
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural constraint at a P–P bond: phosphinophosphination of alkenes, alkynes, and carbonyls by a concerted mechanism†
Lijun
You
 ,
Daniel
Roth
and
Lutz
Greb
*
,
Daniel
Roth
and
Lutz
Greb
*
Anorganisch-Chemisches Institut Ruprecht-Karls-Universität Heidelberg, Im Neuenheimer Feld 270, 69120 Heidelberg, Germany. E-mail: greb@uni-heidelberg.de
First published on 5th November 2024
Abstract
Structurally constraining p-block elements has become a powerful strategy for bond activation chemistry with main group compounds. Traditionally, this approach focuses on mononuclear centers, yet applying structural constraints to systems with element–element bonds remains underexplored. In this study, we introduce a cation featuring a structural constraint-elongated P–P bond that spontaneously adds to unactivated alkynes, alkenes, aldehydes, and ketones. Despite its positive charge, the surprisingly apolar P–P+ bond promotes phosphinophosphination via a concerted, highly regio- and diastereoselective mechanism. This unique reactivity opens pathways to novel seven-membered phosphorus heterocycles with customizable optical properties and a structurally varied array of ligands for transition metal coordination.
Introduction
Double functionalization of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C or CO groups by the addition of element–element bonds, e.g., diboration, disilylation or diphosphination, is a straightforward tool for expanding molecular functionality and complexity.1–3 In particular, adding two phosphorus groups across alkenes and alkynes provides manifold products of interest for catalysis, materials science, pharmaceuticals and biological research.4,5 Usually, transition metal-, Lewis acid-, or light-induced reactivity via phosphinyl radicals is required for P–P to C–C multiple bond additions.6–9 While spontaneous diphosphination has been described with a polarized P–P bonded system A, this reactivity is limited to electron-deficient alkenes and alkynes (Scheme 1a).10–14 More recently, phosphino-phosphenium cations B were shown to undergo FLP-type addition to alkynes, but limited to terminal electron-rich C–C triple bonds and diazo compounds (Scheme 1a).15,16 In this context, P–P bond hydrogenation and its reverse phosphine dehydrocoupling in a FLP manner are to be mentioned.17,18 The addition of P–P bonds across carbonyls leading to P–O–C–P motifs has been reported, but only for the electron-deficient bisphosphine (CF3)2P–P(CF3)2 under Lewis base catalysis.19
C or CO groups by the addition of element–element bonds, e.g., diboration, disilylation or diphosphination, is a straightforward tool for expanding molecular functionality and complexity.1–3 In particular, adding two phosphorus groups across alkenes and alkynes provides manifold products of interest for catalysis, materials science, pharmaceuticals and biological research.4,5 Usually, transition metal-, Lewis acid-, or light-induced reactivity via phosphinyl radicals is required for P–P to C–C multiple bond additions.6–9 While spontaneous diphosphination has been described with a polarized P–P bonded system A, this reactivity is limited to electron-deficient alkenes and alkynes (Scheme 1a).10–14 More recently, phosphino-phosphenium cations B were shown to undergo FLP-type addition to alkynes, but limited to terminal electron-rich C–C triple bonds and diazo compounds (Scheme 1a).15,16 In this context, P–P bond hydrogenation and its reverse phosphine dehydrocoupling in a FLP manner are to be mentioned.17,18 The addition of P–P bonds across carbonyls leading to P–O–C–P motifs has been reported, but only for the electron-deficient bisphosphine (CF3)2P–P(CF3)2 under Lewis base catalysis.19
 | ||
| Scheme 1 (a) Phosphinophosphination of electron deficient (with A) or electron-rich terminal alkynes and diazo compounds (with B). (b) Structurally constrained phosphenium ions applied for E–H, C–H, and H–H bond activation (E = N, O, Si, B). (c) This work: phosphinophosphination of unsaturated compounds by a concerted mechanism. | ||
Inspired by the continuing interest in structurally constrained compounds,20–23 we reasoned whether applying this approach to a P–P bond might facilitate the extension of corresponding diphosphination reactivities. Structurally constrained phosphenium ions have been utilised for the activation of E–H,24,25 C–H,26 and H–H bonds27 (Scheme 1b), while reactions with π-bonds remained underexplored. Moreover, neutral diazadiphosphapentalene featuring a constrained P–P bond was explored for NH3 activation via σ-bond metathesis.28
Here, we describe the phosphanylphosphenium ion 2+ and its reactivity towards non-activated alkynes, alkenes, aldehydes, and ketones (Scheme 1c). Comparisons and computations reveal that a surprisingly apolar P–P+ bond in 2+ leverages high selectivity by a concerted elementary step, while only the structural constraint unlocks the thermodynamic and kinetic feasibility.
Results and discussion
We initiated our work by synthesizing the phosphine-appended phosphenium salts 2a/b (Fig. 1). The required diphenylphosphanylaminophenol (PNOH2) ligand was obtained at a multi-gram scale according to the reported procedure (Fig. 1a).29–31 Treatment of a solution of PNOH2 in toluene with PCl3 furnished the phosphorus chloride 1 in 88% yield (Fig. 1a). Analysis by 31P NMR spectroscopy showed two doublets at 156.4 and −15.8 ppm with equal coupling constants of 4JPP = 61 Hz. Single crystals suitable for X-ray diffraction grown from a concentrated solution in dichloromethane at −40 °C confirmed the installation of a P–Cl unit at the amidophenolate (Fig. 1b). The N-phenyl group is nearly perpendicular to the NPO plane with no direct P–P interaction (see the ESI† for more scXRD information). Chloride abstraction using Na[B(C6F5)4] or Li[Al(OC(CF3)3)4] gave respective phosphinophosphenium salts 2a/b in excellent yields (Fig. 1a). An increased coupling constant of 1JPP = 405 Hz in the corresponding 31P NMR spectra indicated the formation of a P–P bond. Single crystals of [2][Al(OC(CF3)3)4] (2b) analyzed by X-ray diffraction revealed a structure where the cation adopts C1 symmetry in the solid state (Fig. 1b). The distance between the two phosphorus atoms of 2.3437(15) Å is substantially longer compared to untethered phosphinophosphenium ions (e.g. 2.230 Å in [Ph3P–PPh2]+)32 and similar to that of the polarized systems A (2.334 Å).10 While the ∠O1–P1–N1 (94.2°) and the ∠O1–P1–P2 angle (104.5°) are less distorted, the acute ∠N1–P1–P2 angle of 87.39° shows significant deviations compared to unconstrained derivatives (see ESI, Section 9.1†).32 This deformation encompassing the P–P unit in 2+ indicated potential effects on its reactivity, which were investigated next. | ||
| Fig. 1 (a) Synthesis of phosphenium salts [2][WCA] with 31P NMR data (WCA = weakly coordinating anion). 2a: [2][B(C6F5)4], 97% yield; 2b: [2][AlO(C(CF3)3)4], 82% yield. (b) ORTEP plots of the solid-state structures of 1 and 2+ in 2b. Thermal ellipsoid plots are at 50% probability. The counterion [Al(OC(CF3)3)4]− and all hydrogen atoms have been omitted for clarity. Selected bond distances (Å) and angles (deg) of 1: d(P1–O1) = 1.6305(2), d(P1–N1) = 1.6837(3), and ∠O1–P1–N1 = 92.14(57) (the structure of 1 contains two crystallographically independent molecules and the mean values with standard deviation are given); 2+: d(P1–O1) = 1.6316(15), d(P1–N1) = 1.7318(17), d(P1–P2) = 2.3437(15), ∠O1–P1–N1 = 94.18(7), ∠O1–P1–P2 = 104.53(5), and ∠N1–P1–P2 = 87.39(5). | ||
Indeed, swift addition reactions towards broad scope of unsaturated functional groups were observed with high selectivity. Upon exposure of 2a to phenylacetylene, a new species with two doublets in the 31P NMR spectra at 145.5 and 24.5 ppm formed selectively (Scheme 2). Combined multinuclear NMR data were consistent with the phosphinophosphination product 3a, as was further confirmed by scXRD (Fig. 3). Notably, 3a represents only one out of four possible diastereomers (see Section 3.1 in the ESI†). Similarly, the reaction with internal alkynes, such as 1-phenyl-1-propyne or tolane afforded the products 3b/c as orange solids in good isolated yields (Scheme 2). This behavior contrasts that of the reported structurally unconstrained A or B (Scheme 1a), which added only to either electron-poor or electron-rich activated terminal alkynes,10–13,15 and encouraged the investigation of more challenging substrates.
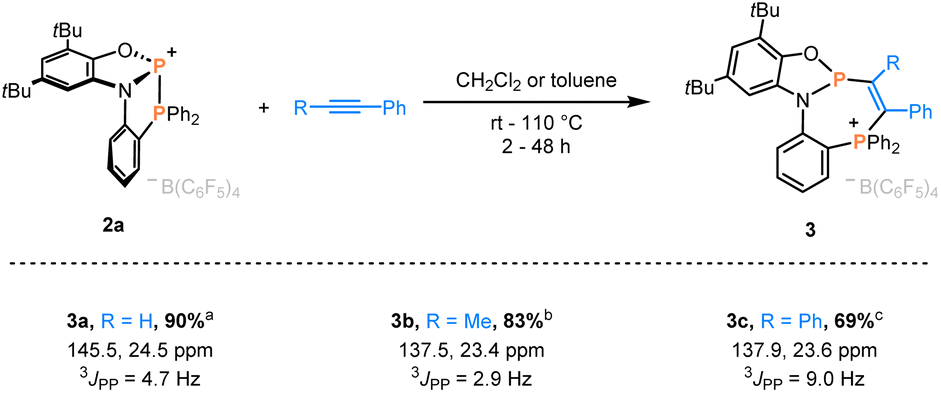 | ||
| Scheme 2 Phosphinophosphination of alkynes affording 3a–c. Isolated yields and 31P NMR data are given. Reaction conditions: a CH2Cl2, rt, 2 h. b CH2Cl2, 60 °C, 18 h. c toluene, 110 °C, 48 h. | ||
On adding styrene to the solution of 2a in CH2Cl2, multinuclear NMR data revealed two diastereomeric products in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. In the 31P NMR spectra, the two doublets of the major product appeared at 160.4 and 29.0 ppm (3JPP = 14.1 Hz), while the minor one showed two doublets at 153.7 and 32.4 ppm (3JPP = 7.2 Hz). Single crystals suitable for X-ray diffraction confirmed the major diastereomer as the racemate of (Sa,S)-4a and (Ra,R)-4a (Scheme 3), while the minor diastereomer corresponded to the racemic mixture of (Sa,R)-4a and (Ra,S)-4a. Increasing the steric demand of the alkene substituent (R1 = tBu) improved diastereoselectivity, and formation of a single racemate ((Ra,R)-4b)/((Sa,S)-4b) was observed (Scheme 3), as confirmed by scXRD (Fig. 3). The larger distance between two phosphorus atoms (d = 3.813 Å) or an unfavorable conformation of 4b could explain the absence of PP coupling in the 31P NMR. Next, the internal alkenes cis-stilbene and trans-stilbene were examined. The reaction of 2a with cis-stilbene at 60 °C for 48 h afforded a single diastereomer with two doublets at 154.0 and 26.4 ppm (3JPP = 4.3 Hz). Single crystal XRD confirmed the structure as the racemate (Sa,R,S)-4c and (Ra,S,R)-4c. Reacting 2a with trans-stilbene under analogous conditions provided two products in a 4:1 ratio, identified as the racemates (Sa,R,R)-4c′/(Ra,S,S)-4c′ (major) and (Sa,S,S)-4c′/(Ra,R,R)-4c′ (minor, see Section 4.1 in the ESI†). Hence, full stereoconservation of cis- and trans-configurations occurred, indicating the absence of long-lived rotatable intermediates but a concerted mechanism. Aliphatic internal alkenes cyclohexene and norbornene provided the phosphinophosphination products 4d/e in good yields with high diastereoselectivities (Scheme 3). To the best of our knowledge, this represents the first spontaneous diphosphination of non-activated CC double bonds. Surprisingly, with the more activated 1,1-diphenylethylene, no reaction occurred at room temperature or 60 °C in CH2Cl2 solution, showcasing a behavior generally different from that of conventional P-cationic Lewis acids that exhibit high local positive charge at phosphorus.33–35
:1 ratio. In the 31P NMR spectra, the two doublets of the major product appeared at 160.4 and 29.0 ppm (3JPP = 14.1 Hz), while the minor one showed two doublets at 153.7 and 32.4 ppm (3JPP = 7.2 Hz). Single crystals suitable for X-ray diffraction confirmed the major diastereomer as the racemate of (Sa,S)-4a and (Ra,R)-4a (Scheme 3), while the minor diastereomer corresponded to the racemic mixture of (Sa,R)-4a and (Ra,S)-4a. Increasing the steric demand of the alkene substituent (R1 = tBu) improved diastereoselectivity, and formation of a single racemate ((Ra,R)-4b)/((Sa,S)-4b) was observed (Scheme 3), as confirmed by scXRD (Fig. 3). The larger distance between two phosphorus atoms (d = 3.813 Å) or an unfavorable conformation of 4b could explain the absence of PP coupling in the 31P NMR. Next, the internal alkenes cis-stilbene and trans-stilbene were examined. The reaction of 2a with cis-stilbene at 60 °C for 48 h afforded a single diastereomer with two doublets at 154.0 and 26.4 ppm (3JPP = 4.3 Hz). Single crystal XRD confirmed the structure as the racemate (Sa,R,S)-4c and (Ra,S,R)-4c. Reacting 2a with trans-stilbene under analogous conditions provided two products in a 4:1 ratio, identified as the racemates (Sa,R,R)-4c′/(Ra,S,S)-4c′ (major) and (Sa,S,S)-4c′/(Ra,R,R)-4c′ (minor, see Section 4.1 in the ESI†). Hence, full stereoconservation of cis- and trans-configurations occurred, indicating the absence of long-lived rotatable intermediates but a concerted mechanism. Aliphatic internal alkenes cyclohexene and norbornene provided the phosphinophosphination products 4d/e in good yields with high diastereoselectivities (Scheme 3). To the best of our knowledge, this represents the first spontaneous diphosphination of non-activated CC double bonds. Surprisingly, with the more activated 1,1-diphenylethylene, no reaction occurred at room temperature or 60 °C in CH2Cl2 solution, showcasing a behavior generally different from that of conventional P-cationic Lewis acids that exhibit high local positive charge at phosphorus.33–35
 | ||
| Scheme 3 Phosphinophosphination of alkenes affording 4a–e. Isolated yields, a NMR conversion and 31P NMR data are given. The dr values are determined from the reaction mixtures by 1H NMR spectroscopy. | ||
The scope was expanded further to carbonyl compounds. Benzaldehyde and phosphenium salt 2a reacted to form two diastereomeric addition products after 5 hours. The major diastereomer appeared as two doublets at 126.0 and 30.3 ppm (3JPP = 5.3 Hz), while the minor diastereomer was observed as two singlets at 116.4 and 29.3 ppm in the 31P NMR spectra (Section S5.1 in the ESI†). Interestingly, only the diastereomer with two singlets remained after storing the sample for a week (Fig. 2), indicating the conversion to a thermodynamic product (vide infra for the mechanism). Single crystals analyzed by scXRD revealed the regioisomer with the oxygen binding to the central P(III) (Fig. 2c). Acetophenone was also reactive towards 2a, giving two species in a diastereomer ratio of 4:1. The solid-state structure of the major diastereomer was determined through scXRD (see the ESI†). Finally, complete conversion of acetone to a single product occurred at room temperature, described by two doublets at 127.0 and 34.6 ppm and a coupling constant of 3JPP = 11.3 Hz in the 31P NMR spectra.
 | ||
| Fig. 2 Phosphinophosphination of (a) benzaldehyde and (b) ketones. Isolated yields and 31P NMR data are given. The dr values are determined from the reaction mixtures by 1H NMR spectroscopy. (c) ORTEP plots of the solid-state structure of 5. Thermal ellipsoid plots are set to 50% probability. The counteranion [B(C6F5)4]− and all hydrogen atoms have been omitted for clarity. Selected bond distances (Å) and angles (deg) of 5: d(P1–O1) = 1.6496(11), d(P1–N1) = 1.7228(13), d(P1–O2) = 1.6442(11), d(C25–O2) = 1.4271(17), d(P2–C25) = 1.8643(14), d(P1–P2) = 3.674, ∠O1–P1–N1 = 90.22(6), ∠O1–P1–O2 = 104.22(5), and ∠N1–P1–O2 = 100.76(5). | ||
 | ||
| Fig. 3 Thermal ellipsoid plots of 3a, 4b, and 4c at 50% probability. The counteranion [B(C6F5)4]− and all hydrogen atoms have been omitted for clarity. Selected bond distances (Å) and angles (deg) of 3a: d(P2–O1) = 1.650(2), d(P2–N1) = 1.717(2), d(P2–C21) = 1.818(3), d(C21–C22) = 1.340(4), d(P1–C22) = 1.832(3), d(P1–P2) = 3.462, ∠O1–P2–N1 = 93.09(11), ∠O1–P2–C21 = 103.61(13), and ∠N1–P2–C21 = 99.06(13); 4b: d(P1–O1) = 1.666(2), d(P1–N1) = 1.725(3), d(P1–C26) = 1.831(3), d(C25–C26) = 1.538(4), d(P2–C25) = 1.852(3), d(P1–P2) = 3.813, ∠O1–P1–N1 = 91.29(12), ∠O1–P1–C26 = 104.21(13), and ∠N1–P1–C26 = 98.48(13); 4c: d(P1–O1) = 1.6436(15), d(P1–N1) = 1.7385(16), d(P1–C26) = 1.8652(19), d(C25–C26) = 1.562(2), d(P2–C25) = 1.8426(18), d(P1–P2) = 3.447, ∠O1–P1–N1 = 92.51(7), ∠O1–P1–C26 = 101.54(7), and ∠N1–P1–C26 = 96.42(8). | ||
The origins of the high regio- and diastereoselectivity were investigated using DFT calculations at the PW6B95-D4/def2-QZVPP(SMD: CH2Cl2)//r2-SCAN-3c(CPCM: CH2Cl2) level of theory (for details, see the ESI†).36–40 For the reaction with phenylacetylene, the computational results suggested that insertion into the P–P bond occurs in a concerted step (Fig. 4). In the lowest transition state, the phosphenium is close to planar, taking full account of its increased electrophilicity.21,35 The formation of a phosphirenium ion (IM1), the proposed intermediate for reactions of B (Scheme 1a),15 was ruled out due to a high computed barrier (30.2 kcal mol−1).
 | ||
| Fig. 4 Computed mechanism at the PW6B95-D4/def2-QZVPP(SMD: CH2Cl2)//r2-SCAN-3c(CPCM: CH2Cl2) level of theory for the addition of phenylacetylene to 2a (black), as well as other higher energy pathways (grey) and transition state structures. | ||
Regioisomer 3a was identified as the kinetic product (22.3 kcal mol−1), while the thermodynamic product 3a′ is disfavored by a higher barrier (26.2 kcal mol−1). The analogous reaction with tolane was predicted to proceed through a rate-determining barrier of 26.8 kcal mol−1, consistent with the experimentally required higher temperature to drive the reaction (Section S10.1.1. in the ESI†). The reaction of styrene with 2a was similarly found to proceed via a concerted addition over a barrier of 20.0 kcal mol−1 (Section S10.1.2.†). For benzaldehyde, the intermediate formation of a phosphaoxirane could also be excluded by prohibitively high reaction barriers, but a lower transition state energy of 16.8 kcal mol−1 was obtained for the concerted addition to the initially observed kinetic product, in agreement with experimental observations.
The thermodynamic product is separated by a barrier of 18.6 kcal mol−1. The experimentally observed interconversion to this more stable diastereomer proceeds either by direct inversion at phosphorus (ΔG‡ = 22.8 kcal mol−1) or by return of the kinetic product back to the reactants (ΔG‡ = 23.7 kcal mol−1). Hence, the observed selectivity relies on the concerted mechanism, which differs from the proposed stepwise process via carbanions during the addition of nucleophilic polar P–P bonds in A to electron-deficient alkenes,13 or via phosphirenium ion intermediates in the case of B and electron-rich alkynes.15
The aptitude for such reactivity could be rationalized by NBO analysis. A surprisingly apolar P–P bond in 2+ (NPA charge at P(N,O) = 1.12 and PPh2 = 1.22) contrasts the far more polar situation in neutral A (charge at P(N,N) = 1.05 and PPh2 = 0.38). This suggests that these apolar features in the P–P bond of 2+ favor the concerted mechanism. At the same time, while other apolar P–P bonds might be reluctant for such reactivity with non-activated substrates, we suggest that feasibility becomes unlocked by the structural constraint imposed by the annulated five-membered rings.
To showcase the value of this new accessible family of phosphorus compounds, their application and properties were considered. Product 5 was treated with [Rh(cod)Cl]2, leading to quantitative formation of complex 7 with 31P NMR resonances at 120.7 (1JRhP = 275.9 Hz) and 27.2 ppm (Fig. 5a). The buried volumes of phosphines 3–6 are spanning a remarkable range (see ESI, Section S7.1†), emphasizing the diverse steric profiles of these easily modificable products. Phosphoramidites are a class of privileged ligands in asymmetric rhodium catalysis,41 qualifying the above-described protocol as a powerful gateway to a library of cationically charged derivatives.
 | ||
| Fig. 5 (a) Coordination chemistry of product 5 with [Rh(cod)Cl]2. 31P NMR data are given. (b) The absorption spectrometry of 2a, 3a, 4a, and 5 in CH2Cl2 (c = 1.0 × 10−4 mol L−1). | ||
Furthermore, we examined the optical properties of phosphenium salt 2a and addition products 3a, 4a, and 5 (Fig. 5b). Gradually shifted absorption wavelength maxima were consistent with the emerging color of corresponding products. In line with the extended π-conjugation in the 7-membered ring in 3a, the absorption is extending into the visible region. This represents an attractive approach for the systematic photophysical evaluation of phosphaheterocylces, which are a topical class of compounds.42–44
Conclusions
Herein, we disclose that structurally constraining a P–P+ unit enables the phosphinophosphination of unsaturated compounds, including alkynes, olefins, and carbonyls. To our knowledge, such spontaneous reactivity towards non-activated C–C multiple bonds has not been reported previously. High regio- and diastereoselectivity originates from a concerted elementary step, contrasting previous addition reactions that occur via stepwise mechanisms and epimerizable intermediates.13,15,45,46 The combinatorial nature allows for the construction of cationic phosphoramidite ligands and P-heterocyclic chromophores. More generally, this study highlights the opportunities of imposing structural constraints on less polar structural motifs and encourages expanding this concept toward other element–element bonds.Data availability
Crystallographic data have been deposited at the CCDC: 2357323–2357331. Further data supporting this study are available in the ESI.†Author contributions
L. Y. and L. G. devised the project and designed the experiments. L. Y. performed the experimental work. D. R. carried out the quantum chemical simulations. L. Y. and D. R. wrote the initial manuscript. All authors contributed to the finalization of the manuscript, and all listed authors agreed to the submitted content.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Dr Manuel Schmitt for help in refinement of scXRD structures. L. Y. is grateful for a Ph.D. fellowship from the China Scholarship Council (CSC) and to M.Sc Christoph Bendel and M.Sc Nils Ansmann for fruitful discussions. We acknowledge support by the state of Baden-Württemberg through bwHPC and the German Research Foundation (DFG) through grant no INST 40/575-1 FUGG (JUSTUS 2 cluster) and GR5007/2-1.Notes and references
- I. Beletskaya and C. Moberg, Chem. Rev., 1999, 99, 3435–3462 Search PubMed.
- I. Beletskaya and C. Moberg, Chem. Rev., 2006, 106, 2320–2354 Search PubMed.
- R. Hua, in Addition Reactions with Unsaturated Hydrocarbons, 2022, ch. 5, pp. 147–182 Search PubMed.
- K. Hirano and M. Miura, Tetrahedron Lett., 2017, 58, 4317–4322 CrossRef CAS.
- E. Gorbachuk, T. Grell, E. Hey-Hawkins and D. Yakhvarov, Eur. J. Inorg. Chem., 2024, 27, e202300751 CrossRef CAS.
- A. Sato, H. Yorimitsu and K. Oshima, Angew. Chem., Int. Ed., 2005, 44, 1694–1696 CrossRef CAS PubMed.
- S.-i. Kawaguchi, S. Nagata, T. Shirai, K. Tsuchii, A. Nomoto and A. Ogawa, Tetrahedron Lett., 2006, 47, 3919–3922 CrossRef CAS.
- Y. Okugawa, K. Hirano and M. Miura, Angew. Chem., Int. Ed., 2016, 55, 13558–13561 CrossRef CAS PubMed.
- Y. Sato, S.-i. Kawaguchi, A. Nomoto and A. Ogawa, Angew. Chem., Int. Ed., 2016, 55, 9700–9703 CrossRef CAS PubMed.
- S. Burck, D. Gudat and M. Nieger, Angew. Chem., Int. Ed., 2004, 43, 4801–4804 CrossRef CAS.
- D. L. Dodds, M. F. Haddow, A. G. Orpen, P. G. Pringle and G. Woodward, Organometallics, 2006, 25, 5937–5945 CrossRef CAS.
- S. Burck, K. Götz, M. Kaupp, M. Nieger, J. Weber, J. Schmedt auf der Günne and D. Gudat, J. Am. Chem. Soc., 2009, 131, 10763–10774 CrossRef CAS.
- S. Burck, I. Hajdók, M. Nieger, D. Bubrin, S. Schulze, D. Gudat and D. Gudat, Organometallics, 2009, 64, 63–72 CAS.
- I. Hajdók, F. Lissner, M. Nieger, S. Strobel and D. Gudat, Organometallics, 2009, 28, 1644–1651 CrossRef.
- H. Kim, Z.-w. Qu, S. Grimme, N. Al-Zuhaika and D. W. Stephan, Angew. Chem., Int. Ed., 2023, 62, e202312587 CrossRef CAS PubMed.
- H. Kim, A. Lough, Z.-w. Qu, S. Grimme and D. W. Stephan, Chem. Commun., 2024, 60, 1031–1034 RSC.
- S. J. Geier and D. W. Stephan, Chem. Commun., 2010, 46, 1026–1028 RSC.
- R. Dobrovetsky, K. Takeuchi and D. W. Stephan, Chem. Commun., 2015, 51, 2396–2398 Search PubMed.
- B. Hoge, C. Thösen and I. Pantenburg, Chem.–Eur. J., 2006, 12, 9019–9024 CrossRef CAS PubMed.
- A. Brand and W. Uhl, Chem.–Eur. J., 2019, 25, 1391–1404 CrossRef CAS.
- J. Abbenseth and J. M. Goicoechea, Chem. Sci., 2020, 11, 9728–9740 RSC.
- A. Maiti, R. Yadav and L. Greb, in Advances in Inorganic Chemistry, ed. K. Meyer and R. van Eldik, Academic Press, 2023, vol. 82, pp. 261–299 Search PubMed.
- T. J. Hannah and S. S. Chitnis, Chem. Soc. Rev., 2024, 53, 764–792 Search PubMed.
- S. Volodarsky and R. Dobrovetsky, Chem. Commun., 2018, 54, 6931–6934 Search PubMed.
- S. Volodarsky, D. Bawari and R. Dobrovetsky, Angew. Chem., Int. Ed., 2022, 61, e202208401 CrossRef CAS.
- D. Roth, A. T. Radosevich and L. Greb, J. Am. Chem. Soc., 2023, 145, 24184–24190 CrossRef CAS PubMed.
- D. Bawari, D. Toami, K. Jaiswal and R. Dobrovetsky, Nat. Chem., 2024, 16, 1261–1266 CrossRef CAS.
- J. Cui, Y. Li, R. Ganguly, A. Inthirarajah, H. Hirao and R. Kinjo, J. Am. Chem. Soc., 2014, 136, 16764–16767 CrossRef CAS.
- D. L. J. Broere, L. L. Metz, B. de Bruin, J. N. H. Reek, M. A. Siegler and J. I. van der Vlugt, Angew. Chem., Int. Ed., 2015, 54, 1516–1520 Search PubMed.
- D. L. J. Broere, D. K. Modder, E. Blokker, M. A. Siegler and J. I. van der Vlugt, Angew. Chem., Int. Ed., 2016, 55, 2406–2410 Search PubMed.
- N. Ansmann, J. Münch, M. Schorpp and L. Greb, Angew. Chem., Int. Ed., 2023, 62, e202313636 CrossRef CAS PubMed.
- N. Burford, T. S. Cameron, P. J. Ragogna, E. Ocando-Mavarez, M. Gee, R. McDonald and R. E. Wasylishen, J. Am. Chem. Soc., 2001, 123, 7947–7948 Search PubMed.
- M. H. Holthausen, R. R. Hiranandani and D. W. Stephan, Chem. Sci., 2015, 6, 2016–2021 RSC.
- J. M. Bayne and D. W. Stephan, Chem. Soc. Rev., 2016, 45, 765–774 RSC.
- D. Roth, J. Stirn, D. W. Stephan and L. Greb, J. Am. Chem. Soc., 2021, 143, 15845–15851 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2005, 109, 5656–5667 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- E. Caldeweyher, S. Ehlert, A. Hansen, H. Neugebauer, S. Spicher, C. Bannwarth and S. Grimme, J. Chem. Phys., 2019, 150, 154122 CrossRef PubMed.
- S. Grimme, A. Hansen, S. Ehlert and J.-M. Mewes, J. Chem. Phys., 2021, 154, 064103 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- J. F. Teichert and B. L. Feringa, Angew. Chem., Int. Ed., 2010, 49, 2486–2528 CrossRef CAS PubMed.
- T. Baumgartner and R. Réau, Chem. Rev., 2006, 106, 4681–4727 CrossRef CAS.
- P. Hindenberg and C. Romero-Nieto, Synlett, 2016, 27, 2293–2300 CrossRef CAS.
- M. A. Shameem and A. Orthaber, Chem.–Eur. J., 2016, 22, 10718–10735 CrossRef CAS PubMed.
- S. S. Chitnis, R. A. Musgrave, H. A. Sparkes, N. E. Pridmore, V. T. Annibale and I. Manners, Inorg. Chem., 2017, 56, 4521–4537 CrossRef CAS.
- S. S. Chitnis, H. A. Sparkes, V. T. Annibale, N. E. Pridmore, A. M. Oliver and I. Manners, Angew. Chem., Int. Ed., 2017, 56, 9536–9540 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data including NMR spectra. CCDC 2357323–2357331. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc06581f |
| This journal is © The Royal Society of Chemistry 2025 |