alter its reactivity with chlorine, leading to changes in DBP formation.39,92,98,116 Furthermore, the reaction of Cl˙ or Cl2−˙ with DOM could produce novel DBPs.116The combination of chlorine and light can alter treated waters by affecting the concentration of DOM compared to water that has only been exposed to chlorine in the dark. Chlorine photolysis of natural waters consistently results in enhanced loss of absorbance in the UV-C region (i.e., absorbance at 254 nm).62,92,98 Loss of absorbance at 254 nm is greater under UV-C irradiation compared to UV-A irradiation, and is greater at lower pH values.92 Loss of absorbance is coupled to enhanced mineralization (i.e., loss of DOC) in most cases using MP UV,117 UV-C,62,118 and UV-A119 light sources at circumneutral pH values and free chlorine concentrations as low as 1.5 mg Cl2 L−1. The loss of absorbance and extent of mineralization is higher for solutions exposed to both chlorine and light compared to light alone98,119 or chlorine alone.62,97,98
Additionally, chlorine photolysis can alter the composition of DOM by changing its structure. The irradiation of natural water in the presence of chlorine results in a loss of chromophoric moieties (i.e., preferential loss of absorbance in the visible and UV-A regions)92,98 and a decrease in fluorescence;92 losses were greater for solutions simultaneously exposed to both chlorine and light relative to chlorine and light separately. This preferential loss of high-molecular weight chromophoric moieties can result in a decrease in the molecular weight of DOM.98 Irradiation of DOM in the presence of chlorine also decreases the specific absorbance at 254 nm (SUVA254), which corresponds to a decrease in aromaticity.92 Similar trends have been observed for DOM exposed to UV irradiation in the absence of chlorine.12,13,88,120
Organic DBPs
The interaction of chlorine with light will alter the concentration and distribution of organic DBPs formed by DOM during the advanced oxidation process. Chlorine photolysis could potentially decrease DBPs by decreasing the concentration of DOM and by allowing for a shorter contact time compared to traditional chlorine disinfection systems.42,52 However, the benefits of a short contact time may be offset by the need for higher chlorine concentrations because depletion of the concentration of chlorine via photolysis (reactions 1–4)39,113,116 and by chain reactions between chlorine and carbon-centered radicals63,121 increases chlorine demand. Finally, the changes in DOM composition (i.e., the decrease in chromophoric, fluorescent, and aromatic moieties) observed during chlorine photolysis will alter the reactivity of DOM with chlorine.The formation of trihalomethanes (THMs), haloacetic acids (HAAs), haloacetonitriles (HANs), and total organic halides (TOX) from DOM during chlorine photolysis has been investigated in multiple studies. It is challenging to make broad conclusions about the trends in DBP formation during chlorine photolysis due to variability in experimental conditions across studies. Variables include the use of different light sources (LP UV, MP UV, UV-A, and Hg lamps), different pH values (6.5–8.5), different chlorine concentrations (1.5–10 mg L−1 as Cl2), and different water sources with a wide range of DOC concentrations (1.5–5 mg C L−1). Available data for experiments in which DBP concentrations change due to reaction of chlorine with DOM in the presence and absence of light are summarized in Fig. 3 and in Tables S1–S4.† The difference between [DBP] measured with chlorine in the light and [DBP] produced by chlorine in the dark enables visualization of the relative increase or decrease in each DBP class. Data points with values <0 represent experiments in which DBP concentrations decreased in the AOP relative to reaction of the same DOM with the same chlorine concentration in the dark, while data points with values >0 represent experiments in which DBP concentrations increased. Note that DBP concentrations increase with time of reaction with chlorine (i.e., post-photolysis); all chlorine reaction times, which range from <1 min to 3 days, are included in the figure to show the relative changes in DBP yield. The trends in DBP formation with respect to concentration, pH, and wavelength are described for individual organic DBP classes below.
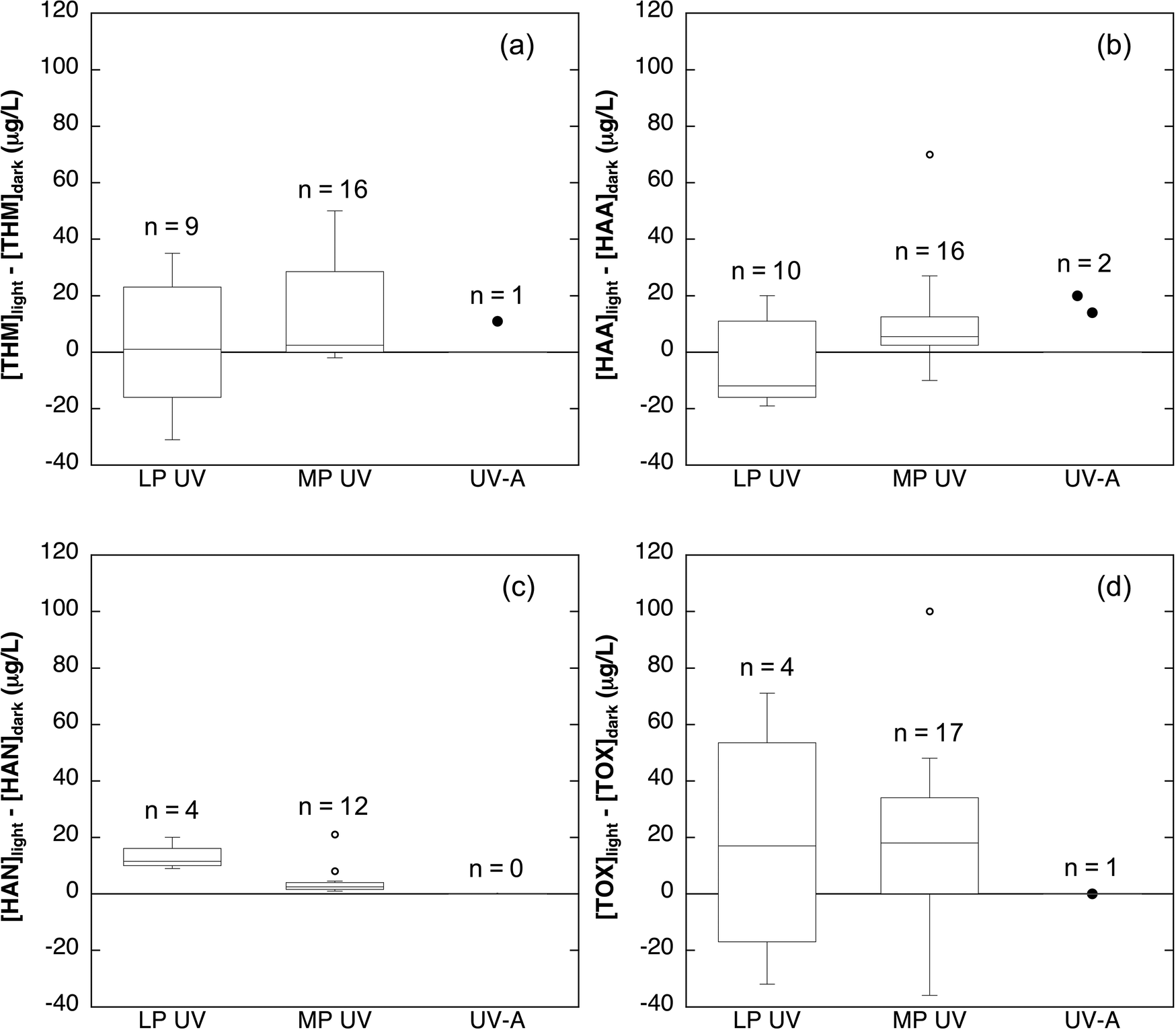 | ||
| Fig. 3 Difference between the concentrations of (a) THMs, (b) total HAAs, (c) HANs, and (d) TOX observed during chlorine photolysis and during reaction of the same source water with the same concentration of chlorine in the dark. Box-and-whisker plots were prepared when sufficient data was available (n ≥ 4). Lines of the boxes represent the first and third quartiles. The line within each box represents the median. Whiskers represent minimum and maximum concentrations. Hollow points represent outliers (i.e., any point less than the lower quartile or greater than the upper quartile by more than 1.5 times the interquartile range). Solid points are individual data points for conditions with insufficient data to construct box-and-whisker plots. Data is summarized from ref. 14, 62, 64, 92, 116 and 124. Specific experimental conditions for each data point are provided in Tables S1–S4.† | ||
The effect of chlorine photolysis on THM yields relative to reaction with chlorine in the dark is highly variable (Fig. 3a; Table S1†). Irradiation of DOM with MP UV14,116 and UV-A92 light in the presence of chlorine generally resulted in higher concentrations of THMs relative to dark controls, while irradiation with a Hg lamp (λ > 300 nm) resulted in lower THM concentrations.50 The use of LP UV light sources both increased14 and decreased62,92 relative THM yields. Despite possible increases in THM production, the reported THM concentrations due to irradiation with chlorine and subsequent reaction with chlorine for up to two hours were always below the maximum contaminant level (MCL) of 80 μg L−1.122,123 Similarly low levels of THM production were observed in additional chlorine photolysis studies that did not report the concentration of THMs in dark reactions.42,113,114 Limited data suggests less THM formation during chlorine photolysis at higher pH values50,116 and with lower wavelengths of light.92 However, these general trends do not hold across all of the available studies, possibly due to differences in experimental parameters or DOM sources.
The production of HAAs during chlorine photolysis is also sensitive to experimental parameters (Fig. 3b; Table S2†). Note that Fig. 3 includes data on the sum of the five regulated HAAs (HAA5), as well as the sum of nine frequently studied HAAs (HAA9 = HAA5 + bromochloro-, bromodichloro-, dibromochloro- and tribromoacetic acid). Total HAA concentrations during chlorine photolysis have been reported to increase,14,92,116 decrease,14,62 or stay the same14 relative to reaction with chlorine in the absence of light. Production of HAAs immediately after chlorine addition was always below the MCL of 60 μg L−1.62,92,113,114,116,122,123 HAA yields in chlorine photolysis systems appear to decrease with increasing pH values.116 With respect to wavelength, UV-C light produced higher yields of HAA5, but lower yields of HAA9 relative to UV-A light.92 However, experiments with four different DOM sources irradiated with LP and MP UV using the same chlorine concentration at the same pH value produced conflicting trends in HAA formation potential, further highlighting the importance of DOM composition in DBP formation potential.14
A limited amount of data is available for nitrogen-containing DBPs (N-DBPs). Two studies on haloacetonitrile (HAN) formation during chlorine photolysis indicate that production of this class of DBPs is enhanced using LP and MP UV (Fig. 3c; Table S3†).62,116 The yield of HANs during chlorine photolysis was higher at pH 6.5 compared to pH 8.5 and increased with increasing chlorine concentrations.116N-Nitrosodimethylamine (NDMA) was not observed in a LP UV-based chlorine photolysis AOP, which is unsurprising given that NDMA is most commonly associated with disinfection by chloramine.42 Finally, elevated levels of cyanogen chloride, dichloroacetonitrile, and chloropicrin were produced during UV-C irradiation of chlorine, but these studies used model amine precursors which may not be representative of DOM in drinking water sources.107,110,125–127
The production of TOX, the total organic halogenated material formed during chlorination, was also quantified during chlorine photolysis in several studies (Fig. 3d; Table S4†). In most cases, TOX decreased or stayed the same in studies using LP UV,64,92,124 MP UV,64,98 or UV-A92 irradiation. Conversely, TOX increased during photolysis relative to dark controls in a limited number of studies using LP UV,64 MP UV,116,124 and UV-A irradiation.49 Yields of TOX during chlorine photolysis were typically lower at higher pH values.64,98,116 Overall, the reported changes in TOX yields were fairly modest (Fig. 3d), suggesting that chlorine photolysis may change the distribution of organic halogens without changing bulk TOX concentrations.
While the above discussion considers the possibility of chlorine photolysis altering DBP production due to changes in DOM reactivity, it is also possible that photolysis could lead to degradation of DBPs after they have formed either through direct or indirect photodegradation. For example, chlorinated THMs and HAAs were predominant in waters irradiated using UV-C light, while their brominated analogues were present in much higher levels in UV-A light and in dark control reactions.92 Similarly, chloroform was the only THM formed in a UV-C/HOCl experiment, while both chloroform and bromodichloromethane were formed with HOCl alone.62 The decreased yield of brominated organics might be due to direct photolysis by UV-C light,92 in agreement with the observation of direct photolysis of brominated THMs and HAAs using a MP UV (i.e., polychromatic) light source.128 Several classes of N-DBPs (e.g., nitrosamines and halonitromethanes) are amenable to direct photolysis129–131 and could also be potentially degraded in the AOP. Additionally, ˙OH formed during chlorine photolysis could lead to the degradation of DBPs;98 this area of research warrants further investigation.
Inorganic DBPs
Chlorine photolysis can produce numerous inorganic products, including chloride (Cl−), chlorate (ClO3−), perchlorate (ClO4−), chlorite (ClO2−), chlorine dioxide (ClO2), and bromate (BrO3−) via O(3P)- and O3-mediated pathways in the presence of chlorine and bromide.45,46,52,87,132,133 With the exception of Cl−, these species are either currently regulated or under consideration for regulation in drinking water by the US Environmental Protection Agency.122,123,134 The major stable inorganic products of chlorine photolysis are Cl− and ClO3−, with yields of free chlorine conversion ranging from 50–91% and 2–30%, respectively.116,121,135,136 Higher yields of chlorate were observed using narrow-band UV-A, -B, and -C irradiation sources121,135,136 compared to polychromatic MP UV light.116 Additionally, the highest yields of chlorate were reported at circumneutral pH values over the pH range 3–10 using UV-B light.136 The rate of chlorate production is first order with respect to [Cl2] and is yield independent of light intensity.136The photolysis of chlorine can produce low levels of ClO4− under some conditions. Reported yields of free chlorine conversion to perchlorate range from 0.09 × 10−3 to 9.2 × 10−3% using a range of chlorine concentrations (70–10000 mg L−1 Cl2), pH values (3–10), and irradiation wavelengths (254, 311, and 365 nm).136 The maximum concentration of perchlorate expected from 7 mg L−1 Cl2, a typical concentration used in the AOP, is on the order of 0.1 μg L−1.136 A second study reported elevated ClO4−generation during OCl− photolysis using 254 nm light relative to dark controls only at very high concentrations of OCl− (i.e., 10000 mg L−1 as Cl2).135 Experimental evidence exists for two proposed mechanisms of perchlorate production which involve either chlorite136 or chlorine dioxide135,137 as intermediates. The underprediction of ClO4− production by a ClO3−-dependent kinetic model in UV-A irradiation experiments,136 as well as enhanced production of ClO4− from ClO2− photolysis at higher wavelengths,135 suggests that multiple intermediates could be responsible for ClO4− production under some conditions. Although two additional studies did not detect ClO4− as a product of chlorine photolysis,116,121 the expected concentrations of ClO4− are very low and it is possible that the anion was below the analytical detection limit.
Chlorine photolysis can also generate ClO2− (reaction 9) and ClO2 (reaction 23), but these species are photolabile and are not expected to accumulate in solution. For example, photoproduction of ClO2− has only been quantified using UV-A irradiation of chlorine (i.e., compared to analogous experiments using UV-B and UV-C irradiation), where it behaved as a transient intermediate.136 This observation is supported by studies on the photolysis of chlorite using 254 nm light, which produces Cl− (68%) and ClO3− (32%) as the major stable species135 and ClO2 as a photolabile intermediate.137 The absence of chlorite116,121 and chlorine dioxide52 in additional studies on chlorine photolysis could be due to either analytical sensitivity issues or the transient nature of the photolabile species.
In bromide-containing waters, the photolysis of chlorine could potentially lead to the production of bromate. There are several possible pathways of bromate formation which require the generation of HOBr or OBr− as intermediates. HOBr/OBr− could be formed from the oxidation of bromide by O3 under conditions in which O3 is generated,86,87 or by oxidation of Br− by free chlorine.88 HOBr/OBr− can subsequently react with either ˙OH or O3 to yield BrO3−, as described in detail by von Gunten.86,87 Although the possible formation of bromate as a DBP during chlorine photolysis has received minimal attention, the oxyanion was detected in one study using MP UV as an irradiation source.116 Approximately 0.01–0.05% of the photolyzed chlorine produced BrO3−, corresponding to concentrations of 0.1–2 μg L−1, with higher formation occurring at lower pH values. The formation of bromate during chlorine photolysis warrants further investigation, particularly in waters with elevated ambient bromide concentrations.
Formation of novel DBPs
The reaction of photochemically generated Cl˙ or Cl2−˙ with DOM could produce novel DBPs. Trends in the chlorination of model compounds during chlorine photolysis provide some insight into the reactivity of reactive halogen species in the AOP. Although ˙OH outcompetes Cl˙ for reaction with many aliphatic compounds (e.g., ethanol and maleic acid),49,98 chlorine photolysis can lead to the production of halogenated products of some compounds (e.g., n-butanol and propionic acid) that do not react with chlorine in the dark.49,103 For example, up to 16% of n-butanol was converted to chlorinated products when the compound was irradiated in the presence of chlorine using UV-A light, with higher yields produced at lower pH values.49 Lower wavelengths of light also produced higher yields of chlorinated n-butanol49 and propionic acid.103 Although these studies used high concentrations of chlorine (Table 4), they indicate that lower pH values and lower wavelengths of light favor the generation of Cl˙ and possible production of organohalogens.The mechanism of halogenated aromatic production during chlorine photolysis is more complex. While chlorinated products of benzoic acid,49,53,98 nitrobenzene,39,98 and metoprolol109 have been observed, the production of organohalogens cannot be solely attributed to reaction with reactive halogen species. As described above, it is also possible for phenolic products generated by ˙OH attack to undergo thermal reaction with chlorine.49,98 The latter halogenation mechanism may be dominant for some compounds that are highly resistant to oxidation (e.g., nitrobenzene).98 Additionally, for compounds that are amenable to direct oxidation by chlorine (e.g., phenol and triclosan), chlorine photolysis can lead to the production of ring cleavage products.54,98
The formation of novel DBPs during chlorine photolysis has not yet been investigated. Studies using high-resolution Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR MS) have identified hundreds of molecular formulas with one, two, or three chlorine or bromine atoms following the chlorination of DOM, many of which had not been previously detected.138–142 Although one study presented the mass spectra of Suwannee River NOM before and after chlorine photolysis,98 the triple quadrupole MS used to generate the mass spectra did not provide sufficient resolving power to identify molecular formulas in complex mixtures of organic molecules (i.e., DOM). More research is needed using high-resolution mass spectrometry techniques to assess whether chlorine photolysis generates novel high molecular weight DBPs compared to reaction of DOM with chlorine alone.
Conclusions and need for future research
This review suggests that photolysis of chlorine could effectively convert existing drinking water treatment systems into advanced oxidation processes. The reaction of HOCl and OCl− with light produces multiple reactive oxidants, including ˙OH, Cl˙, and O3. Chlorine photolysis is able to transform recalcitrant organic compounds through three mechanisms: direct reaction with chlorine, direct photolysis, and reaction with reactive oxidants (e.g., ˙OH). In the case of the two studied chlorine-resistant pathogens, the presence of multiple oxidants can lead to synergistic disinfection mechanisms. Chlorine photolysis is able to outcompete the commonly used UV/H2O2 AOP on the basis of ˙OH production, energy usage, and cost under some conditions.The production of reactive oxidants and transformation of organic compounds during chlorine photolysis is dependent on solution and irradiation conditions. The effect of solution pH on the efficacy of chlorine photolysis is clear; lower pH values result in higher steady-state concentrations of ˙OH, Cl˙, and Cl2−˙, leading to enhanced contaminant removal. This trend can be attributed to the increased efficiency of ˙OH production by HOCl photolysis and decreased rate of reaction between ˙OH and HOCl compared to OCl−. As observed with other AOPs, the presence of other water constituents (e.g., DOM and bicarbonate/carbonate) decreases the efficiency of chlorine photolysis for target contaminant transformation. Finally, the effect of wavelength on oxidant production and contaminant transformation during chlorine photolysis has not been systematically investigated. Limited data suggests that ˙OH production is wavelength-dependent at pH < 6.5 and that lower wavelengths of light favor the generation of Cl˙.
A major concern about the use of chlorine photolysis is its potential impact on organic and inorganic DBP formation. Chlorine photolysis alters the composition and reactivity of DOM by decreasing its concentration and by preferentially removing aromatic and high molecular weight material. The resulting impact on organic DBP yields, such as THMs, HAAs, and HANs, is sensitive to experimental parameters. Although organic DBP yields tend to be lower at higher pH values, it is difficult to compare results across different studies due to differences in experimental conditions. In general, chlorine photolysis can either increase or decrease DBP concentrations compared to reaction with chlorine in the dark, but the effect is modest. The main inorganic products of chlorine photolysis are Cl− and ClO3−, with trace levels of ClO4−, ClO2−, and ClO2 observed in some studies.
This systematic review of chlorine photolysis studies for water treatment applications reveals several limitations associated with the current body of knowledge.
(1) Many studies assume that ˙OH is the dominant oxidant without providing mechanistic evidence. More work is needed to assess the potential importance of reactive halogen species (i.e., Cl˙ and Cl2−˙) and O3 as oxidants. The role of specific oxidants could be assessed through the careful selection of probes and quenchers, or by identifying and quantifying the products of probe compound transformation.
(2) Fundamental research demonstrates that the quantum yields of reactions 1–4 are wavelength-dependent. However, most studies utilize light in the UV-C region and the effect of irradiation wavelength on the production of oxidants from chlorine photolysis has not been comprehensively studied. Consideration of irradiation by wavelengths within the actinic spectrum is critical for certain applications of chlorine photolysis (e.g., enhanced solar disinfection).
(3) Available data on model organism and pathogen inactivation during chlorine photolysis is very limited. This area should be expanded to include both conventional and chlorine-resistant pathogens, both of which are likely to undergo enhanced inactivation.
(4) The effect of chlorine photolysis on DBP yields is unclear; some studies show a modest enhancement in DBP production, while others show a decrease in DBP yields. More systematic work on the effect of experimental parameters on DBP production is needed, with an emphasis on both regulated and unregulated (e.g., N-DBPs) compounds. Analysis for novel DBPs using high-resolution mass spectrometry techniques could provide needed insight into the effect of chlorine photolysis on DOM reactivity with chlorine and subsequent formation of DBPs.
In summary, a complete understanding of the chemistry of chlorine photolysis is necessary to optimize conditions for the AOP in water treatment applications in order to simultaneously enhance pathogen inactivation and contaminant transformation while limiting possible negative effects on DBP yields.
Acknowledgements
This work was supported by an NSF CAREER award (CBET-1451932).References
- J. K. Edzwald, Water Quality & Treatment: A Handbook on Drinking Water, McGraw Hill, 6th edn, 2011 Search PubMed.
- Alternative Disinfectants and Oxidants Guidance Manual, United States Environmental Protection Agency, Washington, DC, 1999 Search PubMed.
- G. V. Korshin, in Aquatic Redox Chemistry, ed. P. G. Tratnyek, T. J. Grundl and S. B. Haderlein, American Chemical Society, Washington, DC, 2011, ch. 11, vol. 1071, pp. 223–245 Search PubMed.
- S. W. Krasner, Philos. Trans. R. Soc. A, 2009, 367, 4077–4095 CrossRef CAS PubMed.
- S. D. Richardson, J. Environ. Monit., 2001, 4, 1–9 RSC.
- S. D. Richardson and T. A. Ternes, Anal. Chem., 2011, 83, 4614–4648 CrossRef CAS PubMed.
- J. L. Rennecker, A. M. Driedger, S. A. Rubin and B. J. Mariñas, Water Res., 2000, 34, 4121–4130 CrossRef CAS.
- B. Corona-Vasquez, A. Samuelson, J. L. Rennecker and B. J. Mariñas, Water Res., 2002, 36, 4053–4063 CrossRef CAS PubMed.
- M. Cho, J.-H. Kim and J. Yoon, Water Res., 2006, 40, 2911–2920 CrossRef CAS PubMed.
- A. M. Driedger, J. L. Rennecker and B. J. Mariñas, Water Res., 2000, 34, 3591–3597 CrossRef CAS.
- M. Cho, V. Gandhi, T.-M. Hwang, S. Lee and J.-H. Kim, Water Res., 2011, 45, 1063–1070 CrossRef CAS PubMed.
- D. A. Reckhow, K. G. Linden, J. Kim, H. Shemer and G. Makdissy, J. - Am. Water Works Assoc., 2010, 102, 100–113 CAS.
- A. D. Dotson, V. O. S. Keen, D. Metz and K. G. Linden, Water Res., 2010, 44, 3703–3713 CrossRef CAS PubMed.
- W. Liu, L.-M. Cheung, X. Yang and C. Shang, Water Res., 2006, 40, 2033–2043 CrossRef CAS PubMed.
- B. A. Lyon, R. Y. Milsk, A. B. DeAngelo, J. E. Simmons, M. P. Moyer and H. S. Weinberg, Environ. Sci. Technol., 2014, 48, 6743–6753 CrossRef CAS PubMed.
- D. W. Kolpin, E. T. Furlong, M. T. Meyer, E. M. Thurman, S. D. Zaugg, L. B. Barber and H. T. Buxton, Environ. Sci. Technol., 2002, 36, 1202–1211 CrossRef CAS PubMed.
- P. E. Stackelberg, E. T. Furlong, M. T. Meyer, S. D. Zaugg, A. K. Henderson and D. B. Reissman, Sci. Total Environ., 2004, 329, 99–113 CrossRef CAS PubMed.
- S. Mompelat, B. Le Bot and O. Thomas, Environ. Int., 2009, 35, 803–814 CrossRef CAS PubMed.
- M. S. Kostich, A. L. Batt and J. M. Lazorchak, Environ. Pollut., 2014, 184, 354–359 CrossRef CAS PubMed.
- H. Gallard, A. Leclercq and J.-P. Croue, Chemosphere, 2004, 56, 465–473 CrossRef CAS PubMed.
- M. Deborde, S. Rabouan, H. Gallard and B. Legube, Environ. Sci. Technol., 2004, 38, 5577–5583 CrossRef CAS PubMed.
- M. C. Dodd and C.-H. Huang, Environ. Sci. Technol., 2004, 38, 5607–5615 CrossRef CAS PubMed.
- M. C. Dodd, A. D. Shah, U. von Gunten and C.-H. Huang, Environ. Sci. Technol., 2005, 39, 7065–7076 CrossRef CAS PubMed.
- K. L. Rule, V. R. Ebbett and P. J. Vikesland, Environ. Sci. Technol., 2005, 39, 3176–3185 CrossRef CAS PubMed.
- M. C. Dodd and C.-H. Huang, Water Res., 2007, 41, 647–655 CrossRef CAS PubMed.
- P. Wang, Y.-L. He and C.-H. Huang, Water Res., 2011, 45, 1838–1846 CrossRef CAS PubMed.
- P. E. Stackelberg, J. Gibs, E. T. Furlong, M. T. Meyer, S. D. Zaugg and R. L. Lippincott, Sci. Total Environ., 2007, 377, 255–272 CrossRef CAS PubMed.
- S. Kleywegt, V. Pileggi, P. Yang, C. Hao, X. Zhao, C. Rocks, S. Thach, P. Cheung and B. Whitehead, Sci. Total Environ., 2011, 409, 1481–1488 CrossRef CAS PubMed.
- P. Westerhoff, Y. Yoon, S. Snyder and E. Wert, Environ. Sci. Technol., 2005, 39, 6649–6663 CrossRef CAS PubMed.
- S. Mompelat, O. Thomas and B. Le Bot, J. Environ. Monit., 2011, 13, 2929–2939 RSC.
- Y. Valcarcel, S. G. Alonso, J. L. Rodríguez-Gil, A. Gil and M. Catalá, Chemosphere, 2011, 84, 1336–1348 CrossRef CAS PubMed.
- M. J. Benotti, R. A. Trenholm, B. J. Vanderford, J. C. Holady, B. D. Stanford and S. A. Snyder, Environ. Sci. Technol., 2009, 43, 597–603 CrossRef CAS PubMed.
- M. Huerta-Fontela, M. T. Galceran and F. Ventura, Water Res., 2011, 45, 1432–1442 CrossRef CAS PubMed.
- G. A. Loraine and M. E. Pettigrove, Environ. Sci. Technol., 2006, 40, 687–695 CrossRef CAS PubMed.
- X. Jin and S. Peldszus, Sci. Total Environ., 2012, 414, 653–663 CrossRef CAS PubMed.
- L. P. Padhye, H. Yao, F. T. Kung'u and C.-H. Huang, Water Res., 2014, 51, 266–276 CrossRef CAS PubMed.
- O. Legrini, E. Oliveros and A. Braun, Chem. Rev., 1993, 93, 671–698 CrossRef CAS.
- J. J. Pignatello, E. Oliveros and A. MacKay, Crit. Rev. Environ. Sci. Technol., 2006, 36, 1–84 CrossRef CAS.
- M. J. Watts and K. G. Linden, Water Res., 2007, 41, 2871–2878 CrossRef CAS PubMed.
- J. Jin, M. G. El-Din and J. R. Bolton, Water Res., 2011, 45, 1890–1896 CrossRef CAS PubMed.
- D. Wang, J. R. Bolton and R. Hofmann, Water Res., 2012, 46, 4677–4686 CrossRef CAS PubMed.
- C. Sichel, C. Garcia and K. Andre, Water Res., 2011, 45, 6371–6380 CrossRef CAS PubMed.
- S. H. Bossmann, E. Oliveros, S. Gob, S. Siegwart, E. P. Dahlen, L. Payawan, M. Straub, M. Worner and A. M. Braun, J. Phys. Chem. A, 1998, 102, 5542–5550 CrossRef CAS.
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 513–886 CrossRef CAS.
- G. V. Buxton and M. S. Subhani, J. Chem. Soc., Faraday Trans., 1972, 68, 958–969 RSC.
- U. K. Kläning, K. Sehested and T. Wolff, J. Chem. Soc., Faraday Trans., 1984, 80, 2969–2979 RSC.
- R. Vogt and R. N. Schindler, J. Photochem. Photobiol., A, 1992, 66, 133–140 CrossRef CAS.
- M. Nakamura and Y. Ogata, Bull. Chem. Soc. Jpn., 1977, 50, 2396–2399 CrossRef CAS.
- B. G. Oliver and J. H. Carey, Environ. Sci. Technol., 1977, 11, 893–895 CrossRef CAS.
- L. H. Nowell and J. Hoigné, Water Res., 1992, 26, 599–605 CrossRef CAS.
- P. Y. Chan, M. G. El-Din and J. R. Bolton, Water Res., 2012, 46, 5672–5682 CrossRef CAS PubMed.
- J. E. Forsyth, P. Zhou, Q. Mao, S. S. Asato, J. S. Meschke and M. C. Dodd, Environ. Sci. Technol., 2013, 47, 12976–12984 CrossRef CAS PubMed.
- J. Fang, Y. Fu and C. Shang, Environ. Sci. Technol., 2014, 48, 1859–1868 CrossRef CAS PubMed.
- W. Ben, P. Sun and C.-H. Huang, Chemosphere, 2015, 1–8 Search PubMed.
- P. Zhou, G. D. Di Giovanni, J. S. Meschke and M. C. Dodd, Environ. Sci. Technol. Lett., 2014, 1, 453–458 CrossRef CAS.
- A. K. Boal, C. Rhodes and S. Garcia, Groundwater Monit. Rem., 2015, 35, 93–100 CAS.
- W. J. Cooper, A. C. Jones, R. F. Whitehead and R. G. Zika, Environ. Sci. Technol., 2007, 41, 3728–3733 CrossRef CAS PubMed.
- Y. Qian, W. Wang, X.-F. Li and S. E. Hrudey, Water Sci. Technol.: Water Supply, 2015, 15, 84–93 CrossRef CAS.
- L. H. Nowell and J. Hoigné, Water Res., 1992, 26, 593–598 CrossRef CAS.
- A. Z. Gu and J. B. Neethling, Water Environ. Res., 2008, 80, 179–185 CrossRef CAS PubMed.
- A. Spiliotopoulou, K. M. S. Hansen and H. R. Andersen, Sci. Total Environ., 2015, 520, 96–105 CrossRef CAS PubMed.
- X. Zhang, W. Li, E. R. Blatchley, X. Wang and P. Ren, Water Res., 2015, 68, 804–811 CrossRef CAS PubMed.
- Y. Feng, D. W. Smith and J. R. Bolton, J. Environ. Eng. Sci., 2007, 6, 277–284 CrossRef CAS.
- Q. Zhao, C. Shang and X. Zhang, Water Sci. Technol., 2009, 9, 627–634 CAS.
- M. J. Molina, T. Ishiwata and L. T. Molina, J. Phys. Chem., 1980, 84, 821–826 CrossRef CAS.
- W. D. McGrath and J. J. McGarvey, Planet. Space Sci., 1967, 15, 427–455 CrossRef CAS.
- M. Deborde and U. von Gunten, Water Res., 2008, 42, 13–51 CrossRef CAS PubMed.
- R. N. Schindler, M. Liesner, S. Schmidt and U. Kirchner, J. Photochem. Photobiol., A, 1997, 107, 9–19 CrossRef CAS.
- U. K. Kläning and T. Wolff, Ber. Bunsenges. Phys. Chem., 1985, 89, 243–245 CrossRef.
- G. V. Buxton and M. S. Subhani, J. Chem. Soc., Faraday Trans., 1972, 68, 970–977 RSC.
- A. Treinin, Isr. J. Chem., 1970, 8, 103–113 CrossRef CAS.
- G. V. Buxton and M. S. Subhani, J. Chem. Soc., Faraday Trans., 1972, 68, 947–957 RSC.
- G. V. Buxton, M. Bydder, G. A. Salmon and J. E. Williams, Phys. Chem. Chem. Phys., 2000, 2, 237–245 RSC.
- K. Kumar and D. W. Margerum, Inorg. Chem., 1987, 26, 2706–2711 CrossRef CAS.
- R. E. Connick, J. Am. Chem. Soc., 1947, 69, 1509–1514 CrossRef CAS.
- B. M. Matthew and C. Anastasio, Atmos. Chem. Phys., 2006, 6, 2423–2437 CAS.
- Z. Zuo, Y. Katsumura, K. Ueda and K. Ishigure, J. Chem. Soc., Faraday Trans., 1997, 93, 1885–1891 RSC.
- G. G. Jayson, B. J. Parsons and A. J. Swallow, J. Chem. Soc., Faraday Trans., 1973, 69, 1597–1607 RSC.
- G. V. Buxton, M. Bydder and G. Arthur Salmon, J. Chem. Soc., Faraday Trans., 1998, 94, 653–657 RSC.
- B. C. Gilbert, J. K. Stell, W. J. Peet and K. J. Radford, J. Chem. Soc., Faraday Trans., 1988, 84, 3319–3330 RSC.
- V. Nagarajan and R. W. Fessenden, J. Phys. Chem., 1985, 89, 2330–2335 CrossRef CAS.
- K. Hasegawa and P. Neta, J. Phys. Chem., 1978, 82, 854–857 CrossRef CAS.
- A. E. Grigorev, I. E. Makarov and A. K. Pikaev, High Energy Chem., 1987, 21, 99–102 Search PubMed.
- U. von Gunten, Water Res., 2003, 37, 1443–1467 CrossRef CAS PubMed.
- M. Deborde, S. Rabouan, J.-P. Duguet and B. Legube, Environ. Sci. Technol., 2005, 39, 6086–6092 CrossRef CAS PubMed.
- U. von Gunten and J. Hoigné, Environ. Sci. Technol., 1994, 28, 1234–1242 CrossRef CAS PubMed.
- U. von Gunten, Water Res., 2003, 37, 1469–1487 CrossRef CAS PubMed.
- B. A. Lyon, A. D. Dotson, K. G. Linden and H. S. Weinberg, Water Res., 2012, 46, 4653–4664 CrossRef CAS PubMed.
- O. Amichai and A. Treinin, Chem. Phys. Lett., 1969, 3, 611–613 CrossRef CAS.
- A. Fischbacher, K. Löppenberg, C. von Sonntag and T. C. Schmidt, Environ. Sci. Technol., 2015, 49, 11714–11720 CrossRef CAS PubMed.
- G. V. Buxton and F. S. Dainton, Proc. R. Soc. London, Ser. A, 1968, 304, 427–439 CrossRef CAS.
- A. N. Pisarenko, B. D. Stanford, S. A. Snyder, S. B. Rivera and A. K. Boal, J. Adv. Oxid. Technol., 2013, 16, 137–150 CAS.
- L. Ashton, G. V. Buxton and C. R. Stuart, J. Chem. Soc., Faraday Trans., 1995, 91, 1631–1633 RSC.
- D. O. Mártire, J. A. Rosso, S. Bertolotti, G. C. Le Roux, A. M. Braun and M. C. Gonzalez, J. Phys. Chem. A, 2001, 105, 5385–5392 CrossRef.
- O. Horvath, J. Photochem. Photobiol., A, 1989, 48, 243–248 CrossRef CAS.
- R. Mertens and C. von Sonntag, J. Photochem. Photobiol., A, 1995, 85, 1–9 CrossRef CAS.
- P. Neta and L. M. Dorfman, Adv. Chem. Ser., 1968, 81, 222–230 CrossRef.
- Q. Zhao, C. Shang, X. Zhang, G. Ding and X. Yang, Water Res., 2011, 45, 6545–6554 CrossRef CAS PubMed.
- N. Getoff, Radiat. Phys. Chem., 1991, 37, 673–680 CrossRef CAS.
- J. E. Chateauneuf, J. Am. Chem. Soc., 1990, 112, 442–444 CrossRef CAS.
- G. W. Klein, K. Bhatia, V. Madhavan and R. H. Schuler, J. Phys. Chem., 1975, 79, 1767–1774 CrossRef CAS.
- C. R. Keenan and D. L. Sedlak, Environ. Sci. Technol., 2008, 42, 1262–1267 CrossRef CAS PubMed.
- Y. Ogata, T. Suzuki and K. Takagi, J. Chem. Soc., Perkin Trans. 2, 1979, 2, 1715–1719 RSC.
- M. Kimura and Y. Ogata, Bull. Chem. Soc. Jpn., 1983, 56, 471–473 CrossRef CAS.
- M. D. Tucker, L. L. Barton, B. M. Thomson and B. M. Wagener, Waste Manage., 1999, 19, 477–482 CrossRef CAS.
- Z. Shu, C. Li, M. Belosevic, J. R. Bolton and M. G. El-Din, Environ. Sci. Technol., 2014, 48, 9692–9701 CrossRef CAS PubMed.
- Z.-B. Guo, Y.-L. Lin, B. Xu, H. Huang, T.-Y. Zhang, F.-X. Tian and N.-Y. Gao, Chem. Eng. J., 2016, 283, 412–419 CrossRef CAS.
- D. Wang, J. R. Bolton, S. A. Andrews and R. Hofmann, Chemosphere, 2015, 136, 239–244 CrossRef CAS PubMed.
- Y. Yoon, D.-J. Choi and K.-D. Zoh, J. Hazard. Mater., 2015, 285, 453–463 CrossRef PubMed.
- L. Qin, Y.-L. Lin, B. Xu, C.-Y. Hu, F.-X. Tian, T.-Y. Zhang, W.-Q. Zhu, H. Huang and N.-Y. Gao, Water Res., 2014, 65, 271–281 CrossRef CAS PubMed.
- N. Kishimoto and H. Nishimura, Environ. Technol., 2015, 36, 2436–2442 CrossRef CAS PubMed.
- Y. Ogata, K. Takagi and T. Suzuki, J. Chem. Soc., Perkin Trans. 2, 1978, 562–567 RSC.
- M. J. Watts, R. Hofmann and E. J. Rosenfeldt, J. - Am. Water Works Assoc., 2012, 104, 47–48 CrossRef CAS.
- E. Rosenfeldt, A. K. Boal, J. Springer and B. Stanford, J. - Am. Water Works Assoc., 2013, 105, 29–33 CrossRef CAS.
- Z. Wang, Y.-L. Lin, B. Xu, S.-J. Xia, T.-Y. Zhang and N.-Y. Gao, Chem. Eng. J., 2016, 283, 1090–1096 CrossRef CAS.
- D. Wang, J. R. Bolton, S. A. Andrews and R. Hofmann, Sci. Total Environ., 2015, 518–519, 49–57 CrossRef CAS PubMed.
- V. Hancil and J. M. Smith, Ind. Eng. Chem. Process Des. Dev., 1971, 10, 515–523 Search PubMed.
- X. R. Zhang, W. G. Li and P. F. Ren, Adv. Mater. Res., 2013, 807–809, 466–471 Search PubMed.
- D. Gonenc and M. Bekbolet, Water Sci. Technol., 2001, 44, 205–210 CAS.
- R. Lamsal, M. E. Walsh and G. A. Gagnon, Water Res., 2011, 45, 3263–3269 CrossRef CAS PubMed.
- Y. Feng, D. W. Smith and J. R. Bolton, Water Environ. Res., 2010, 82, 328–334 CrossRef CAS PubMed.
- National Primary Drinking Water Regulations, Disinfectants and Disinfection Byproducts Rule, U.S. Environmental Protection Agency, Washington DC, 1998 Search PubMed.
- National Primary Drinking Water Regulations, Stage 2 Disinfectants and Disinfection Byproducts Rule, U.S. Environmental Protection Agency, Washington DC, 2006 Search PubMed.
- M. J. Plewa, E. D. Wagner, D. H. Metz, R. Kashinkunti, K. J. Jamriska and M. Meyer, Environ. Sci. Technol., 2012, 46, 7811–7817 CrossRef CAS PubMed.
- S. Weng, J. Li and E. R. Blatchley, Water Res., 2012, 46, 2674–2682 CrossRef CAS PubMed.
- S. Weng and E. R. Blatchley, Environ. Sci. Technol., 2013, 47, 4269–4276 CrossRef CAS PubMed.
- L. Deng, C.-H. Huang and Y.-L. Wang, Environ. Sci. Technol., 2014, 48, 2697–2705 CrossRef CAS PubMed.
- K. M. S. Hansen, R. Zortea, A. Piketty, S. R. Vega and H. R. Andersen, Sci. Total Environ., 2013, 443, 850–856 CrossRef CAS PubMed.
- N. Fiz, J. L. Usero and J. Casado, Int. J. Chem. Kinet., 1993, 25, 341–351 CrossRef CAS.
- J.-Y. Fang, L. Ling and C. Shang, Water Res., 2013, 47, 1257–1266 CrossRef CAS PubMed.
- N. Dai and W. A. Mitch, Environ. Sci. Technol., 2015, 49, 8878–8886 CrossRef CAS PubMed.
- J. Hoigné and H. Bader, Water Res., 1994, 28, 45–55 CrossRef.
- W. R. Haag and J. Hoigné, Water Res., 1983, 17, 1397–1402 CrossRef CAS.
- U.S. Environmental Protection Agency, Contaminant Candidate List 3, 2009. Retrieved from http://www2.epa.gov/ccl/contaminant-candidate-list-3-ccl-3 (accessed December 1, 2015) Search PubMed.
- N. Kang, T. A. Anderson and W. Andrew Jackson, Anal. Chim. Acta, 2006, 567, 48–56 CrossRef CAS PubMed.
- B. Rao, N. Estrada, S. McGee, J. Mangold, B. Gu and W. A. Jackson, Environ. Sci. Technol., 2012, 46, 11635–11643 CrossRef CAS PubMed.
- N. Kang, T. A. Anderson, B. Rao and W. A. Jackson, Environ. Chem., 2009, 6, 53–59 CrossRef CAS.
- H. Zhang, Y. Zhang, Q. Shi, J. Hu, M. Chu, J. Yu and M. Yang, Environ. Sci. Technol., 2012, 46, 4396–4402 CrossRef CAS PubMed.
- H. Zhang, Y. Zhang, Q. Shi, S. Ren, J. Yu, F. Ji, W. Luo and M. Yang, Water Res., 2012, 46, 5197–5204 CrossRef CAS PubMed.
- H. Zhang, Y. Zhang, Q. Shi, H. Zheng and M. Yang, Environ. Sci. Technol., 2014, 48, 3112–3119 CrossRef CAS PubMed.
- E. E. Lavonen, M. Gonsior, L. J. Tranvik, P. Schmitt-Kopplin and S. J. Köhler, Environ. Sci. Technol., 2013, 47, 2264–2271 CrossRef CAS PubMed.
- M. Gonsior, P. Schmitt-Kopplin, H. Stavklint, S. D. Richardson, N. Hertkorn and D. Bastviken, Environ. Sci. Technol., 2014, 48, 12714–12722 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ew00029k |
| This journal is © The Royal Society of Chemistry 2016 |