
Photoswitchable inhibitors: temporally regulated inhibition of the IDO1 enzyme using photoactive merocyanine derivatives
Niku Moni
Das
a,
Sayantani
Biswas
b,
Suravi
Chauhan
c,
Adyasa
Sahoo
a,
Debdas
Dhabal
 a and
Debasis
Manna
*ab
a and
Debasis
Manna
*ab
aIndian Institute of Technology Guwahati, Chemistry, Guwahati, Assam, India. E-mail: dmanna@iitg.ac.in
bIndian Institute of Technology Guwahati, Biosciences and Bioengineering, Guwahati, Assam, India
cIndian Institute of Technology Guwahati, Center for the Environment, Guwahati, Assam, India
First published on 1st December 2025
Abstract
Developing photoactive molecules represents an emerging strategy for temporally regulated inhibition of the indoleamine 2,3-dioxygenase 1 (IDO1) enzyme. Interestingly, only the photoactive merocyanine isomer of a potent compound effectively binds to both the apo-IDO1 protein and heme cofactor, enabling photoisomer-specific enzyme inhibition. These phototunable molecules hold significant promise for advancing targeted immunotheranostics.
Immunotherapy is an innovative approach to combat cancer and Alzheimer's disease by rejuvenating the immune system to target and eliminate disease-causing cells or pathological markers. In cancer immunotherapy, immune checkpoint inhibitors rejuvenate antitumor immunity by obstructing the ‘off’ signals from immune checkpoint proteins, thereby allowing T cells to target and destroy cancer cells effectively.1–3 In Alzheimer's disease, immunomodulation focuses on neuroinflammatory processes and abnormal protein deposits, such as amyloid-beta and tau, to slow the progression of the disease.2 Cytokine (such as IFN-γ, TNF-α)-mediated overexpression of the indoleamine 2,3-dioxygenase 1 (IDO1) enzyme under pathogenic conditions leads to the uncontrolled metabolism of L-tryptophan (L-Trp), which significantly contributes to immune suppression, tumour growth, neurodegeneration, and other pathological processes.4 Therefore, IDO1 is a promising target for combating cancer, Alzheimer's disease, and other pathological conditions.
In the last decade, various inhibitors, including indoximod, linrodostat, and epacadostat, have been developed to inhibit the hemin-containing IDO1 enzyme selectively.5 However, these conventional IDO1 enzyme inhibitors often cause systemic effects that result in widespread immune suppression, potentially leading to adverse side effects and reduced therapeutic benefits. In addition, conventional IDO1 enzyme inhibitors lack the ability to target IDO1 specifically in tissues where its role is most adverse, such as tumours or areas of inflammation.6 This lack of tissue specificity and the systemic effects might have contributed to the failure of the ECHO-301 Phase III clinical trials, which tested the combination of the IDO1 inhibitor epacadostat with the anti-PD-1 antibody Keytruda for treating metastatic melanoma.6,7 Recently, inhibitors targeting apo-IDO1 have been developed as an alternative strategy to overcome the limitations of conventional IDO1 inhibitors and to achieve more sustained and potent inhibition.8–11 These findings indicate that a targeted and dynamic approach to IDO1 inhibition is necessary to enhance the precision and efficacy of treatment.
The use of photoswitchable compounds presents a promising approach in addressing the challenges of targeted IDO1 inhibition, as it allows for temporal control, potentially enhancing therapeutic benefits while minimizing systemic side effects.12,13 Spiropyrans are known to undergo reversible isomerization when exposed to light, transitioning between the ring-closed spiropyran (SP) and ring-opened merocyanine (MC) form, which makes them useful in applications such as chemical sensors, drug delivery systems, and smart materials.14,15 We hypothesize that the appropriately substituted MC form could selectively bind and inhibit IDO1 in the targeted tissues, only after photoirradiation (Fig. 1A). This strategy could allow us to control the formation of the active inhibitor in a temporal manner and regulate IDO1 activity in the targeted cells and tissues.
 | ||
| Fig. 1 (A) Schematic diagram representing the photoactive MC-3e-mediated inhibition of IDO1 enzyme activity via apo-IDO1 binding. (B) Absorbance spectra demonstrating the photoinduced conversion of SP-3e to MC-3e, both before and after visible light treatment (400 nm). (C) Absorbance spectra of SP-3e, MC-3e, apo-IDO1, and MC-3e-bound apo-IDO1 at different time intervals. | ||
Inspired by the benefits of photopharmacology in drug discovery, we designed and synthesized a series of spiropyran derivatives from N-alkylated 2,3,3-trimethylindolenine derivatives.12,13 The cyclocondensation of N-alkylated 2,3,3-trimethylindolenine with substituted 2-hydroxy benzaldehyde or 2-hydroxy-1-napthaldehyde under basic conditions provided the targeted spiropyran derivatives 2a–2e and 3a-3e (Scheme S1). Spectral studies of 3e demonstrated photo-responsive (λex = 400 nm) isomerization, transitioning from the closed-ring spiropyran to the open-ring merocyanine isomer and its reversal upon photoirradiation (λex = 545 nm) or under dark ambient conditions (Fig. 1B). The spectral studies also revealed the pH-dependent reversible isomerization of 3e (Fig. S1).15
The initial screening of the synthesized compounds using the HPLC-based kynurenine assay revealed that the merocyanine (MC) form of 3e (MC-3e) strongly inhibits the activity of the IDO1 enzyme, with an IC50 value of 53.9 ± 2 nM (Fig. 2A, B, Table 1, and Table S2). The MC-3e also showed stronger IDO1 inhibitory activity under an acidic environment (Fig. S2). In contrast, the spiropyran derivative isomer of 3e (SP-3e) exhibited significantly weaker IDO1 inhibitory activity (IC50 > 10 µM). The other synthesized merocyanine derivatives showed much lower inhibitory activities against IDO1. The potent MC-3e exhibited no significant inhibitory activity against the tryptophan 2,3-dioxygenase (TDO) enzyme, which belongs to the same enzyme family as IDO1 (Table S3).
 | ||
| Fig. 2 Inhibitory activities of IDO1 (A) at different concentrations of MC-3e at pH 6.5, and (B) after photoirradiation (using LED light) of MC-3e with 400 nm light at different time intervals. (C) IDO1 inhibitory activities at different temperatures and time intervals with MC-3e at pH 6.5. (D) The activity of apo-IDO1 in the absence and presence of the compound and/or hemin. | ||
| Compounds | IC50 values (nM) | Inhibition (%) | |
|---|---|---|---|
| 0.25 µM | 1 µM | ||
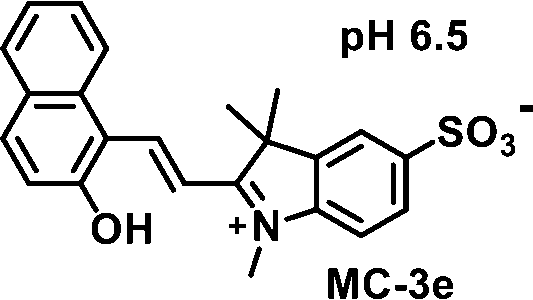
|
54 ± 2 | 83 ± 1.5 | 97 ± 1 |
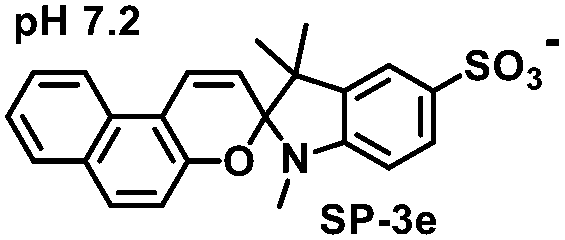
|
>10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
9 ± 1 | 21 ± 9 |
| pH 6.5 | 61 ± 6 | 76 ± 4 | 96 ± 2 |
| Epacadostat | |||
The activity of MC-3e encouraged us to explore its detailed inhibitory mechanism on IDO1. A variation in the pre-incubation time period of MC-3e with IDO1 enzyme at 37 °C demonstrated a significant increase in its inhibitory activity over time, reaching maximum activity (>90%) at 60 minutes (Fig. 2C). The temperature-dependent activity assays showed that the inhibition of IDO1 by MC-3e increased when the temperature was raised from 20 °C to 37 °C. These results indicate that the inhibition process may involve time and temperature-sensitive events, such as the dissociation of the heme cofactor from the IDO1 enzyme (Fig. 2C). The binding of MC-3e to “pocket C”, the dynamic JK-loop region (360–380 AA) of the IDO1 enzyme, could induce heme release. The flexibility and plasticity of this loop are crucial for the activity and substrate selectivity of the enzyme.16,17
To determine whether the loss of the heme-cofactor is a decisive step in the inhibition process of the IDO1 enzyme, we performed UV-Vis spectroscopic analysis both in the presence and absence of MC-3e. The results showed a decrease in the Soret peak in the presence of MC-3e, indicating that heme was released from IDO1.11 Notably, no significant changes were observed in the Soret peaks of TDO or other heme-containing proteins, such as haemoglobin and myoglobin, suggesting that the hemin loss induced by MC-3e is selective for the IDO1 enzyme (Fig. S3). For further investigation of the MC-3e-mediated release of the heme cofactor from the IDO1 enzyme, a protoporphyrin IX (PPIX)-based fluorescence quenching assay was performed. A significant decrease in the PPIX fluorescence upon the addition of MC-3e indicated the interaction between PPIX and hemin. In contrast, no observable change in the PPIX fluorescence signal was detected in the presence of SP-3e (Fig. S4 and S5). Thus, these UV-Vis and fluorescence spectral changes of the IDO1 enzyme and PPIX suggest the potential for MC-3e-mediated release of the heme from IDO1. Additionally, we did not observe any significant changes in the circular dichroism (CD) spectra of IDO1 before and after incubation with MC-3e, indicating that the compound-mediated release of the heme cofactor from the IDO1 active site is not caused by any significant alteration in its structural integrity (Fig. S6 and Table S4). Therefore, removing the heme-cofactor from the IDO1 enzyme and generating apo-IDO1 are crucial steps in the IDO1 inhibitory activity of MC-3e.
A concentration-dependent activity assay was performed to investigate the rate-limiting process of MC-3e-mediated IDO1 inhibition. The activity assay was performed after incubating the IDO1 enzyme with MC-3e for 60 minutes. The IDO1 activity at various concentrations of MC-3e exhibited a non-linear relationship. The IDO1 inhibition assay in the presence of L-Trp and the reducing agents (ascorbate, methylene blue, and catalase) against different concentrations of MC-3e also showed a nonlinear inhibition profile, suggesting that the oxidation state of iron does not influence the MC-3e-mediated IDO1 inhibitory activity in the hemin group. Therefore, releasing the heme cofactor or generating the apo-IDO1 protein is the rate-limiting step of the MC-3e-mediated IDO1 inhibition mechanism. The UV-Vis spectral analysis showed that the extent of reduction of the Soret peak was quite similar for both the catalytically active and inactive IDO1 enzyme, indicating that the release capability of the heme cofactor was nearly the same (Fig. S7). The apo-IDO1 protein equilibrated with external hemin regained its catalytic activity, whereas the apo-IDO1 protein equilibrated with MC-3e failed to show any significant activity even after adding external hemin (Fig. 2D).
Recent studies indicate that free hemin is necessary for cellular homeostasis and plays a dual role in cancer, where its accumulation and metabolic dysregulation promote tumour growth by generating oxidative stress and inflammation. However, hemin deficiency also impairs critical cellular functions, necessitating careful regulation of hemin metabolism. Various spectrometric techniques were employed to investigate whether MC-3e exhibits hemin-binding properties. The concentration-dependent decrease of the Soret peak indicated that MC-3e also interacts with the free heme cofactor. The binding affinities of these MC-3e are similar to quinine, a known hemin-binding molecule (Fig. S8). The time-dependent decrease in PPIX fluorescence in the presence of hemin indicates the interaction of PPIX with hemin. However, the addition of MC-3e, in combination with hemin, significantly alters the degree of PPIX fluorescence quenching induced by hemin, indicating the potential interaction of MC-3e with hemin (Fig. S9). In contrast, the addition of SP-3e, in combination with hemin, did not result in any significant alterations in the PPIX fluorescence quenching caused by hemin. Thus, the PPIX-based fluorescence studies indicate that MC-3e could inhibit the activity of overproduced labile hemin, whereas SP-3e failed to do so. The photo- or pH-induced conversion of SP-3e to MC-3e, as well as the interaction between MC-3e and apo-IDO1, was examined using various spectroscopic methods. The steady-state fluorescence anisotropy measurements of MC-3e showed a significant increase in anisotropy with increasing concentrations of apo-IDO1, resulting in a binding affinity (Kd) of 3.9 ± 0.58 µM (Fig. 3A and Fig. S10A).18 Additionally, the introduction of NaCl led to a concentration-dependent decrease in the steady-state anisotropy value, indicating that the interaction between MC-3e and apo-IDO1 is reversible (Fig. 3A and Fig. S10B). The time-dependent UV-Vis spectral studies of MC-3e in equilibrium with apo-IDO1 were conducted under neutral and dark conditions. The absorbance profiles of MC-3e in the presence of apo-IDO1 exhibited minimal variation over time. This indicates that the MC-3e isomer remains stable within the binding site of apo-IDO1 and does not experience significant photochemical or structural changes during a prolonged incubation period of up to 10 h (Fig. 1C and Fig. S11A, B). However, the control study without apo-IDO1 showed that the merocyanine (MC-3e) spontaneously converted to the spiropyran isomer (SP-3e) under similar experimental conditions (Fig. S11C, D). Hence, these spectral studies suggest a strong and persistent interaction between MC-3e and the apo-IDO1, which is crucial for the MC-3e-mediated inhibitory activity of IDO1.
 | ||
| Fig. 3 (A) Steady-state fluorescence anisotropy measurements of MC-3e (500 nM) in the presence of various concentrations of apo-IDO1 protein (0–12 µM) and in the absence and presence of different concentrations of NaCl (0–75 mM). (B) The variation in RMSD obtained for apo IDO1 (orange) and MC-3e (blue). (C) IDO1 inhibition activity in cells with and without IFN-γ treatment in the presence and absence of the isoforms of 3e. (D) Confocal microscopy image of MC-3e-treated HeLa cells. | ||
To investigate the molecular determinants responsible for the binding of MC-3e to the apo-IDO1, molecular docking and molecular dynamics (MD) simulations were performed. The molecular docking analysis of MC-3e with the X-ray co-crystal structure of the apo-IDO1 protein (PDB ID: 6AZW) revealed that MC-3e is located within the binding pocket of the apo-IDO1 (Fig. S12A).19 The MD simulation studies (100 ns) showed that MC-3e remains strongly bound to the apo-IDO1, with an RMSD of 0.2 nm, while MC-3e stabilized at 0.1 nm (Fig. 3B).
The viability assay of MC-3e showed low toxicity in HeLa and HEK-293 cells (after 48 h), indicating that this compound is suitable for IDO1 inhibitory studies (Fig. S13). The calculated IC50 value for MC-3e using the IFN-γ-treated HeLa cells was found to be 5.8 ± 0.7 nM. The cellular IDO1 activity assays demonstrated a considerable increase in IDO1 activity when treated with external IFN-γ and hemin, suggesting that a notable amount of apo-IDO1 is present in the cellular environment. Under similar experimental conditions, hemin, which was equilibrated with the potent compound, demonstrated a significant decrease in IDO1 activity. This suggests that the complexation of hemin with the MC-3e hinders the ability of apo-IDO1 to rebind hemin (Fig. 3C and Fig. S14). Therefore, compound binding to the apo-IDO1 and the formation of the hemin-MC-3e complex could inhibit the cellular IDO1 activity. The confocal microscopic images revealed distinct red fluorescent puncta in treated HeLa cells, confirming internalization of MC-3e, while untreated control cells showed no fluorescence (Fig. 3D and Fig. S16).
The ability to respond to visible light and changes in pH provides a mechanism for the controlled inhibition of the IDO1 enzyme, offering the prospect of targeted therapeutic intervention with minimized off-target effects. The acidic pH-dependent isomerization of 3e from inactive (SP-3e) to active (MC-3e) IDO1 enzyme inhibitor offers a significant advantage, because a lower extracellular pH typically characterizes the tumour microenvironment compared to healthy tissues.20 The visible light can serve as an external stimulus to convert an inactive inhibitor of the IDO1 enzyme into an active one. This approach also offers significant advantages for biomedical applications, as it has relatively low phototoxicity and allows for deeper tissue penetration compared to ultraviolet light.21 Therefore, the inherent properties of spiropyran derivatives make them a promising scaffold for the development of smart drugs that effectively respond to specific stimuli in a temporally controlled manner, particularly in the field of immunotherapy.
In summary, we synthesized a series of spiropyran derivatives and identified 3e as a potent and stimuli-responsive inhibitor of IDO1. Upon exposure to light, 3e undergoes conversion to the photoactive MC-3e isoform, which exhibits strong IDO1 inhibitory activity in comparison to that of the photoinactive SP-3e isoform. By facilitating the release of heme from holo-IDO1 and transforming it into the apo form, the MC-3e exhibits the ability to bind both apo-IDO1 protein and free hemin. These findings highlight a novel strategy for the temporal regulation of IDO1 activity by utilizing light and pH as external stimuli. Compared to traditional IDO1 inhibitors, this photoresponsive approach offers significant potential for the development of temporally regulated therapies for cancer, Alzheimer's disease, and other pathological conditions, enabling greater specificity and safety.
The authors acknowledge the Science & Engineering Research Board, Government of India (CRG/2021/000306) for financial support, the ICMR Centre for Excellence (5/3/8/20/2019-ITR), MeitY (5(1)/2021-NANO), and INUP for the instrumental facility.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: detailed experimental procedures, compound characterization, ion transport activity data, and catalytic activity data. See DOI: https://doi.org/10.1039/d5cc05796e.References
- A. C. Newman, M. Falcone, A. H. Uribe, T. Zhang, D. Athineos, M. Pietzke, A. Vazquez, K. Blyth and O. D. K. Maddocks, Mol. Cell, 2021, 81, 2290–2302.e2297 CrossRef CAS PubMed.
- P. S. Minhas, J. R. Jones, A. Latif-Hernandez, Y. Sugiura, A. S. Durairaj, Q. Wang, S. D. Mhatre, T. Uenaka, J. Crapser and T. J. S. Conley, Science, 2024, 385, eabm6131 CrossRef CAS PubMed.
- S. Panda, N. Pradhan, S. Chatterjee, S. Morla, A. Saha, A. Roy, S. Kumar, A. Bhattacharyya and D. J. S. R. Manna, Sci. Rep., 2019, 9, 18455 CrossRef CAS PubMed.
- K. Watcharanurak, L. Zang, M. Nishikawa, K. Yoshinaga, Y. Yamamoto, Y. Takahashi, M. Ando, K. Saito, Y. Watanabe and Y. Takakura, Gene Ther., 2014, 21, 794–801 CrossRef CAS PubMed.
- R. M. Al-Zoubi, M. Elaarag, A. R. Al-Qudimat, E. A. Al-Hurani, Z. E. Fares, A. Farhan, S. R. Al-Zoubi, A. Khan, A. Agouni, M. Shkoor, H. Bawadi, Z. Z. Zakaria, M. Al Zoubi and K. Alrumaihi, Front. Pharmacol., 2025, 16, 1632446 CrossRef PubMed.
- K. Tang, Y. H. Wu, Y. H. Song and B. Yu, J. Hematol. Oncol., 2021, 14, 68 CrossRef CAS PubMed.
- S. Rossini, S. Ambrosino, C. Volpi, M. L. Belladonna, M. T. Pallotta, E. Panfili, C. Suvieri, A. Macchiarulo, G. Mondanelli and C. Orabona, Front. Immunol., 2024, 15, 1346686 CrossRef CAS PubMed.
- L. Cen, Y. Wu, M. He, J. Huang, W. Ren, B. Liu, L. Meng, L. Huang, H. Gu and Y. Xu, J. Med. Chem., 2025, 68, 6633–6655 CrossRef CAS PubMed.
- M. T. Nelp, P. A. Kates, J. T. Hunt, J. A. Newitt, A. Balog, D. Maley, X. Zhu, L. Abell, A. Allentoff and R. Borzilleri, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3249–3254 CrossRef CAS PubMed.
- N. M. Das, B. M. Prusty, N. Pradhan, A. Gupta, M. Carmena-Bargueño, R. Karn, H. Pérez-Sánchez, S. Kumar and D. Manna, Eur. J. Med. Chem. Rep., 2023, 9, 100110 CAS.
- N. Pradhan, N. Akhtar, B. Nath, J. Peña-García, A. Gupta, H. Pérez-Sánchez, S. Kumar and D. J. C. C. Manna, Chem. Commun., 2021, 57, 395–398 RSC.
- M. J. Fuchter, J. Med. Chem., 2020, 63, 11436–11447 CrossRef CAS PubMed.
- Y. Liu, T. Wang and W. Wang, Chem. Soc. Rev., 2025, 54, 5792–5835 RSC.
- C. L. Fleming, S. Li, M. Grøtli and J. Andréasson, J. Am. Chem. Soc., 2018, 140, 14069–14072 CrossRef CAS PubMed.
- M. Reifarth, M. Bekir, A. M. Bapolisi, E. Titov, F. Nußhardt, J. Nowaczyk, D. Grigoriev, A. Sharma, P. Saalfrank and S. Santer, Angew. Chem., Int. Ed., 2022, 61, e202114687 CrossRef CAS PubMed.
- K. N. Pham, A. Lewis-Ballester and S. R. Yeh, J. Am. Chem. Soc., 2019, 141, 18771–18779 CrossRef CAS PubMed.
- U. F. Rohrig, O. Michielin and V. Zoete, J. Med. Chem., 2021, 64, 17690–17705 CrossRef CAS PubMed.
- L. F. Cheow, R. Viswanathan, C. S. Chin, N. Jennifer, R. C. Jones, E. Guccione, S. R. Quake and W. F. Burkholder, Anal. Chem., 2014, 86, 9901–9908 CrossRef CAS PubMed.
- N. M. Das, B. M. Prusty, A. Sahoo, P. Mazumder, S. Chauhan, G. Hazarika, S. Kumar, D. Dhabal and D. J. R. M. C. Manna, RSC Med. Chem., 2025, 16, 3240–3250 RSC.
- S. Yamamoto, K. Nishimura, K. Morita, S. Kanemitsu, Y. Nishida, T. Morimoto, T. Aoi, A. Tamura and T. J. B. Maruyama, Biomacromolecules, 2021, 22, 2524–2531 CrossRef CAS PubMed.
- M. Shamsipur, A. Ghavidast and A. Pashabadi, Acta Pharm. Sin. B, 2023, 13, 2844–2876 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |