DOI:
10.1039/D5CC00655D
(Feature Article)
Chem. Commun., 2025,
61, 5563-5576
Recent strategy for the synthesis of indole and indoline skeletons in natural products
Received
6th February 2025
, Accepted 17th March 2025
First published on 18th March 2025
Abstract
Indole alkaloids are one of the most important classes of natural products found in nature, particularly in a wide variety of plants. These compounds have compact polycyclic systems with at least one nitrogen atom. Several of these alkaloids are bioactive and have raised hopes for the development of new drugs. Their biosynthesis involves tryptophan as an amino acid precursor, since the indole or indoline moiety is the main heterocycle of these natural products. However, in their quest to synthesize such complex architectures, chemists have developed several different strategies to produce this key heterocycle quickly and in an unnatural way. This review focuses on the recent total synthesis methods used to prepare the indole and indoline core of these important alkaloids. Novel and older methods that allow the rapid formation of this heterocycle are described as key steps in the total synthesis of these fascinating structures designed by Mother Nature.
Carl Bowman
| Carl Bowman was born in Montreal. Carl will obtain his BSc in Chemistry in May 2025 from l’Université du Québec à Montréal (UQAM), graduating with honors. In the summer of 2023, he received an NSERC-USRA, which allowed him to be a part-time researcher in Professor Nazemi's group studying mesoionic N-heterocyclic carbenes (mNHCs). In the summer of 2024, as part of his second NSERC-USRA, Carl was a part-time researcher in the research group of Professor Canesi of UQAM, studying the oxidative dearomatization of phenols. In the summer of 2025, Carl will be doing his master's in the Canesi group. |

Maxime Denis
| Maxime Denis has obtained his bachelor's degree in chemistry from the Université de Bordeaux in France in 2019. He then continued his studies by obtaining his master's degree in total synthesis in 2022 under the supervision of Prof. S. Canesi at Université du Québec à Montréal (UQAM), He worked on advanced synthesis of Strychnos intermediates and total synthesis of alkaloids belonging to the lycoricidine family. Following his master's degree, he continued his research as a PhD student. His current research is directed toward the use of hypervalent iodine reagent in the asymmetric total synthesis of gracilamine. |

Sylvain Canesi
| Sylvain Canesi was born in Corsica and obtained his BSc level in chemistry at the University of Corsica. Subsequently, he moved to Lyon to obtain an advanced degree from the “Ecole Superieure de Chimie, Physique et Electronique de Lyon” and went on to become a graduate student in the group of Professor Ciufolini. There, he did the synthesis of (−)-Cylindricine and obtained his PhD degree. After postdoctoral studies in the group of Professor Deslongchamps in Sherbrooke, where he worked on the synthesis of a paclitaxel macrocyclic model, he accepted, in 2006, a faculty position at Université du Québec à Montréal. |
1. Introduction
Alkaloids are one of the classes of natural products that have fascinated the scientific community, both for their elaborate architecture and for their biological activities. These compounds have represented a hope to develop numerous cures against contemporary diseases.1 Among the broad class of alkaloids, the indole alkaloids are among the most known and abundant. Such compounds carry an indole or indoline core incorporated in a complex polycyclic structure containing one or more nitrogen atoms. Several of these indole alkaloids are biologically active, and some of them are used in medicine, such as vinblastine, for the treatment of cancer.2 The biosynthesis of these compounds involves tryptophan as an amino acid precursor, since the indole or indoline moiety is the main subunit of these alkaloids.3 The most famous alkaloid family is probably the aspidosperma family,4 which includes many known natural products, not counting those that have not yet been isolated and identified. In addition, several indole alkaloids belong to the corynanthe,5 and iboga6 classes. It appears that this important motif is very present in nature, probably due to its strong bioactivity (Fig. 1).
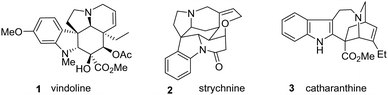 |
| | Fig. 1 Some natural products of the indole alkaloids. | |
Indole alkaloids have aroused the curiosity of chemists, and the preparation of their main heterocycle has often been reported as the key synthetic step in their total synthesis. Consequently, the development of new methodologies for the rapid formation of such heterocycles has been considered a critical goal by chemists involved in total synthesis. These strategies have laid the foundation for rapid access to these important scaffolds. In this review, we report on the most recent approaches over the past eleven years from 2014 that have been used by chemists to achieve the synthesis of the main indole or indoline core present in the total synthesis of complex natural products. The heterocycle formation of more than 40 syntheses is described as a key step in the development of these complex natural products. However, this review does not report on the biosynthetically inspired total syntheses starting from an indole or indoline precursor as starting materials, and some elegant syntheses of these complex alkaloids are not reported and would constitute another review topic that would include, for example, the work of Vincent, Zhu, and many other remarkable total syntheses carried out by leading groups in this field, which unfortunately cannot all be cited in this review.7
2. Indole formation in the total synthesis of natural products
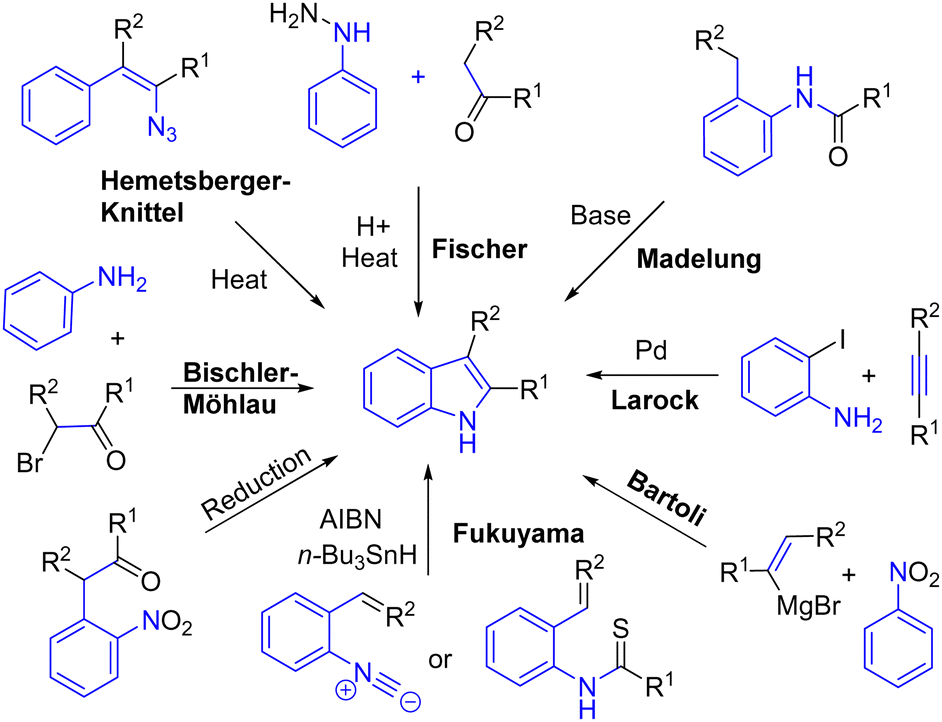
Indole is a heterocycle found in several natural products and bioactive compounds. This subunit is involved in the bioactivity of these alkaloids, which could act as a cure for several diseases. Consequently, several transformations have been developed to rapidly produce indoles. The best known of which is probably the Fischer indole process,8 but many others have been developed, such as the Madelung,9 Larock,10 Bartoli,11 Fukuyama,12 Bischler–Möhlau,13 Hemetsberger–Knittel14 indole syntheses, as well as several reductive nitro processes, to name a few methods to produce this crucial heterocycle (Scheme 1).
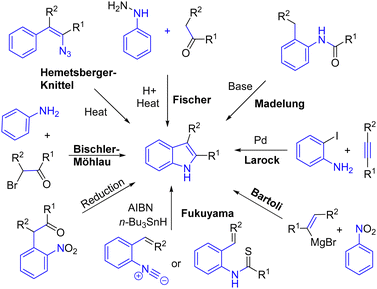 |
| | Scheme 1 Some known processes for the production of indoles. | |
Most of these processes were developed a long time ago. However, some are still in use today, and new approaches have been developed in recent decades. In this chapter, we present the recent processes used in the total synthesis of alkaloids to generate the key indole heterocycle.
2.1 Fischer indole formation
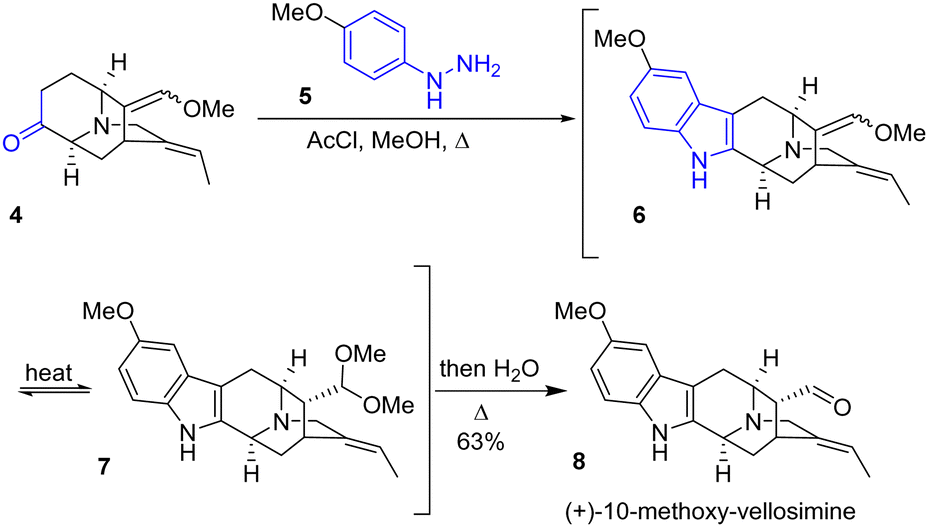
The Fischer indole synthesis was reported in 1900 by Nobel Laureate Emil Fischer. The efficiency of this reaction is due to the fact that it allows the formation of indole rings from simple ketones. However, it requires harsh conditions as it involves a [3,3]-sigmatropic rearrangement, and therefore this process is not compatible with certain sensitive functionalities. Although it is an old transformation, this approach is still very much used to produce the indole moiety present in natural products, as exemplified below. In 2014, the Gaich group reported an asymmetric protecting-group-free total synthesis of sarpagine alkaloids N-methylvellosimine, vellosimine, and 10-methoxyvellosimine 8 (Scheme 2).15 Their strategy involves the synthesis of the privileged intermediate 4, followed by a late Fischer indolization to form the corresponding natural product. Introducing the indole moiety late into the synthesis presents two advantages for the synthesis: it prevents the requirement of modifying existing substituents throughout the synthesis, causing a rework of the synthetic route, and it allows for varying the substitution of the aryl hydrazine used, which opens the possibility for other derivatives of the sarpargine alkaloid family. Fischer indolization of the ketone 4 with hydrazine 5, followed by hydrolysis, generated (+)-10-methoxy-vellosimine 8 with a yield of 63%. Four years later, the group optimized the synthesis of the key intermediate ketone 4 at gram scale.16 This was used in the synthesis of various sarpagine alkaloids.
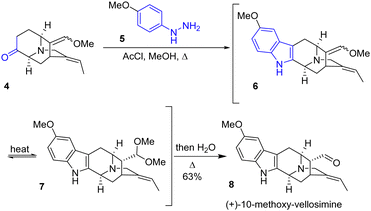 |
| | Scheme 2 Total synthesis of (+)-10-methoxy-vellosimine 8 by Fischer indolization. | |
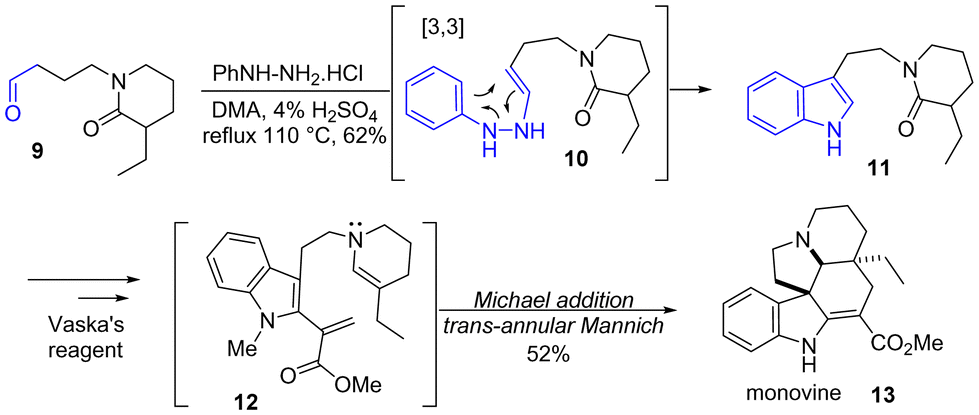
In 2016, Dixon and coworkers reported a short and divergent total synthesis of several vincadifformine-type, quebrachamine-type, and iboga-type alkaloids such as minovine 13 (Scheme 3).17 In their approach, a Stork-modified Fischer indolization was used to synthesize the indole skeleton. Using the hydrochloride salt of phenylhydrazine 10, the authors reacted lactam-containing aldehyde 9 to Fischer indolization conditions to generate indole 11 in 62% yields. Treatment of the lactam moiety with Vaska's reagent18 resulted in intermediate 12 and a remarkable formal Diels–Alder, comprising an enamine-Michael addition, followed by a Mannich-reaction-type cascade to form the pentacyclic alkaloid, minovine 13, in a yield of 52%.
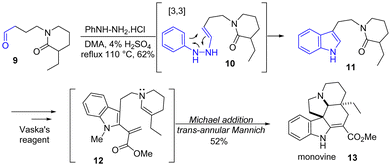 |
| | Scheme 3 Stork-modified Fischer indolization to indole 11, followed by a formal Diels–Alder-type cascade to minovine 13. | |
In 2019, Zhang and coworkers developed a fascinating green approach for the synthesis of (−)-suaveoline 18 (Scheme 4).19 Using a copper(II) catalyst, they were able to perform an oxidation-cyclization cascade of a derivative of cyclopropanol 14 to generate the third ring of the natural product with minimal modification of functional groups. Afterwards, the group introduced the indole moiety into 16 by Fischer indolization, obtaining indole 17 with a yield of 89%. Finally, removal of the PMP group with orthoperiodic acid (H5IO6) obtained (−)-suaveoline 18 with a yield of 75%.
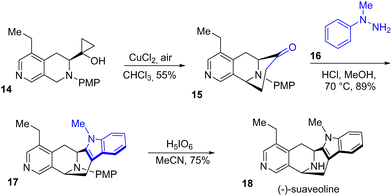 |
| | Scheme 4 Synthesis of (−)-suaveoline 18 by Fischer indolization. | |
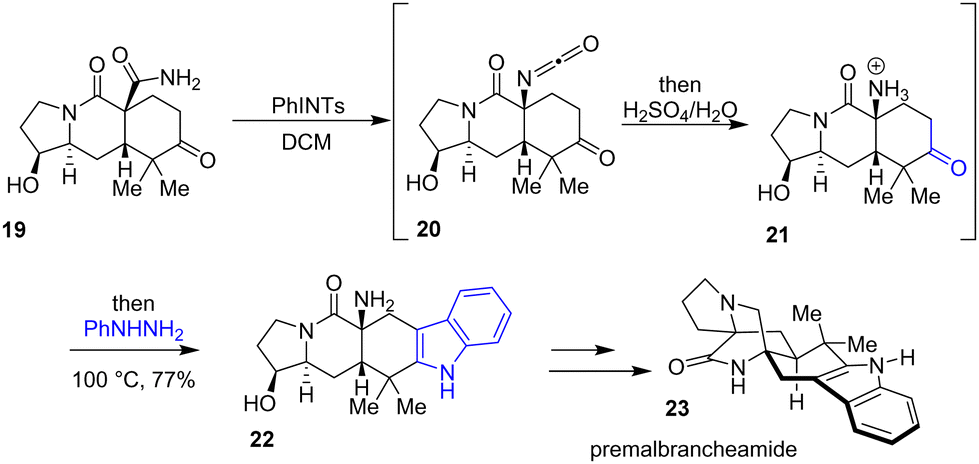
In 2020, Sarpong and colleagues reported a divergent route for the total synthesis of preparaherquamide, premalbrancheamide 23, and (+)-VM-55599, which each feature a complex bicyclo[2.2.2]diazaoctane core (Scheme 5).20 Their strategy involves a one-pot Hofmann rearrangement followed by a Fischer indolization to generate the pentacyclic indole scaffold. In this process, amide 19 reacts with a hypervalent-iodine reagent, (tosylimino)phenyl-λ3-iodane (PhINTs), which promotes the formation of an isocyanate 20, which is subsequently hydrolyzed to the ammonium bisulfate salt intermediate 21. Finally, the addition of phenylhydrazine gave indole 22 in 77% yields. This intermediate was then used for the synthesis of premalbrancheamide 23.
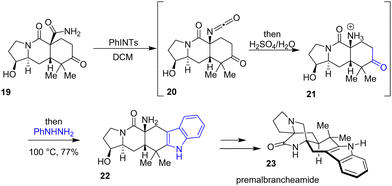 |
| | Scheme 5 One-pot Hofmann rearrangement and Fischer indolization to 22 in the synthesis of premalbrancheamide 23. | |
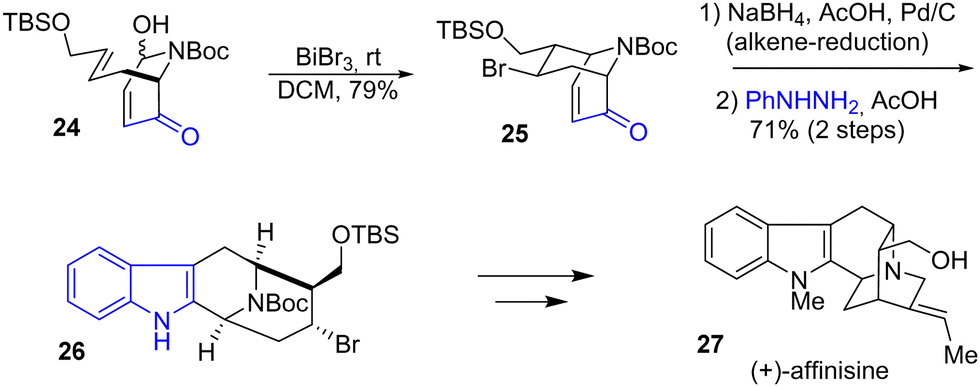
In 2023, Tong and coworkers developed a concise synthesis of several suaveoline and other sarpagine alkaloids (Scheme 6).21 These natural products have attracted synthetic interest due to their unusual indole-fused 9-azabicyclo[3.3.1]nonane structure (9-ABN). Their strategy involves a Lewis acid-mediated intramolecular aza-Prins cyclization mediated by BiBr3 to generate the bicyclic piperidine rings. After this step, the alkene of the bicyclic enone 25 was reduced, and a Fischer indolization in two steps generated the 9-ABN system 26 with a yield of 71%. Several steps later, this allowed the synthesis of several natural products such as (+)-affinisine 27.
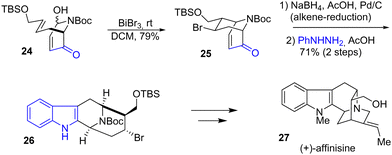 |
| | Scheme 6 Synthesis of (+)-affinisine 27 by Fischer indolization. | |
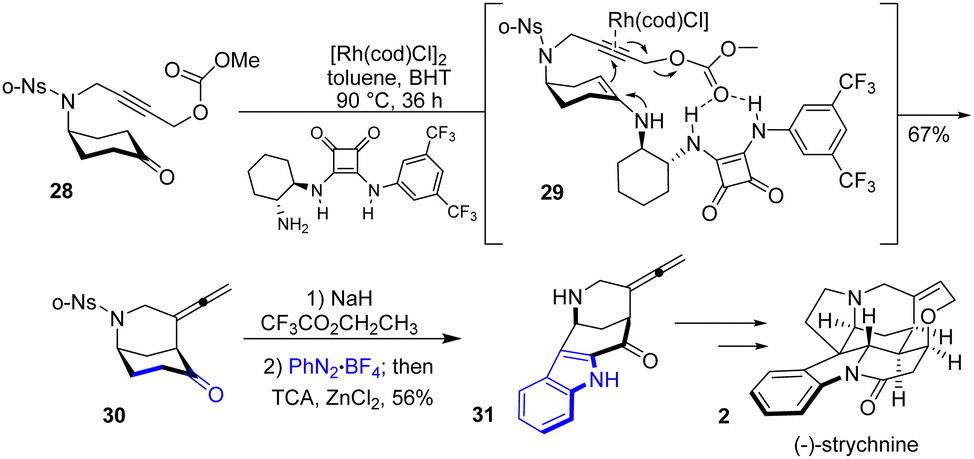
The same year, Zhang and colleagues reported a total synthesis of several strychnan alkaloids22 using a fascinating bridge backbone strategy (Scheme 7). Their work involves an asymmetric α-allenylation of ketones using a cyclohexanone 28 as the precursor in the presence of a metal catalyst and an amine-containing organocatalyst. This allows for the simultaneous formation of a piperidine ring and an allene to bicyclic 30. After having access to this precursor, the authors were interested in applying their methodology to total synthesis. The next step involves installing the indoline moiety through a Japp–Klingemann reaction and Fischer indole-type process. Treatment of precursor 30 with NaH and CF3CO2CH2CF3 followed by PhN2BF4 in the presence of trichloroacetic acid (TCA) and ZnCl2 produced indole 31 in 56% yields. This was then carried out on various strychnan alkaloids, such as (−)-strychnine 2.
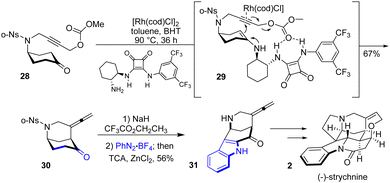 |
| | Scheme 7 Zhang's synthesis of (−)-strychnine 2. | |
2.2 Palladium (Pd) coupling processes
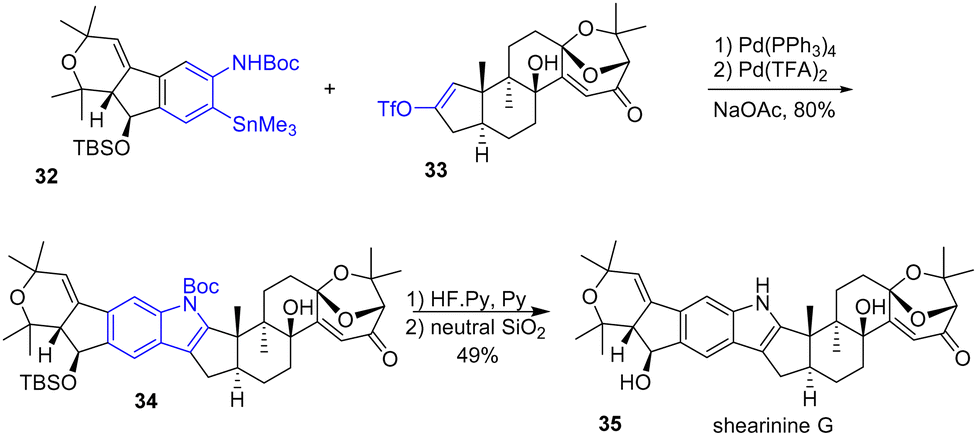
Palladium has also proven to be a powerful reagent in synthesis, enabling many transformations. One of these outstanding applications allows the rapid formation of heterocycles such as indoles. This approach starts with a cross-coupling process followed by an Aza-Wacker type process. In 2022, Carreira and coworkers reported the first total synthesis of indole diterpenoids (+)-shearinine G 35 (Scheme 8).23 Their strategy involves the late Pd-oxidative Stille-coupling of an ortho N-protected arylstannane 32 with a triflyl enol ether 33 in tandem with an aza-Wacker process to form the N-protected indole 34 of the (+)-shearinine. This two-step process, mediated by the presence of Pd(PPh3)4 and Pd(TFA)2 was achieved in 80% yield. Deprotection of the TBS group with Olah's reagent and deprotection of the Boc group by adsorption on silica gel finished the total synthesis of shearinine D 35.
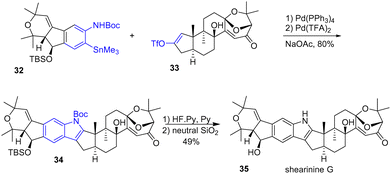 |
| | Scheme 8 Total synthesis of shearinine G 35 using a Stille and aza-Wacker tandem process. | |
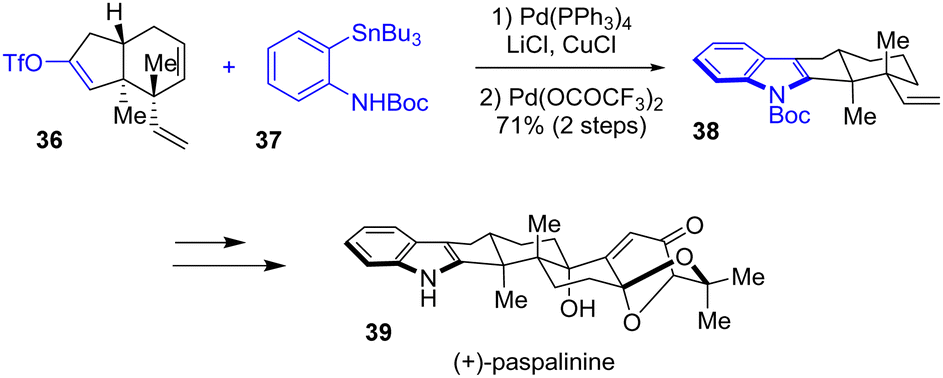
In the meantime, Tong and coworkers published an asymmetric total synthesis of indole diterpenes, paspalinine 39 and paspalinine-13-ene, with a similar strategy (Scheme 9).24 These structures contain a complex, 6,8-dioxabicyclo[3.2.1]octane motif. To synthesize the indole portion of these natural products, the authors utilized a Stille-coupling reaction between triflyl enol ether 36 and ortho-N-Boc protected aniline 37 as well as a PdII-mediated aza-Wacker reaction. This generated tetracyclic indole 38 in a yield of 71%, which served as the building block for e.g., paspalinine 39.
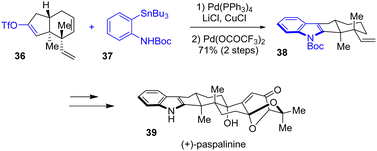 |
| | Scheme 9 Synthesis of indole 38 by a Stille-coupling-aza-Wacker reaction in the synthesis of (+)-paspalinine 39. | |
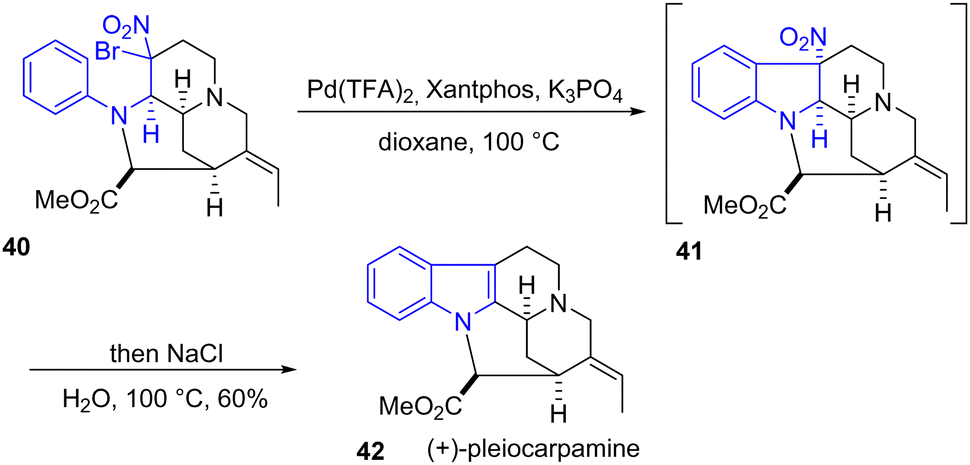
In 2023, Tokuyama and coworkers reported the total synthesis of (+)-pleiocarpamine for the convergent total syntheses of (+)-voacalgine A and (+)-bipleiophylline through an oxidative coupling (Scheme 10).25 During the total synthesis of (+)-pleiocarpamine 42, an indole formation was required to prepare the pentacyclic skeleton. Their approach involves a one-pot palladium-catalyzed aromatic C–H alkylation of the geminal bromo-nitro aniline 40 to form the indoline intermediate 41, then elimination of the nitro group followed by aromatization using excess NaCl and H2O gave the desired indole product 42 in a yield of 60%. It should be noted that a similar palladium strategy was reported for the synthetic studies of jerantinine E26 and eleganine A,27 demonstrating the potential of this approach.
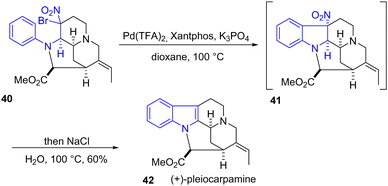 |
| | Scheme 10 One-pot Pd-catalyzed aromatic C–H alkylation of aniline 40 to indole 41 in the synthesis of (+)-pleiocarpamine 42. | |
In 2024, Stoltz and coworkers reported a divergent total synthesis of pyrroloiminoquinone alkaloids makaluvamines A, C, D, and N, as well as isobatzelline B, which feature an interesting pyrrolo[4,3,2-de]quinoline ring skeleton (Scheme 11).28 Their strategy to prepare these alkaloids utilizes a tandem Larock/Buchwald–Hartwig annulation reaction, followed by acidification. Palladium coupling of TES-protected alkyne 43 with pentasubstituted arene 44 in the presence of ligands XPhos and 1,1′-bis(di-tert-butylphosphino)ferrocene (D-t-BPF) using K2CO3 and followed by acidification gave indole 45 in 48% yields. Afterwards, this indole core was carried out to makaluvamine N 46, completing the first total synthesis of this product.
 |
| | Scheme 11 Tandem Larock annulation/Buchwald–Hartwig coupling to indole 45, in the total synthesis of makaluvamine N 46. | |
2.3 Reductive cyclization processes
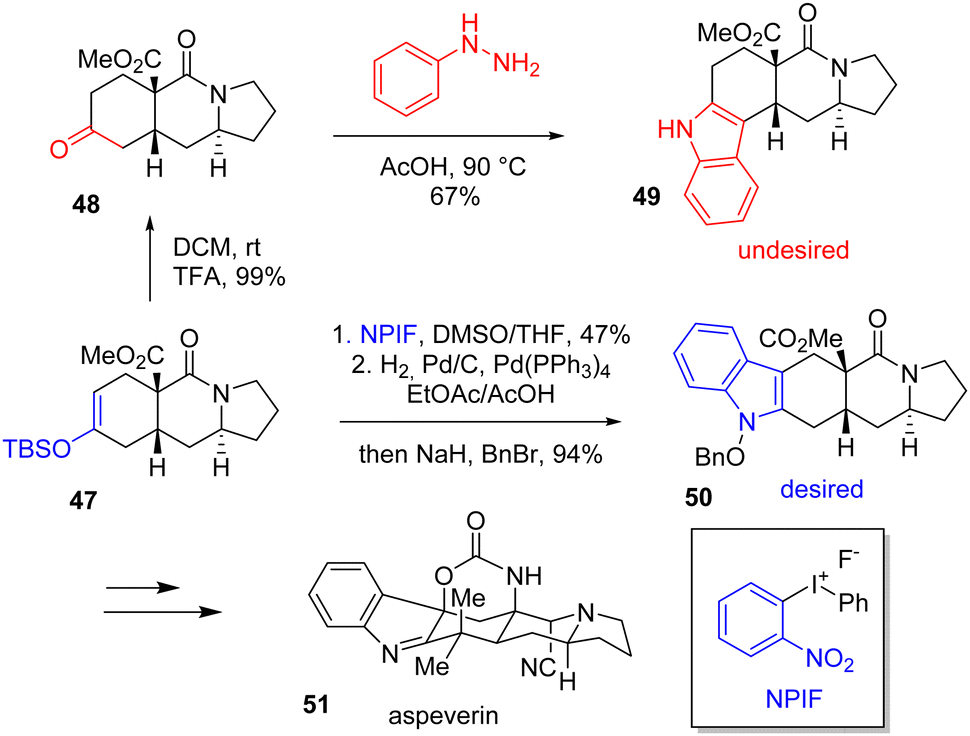
A simple and effective way of making indole is to reduce a nitroaryl segment close to a carbonyl group, producing an imine by condensation first with the resulting carbonyl and then by isomerization to the desired heterocycle. To produce indoles, this is still the simplest, most common, and most efficient route. In 2014, Levinson published a total synthesis of aspeverin 51, a prenylated indole alkaloid (Scheme 12).29 At the indolization step, the author initially proposed to generate the indole moiety into the skeleton of 48 by a Fischer indolization to obtain indole 50. However, he observed the wrong regioselectivity of the heterocycle formation and obtained isomer 49 instead. To remediate this issue, Levinson reversed the regioselectivity by using an iodonium reagent. Arylation using silyl enol ether 47 with ortho-nitrophenyliodonium fluoride (NPIF) followed by reductive cyclization obtained desired regioisomer 50 with a yield of 94%. This was then carried out to aspeverin 51.
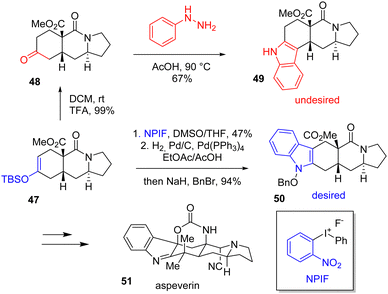 |
| | Scheme 12 Iodonium arylation and reductive cyclization in the synthesis of aspeverin 51. | |
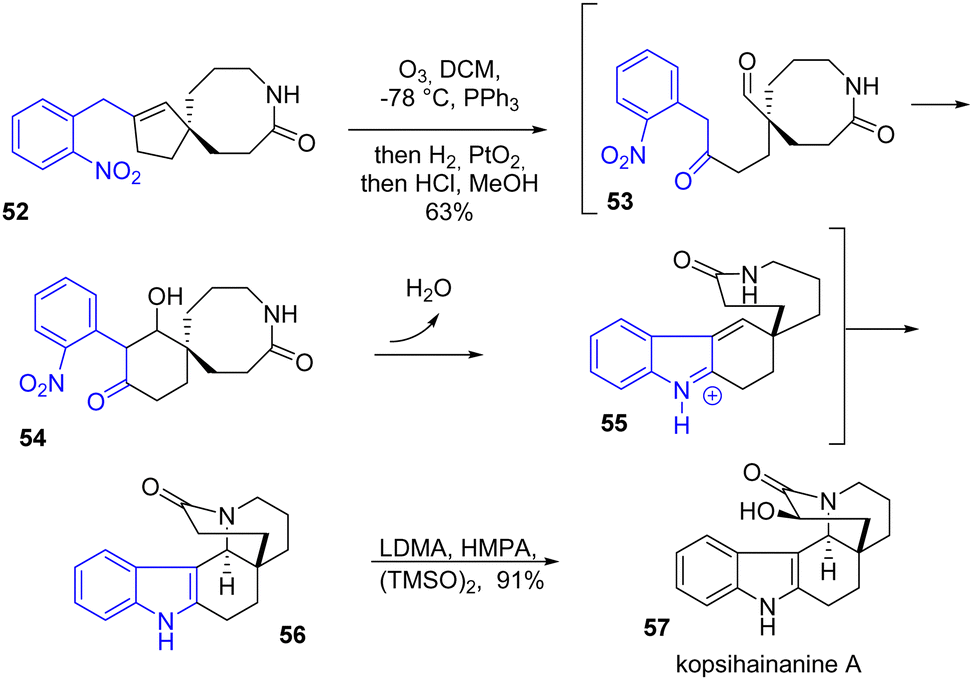
The same year, Zhu and coworkers reported the synthesis of kopsihainanine A 57 and several other monoterpene indole alkaloids (Scheme 13).30 This natural product is interesting synthetically due to its unusual [6.5.6.6.6] pentacyclic structure. The synthesis features a late-stage indole formation by a one-pot domino integrated oxidation/reduction/cyclization (iORC) sequence. At a certain point in the synthesis, the alkene of spirolactam 52 is cleaved by ozonolysis followed by an aldolization to generate intermediate 54, a reduction of the nitroarene moiety, and an indole formation followed by an E1cB elimination led to 55, an Aza-Michael process mediated under acidic conditions afforded the indole 56 in 63% yield. Finally, the addition of a hydroxyl group achieved kopsihainanine A 57.
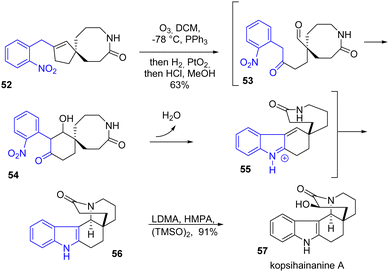 |
| | Scheme 13 One-pot domino cyclization process in the synthesis of kopsihainanine A 57. | |
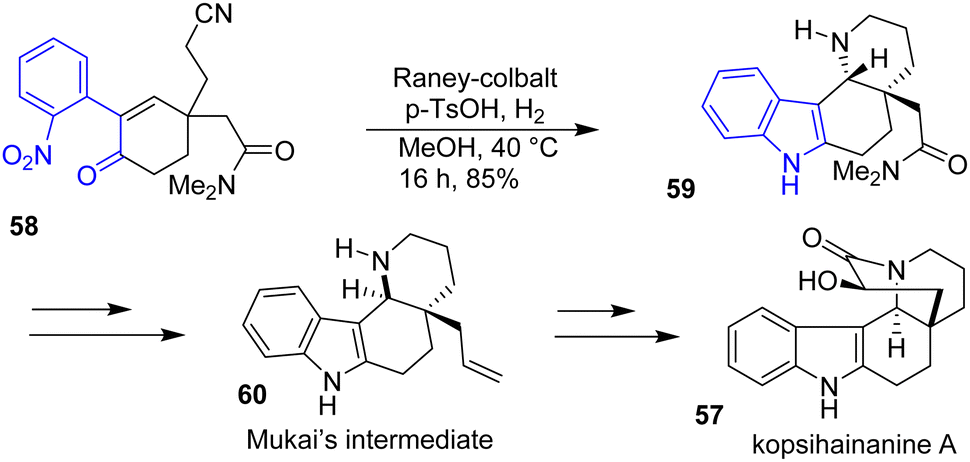
Two years later, the Banwell group sought to improve the indole formation step of kopsihainanine A 57 (Scheme 14).31 Rather than going towards an ozonolysis-aldolization cyclization route to generate the main 6-membered core, they cyclized the indole by a RANEY®-cobalt hydrogenation on a substituted enone precursor 58 in the presence of para-toluene sulfonic acid (p-TsOH) in methanol. This approach allowed for the formation of the indole moiety 59 as well as the piperidine ring of kopsihainanine with a yield of 85%. However, an additional step was required in the synthesis to epimerize the stereochemistry of the piperidine ring junctions for the synthesis of an advanced intermediate 60 reported in the approach of Mukai and coworkers.32
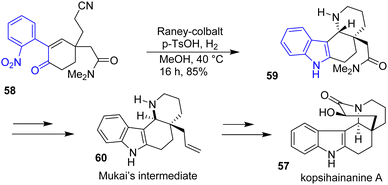 |
| | Scheme 14 Reductive cyclization in the synthesis of kopsihainanine A 57. | |
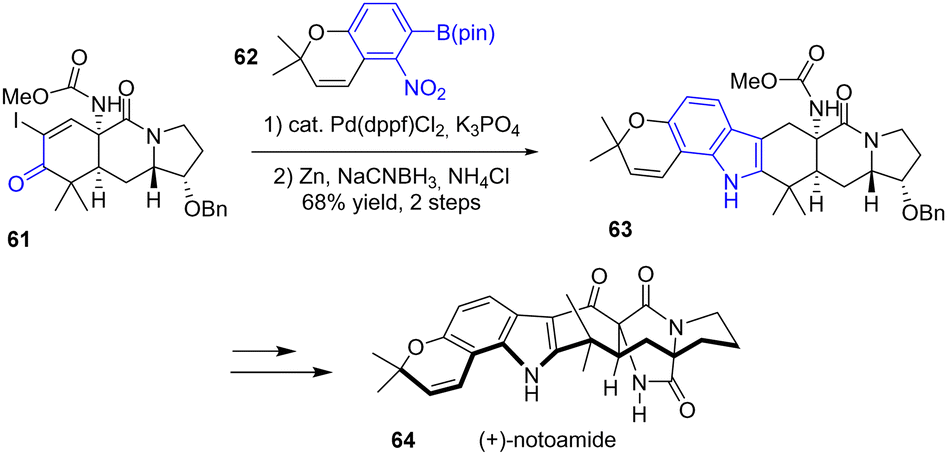
In 2015, Sarpong and coworkers published a unified approach for the total syntheses of (−)-17-hydroxy-citrinalin B, (+)-stephacidin A, and (+)-notoamide 64, which each feature a complex bicyclo[2.2.2]diazaoctane moiety (Scheme 15).33 For this synthesis, the authors utilized a Suzuki cross-coupling reaction of 61 with aryl boronate 62. In the second step, reductive cyclization with zinc metal and sodium cyanoborohydride (NaBH3CN) and a weak acid (NH4Cl) generated indole 63 with a yield of 68%. This indole was then used as an intermediate for each of the natural products, such as in the total synthesis of (+)-notoamide 64.
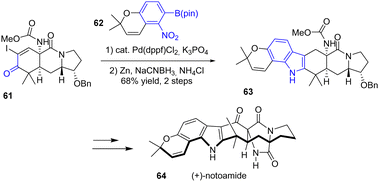 |
| | Scheme 15 Indole formation for the synthesis of (+)-notoamide 64. | |
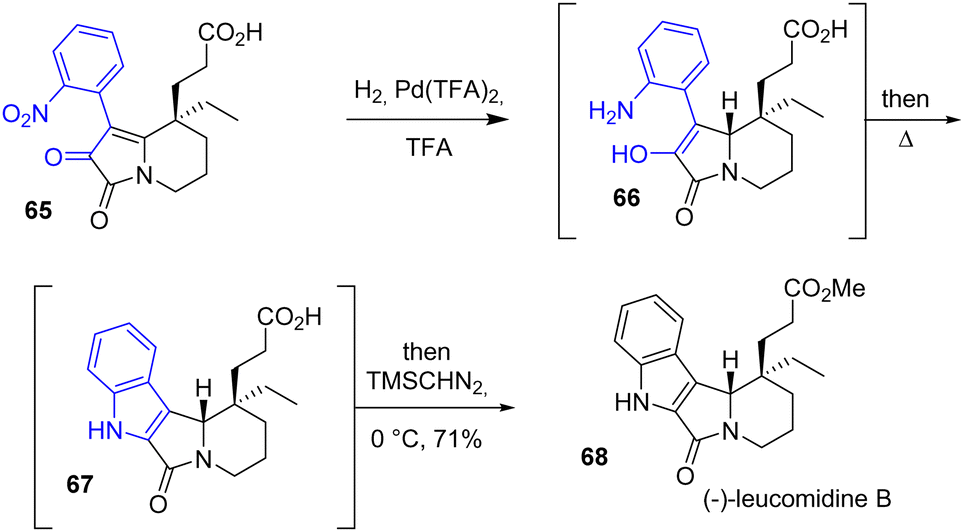
In 2016, Zhu and coworkers published the divergent enantioselective synthesis of (−)-rhazinilam, (−)-leucomidine B 68, and (+)-leuconodine F, which are monoterpene indole alkaloids that feature an axially chiral tetracyclic structure (Scheme 16).34 Their structures have gained significant synthetic interest due to their unusual [5.5.6.6]diazafenestrane skeleton. Among these natural products, (−)-leucodinine B features an indole functional group. Hydrogenation of the nitro group of arene 65 in the presence of Pd(TFA)2 resulted in a mixture of aniline 66 and indole 67. Refluxing the reaction induced the full conversion of aniline into the corresponding indole. After methylation of the carboxylic acid group, the natural (−)-leucomidine B 68 is produced with a yield of 71%.
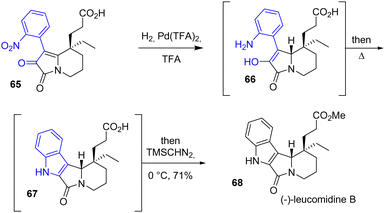 |
| | Scheme 16 Synthesis of (−)-leucomidine B 68 by a Pd-catalyzed hydrogenation. | |
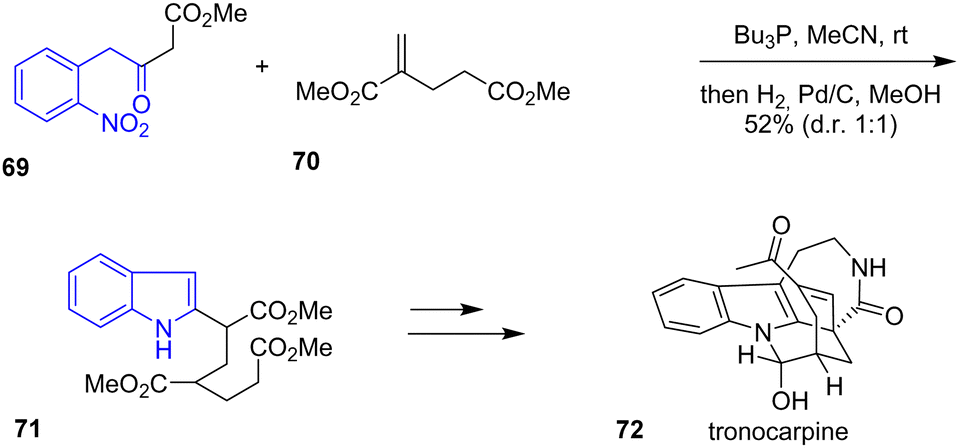
In 2020, Namba and coworkers completed a concise total synthesis of tronocarpine 72, a chippiine-type alkaloid.35 The synthesis consists of a one-pot construction of an azabicyclo[3.3.1]nonane core, a pentacyclic structure (Scheme 17). The beginning of the synthesis starts with a Michael addition of 1,3-dicarbonyl 69 with an α,β-unsaturated diester 70 mediated by tributylphosphine, followed by hydrogenation of the nitro group in the presence of MeOH and Pd/C to give the indole 71 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereoselectivity ratio and a 52% yield. Working from the indole framework, the authors assembled the piperidine ring and a cyclohexene motif to synthesize tronocarpine 72.
:1 diastereoselectivity ratio and a 52% yield. Working from the indole framework, the authors assembled the piperidine ring and a cyclohexene motif to synthesize tronocarpine 72.
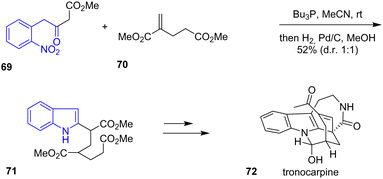 |
| | Scheme 17 Synthesis of tronocarpine 72. | |
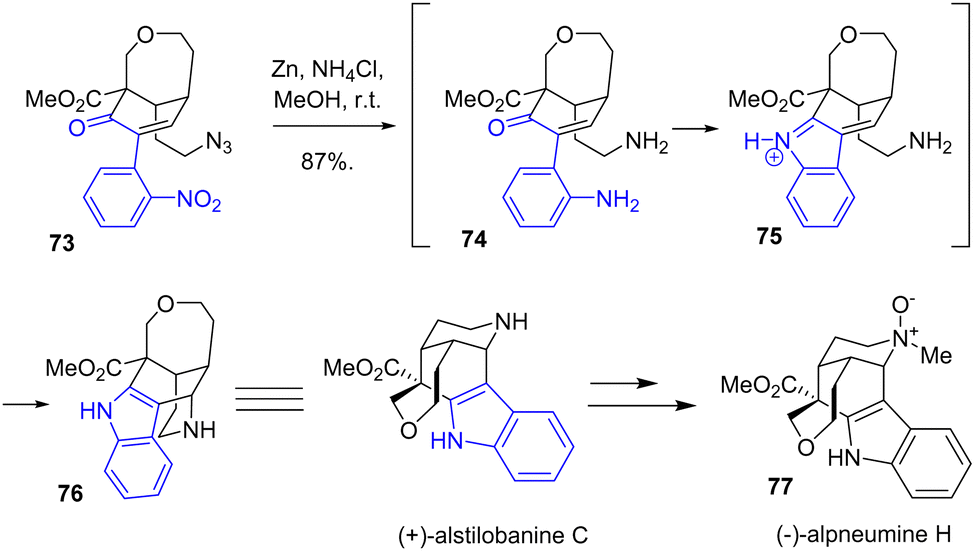
In 2021, Zhu and coworkers reported an enantioselective total synthesis of (+)-alstilobanine C, (+)-undulifoline, and (−)-alpneumine H 77, which are monoterpene indole alkaloids that feature a tetrahydropyran ring (Scheme 18).36 The synthesis consisted of a late-stage indole formation using reductive cyclization conditions. This process was achieved by a one-pot cyclization process of the indole moiety and the piperidine ring using reductive cyclization conditions. Reduction of both the nitro group of the arene and the azide of 73 using zinc and ammonium chloride in methanol at room temperature cyclized to (+)-alstilobanine C 76 with a yield of 87%. This was then used in the synthesis of (−)-alpneumine H 77.
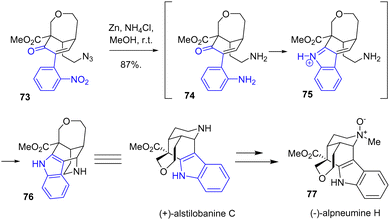 |
| | Scheme 18 Zinc reductive cyclization to the syntheses of (+)-alstilobanine C 76 and (−)-alpneumine H 77. | |
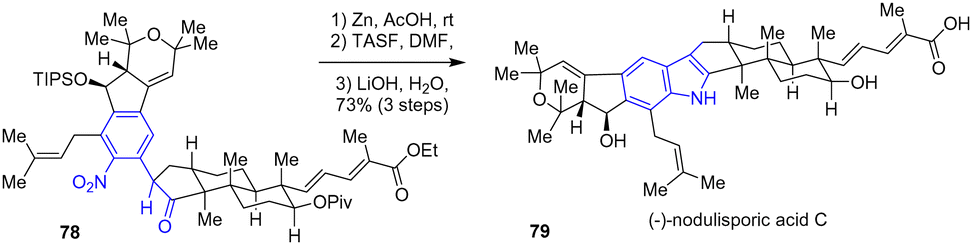
The same year, Pronin and coworkers developed a synthesis of the complex congener nodulisporic acid C 79 with excellent diastereoselectivity (Scheme 19).37 For example, this synthesis featured a radical-polar crossover cascade guided by pseudodiaxial interactions to produce an intermediate aldol. After coupling of an aryl chloride with the tricyclic ketone, the authors joined the polycyclic motifs with a late-stage indole formation. Reduction of the complex nitroarene 78 with zinc in the presence of acetic acid followed by a desilylation with tris(dimethylamino)sulfonium difluorotrimethylsilicate (TASF) and a double saponification afforded nodulisporic acid C 79 with a yield of 73%.
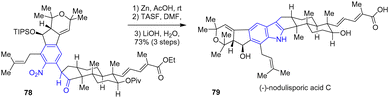 |
| | Scheme 19 Completion of (−)-nodulisporic acid C 79 by reductive cyclization. | |
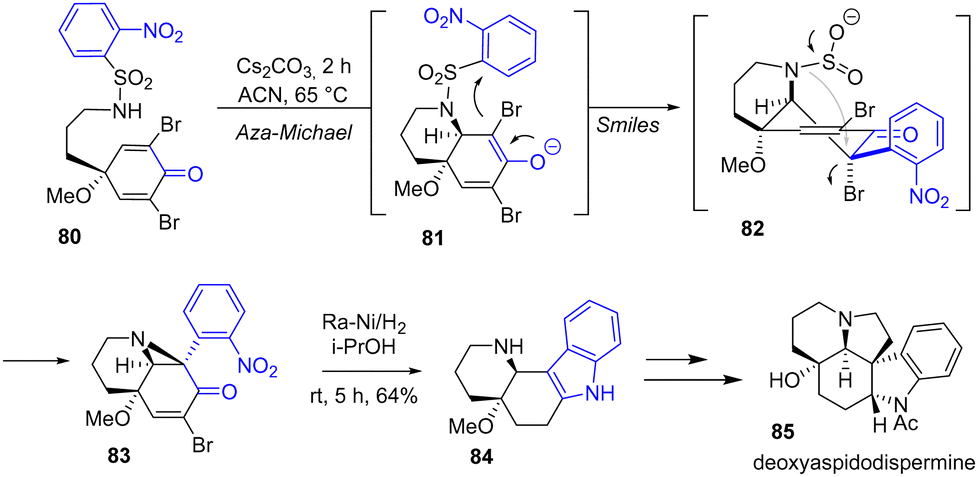
In 2022, Canesi and coworkers reported a total synthesis of deoxyaspidodispermine 85 using an ortho-nosyl functional protecting group to introduce the indole moiety into the natural product (Scheme 20).38 This synthesis highlights an aza-Michael-Smiles ring-closure cascade, which enables the formation of a tetracyclic system from a nosylamide functional protecting group. In the fourth step of the synthesis of deoxyaspidodispermine 85, an indole formation step was required to form the tetracyclic system. This was achieved by reduction of the nitro group, the bromide, and the double bond of bicyclic enone 83 using RANEY® nickel and cyclization to indole intermediate 84 with a yield of 64%. This key compound was then transformed into the natural product 85.
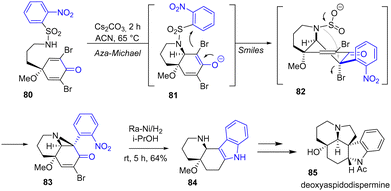 |
| | Scheme 20 RANEY®-Ni cyclization for indole formation in the synthesis of deoxyaspidodispermine 85. | |
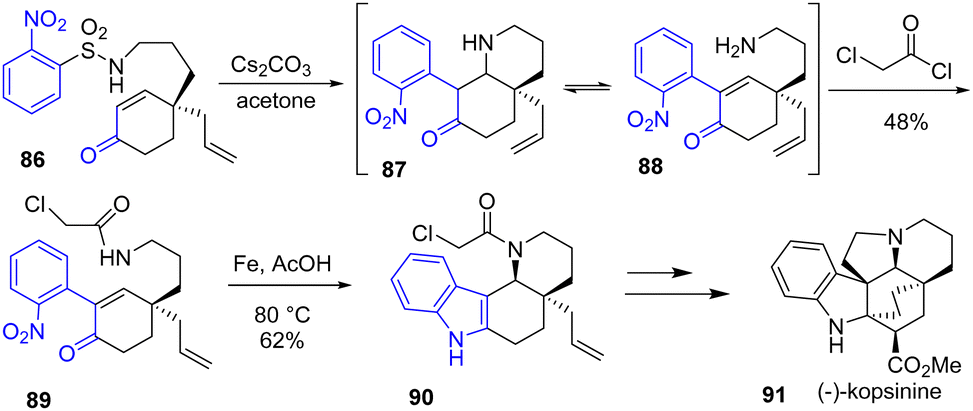
In the meantime, Ruijter and coworkers published an enantioselective total synthesis of monoterpene indole alkaloids (−)-limaspermidine, (−)-kopsinilam, (−)-kopsinine 91, and tetrahydrokopsifoline D39 with an aza-Michael-Smiles strategy that is similar to Canesi's strategy,40 but with a notable asymmetric variant (Scheme 21). Their approach involves the reductive cyclization of nitroarenes using a source of iron and acetic acid to form the D-ring of the skeleton of these alkaloids. Treatment of nitroarene 89 with these conditions promoted the reduction-cyclization to intermediate tetracyclic indole and piperidine ring 90 with a yield of 62%. This was then used as the precursor to the indoline moiety of (−)-kopsinine 91 and other alkaloids.
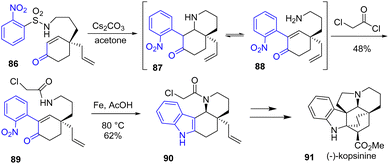 |
| | Scheme 21 Reductive cyclization from nitroarene 89 to indole 90 in the synthesis of (−)-kopsinine 91. | |
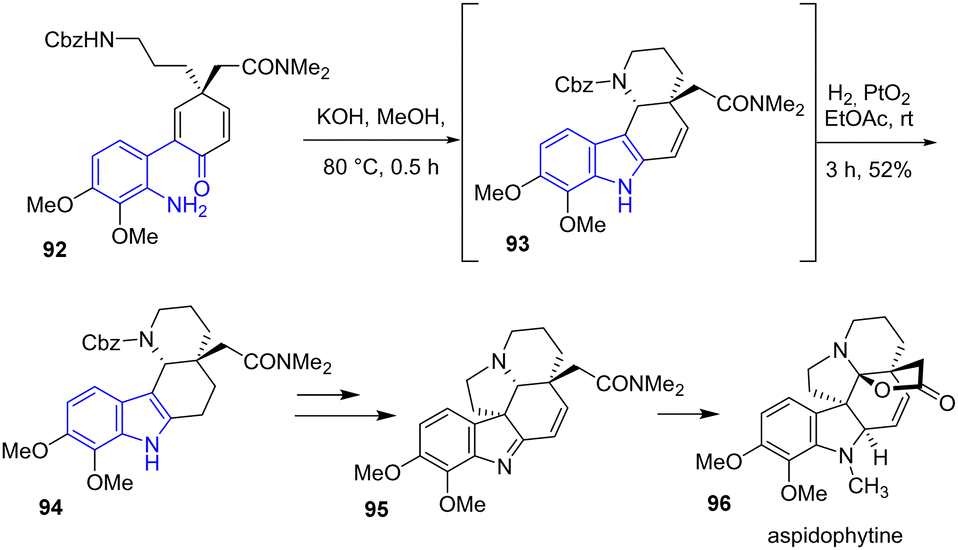
The same year, Banwell, Ye, and coworkers developed formal asymmetric syntheses of aspidophytine 96, a pentacyclic indole alkaloid (Scheme 22).41 To generate the indole ring and piperidine rings of aspidophytine, the authors performed a Schiff base condensation-hetero-Michael-type cascade of 92 to generate an unstable indole intermediate 93. After hydrogenating using Adam's catalyst, they isolated indole 94 in a yield of 52%, which was subsequently used in later steps for the formation of 95.
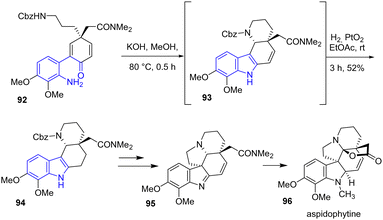 |
| | Scheme 22 Indole synthesis by base-promoted cyclization-hydrogenation in the synthesis of aspidophytine 96. | |
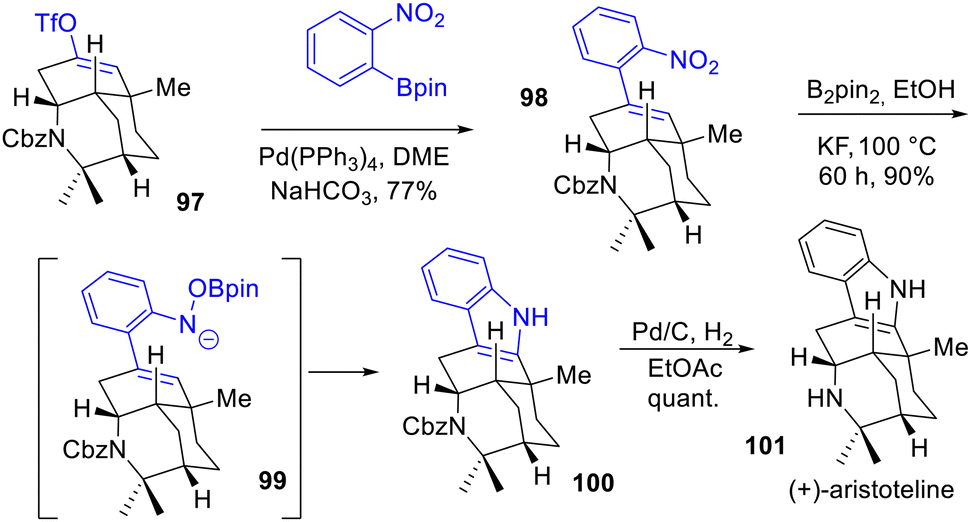
In 2023, Cho and coworkers prepared a divergent asymmetric synthesis of (−)-alloaristoteline 106 and (+)-aristoteline 101, which features a highly complex fused-pentacyclic indole skeleton (Scheme 23).42 Their method consists of using a tricyclic enol triflate 97 to build the pentacyclic framework. After coupling of the enol triflate with an aryl boronate, indole 100 was accessed by a remarkable reductive cyclization of nitroarene (6π-electron-5-atom electrocyclization) developed by Song, Driver et al.43 with bis-pinacolborane with a yield of 90%. Finally, removal of the Cbz-protecting group completed the total synthesis of (+)-aristoteline 101.
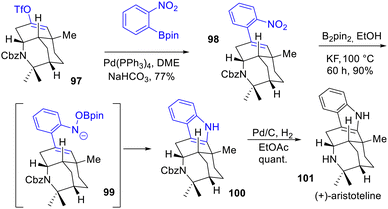 |
| | Scheme 23 Reductive cyclization of nitroarene 98 to indole 100 in the synthesis of (+)-aristoteline 101. | |
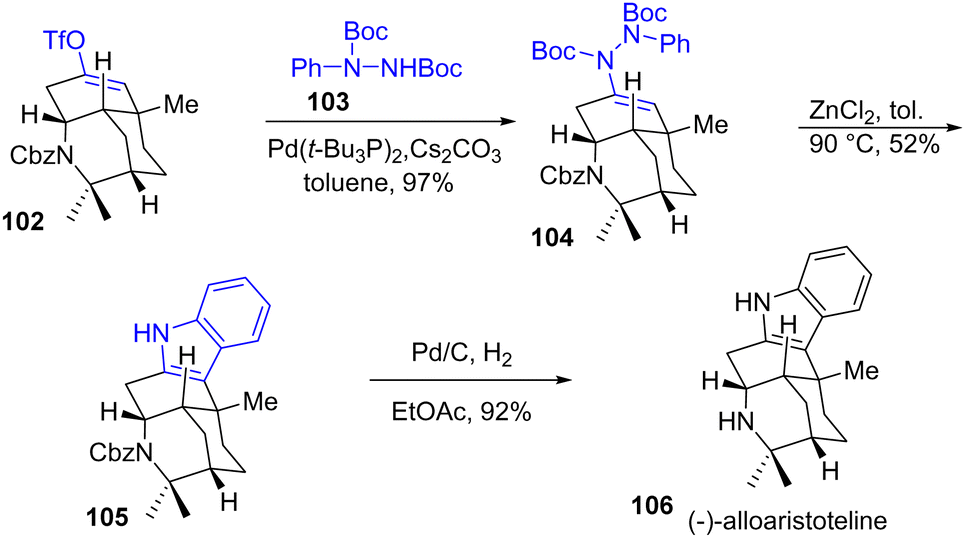
Interestingly, the structure of (−)-alloaristoteline 106, which is a constitutional isomer at the indole position of (+)-aristoteline 101, was prepared by a reminiscence of the Fischer process (Scheme 24). First, an N-coupling with N-Boc-phenyl hydrazide and enol triflate 102 led to the hydrazone 104 in 97% yield, which was then converted to (−)-alloaristoteline 106 in 52% yield by a subsequent ZnCl2-mediated indolization process followed by a hydrogenation.
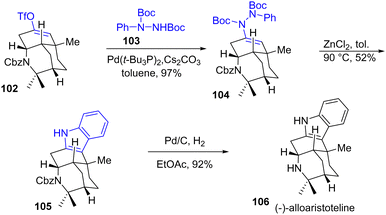 |
| | Scheme 24 Synthesis of the indole core of (−)-alloaristoteline 106. | |
2.4 Gold (Au) catalyst processes
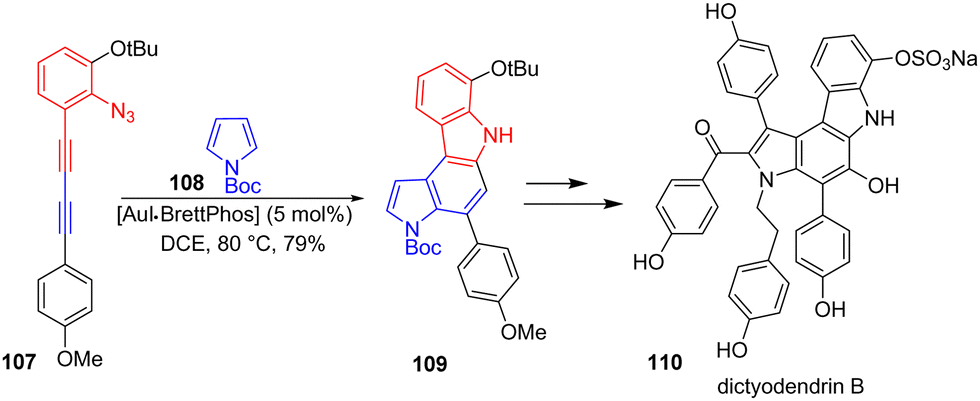
The chemistry of gold has developed considerably in the early 21st century. Since it has made it possible to selectively react nucleophilic sites such as a nitrogen with an alkyne in a catalytic manner, it is not surprising that it has been applied to the formation of indole moieties. In 2017, Ohno and coworkers reported the total synthesis of several dictyodendrins, which are marine indole alkaloids that feature a highly substituted pyrrolo[2,3-c]carbazole core (Scheme 25).44 Their strategy involves the cyclization of 1,3-diynes with substituted pyrroles in the presence of an axially chiral gold catalyst, AuIBrettPhos, to govern the regioselectivity. For the synthesis of the skeleton of dictyodendrin B 110, 1,3-diyne 107 was cyclized with pyrrole 108 in the presence of AuIBrettPhos to form the corresponding bis-indole 109 with a regioselectivity of 84:16 and a yield of 79%. This was then carried out to dictyodendrin B 110.
 |
| | Scheme 25 Au-catalyzed indole synthesis by cyclization of 1,3-diyne 107 and pyrrole 108 in dictyodendrin B 110. | |
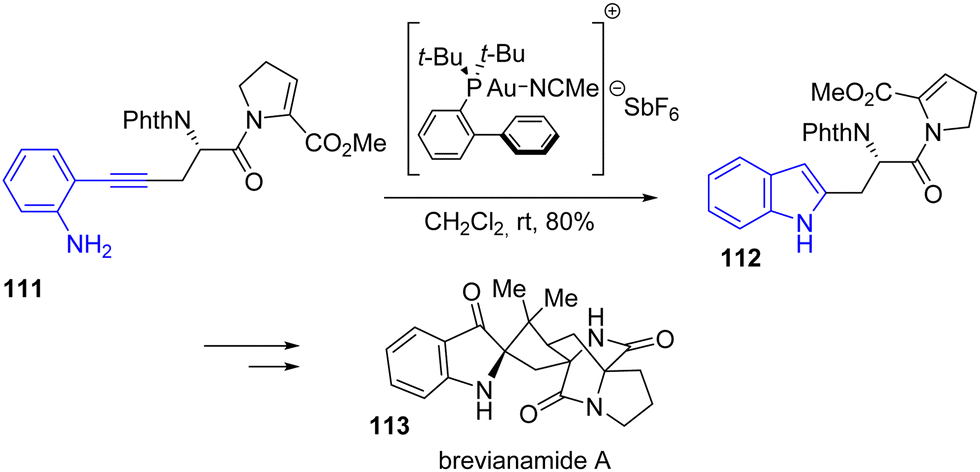
In 2021, Smith and coworkers published a general approach to the total synthesis of brevianamide A 113 and trigonoliimine C (Scheme 26).45 Among these two natural products synthesized in this article, brevianamide A is synthetically interesting due to its spiranic-type nature. For this synthesis, the authors proposed an Au-catalyzed cyclization of an alkyne attached to an aniline to generate the indole as a precursor to the indoline moiety found in the natural product. Cyclization of ortho-alkynyl aniline 111 using JohnPhosAu(MeCN)SbF6 led to the formation of indole intermediate 112 with a yield of 80%. This was carried out in multiple steps to brevianamide A 113.
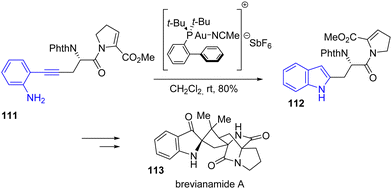 |
| | Scheme 26 Au-catalyzed indole cyclization in the synthesis of brevianamide A 113. | |
2.5 Miscellaneous processes
This section discusses some mixed processes that have been used on an ad hoc basis in recent years to produce indoles. In 2019, Zhou and coworkers reported a scalable, enantioselective synthesis of (−)-goniomitine 115, a monoterpene indole alkaloid that features a complex octahydroindolo[1,2-α][1,8]naphthyridine core (Scheme 27). To construct the main skeleton of (−)-goniomitine, the authors utilized Zhu and coworkers one-pot integrated-oxidation–reduction-cyclization (iORC) process.46 This method allowed for the formation of (−)-goniomitine 115 by a cascade formation of both the indole and piperidine rings from 114 with a yield of 74%.
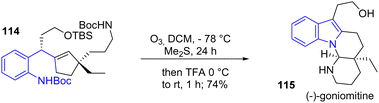 |
| | Scheme 27 Ozonolysis-initiated one-pot indole formation process for the synthesis of (−)-goniomitine 115. | |
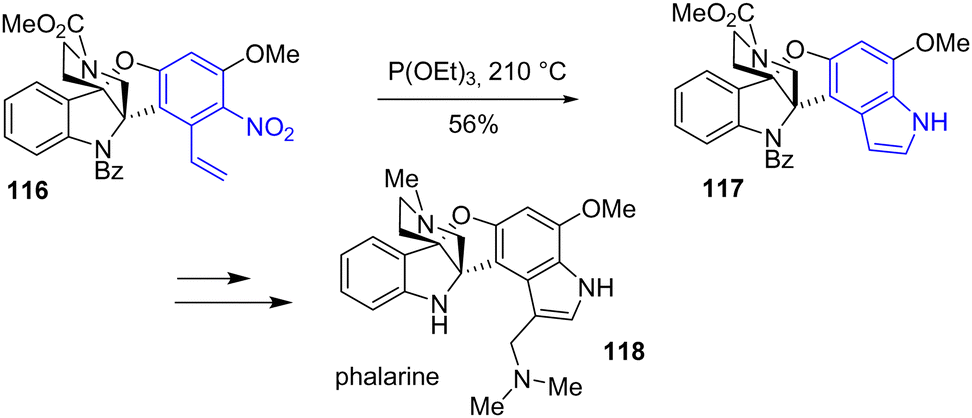
The same year, Jia and coworkers completed an eight-step total synthesis of phalarine 118, a furanobisindole alkaloid that contains a unique structure, a benzofuro[3,2-b]indoline framework (Scheme 28).47 This synthesis was the first regioselective oxidative coupling of a 2,3-disubstituted indole with a phenol using a hypervalent iodine reagent (PIDA) to generate the benzofuro[3,2-b]indoline framework. Later into the synthesis, the authors introduced the indole moiety into the natural product by a Cadogan reductive cyclization with nitroarene 116 using triethyl phosphite to form indole 117 with a yield of 56%. Two steps later, phalarine 118 was obtained.
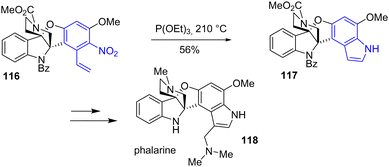 |
| | Scheme 28 Cadogan indolization for the synthesis of phalarine 118. | |
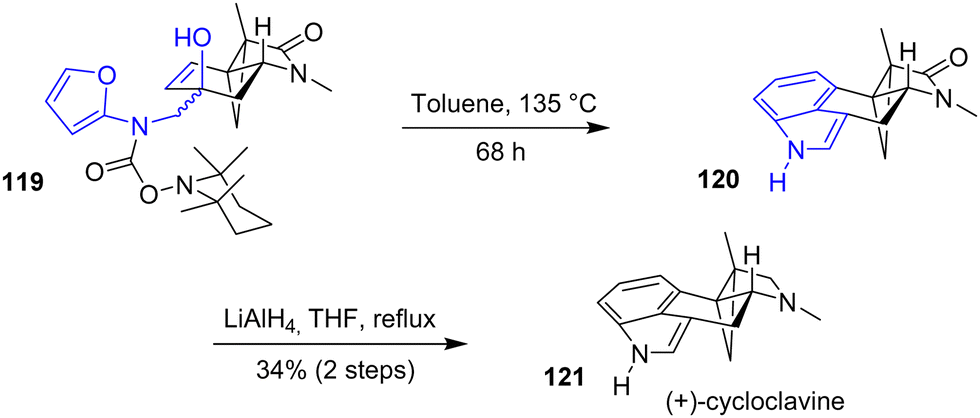
In 2019, the Wipf group reported the first asymmetric synthesis of (+)-cycloclavine 121, a cyclopropane-containing ergot alkaloid (Scheme 29).48 This synthesis features an intramolecular Diels–Alder reaction (IMDA) of a substituted furan motif containing a TEMPO-protected carboxylic acid. Cleavage of the TEMPO group by heating the major α-epimer 119 in the presence of toluene in a sealed tube allowed for the cyclization to indole 120. Then, reduction of the lactam with lithium aluminum hydride generated (+)-cycloclavine 121 in 34% yields over two steps. Unlike the α-epimer, the minor β-epimer did not proceed towards cyclization, possibly due to pseudo-axial interactions in the transition state.
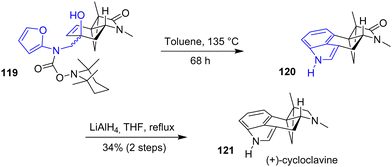 |
| | Scheme 29 Asymmetric IMDA reaction in the synthesis of (+)-cycloclavine 121. | |
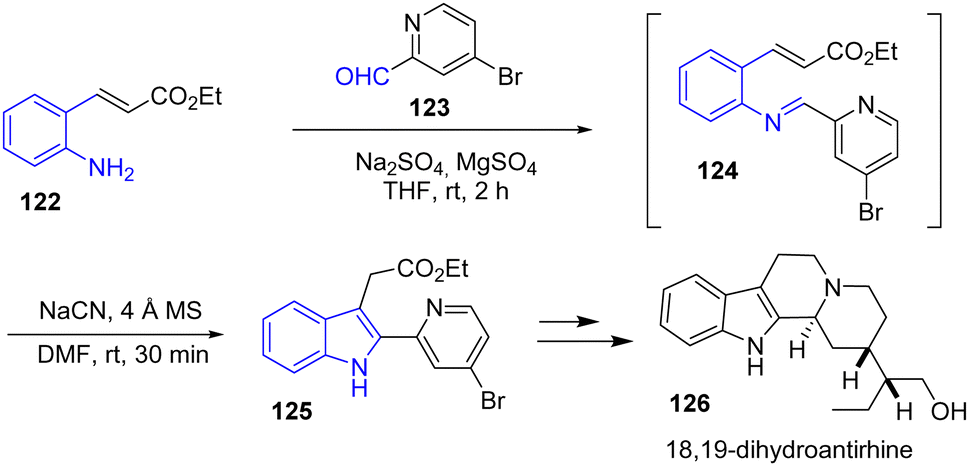
In 2020, Cheon and coworkers reported the divergent total synthesis of antirhine alkaloids antirhine, 18,19-dihydroantirhine 126, and their C-20 epimers (Scheme 30).49 To rapidly access their tetracyclic scaffold, the authors utilized a cyanide-catalyzed imino-Stetter reaction to prepare the indole moiety. Their one-pot procedure involves the in situ generation of aldimine 124 from the condensation of an α,β-unsaturated aniline ester 122 with an aldehyde-disubstituted pyridine 123. Then, the addition of NaCN in the presence of molecular sieves using DMF as the solvent afforded indole 125 in 92% yields. Afterwards, this was subsequently carried out to 18,19-dihydroantirhine 126.
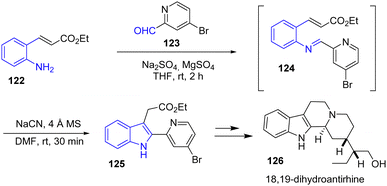 |
| | Scheme 30 One-pot cyanide-catalyzed imino-Stetter reaction in the synthesis of 18,19-dihydroantirhine 126. | |
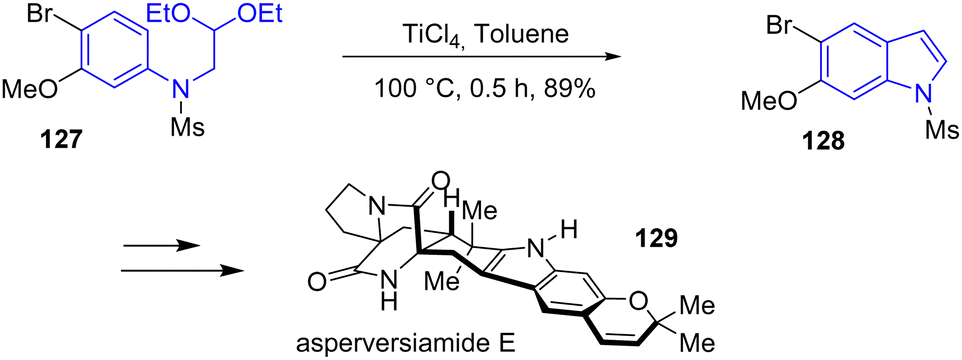
In 2024, Banwell and coworkers reported the first and concise total syntheses of the linearly fused pyrano[3,2-f] indole alkaloids asperversiamide B, D, E, G, and J, as well as dihydrocarneamide A (Scheme 31).50 Their approach involves a titanium-promoted intramolecular Friedel–Crafts alkylation to form the indole motif of these natural products. Reacting the N-mesylated acetal-containing aniline 127 with TiCl4 allowed for the cyclization and subsequent elimination reaction to the indole motif 128 with a yield of 89%. Next, the authors carried out the indole to asperversiamide E 129.
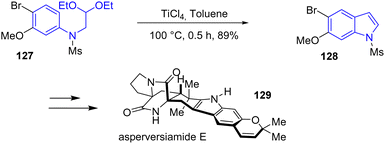 |
| | Scheme 31 Titanium-promoted Friedel–Crafts reaction forming indole skeleton in the synthesis of asperversiamide E 129. | |
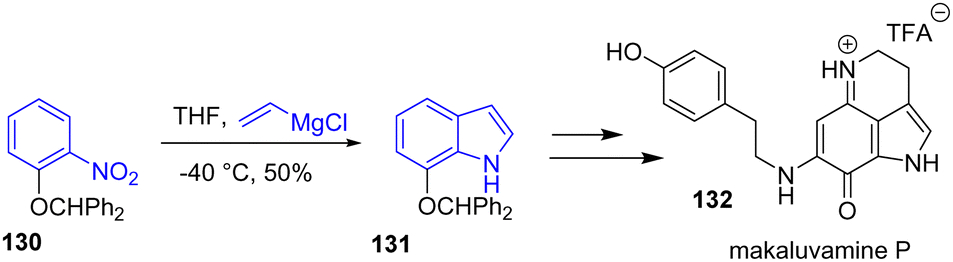
The same year, Kotoku and coworkers reported the unified synthesis and biological evaluation of several makaluvamine alkaloids (Scheme 32).51 To prepare the scaffold of these natural products, a Bartoli indole synthesis was used. Protection of the phenol with benzhydryl bromide followed by a Grignard reaction with vinylmagnesium chloride underwent a Bartoli reaction with a yield of 50%. Afterwards, the indole 131 was used directly for the synthesis of the TFA iminium salt form of makaluvamine P 132.
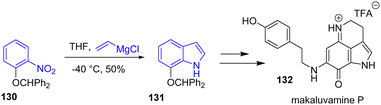 |
| | Scheme 32 Bartoli indole synthesis in the synthesis of makaluvamine P 132. | |
3. Indoline formation in the synthesis of natural products
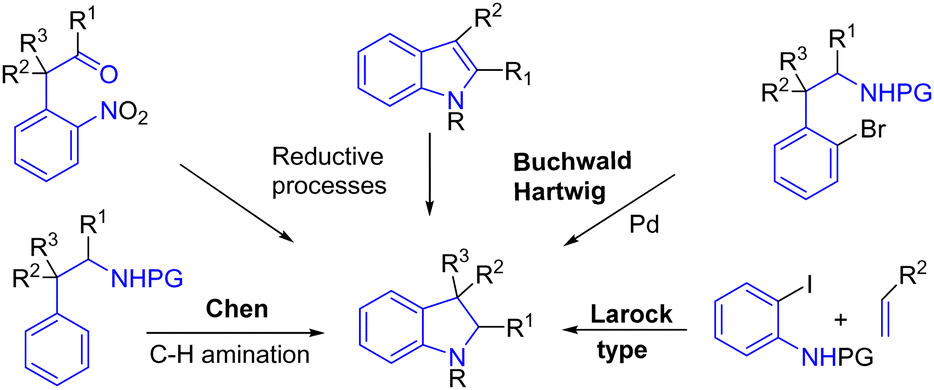
Indole is an aromatic system with ten π electrons. Unlike the indole functional group, indoline is an aromatic derivative with six π electrons where the alkene moiety of indole has been reduced. This subunit is often more elaborate as it could carry asymmetric and quaternary carbon centers. However, their formation is less developed than that of indole, and their synthesis methods are less varied, despite their structure being present in many bioactive natural products. They could be synthesized from an indole or by another strategy such as an intramolecular Buchwald process,52 using palladium and hypervalent iodine reagents53 to promote a remarkable C–H amination, as shown by Chen and coworkers,53a a Larock-type approach,54 or several reductive processes as examples among many other transformations (Scheme 33).
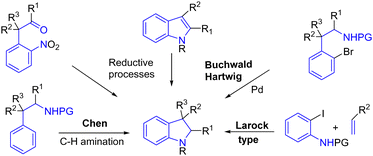 |
| | Scheme 33 Some strategies for indoline synthesis. | |
3.1 Fischer indole processes
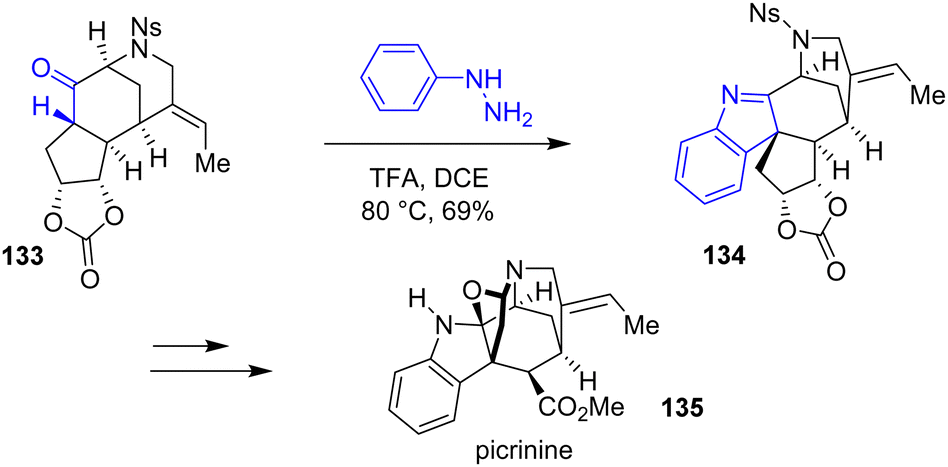
In 2014, Garg and coworkers reported the first total synthesis of picrinine 135 using Fischer indolization (Scheme 34).55 Using a similar approach, they were able to synthesize two other related alkaloids.56 Initially, they proposed an interrupted indole formation using a cyclopentene substrate. However, they were not able to proceed any further with the synthesis because the oxidation step afterwards recovered the starting material or led to decomposition. Instead, the authors proposed a different substrate, containing a cyclic carbonate 133, which underwent Fischer indolization with TFA to a mixture of indolenines 134 and indolines, respectively, in a combined yield of 69% and carried out the synthesis of picrinine 135.
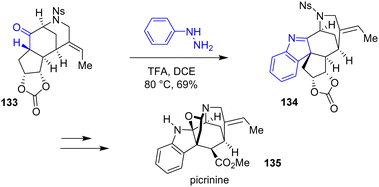 |
| | Scheme 34 Fischer indolization in the synthesis of picrinine 135. | |
3.2 Coupling processes
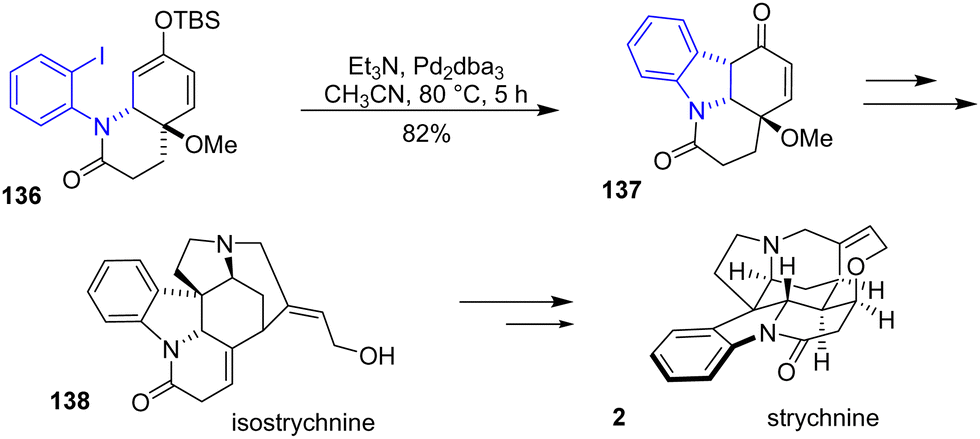
In 2015, Canesi and coworkers reported an unexplored synthetic route of isostrychnine 138, a complex hexacyclic indole alkaloid mediated by a hypervalent iodine reagent (Scheme 35).57 During the synthesis, an indoline formation was required to form the tetracyclic skeleton of isostrychnine. Treatment of ortho-iodoarene 136 by Pd-catalyzed Heck cyclization prompted the formation of indoline intermediate 137 in an 82% yield. This can then be used as the precursor to the synthesis of isostrychnine 138 or even strychnine 2.
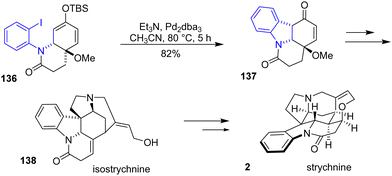 |
| | Scheme 35 Pd-catalyzed Heck cyclization of the indoline skeleton in the synthesis of isoctrychnine 138. | |
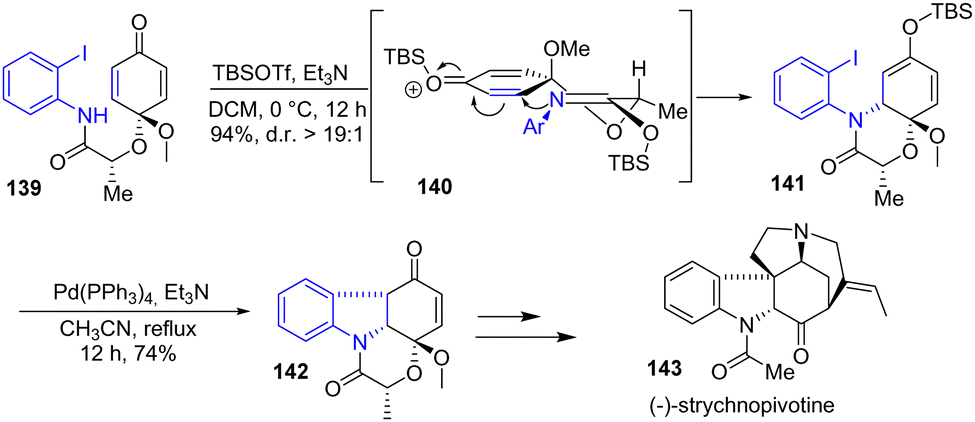
Shortly after, Canesi's group reported a new strategy for the synthesis of strychnos indole alkaloids via a common tetracyclic subunit 142 (Scheme 36).58 To generate this subunit, a Pd-catalyzed Heck cyclization was used to join the amide-anchored aryl iodide with the TBS-protected dienone 141 to introduce the indoline moiety with good diastereoselectivity. The stereoselectivity result was explained by silicon Lewis acid activation leading to transition state 140, where minimized 1,3-diaxial half-chair interactions controlled stereoselectivity, forming TBS-protected enol ether 141 with a yield of 94% and a 19:1 diastereomeric excess. Afterwards, a Pd-catalyzed Heck cyclization gave the desired tetracyclic subunit 142 with a 74% overall yield over two steps. This was then used for the synthesis of (−)-strychnopivotine 143.
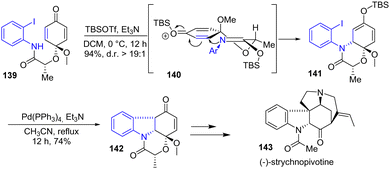 |
| | Scheme 36 Indoline synthesis by Heck cyclization with silyl enol ether in the synthesis of (−)-strychnopivotine 143. | |
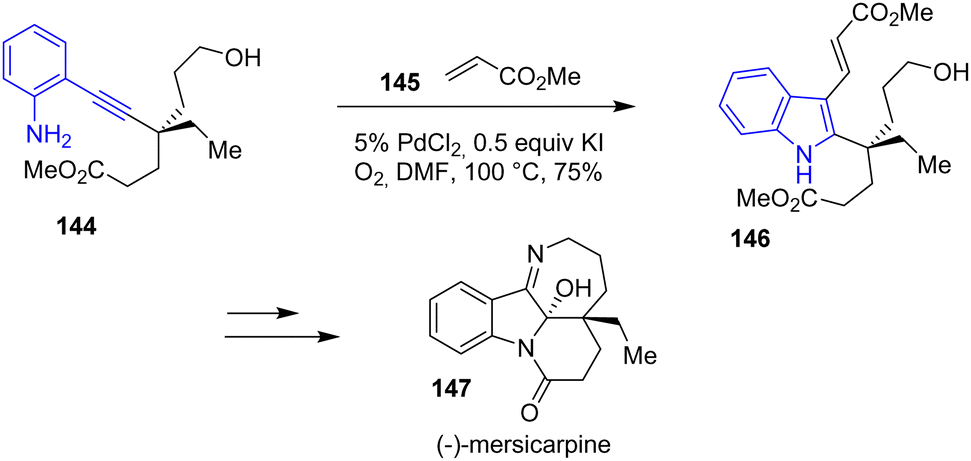
In 2017, Luo and coworkers published a total synthesis of (−)-mersicarpine 147 through an unexpected aziridination/rearrangement oxidation tandem reaction (Scheme 37).59 One of the steps for the preparation of (−)-mersicarpine involves an indole formation through a Pd-catalyzed amine cyclization that could be assimilated to an aza-Wacker/Heck tandem process. Reacting alkynylaniline 144 with PdCl2-KI in the presence of oxygen followed by the addition of methyl acrylate generated indole 146, a precursor to the indoline of (−)-mersicarpine 147 in a yield of 75%.
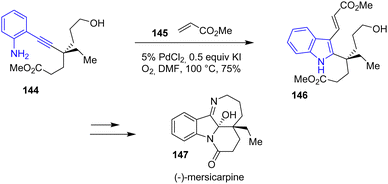 |
| | Scheme 37 Pd-catalyzed amine cyclization of alkynylaniline 144 to indole 146 in the synthesis of (−)-mersicarpine 147. | |
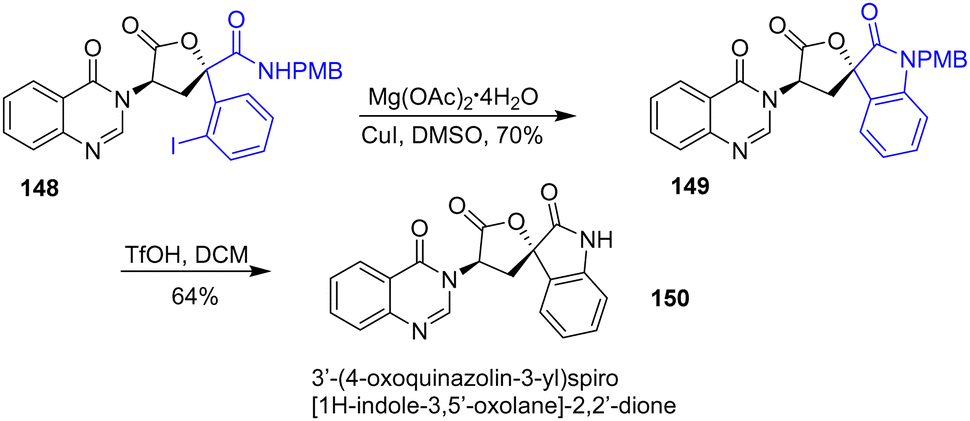
In 2018, Dixon and coworkers published the total synthesis of the tryptoquivaline alkaloid (+)-3′-(4-oxoquinazolin-3-yl)spiro[1H-indole-3,5′-oxolane]-2,2′-dione 150 (Scheme 38).60 At the end of the synthesis, the researchers utilized a copper-(I) catalyzed Buchwald–Hartwig C–N coupling to form the final ring of the natural product. Treatment of aryl iodide 148 with copper iodide and magnesium acetate tetrahydrate in DMSO afforded spiro-oxindole 149 in a yield of 70%. Finally, deprotection of the PMB group afforded natural product 150.
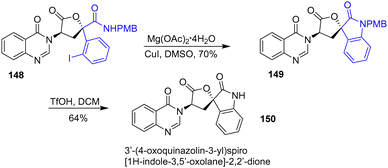 |
| | Scheme 38 Buchwald–Hartwig C–N coupling in the synthesis of 3′-(4-oxoquinazolin-3-yl)spiro[1H-indole-3,5′-oxolane]-2,2′-dione 150. | |
3.3 Reductive cyclization processes
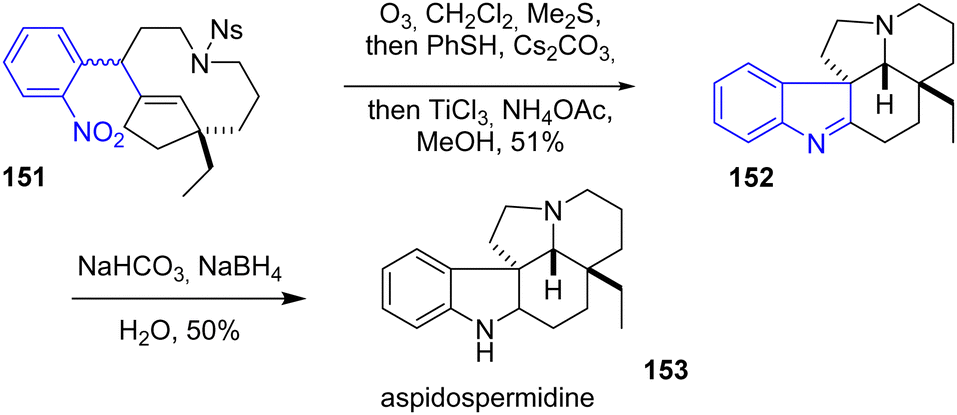
In 2014, Zhu and coworkers reported the synthesis of goniomitine, 1,2-dehydroaspidospermidine, aspidospermidine, vincadifformine, and kopsihainanine A (Scheme 39).30,61 Among these alkaloids, aspidospermidine 153 is interesting synthetically due to its pentacyclic system and the presence of an indoline motif. Their strategy used in Zhu's group involves using an integrative oxidation–reduction–cyclization (iORC) process to form the main core of aspidospermidine 153. Ozonolysis of the double bond of molecule 151, followed by the deprotection of the N-nosyl group with PhSH and Cs2CO3 (Fukuyama process) and reduction of the nitro group with TiCl3 in the presence of an ammonium acetate buffer, afforded the indolenine intermediate 1,2-dehydroaspidospermidine 152 as a single diastereomer with a yield of 51%. Their target was obtained by subsequent reduction.
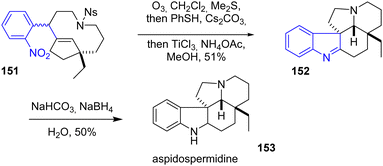 |
| | Scheme 39
iORC process for the synthesis of aspidospermidine 153. | |
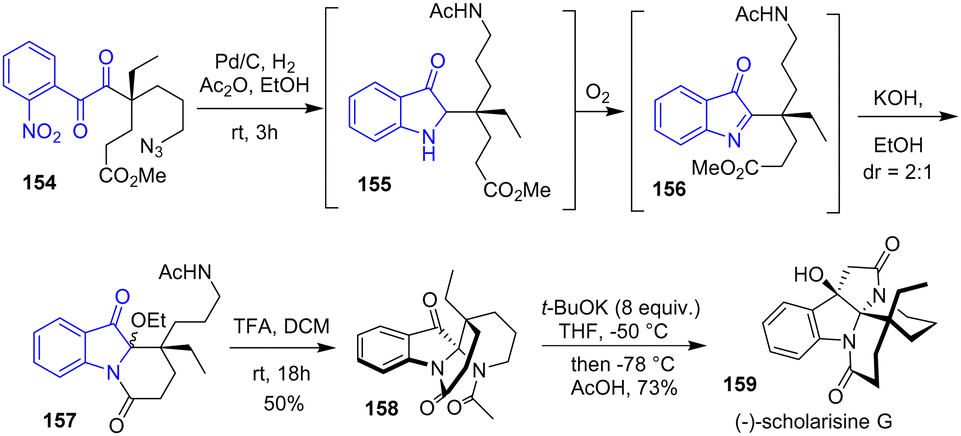
In 2015, the same group achieved the total syntheses of (−)-mersicarpine, (−)-scholarisine G 159, (+)-melodinine E, (−)-leuconoxine, (−)-leuconolam, (−)-leuconodine A, (+)-leuconodine F, and (−)-leuconodine C (Scheme 40).62 Their strategy involves a one-pot reduction-cyclization using hydrogenation and acetic anhydride in 3 steps: (1) simultaneous reduction of both the nitro group and the azido group of substrate 154, (2) condensation of the free amine and the hindered ketone to an imine followed by reduction, and (3) selective N-acetylation of the side-chain amine to intermediate 155. Then, without isolation, the authors exposed precursor 155 to atmospheric oxygen, forming intermediate 156. Then, the addition of potassium hydroxide prompted the lactamization of the tricyclic compound 157, affording a mixture of diastereomers. To resolve the diastereoselectivity, the authors acidified tricyclic compound 157 in a solution of 1:1 TFA in DCM through an N-acyliminium ion to afford spiro indolinone 158 as a single diastereomer in a 50% yield. This was then carried out to (−)-scholarisine G 159.
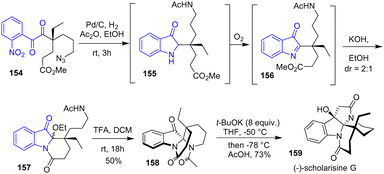 |
| | Scheme 40 Reductive indoline cyclization-N-acetylation cascade in the synthesis of (−)-scholarisine G 159. | |
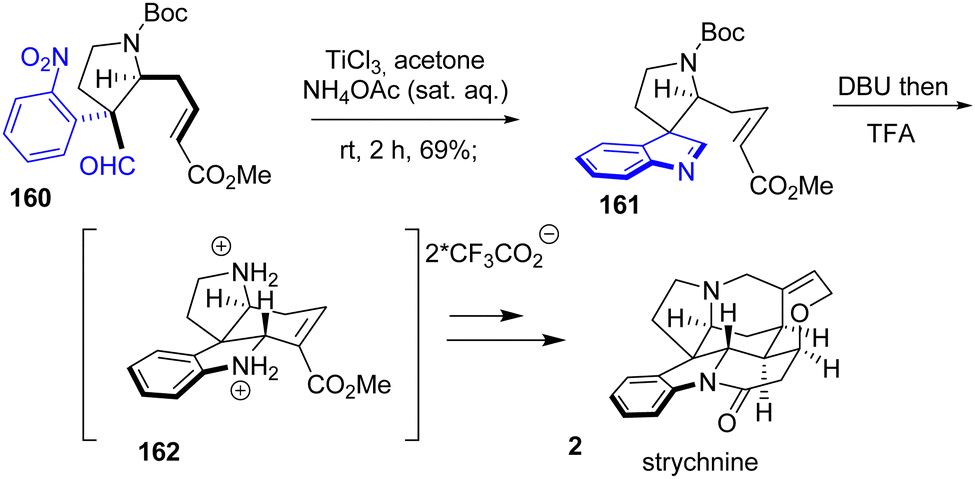
In 2017, Chen and coworkers reported the total synthesis of strychnine 2, a strychnos indole alkaloid (Scheme 41).63 Their strategy involves the reduction of a nitroarene followed by the condensation of an aldehyde to generate an indolenine, an intermediate in the introduction of the indoline moiety. However, the in situ reaction of aniline and aldehyde alone was not able to produce indolenine 161. This was only achieved by treating nitroarene 160 with TiCl3 in a 1:1 solution of ammonium acetate in acetone with a yield of 69%. Subsequent treatment with DBU and then TFA resulted in the formation of their key tetracyclic system 162, which was converted to strychnine 2.
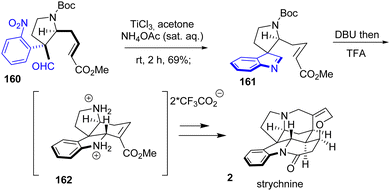 |
| | Scheme 41 Indoline synthesis by imine condensation in the synthesis of strychnine 2. | |
3.4 Radical cyclization processes
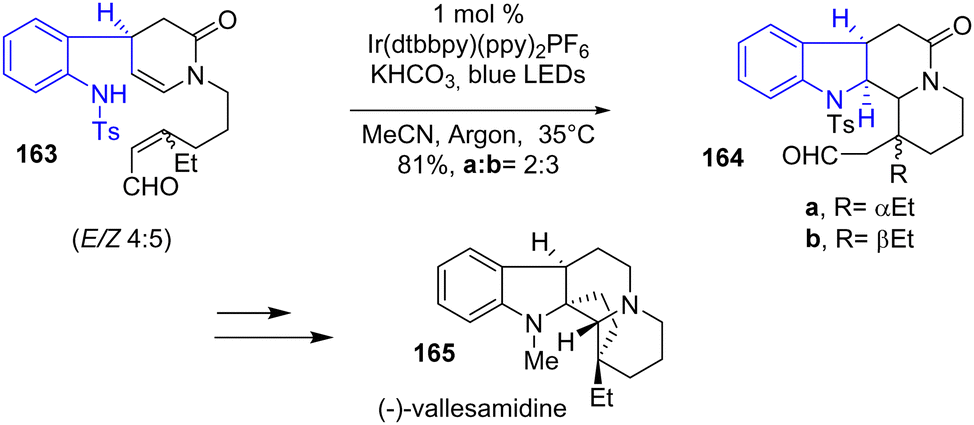
In 2019, Qin and coworkers published the synthesis of 42 different indole alkaloids of 7 different families using a green and economic iridium (Ir)-catalyzed photoredox ring-forming cascade (Scheme 42).64 This process uses Ir(dtbbpy)2PF6 as the photocatalyst in the presence of a weak base and blue LEDs to induce radical cyclization. For example, this process was used in the synthesis of the eburnane alkaloid, (−)-vallesamidine 165, by the reaction of 163 with the photocatalyst to generate a tetracyclic intermediate as a mixture of diastereomers 164 with a yield of 81%.
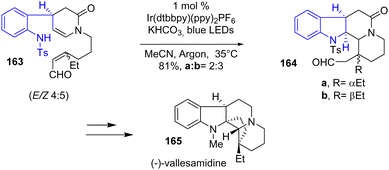 |
| | Scheme 42 Ir-catalyzed photoredox ring-forming cascade reaction in the synthesis of (−)-vallesamidine 165. | |
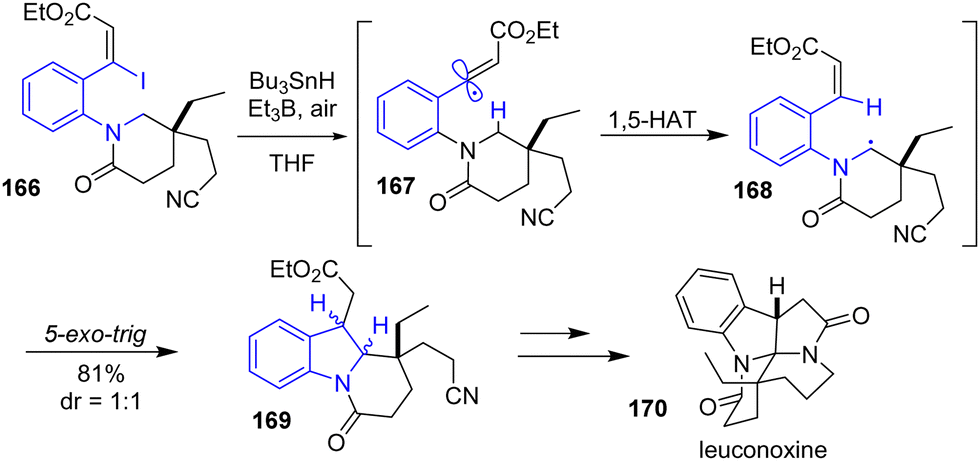
The same year, Beaudry and colleagues reported the total synthesis of leuconoxine 170, melodinine E, and mersicarpine using a radical translocation-cyclization cascade reaction (Scheme 43).65 Their strategy involves using an aryl-vinyl iodide in the presence of an organotin reagent, an alkylborane, and air to promote a 1,5-Heteroatom Transfer (1,5-HAT) radical cyclization to the indoline 169. This approach was used to synthesize leuconoxine 170 using tributyltin hydride and triethylborane from aryl-vinyl iodide 166 through radical intermediates 167 and 168 in 81% yields, Scheme 43.
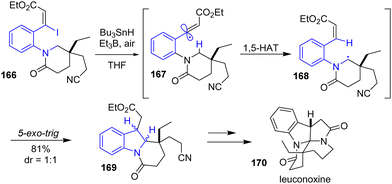 |
| | Scheme 43 Radical translocation-indoline cyclization cascade of aryl-vinyl iodide 166 to leuconoxine 170. | |
3.5 Miscellaneous processes
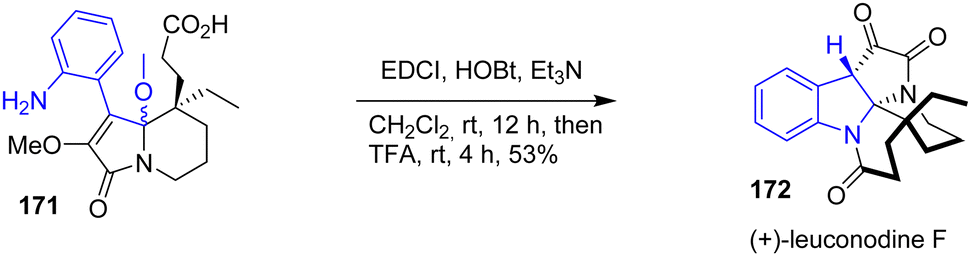
In 2016, Zhu and coworkers published an enantioselective total synthesis of (−)-rhazinilam, (−)-leucomidine B, and (+)-leuconodine F (Scheme 44).34 Among these natural products, (+)-leuconodine F 172 is synthetically intriguing due to the presence of the indoline moiety in its structure. To implement the indoline moiety, the authors proposed a lactamization followed by an addition of an acid to promote a diastereoselective transannular cyclization of two rings simultaneously. EDCI coupling of 171 with HOBt followed by the addition of TFA led to (+)-leuconodine F 172 with a yield of 53% as a single diastereomer.
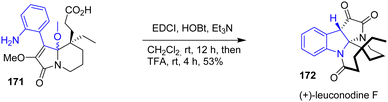 |
| | Scheme 44 Enantioselective total synthesis of (+)-leuconodine F 172 through acid-promoted indoline-ring formation. | |
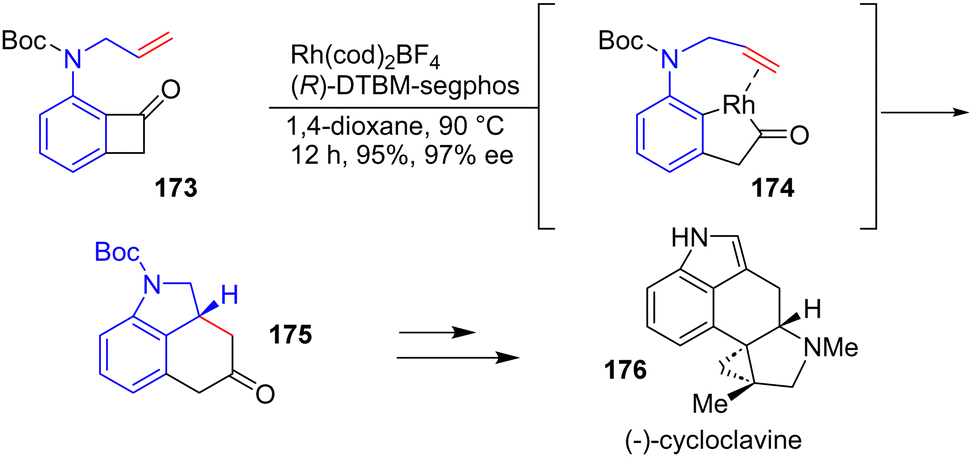
In 2018, Dong and coworkers reported an enantioselective total synthesis of ergot alkaloids (−)-cycloclavine 176via a transition metal-catalyzed C–C bond formation (Scheme 45).66 Their “cut-and-sew” approach involves an Rh-catalyzed C–C activation of nitrogen-tethered benzocyclobutanones with olefins to promote intramolecular indoline formation and the ring expansion of cyclobutanones to form the framework of (−)-cycloclavine 176. Reacting Boc-protected N-allyl benzocyclobutanone 173 in the presence of a Rh(cod)2BF4 precatalyst using (R)-DTBM-segphos as the ligand allowed the enantioselective formation of tricyclic indoline 175 in 95% yields and 97% enantiomeric excess. This was then carried out to (−)-cycloclavine 176.
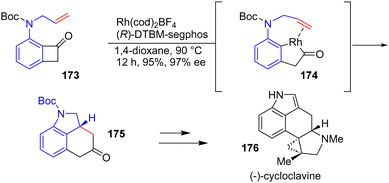 |
| | Scheme 45 Rh-Catalyzed asymmetric cut-and-sew reaction for the formation of indoline 175 in the synthesis of (−)-cycloclavine 176. | |
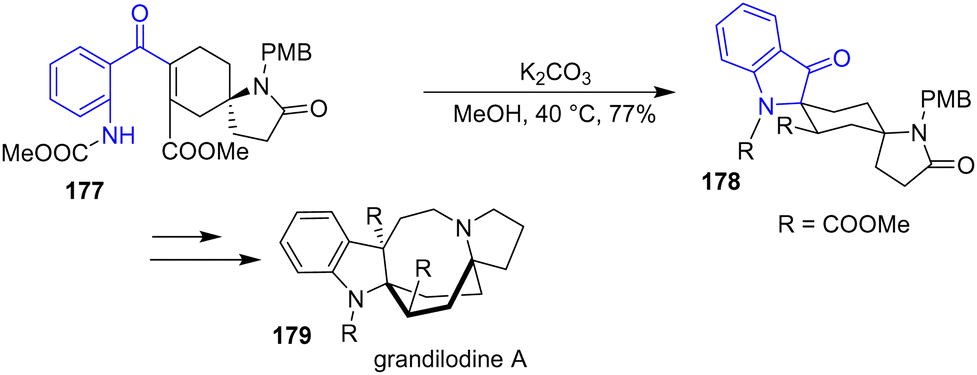
In 2022, the Zu group reported an enantioselective and regioselective formal synthesis of spiro[cyclohexane-2-indoline] alkaloids lundurine A, lapidilectine B, and grandilodine C, as well as the synthesis of grandilodine A 179 (Scheme 46).67 These compounds are intriguing due to their rigid cyclohexane boat conformation motif within their structures. Treatment of 177 with K2CO3 in methanol produced indolinone intermediate 178 with a yield of 77%. Using this intermediate, they synthesized grandilodine A 179.
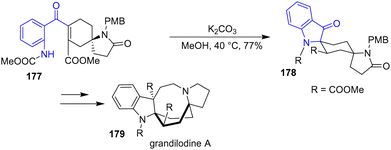 |
| | Scheme 46 Base-promoted indoline synthesis in the synthesis of grandilodine A 179. | |
4. Conclusion
In summary, we report an overview of several recent strategies that allow the formation of indoles and indolines present in complex alkaloids. It appears that although it is an old reaction, the Fischer indole process is still used as a crucial step. Palladium is also involved in several coupling processes to elaborate these heterocycles. Most strategies involve a reductive cyclization of a nitroaryl segment on a ketone or aldehyde. Some gold catalyst processes have also been reported. Finally, other processes have also been used occasionally. Throughout the methods mentioned in this review, we observed that the indole moiety is often introduced early into the total synthesis, possibly to prevent the incompatibility with other functional groups during its formation. This shows that the late-stage indole/indoline introduction into natural products remains an important synthetic challenge to this day. Surprisingly, despite the numerous methods known for the preparation of indoles, only a few strategies have been used extensively, demonstrating that they are robust processes and tools for the synthesis of complex alkaloids. This shows that the formation of indoles and indolines in natural products is a topic of high interest.
Data availability
No primary research results, software, or code have been included, and no new data were generated or analyzed as part of this review.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful to the Natural Sciences and Engineering Research Council of Canada (NSERC) and the provincial government of Quebec FRQNT (CCVC-CGCC Strategic Clusters RS-265155) for their financial support of this research.
Notes and references
- A. Kumari and R. K. Singh, Bioorg. Chem., 2019, 89, 103021 Search PubMed.
-
H. L. Pearce, In The Alkaloids, ed. Brossi, A., Suffness, M., Academic, San Diego, CA, 1990, vol. 37, p. 145 Search PubMed.
- S. Tang and G. Vincent, Chem. – Eur. J., 2021, 27, 2612 CrossRef CAS.
-
J. E. Saxton, In The Alkaloids: Chemistry and Biology, ed., Geoffrey, A. C., Academic Press, 1998, vol. 51, p. 1 Search PubMed.
-
P. M. Dewick, Medicinal Natural Products, Wiley, 2009 Search PubMed.
- C. Lavaud and G. Massiot, Prog. Chem. Org. Nat. Prod., 2017, 105, 89 CAS.
-
(a) D. Lachkar, N. Denizot, G. Bernadat, K. Ahamada, M. A. Beniddir, V. Dumontet, J.-F. Gallard, R. Guillot, K. Leblanc, E. Otogo N'nang, V. Turpin, C. Kouklovsky, E. Poupon, L. Evanno and G. Vincent, Nat. Chem., 2017, 9, 793 CrossRef CAS PubMed;
(b) M. Jarret, V. Turpin, A. Tap, J. F. Gallard, C. Kouklovsky, E. Poupon, G. Vincent and L. Evanno, Angew. Chem., Int. Ed., 2019, 58, 9861 CrossRef CAS PubMed;
(c) Y. Dou, C. Kouklovsky, V. Gandon and G. Vincent, Angew. Chem., Int. Ed., 2020, 59, 1527 CrossRef CAS PubMed;
(d) C. Wei, Y. Dou, C. Kouklovsky and G. Vincent, Angew. Chem., Int. Ed., 2022, 61, e202209135 CrossRef PubMed;
(e) A. Mauger, M. Jarret, A. Tap, R. Perrin, R. Guillot, C. Kouklovsky, V. Gandon and G. Vincent, Angew. Chem., Int. Ed., 2023, 62, e202302461 CrossRef CAS PubMed;
(f) J. Xu, L.-D. Shao, D. Li, X. Deng, Y.-C. Liu, Q.-S. Zhao and C. Xia, J. Am. Chem. Soc., 2014, 136, 17962 CrossRef CAS PubMed;
(g) J. Ren, S.-H. Ding, X.-N. Li and Q.-S. Zhao, J. Am. Chem. Soc., 2024, 146, 7616 CrossRef CAS PubMed;
(h) R. Lavernhe, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2025, 64, e202414612 CrossRef CAS PubMed;
(i) G. V. Ramakrishna, Z. Latif and F. Romiti, J. Am. Chem. Soc., 2025, 147, 4613 CAS;
(j) M. Ghosh, S. Saha and M. S. Maji, Chem. – Eur. J., 2025, 31, e202403966 CAS;
(k) D. Hiruma, A. Yoshidome, K. Rakumitsu, M. Kitajima, Y. Hitora, S. Tsukamoto, J. Schinnerl, L. Brecker and H. Ishikawa, Chem. – Eur. J., 2025, 31, e202500069 CrossRef PubMed;
(l) L. Shivers, J. Goodyear and S. D. Taylor, Org. Lett, 2025, 27, 450 CAS.
- E. Fischer and F. Jourdan, Chem. Ber., 1883, 16, 2241 Search PubMed.
- W. Madelung, Ber. Dtsch. Chem. Ges., 1912, 45, 1128 Search PubMed.
- R. C. Larock and E. K. Yum, J. Am. Chem. Soc., 1991, 113, 6689 CAS.
- G. Bartoli, R. Dalpozzo and M. Nardi, Chem. Soc. Rev., 2014, 43, 4728 CAS.
-
(a) T. Fukuyama, X. Chen and G. Peng, J. Am. Chem. Soc., 1994, 116, 3127 CrossRef CAS;
(b) H. Tokuyama, T. Yamashita, M. T. Reding, Y. Kaburagi and T. Fukuyama, J. Am. Chem. Soc., 1999, 121, 3791 CrossRef CAS.
- J. R. Henry and J. H. Dodd, Tetrahedron Lett., 1998, 38, 8763 CrossRef.
- H. Hemetsberger and D. Knittel, Monatsh. Chem., 1972, 103, 194 CrossRef CAS.
- S. Krüger and T. Gaich, Angew. Chem., Int. Ed., 2014, 54, 315 CrossRef PubMed.
- H. Rebmann, C. K. G. Gerlinger and T. Gaich, Chem. – Eur. J., 2019, 25, 2704 CrossRef CAS PubMed.
- P. W. Tan, J. Seayad and D. J. Dixon, Angew. Chem., Int. Ed., 2016, 55, 13436 CrossRef CAS PubMed.
- A. W. Gregory, A. Chambers, A. Hawkins, P. Jakubec and D. J. Dixon, Chem. – Eur. J., 2015, 21, 111 CrossRef CAS PubMed.
- Q. Tan, Z. Yang, D. Jiang, Y. Cheng, J. Yang, S. Xi and M. Zhang, Angew. Chem., Int. Ed., 2019, 58, 6420 CrossRef CAS PubMed.
- J. B. Roque, E. V. Mercado-Marin, S. C. Richter, D. P. de Sant'Ana, K. Mukai, Y. Ye and R. Sarpong, Chem. Sci., 2020, 11, 5929 RSC.
- W. F. Cheng, S. Ma, Y. T. Lai, Y. T. Cheung, K. Akkarasereenon, Y. Zhou and R. Tong, Angew. Chem., Int. Ed., 2023, 62, e202311671 CrossRef CAS PubMed.
- W. Zhou, S. Xi, H. Chen, D. Jiang, J. Yang, S. Liu, L. He, H. Qiu, Y. Lan and M. Zhang, Nat. Chem., 2023, 15, 1074 CrossRef CAS PubMed.
- N. Hauser, M. A. Imhof, S. S. Eichenberger, T. Kundig and E. M. Carreira, Angew. Chem., Int. Ed., 2022, 61, e202112838 CrossRef CAS PubMed.
- L.-D. Guo, Z. Xu and R. Tong, Angew. Chem., Int. Ed., 2022, 134, e202115384 CrossRef.
- K. Koada, K. Ojima, H. Ueda and H. Tokuyama, J. Am. Chem. Soc., 2023, 145, 16337 CrossRef PubMed.
- T. Huber, T. A. Preuhs, C. K. G. Gerlinger and T. Magauer, J. Org. Chem., 2017, 82, 7410 CrossRef CAS PubMed.
- C. F. Cain, E. H. Howard, J. A. Goodwin and J. R. Del Valle, Arkivoc., 2019, iv, 80 Search PubMed.
- S. P. Rezgui, J. Farhi, H. Yu, Z. P. Sercel, S. C. Vergil and B. M. Stoltz, Chem. Sci., 2024, 15, 12284 CAS.
- A. M. Levinson, Org. Lett., 2014, 16, 4904 CAS.
- O. Wagnieres, Z. Xu, Q. Wang and J. Zhu, J. Am. Chem. Soc., 2014, 136, 15102 CAS.
- S. H. Tan, M. G. Banwell and A. C. Willis, J. Org. Chem., 2016, 81, 8022 CAS.
- M. Mizutani, S. Yasuda and C. Mukai, Chem. Commun., 2014, 50, 5782 CAS.
- E. V. Mercado-Marin and R. Sarpong, Chem. Sci., 2015, 6, 5048 CAS.
- D. Dagoneau, Z. Xu, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2016, 55, 760 CAS.
- A. Nakayama, T. Nakamura, T. Zaima, S. Fujimoto, S. Karanjit and K. Namba, Angew. Chem., Int. Ed., 2021, 60, 635 CAS.
- G. Li, N. Gaeng, C. Piemontesi, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2021, 60, 12392 CAS.
- W. P. Thomas and S. V. Pronin, Acc. Chem. Res., 2021, 54, 1347 CAS.
- K. Signo and S. Canesi, Org. Lett., 2022, 24, 4939 CAS.
- B. Horst, D. S. Verdoorn, S. Hennig, G. van der Heijden and E. Ruijter, Angew. Chem., Int. Ed., 2022, 61, e202210592 CAS.
- S. Coulibali, T. Godou and S. Canesi, Org. Lett., 2016, 18, 4348 CAS.
- M. G. Banwell, L. V. White and S. Y. Ye, J. Org. Chem., 2022, 87, 14407 CAS.
- T.-H. Jeon and C.-G. Cho, Org. Lett., 2023, 25, 3755 CAS.
- K. Yang, F. Zhou, Z. Kuang, G. Gao, T. G. Driver and Q. Song, Org. Lett., 2016, 18, 4088 CAS.
- J. Matsuoka, Y. Matsuda, Y. Kawada, S. Oishi and H. Ohno, Angew. Chem., Int. Ed., 2017, 56, 7444 CAS.
- F. Xu and M. W. Smith, Chem. Sci., 2021, 12, 13756 CAS.
- H.-Y. Bin, K. Wang, D. Yang, X.-H. Yang, J.-H. Xie and Q.-L. Zhou, Angew. Chem., Int. Ed., 2019, 58, 1174 CrossRef CAS PubMed.
- L. Li, K. Yuan, Q. Jia and Y. Jia, Angew. Chem., Int. Ed., 2019, 58, 6074 CrossRef CAS PubMed.
- S. R. McCabe and P. Wipf, Synthesis, 2019, 213 CAS.
- C. Bae, E. Park, C.-G. Chu and C.-H. Cheon, Org. Lett., 2020, 22, 2354 CrossRef CAS PubMed.
- Z. Xu, X.-T. Liang, P. Lan and M. G. Banwell, Org. Chem. Front., 2024, 11, 2433 RSC.
- Y. Kiichi, K. Fukuoka, A. Kitano, K. Ishino and N. Kotoku, Molecules, 2024, 29, 1389 Search PubMed.
-
(a) B. H. Yang and S. L. Buchwald, Org. Lett., 1999, 1, 35 CrossRef CAS PubMed;
(b) B. Haffemayer, M. Gulias and M. J. Gaunt, Chem. Sci, 2011, 2, 312 Search PubMed.
-
(a) G. He, C. Lu, Y. Zhao, W. A. Nack and G. Chen, Org. Lett., 2012, 14, 2944 CrossRef CAS PubMed;
(b) T.-S. Mei, D. Leow, H. Xiao, B. N. Laforteza and J.-Q. Yu, Org. Lett., 2013, 15, 3058 Search PubMed;
(c) C. Rocq, M. Denis and S. Canesi, Chem. Commun., 2023, 59, 6495 Search PubMed.
- S. Z. Tasker and T. F. Jamison, J. Am. Chem. Soc., 2015, 137, 9531 Search PubMed.
-
(a) J. M. Smith, J. Moreno, B. W. Boal and N. K. Garg, J. Am. Chem. Soc., 2014, 136, 4504 Search PubMed;
(b) J. M. Smith, J. Moreno, B. W. Boal and N. K. Garg, J. Org. Chem., 2015, 80, 8954 Search PubMed.
-
(a) J. Moreno, E. Picazo, L. A. Morrill, J. M. Smith and N. K. Garg, J. Am. Chem. Soc., 2016, 138, 1162 Search PubMed;
(b) E. Picazo, L. A. Morrill, R. B. Susick, J. Moreno, J. M. Smith and N. K. Garg, J. Am. Chem. Soc., 2018, 140, 6483 Search PubMed.
- G. Jacquemot, G. Maertens and S. Canesi, Chem. – Eur. J., 2015, 21, 7713 Search PubMed.
-
(a) G. Maertens and S. Canesi, Chem. – Eur. J., 2016, 22, 7090 Search PubMed;
(b) G. Maertens, E. Deruer, M. Denis and S. Canesi, J. Org. Chem., 2020, 85, 6098 Search PubMed.
- Y. Zhang, Y. Xue and T. Luo, Tetrahedron, 2017, 73, 4201 Search PubMed.
- T. Wei and D. J. Dixon, Chem. Commun., 2018, 54, 12860 CAS.
- B. Delayre, C. Piemontesi, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2020, 59, 13990 Search PubMed.
- Z. Xu, Q. Wang and J. Zhu, J. Am. Chem. Soc., 2015, 137, 6712 CAS.
- G. S. Lee, G. Namkoong, J. Park and D. Y.-K. Chen, Chem. – Eur. J., 2017, 23, 16189 CAS.
- X.-Y. Liu and Y. Qin, Acc. Chem. Res., 2019, 52, 1877 Search PubMed.
- R. Kim, A. J. Ferreira and C. M. Beaudry, Angew. Chem., Int. Ed., 2019, 58, 12595 CAS.
- L. Deng, M. Chen and G. Dong, J. Am. Chem. Soc., 2018, 140, 9652 CAS.
- L. Chen, K. Xie, J. Zhang and L. Zu, Angew. Chem., Int. Ed., 2022, 61, e202212042 CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Maxime
Denis
and
Sylvain
Canesi
,
Maxime
Denis
and
Sylvain
Canesi