
Uncovering tetrazoles as building blocks for constructing discrete and polymeric assemblies
Soumya
Dey†
 ,
Medha
Aggarwal†
,
Debsena
Chakraborty
and
Partha Sarathi
Mukherjee
*
,
Medha
Aggarwal†
,
Debsena
Chakraborty
and
Partha Sarathi
Mukherjee
*
Department of Inorganic and Physical Chemistry, Indian Institute of Science, Bangalore-560012, India. E-mail: psm@iisc.ac.in
First published on 1st May 2024
Abstract
Metal–organic self-assembly with flexible moieties is a budding field of research due to the possibility of the formation of unique architectures. Tetrazole, characterised by four nitrogen atoms in a five-member ring, exhibits immense potential as a component. Tetrazole offers four coordination sites for binding to the metal centre with nine distinct binding modes, leading to various assemblies. This review highlights different polymeric and discrete tetrazole-based assemblies and their functions. The meticulous manipulation of stoichiometry, ligands, and metal ions required for constructing discrete assemblies has also been discussed. The different applications of these architectures in separation, catalysis and detection have also been accentuated. The latter section of the review consolidates tetrazole-based cage composites, highlighting their applications in cell imaging and photocatalytic applications.
 Clockwise from the top left: Partha Sarathi Mukherjee, Medha Aggarwal, Debsena Chakraborty and Soumya Dey | Soumya Dey received his BSc in 2020 from Midnapore College under Vidyasagar University and his MSc in Chemistry in 2022 from IIT Kanpur. He is currently pursuing his PhD at the Indian Institute of Science, Bangalore, under the supervision of Prof. P. S. Mukherjee. His research interests include chiral molecular cages and various chemical reactions under confinement. Medha Aggarwal received her BSc degree, majoring in chemistry, in 2021 from Doon University and her MS in chemistry in 2023 from the Indian Institute of Science, Bangalore. She is currently pursuing her PhD degree, working with Prof. P. S. Mukherjee at the Indian Institute of Science, Bangalore. Her principal research interests are on stimuli-responsive materials and exploring photophysical and electrochemical aspects of coordination cages. Debsena Chakraborty received his BSc in 2017 from the University of Burdwan and MS in chemistry in 2019 from the Indian Institute of Science, Bangalore. He is currently pursuing his PhD degree, working with Prof. P. S. Mukherjee at the Indian Institute of Science, Bangalore. His principal research interests concern various applications of water-soluble metal–organic architectures. Partha Sarathi Mukherjee worked at IACS (Kolkata) for his PhD on magnetic studies of Cu(II) coordination polymers. Subsequently, he worked as a postdoctoral fellow at the University of Utah. He was an Alexander von Humboldt fellow at the University of Göttingen before joining the Indian Institute of Science (IISc) as a faculty in 2005. Partha is currently a professor of chemistry at IISc (Bangalore), where his group is mainly focused on chemistry inside confined space of self-assembled molecular cages. He is a recipient of S. S. Bhatnagar prize and the Swarnajayanti fellowship of the Govt. of India. He is an elected fellow of the Indian Academy of Sciences, and the World Academy of Science (TWAS). Partha is/was in the editorial advisory boards of Inorganic Chemistry, Inorganica Chimica Acta, Inorganic Chemistry Frontiers, The Chemical Records, and Scientific Reports. He is currently serving as an Associate Editor of Inorganic Chemistry. |
1. Introduction
Self-assembly is the process through which multiple components can associate to give an ordered structure. It is like a Lego game where pieces combine to manifest into various intricate structures. The self-assembly process is ubiquitous in nature and is found in atomic interactions, cellular replications and in RNA, DNA, and protein folding.1 Intrigued by the extensive use of self-assembly in nature and the desire to replicate such process in artificial systems, the field of supramolecular chemistry has since then been rapidly developing.Supramolecular chemistry dates its origin to the discovery of crown ethers, spherands, and cryptands by Pederson,2 Cram3 and Lehn,4 respectively. Lehn defined supramolecular chemistry as the “chemistry beyond the molecule”. In other words, it describes how individual species can communicate through various non-covalent interactions (ion–ion, ion–dipole, hydrogen bonding, cation–π, anion–π, π–π, hydrophobic and van der Waals interactions). These interactions help to develop complex structures that can be challenging to construct using conventional covalent chemistry.5–10 Since the final architecture is made from the association of multiple building blocks, the properties of these blocks are often translated into the final structure.11–15 This brings us closer to developing artificial analogues of naturally occurring systems.16,17
As mentioned earlier, the self-assembly of supramolecular architectures can take place by utilising various interactions. For example, hydrogen bonding is the primary mode of interaction between DNA strands (complementary purine and pyrimidine bases). Rebek and co-workers applied this principle of complementarity to form different hydrogen-bonded capsules.18,19 Another approach can be the use of electrostatic and aromatic interactions.20,21 However, the outcome of these electrostatic and aromatic interactions is often unpredictable due to a lack of directionality. This makes it challenging to postulate the consequences, and thus, such interactions are less commonly used to make self-assembled architectures.22
Coordination-driven self-assembly is an alternative, robust method that allows predictability and certainty in designing molecular assemblies.23–25 This approach also has a self-correcting trait due to the dynamic nature of the metal–ligand bonds. This allows concomitant bond cleavage and formation, which leads to the development of thermodynamically stable product(s).26,27 The coordination-driven self-assembly field has evolved over the years, starting from the seminal work by Lehn28 and Sauvage29 to develop metal–organic helicates,30–32 catenates,33–35 grids,36 knots37,38 and rotaxanes.39–41 The coordination-driven approach is high-yielding, and the ease of such synthesis has led to the development of a plethora of assemblies.42 Such assemblies can be either polymeric or discrete, depending on the binding of ligands and metals. Polymeric assemblies mainly constitute coordination polymers (CPs)43 and their subclass metal–organic frameworks (MOFs).44 Both CPs and MOFs comprise metal/metal clusters as the node and organic units as the linkers, forming infinite and versatile porous networks. The growth of these polymeric assemblies has been exponential, and such materials have been used in gas adsorption, sensing, and biomedical uses.45 Nonetheless, these assemblies suffer from a significant drawback of solution-phase processibility due to their low solubility in most polar and non-polar solvents. On the other hand, discrete coordination assemblies, which are comprised of metal–organic cages (MOCs) and metallacycles (MCs), exhibit better solubility in polar and non-polar solvents.46–49 These structures have diverse applications ranging from catalysis,50–57 selective separation,58–62 fluorescence tuning,63–68 sensing,69–71 to photochromism.72,73
Most self-assembled coordination assemblies use pyridines, carboxylates and imidazoles as the primary linking motifs to the metal centres. All these motifs provide a rigid mono- or bi-dentate binding site. The architectures developed by these motifs using coordination self-assembly are generally predictable and lead to the formation of only a certain number of structures.74 Tuning the motif using ligands with additional donor sites can open new possibilities for constructing unprecedented structures. This makes tetrazoles a prime candidate for the formation of unusual structures.
Tetrazole is a five-membered aromatic heterocyclic ring with 6-π electrons. It consists of four nitrogen atoms and one carbon atom, where all the nitrogen atoms are linked contiguously. Tetrazoles exist in two tautomeric forms (Fig. 1), where 1H is predominant in the solution state, and 2H tautomer predominates in the gaseous phase.75 They are a class of stable heterocycles with the most contiguous nitrogen atoms (as pentazoles are explosive even at low temperatures). The tetrazole motif possesses important traits in terms of low basicity, low pKa values, higher nitrogen count, high formation enthalpy, good stability, high dipole moment and the presence of a mobile N–H hydrogen donor. These properties have led to the fusion of tetrazoles in profuse applications such as antihypertensive drugs, energy materials, efficient anticancer and antitumour agents, starting materials for propellants, analytical reagents in material science, and as ligands in organisation of complexes. Tetrazoles are bioequivalent of carboxylic acid due to their metabolic stability, so they are also a valuable substitute in drug designs.76
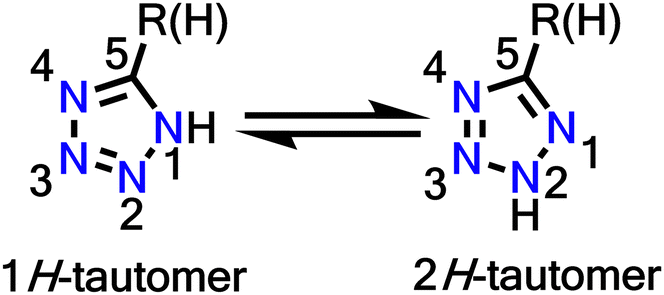 | ||
| Fig. 1 The two stable tautomers of tetrazole. | ||
The development of tetrazole-based ligands has been a fascinating topic because of their strong coordination abilities. The four nitrogens of tetrazole have four lone pairs oriented in different directions, which provides diversity in binding. Tetrazole can participate in nine diverse binding modes (Fig. 2), including monodentate (two modes), bidentate (four modes), tridentate (two modes) and a tetradentate mode.77 Tetrazole can act as a multidentate or bridging ligand, depending on the modes. The nitrogen atom of tetrazole can also bind to metal as a neutral ligand or anionic ligand through the deprotonation of the N–H bond.
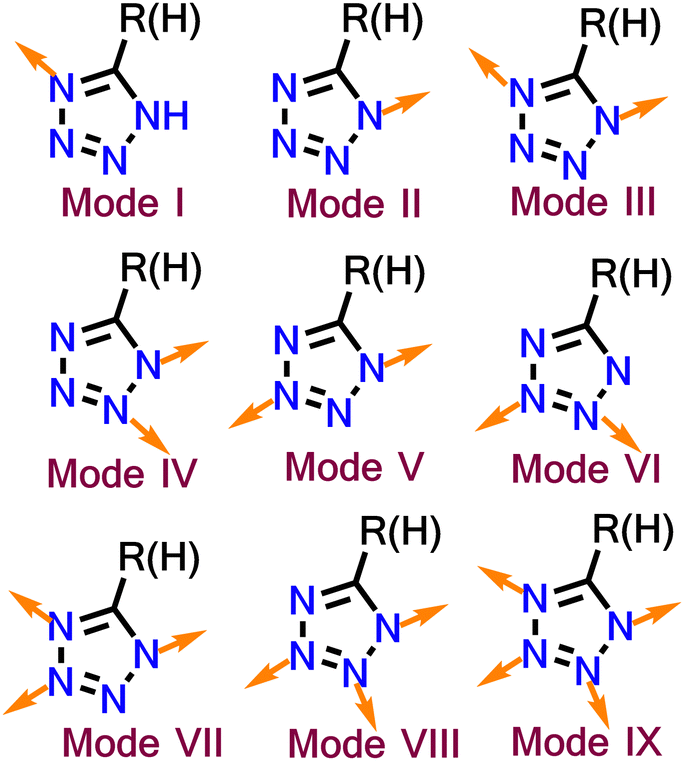 | ||
| Fig. 2 Different possible binding modes of tetrazole. | ||
This review will focus on the use of tetrazoles as the building units. The aim is to highlight the binding affinity of tetrazole to develop molecular assemblies via coordination-driven self-assembly. The initial part of the review will present a short discussion on tetrazole-based coordination polymers and their subclass metal–organic frameworks, followed by details on the formation of tetrazole-based discrete assemblies and their various applications.
2. Tetrazole-based polymeric assemblies
The tetrazole core has diverse binding motifs and has often been used to construct polymeric assemblies by bonding with different metal ions. These metal-derived polymeric assemblies belong to the category of “coordination polymers”. Coordination polymers (CPs) can form infinite 1D chains, 2D sheets or 3D networks composed of metal ions/clusters and organic ligands connected via coordination bonds.78 MOFs (metal–organic frameworks) are a subclass of coordination polymers with well-defined pores or voids.79,80 The scope of tetrazole-based CPs has been discussed in other reviews.77,80,81 Thus, this review will only provide a glimpse of the use of tetrazole in polymeric assemblies (CPs and MOFs), detailing a few unique examples and applications.In the first example, Zhong et al. showed the formation of different coordination polymers (CPs) with distinct topologies using the reaction of Cu(II) or Zn(II) salt and ethyl tetrazolate-5-carboxylate (H-1-COOEt). This reaction resulted in the formation of three CPs [Zn3(1)6(H2O)2]n (2), [Cu2(1)3(OH)]n (3), and {[Cu5(1)9](NO3)3·8H2O}n (4) (Fig. 3). The tetrazolate ligand 1 was generated by an in situ decarboxylation reaction of H-1-COOEt.
 | ||
| Fig. 3 Schematic representation for the formation of coordination polymers 2 (kgd layer), 3 (fsc net) and 4 (acs net). Crystal structures of 2,3,4 obtained from ref. 83 [color code: blue, N; black, C; brown, Cu; purple, Zn (H atoms and anions omitted for clarity)]. | ||
Coordination polymer 2 ([Zn3(1)6(H2O)2]n) had a rare Kagome dual 2D topological layer structure. In this polymer, the tetrazolate ligand 1 displayed a bidentate nature with mode III (Fig. 2) binding in the network. Coordination polymer 3 had a different 3D pillared layered structure with fcs topology net. The ligand 1 showed both bidentate (mode IV) and tridentate (mode VII) binding to the metal (Cu(II)) centre. The coordination polymer 4 exhibited a 3D porous network with an acs topology net, and 1 exhibited both tridentate (mode VIII) and tetradentate (mode IX) binding to the metal. Owing to its unique structure, coordination polymer 4 could selectively adsorb CO2 and H2 over N2. Coordination polymer 2, on the other hand, displayed photoluminescence upon excitation at 292 nm due to LMCT between the zinc centre and the tetrazolate ligand 1.82 Similar polymeric materials with other metals like Cobalt (Co(II)) have also been reported.83
Micro/nano infinite coordination polymers (ICPs) have recently drawn considerable attention because of their unique physical and chemical properties. Recently, a tetrazole-based ICP was reported to encapsulate TiO2 and form a distinct kind of ZnO@TiO2 core–shell structure (Fig. 4). The reaction of Zn(NO3)2·6H2O (5) with 1,3-bis(tetrazol-5-yl methyl) benzene (6), rhodamine B (7) and TiO2 (8) in 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100:3.3:1.25 molar ratio in DMF resulted in the formation of a coordination polymer-dye-nanoparticle composite [7/8@Zn(6)] (9). 7 was firmly bound to the surface of TiO2 due to esterification between the carboxylic group of 7 and the –OH group present on the surface site of TiO2. The various experimental techniques, such as SEM, HRTEM, EDS, and XRD analyses, confirmed the formation of 9. It could be used as a template for the synthesis of ZnO@TiO2 (10) core–shell nanoparticles upon calcination between 400 °C and 500 °C.84
:100:3.3:1.25 molar ratio in DMF resulted in the formation of a coordination polymer-dye-nanoparticle composite [7/8@Zn(6)] (9). 7 was firmly bound to the surface of TiO2 due to esterification between the carboxylic group of 7 and the –OH group present on the surface site of TiO2. The various experimental techniques, such as SEM, HRTEM, EDS, and XRD analyses, confirmed the formation of 9. It could be used as a template for the synthesis of ZnO@TiO2 (10) core–shell nanoparticles upon calcination between 400 °C and 500 °C.84
 | ||
| Fig. 4 Schematic representations for the formation of coordination polymer composite RB/TiO2@Zn(btb) (9) [conditions: (a) 5 min sonication, (b) stirring at room temperature for 12 h, (c) methanolic solution of 5 was added with stirring at room temperature]. | ||
Tetrazoles can also be used to construct MOFs.85,86 In 2016, Biswas and co-workers reported the formation of a 3D-amide functionalised Cd(II) MOF with a tetrazole-based organic ligand. This MOF [Cd5Cl6(12)(H-12)2]·7H2O (13), (H2-12 = 4-(1H-Tetrazol-5-yl)-N-[4-(1H-tetrazol-5-yl)phenyl]benzamide) (12), was synthesised by the reaction of CdCl2·H2O (11) and 12 in 2:1 molar ratio in a DMF–MeOH (1:1) solution at 80 °C (Fig. 5(a)). The crystal structure of 13 showed that the ligand 12 acted as a tridentate ligand (via mode VIII, Fig. 5(b)). The metal Cd2+ (d10-electronic configuration), and π-conjugated electron-rich aromatic ligands endowed 13 with excellent luminescence properties. It was thus used to detect NAEs (nitroaromatic explosives). The steady-state fluorescence titration experiment of activated 13 showed that it could selectively detect 2,4,6-trinitrophenol (TNP) with a detection limit of 1.87 × 10−7 M (42.84 ppb). Gradual fluorescence quenching of 13 was observed with increasing concentration of NAEs with a significantly higher quenching (92%) for TNP. The sensing mechanism revealed that other NAEs were only adsorbed on the surface, while TNP was selectively adsorbed inside the pores of the MOF (13) due to proper size matching. UV-visible spectroscopy showed that the absorption band of TNP overlapped with the emission band of 13, unlike other NAEs, which had almost no overlap. Further, the LUMO of TNP was closer in energy to the HOMO of the ligand 12, making electron transfer more efficient for TNP than other NAEs. Hence, both the energy and electron transfer mechanisms favoured the highest quenching efficiency of 13 by TNP. Moreover, the recyclability of 13 suggested high photostability and reusability, which made it suitable for detecting explosive materials for a longer duration.87
 | ||
| Fig. 5 (a) Schematic representation for the formation of 13. Crystal structure of 13 was obtained from ref. 87 [color code: blue, N; black, C; magenta, Cd; red, O; green, Cl (H atoms and anions omitted for clarity)]. (b) Binding mode of ligand 12. | ||
Recently, Huang and co-workers also reported the synthesis of AIE (aggregation-induced emission) active tetrazole-based MOF (17). The solvothermal reaction of tetrakis[(4-cyanyl)- phenyl]ethylene (14), CdCl2·H2O (11), and NaN3 (15) in 1:3:4 molar ratio in a solution of DMA and H2O resulted in the formation of {[Cd4(16)2Cl5]·(N3)3} (17) (Fig. 6(a)). The crystal structure of 17 displayed the bidentate binding mode (via mode VI, Fig. 6(b)) of in situ generated ligand 16 ([4-(1H-tetrazol-5-yl) phenyl]ethylene). 17 showed excellent AIE properties in the methanol/acetonitrile system with a 16-fold increase in fluorescence quantum yield on going from a non-aggregated state (only methanol solvent) to an aggregated state [90% (v/v) acetonitrile/methanol solvent]. The emulsion of 17 in water was also used for NAE detection. 17 was able to detect TNP selectively over other NAEs with a detection limit of 6.023 × 10−8 M through fluorescence quenching.88 Other groups have also reported different tetrazole-based luminescent MOFs, which exhibit different applications.89–91
 | ||
| Fig. 6 (a) Schematic representation for the formation of 17. Crystal structure of 17 obtained from ref. 88 [color code: blue, N; black, C; magenta, Cd; green, Cl; (H atoms and anions omitted for clarity.)] (b) Binding mode of ligand 16. | ||
The central utility of MOFs lies widely in gas adsorption and its capability for selective separation of gases.92–95 In 2015, Chen and co-workers reported a microporous MOF (20) which was prepared by treating CuCl2·2H2O (18) and 19 (5-(5-Amino-1H-tetrazol-1-yl)-1,3-benzenedicarboxylic acid) in 1:1 molar ratio with HBF4 in a mixture of DMF–MeOH at 80 °C (Fig. 7(a)). The tetrazole ligand 19 had binding via mode III (Fig. 7(b)) in the framework. The crystal structure also confirmed the existence of well-defined pores. Adsorption experiments of N2 gas on activated 20 showed a surface area of 970 m2 g−1 and a specific pore volume of 0.399 cm3 g−1. Owing to these unique pore structures and properties, 20 was used to separate acetylene and ethylene. Such separation is essential because acetylene is an impurity obtained in the industrial production of ethylene via the hydrocarbon cracking method. It is necessary to separate acetylene from the mixture of ethylene/acetylene as both pure ethylene and pure acetylene are widely employed as raw chemicals in various industries. Further, the presence of acetylene has detrimental effects on the end products formed from ethylene.96 The gas sorption isotherm of activated 20 indicated that the uptake ability for acetylene was 95.6 cm3 g−1, and that of ethylene was 37.2 cm3 g−1 at 296 K. The uptake ratio was reported as 2.57 from a mixture of acetylene/ethylene. The authors concluded that the availability of a suitable pore size and selective window allowed 20 to display such a high sieving effect and high adsorption selectivity for acetylene over ethylene gas. They also hypothesised that the weak acid–base interactions between the –NH2 groups of the ligand 19 and acetylene molecules also played an essential role in this preferential binding.97 Co-based mixed tetrazole MOFs are also known to exhibit similar gas adsorption properties.98
 | ||
| Fig. 7 (a) Schematic representation for the formation of MOF 20. Crystal structure 20 was obtained from ref. 97 [color code: blue, N; black, C; brown, Cu; green, Cl; red, O (H atoms and anions omitted for clarity.)] (b) Binding mode of ligand 19. | ||
Recently, two Co-based isorecticular MOFs, 24 (Co3(22)3(DMF)3(H2O)6) and 25 (Co3(23)3(DMF)3(H2O)6) have been reported. The reaction between CoCl2·6H2O (21) and ligands 22 (2,6-di(1H-tetrazol-5-yl)naphthalene) and 23 (2,6-bis(4-(1H-tetrazol-5-yl)phenyl)naphthalene) in a 1:1 molar ratio in H2O/DMF led to the formation of 24 and 25, respectively (Fig. 8(a)). The ligands bind with the Co centre through the tetrazole moiety in a bidentate fashion (via mode VI, Fig. 8(b)). The surface area calculated for the activated MOFs 24 and 25 using BET isotherm for N2 gas was 43.0 m2 g−1 and 4.47 m2 g−1, respectively. However, the CO2-sorption studies with activated 24 showed a Dubinin–Astakhov CO2-specific surface area of 636 m2 g−1 and 308 m2 g−1 for activated 25. The CO2 uptake for 24 and 25 was 2.35 mmol g−1 and 1.31 mmol g−1, respectively. The higher CO2 uptake compared to that of N2 was attributed to the presence of the tetrazole motifs, which display strong dipole–dipole and acid–base interactions between the MOFs and CO2. Moreover, this CO2 uptake property of 24 was used for the heterogeneous catalysis of tandem epoxidation carboxylation of styrene (Fig. 9). This conversion of CO2 to a valuable product showed an excellent approach to tackle the emission of greenhouse gases. This reaction resulted in the formation of cyclic lactones, which are essential chemicals in polymer and pharmaceutical industries.99 Interestingly, ligand 22 is very versatile and could be used to construct tetrazole-based MOF using other metal units as well.100
 | ||
| Fig. 8 (a) Schematic representation for the formation of 24 & 25. Crystal structures of 24 and 25 were obtained from ref. 99 [color code: blue, N; black, C; violet, Co; green, Cl; red, O (H atoms and anions omitted for clarity.)] (b) Binding mode of ligands 22 & 23. | ||
 | ||
| Fig. 9 (a) Reaction scheme for styrene epoxidation followed by cycloaddition. (b) Proposed mechanism. | ||
Using chiral tetrazole ligands, chiral self-assembled structures can also be created. Zhang and co-workers reported a homochiral mixed-valent MOF {[Cu2ICuII(26)2(CN−)(H2O)]+ [NO3−]}·[DMF] (29) by using an enantiopure ligand 26 ((1S)-1-(5-tetrazolyl)-ethylamine). The reaction of 26, K3[Fe(CN)6] (27), Cu(NO3)2 (28) and NaOH in the solvent mixture DMF, H2O and EtOH at 100 °C for two days resulted in the formation of 29 (Fig. 10(a)). The crystal structure of 29 showed a tridentate binding mode of the tetrazole ligand 26 (via mode VII, Fig. 10(b)). Since chiral sites can be employed to recognise and separate enantiomers, the ability of 29 to separate chiral alcohols was examined. 29 was able to separate (R/S)-1-phenyl ethanol and (R/S)-1-phenyl propanol with a moderate ee (enantiomeric excess) of 42% and 48%, respectively, favouring the R-isomers.
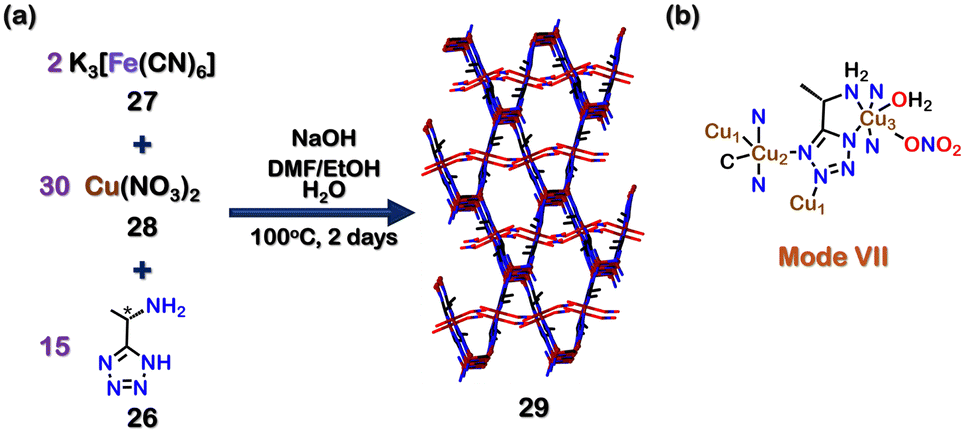 | ||
| Fig. 10 (a) Schematic representation for the formation of MOF 29. Crystal structure of 29 was obtained from ref. 101 [color code: blue, N; black, C; red, O; brown, Cu (H atoms and anions omitted for clarity.)] (b) Binding mode of ligands 24 & 25. | ||
The interactions (hydrogen-bonding, π–π, and stereochemical interactions) between the chiral surface of the MOF and the chiral carbon were mainly responsible for separating the enantiomers.101
3. Tetrazole-based discrete assemblies
As seen in the previous section, most of the applications of polymeric assemblies are related to the separation of fluids using amorphous/crystalline solids99,101 or as an emulsion in different solvents.88 These polymeric architectures suffer from solution processibility, significantly limiting their applications. To tackle this hardship, other discrete self-assembled architectures soluble in organic and polar solvents are needed. The diversity in binding modes of tetrazole often leads to the formation of polymeric assemblies. However, precise mapping of the coordination sites is vital to forming discrete assemblies, which makes such systems rare. The preference of different metal ions towards different binding modes has been detailed in Table 1. This section will discuss such assemblies and their uses in detail.In 2015, Stang and coworkers reported the selective formation of four self-assembled molecules through stoichiometric control. The combination of metal acceptor [(p-cymene) RuCl2]2 (30), ligand (5-(2-hydroxyphenyl)-1H-tetrazole) (31) and silver triflate (32) in different ratios formed different homometallic and heterometallic architectures (Fig. 11(a)). The combination of 30, 31 and 32 in an equimolar ratio led to formation of 1D homometallic complex (33). When 30, 31 and 32 were combined in a 3:4:12 ratio, a 2D rectangular macrocycle (34) was formed. In the complex (33), ligand 31 acted as a bridging ligand (mode VI, Fig. 2) by binding in a bidentate (Fig. 11(b)) fashion through μ2-mode. In 34, the ligand 31 also coordinates in a bidentate fashion but via mode V, thus forming a new assembly 34. 34 contains two types of Ru centres. Four Ru(II) centres form the rectangle by coordinating with the tetrazole moiety's N and O from the phenyl ring. The remaining two Ru centres coordinate to the η6-of the benzene ring (Fig. 11(b)). The tetrazole moiety of ligand 31 in the dimer (33) and rectangle (34) still contained free N-atoms with a lone pair of electrons. Adding additional metal ions enabled the tetrazole motif to utilise these binding sites. The introduction of excess silver in a ratio of 1:2:6 and 3:6:20 resulted in two new heterometallic 3D architectures (35, 36). In a rare mode, both 35 and 36 utilised 31 as a tetradentate ligand (via mode IX, Fig. 2). The attainment of mode IX of tetrazole in a discrete assembly without polymer formation was noteworthy. Compound 35 had a cylindrical shape consisting of 6 Ru centres, 6 Ag ions and 6 ligands (Fig. 11(b)). The new heterometallic cylinder (35) allows Ru and Ag to bind in a tetra-coordinated fashion. The second heterometallic structure 36 was a double cone architecture due to the presence of excess Ag ions. The binding of the Ru centres was similar to that in 35; the only difference lied in the additional Ag ions in 36. The double cone architecture 36 consists of two types of Ag ions. One type of Ag was bonded to the ligand (31), whereas the other type bonded to the benzene ring (Fig. 11(b)). This study demonstrated the importance of binding modes in determining the structure of the final assembly. It also showed the dependence of the tetrazole moiety on stoichiometry and the relative abundance of metal centres.102
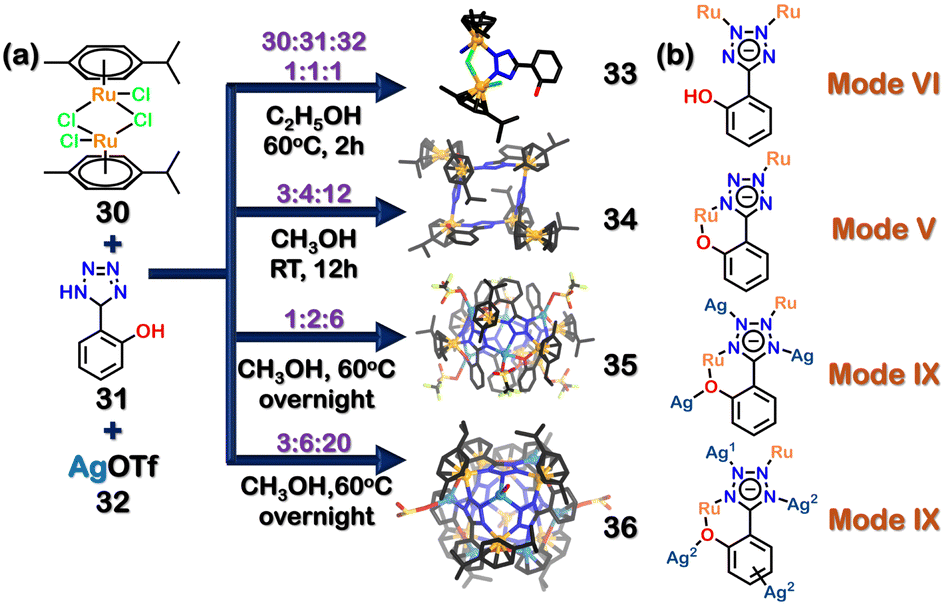 | ||
| Fig. 11 (a) Schematic representation of the formation of the homometallic dimer (33) and 2D-rectangle (34), heterometallic cylinder (35) and double cone (36). Crystal structures of 33, 34, 35, and 36 were adopted from ref. 102 [color code: blue, N; black, C; orange, Ru; pale blue, Ag (H atoms and anions omitted for clarity.)] (b) Representation of multiple binding modes of 31. | ||
Mukherjee and co-workers reported two water-soluble tetrahedral nano-cages with tetrazolate ligands (Fig. 12(a)). The combination of 1,4-di(1H-tetrazol-5-yl) benzene (37) and 1,3,5-tri(1H-tetrazol-5-yl) benzene (38) with cis-(tmeda)Pd(NO3)2 [tmeda = N,N,N′,N′-tetramethylethane-1,2-diamine] (39) in 1:1 and 3:2 molar ratios in water or DMSO formed soft metallogels. The formation of metallogels (a form of polymeric assembly) arose due to the versatile binding nature of the tetrazole motif.
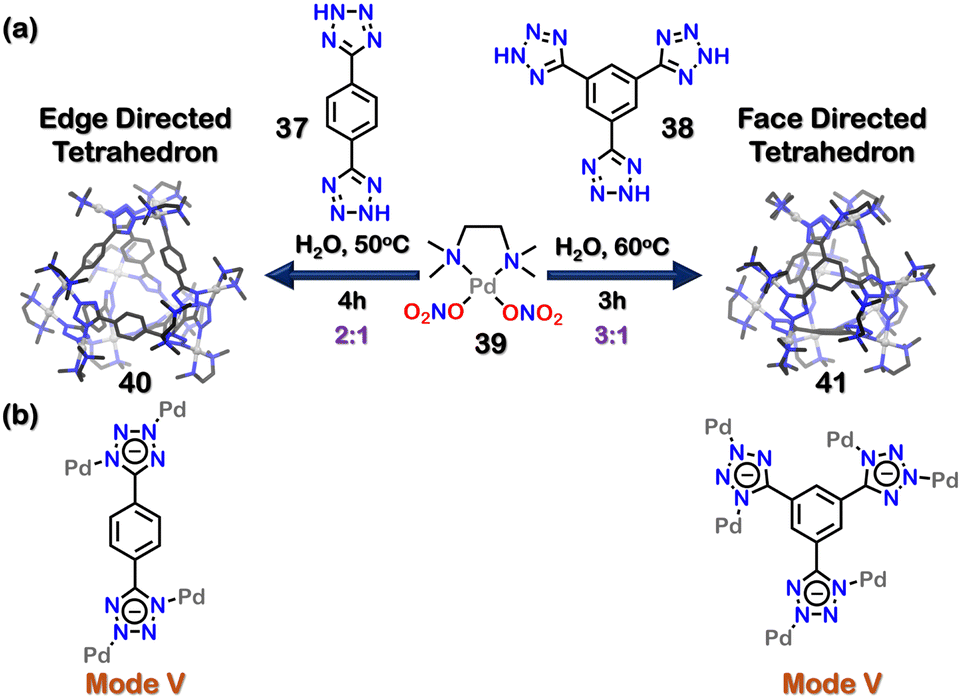 | ||
| Fig. 12 (a) Schematic representation of the formation of tetrahedron cages (40, 41). Crystal structures of 40, 41 reproduced from ref. 103 [color code: blue, N; black, C; grey, Pd (H atoms and anions omitted for clarity.)] (b) Binding modes of 37 and 38. | ||
However, when the ratio of 37 to cis-(tmeda) Pd(NO3)2 (39) was changed to 1:2, a discrete edge-directed tetrahedral cage 40 was obtained. In this cage, ligand 37 occupied the edges of a tetrahedron and the corners were made of three tetrazole moieties connected to three cis-(tmeda)Pd(NO3)2 units. The ligand 37 bonded to the Pd centres in a bidentate manner (via mode V, Fig. 12(b)). Similarly, when the ratio of ligand 38 to acceptor cis-(tmeda)Pd(NO3)2 was changed to 1:3, a discrete face-directed tetrahedral cage (41) was formed. In 41, ligand 38 occupied the tetrahedron's 4 faces, and the corners were formed by bonding three tetrazole moieties with three acceptor units. The denticity and binding mode for ligand 38 were the same as that observed in ligand 37. The unique 1,3-binding mode (mode V, Fig. 12(b)) of tetrazole moieties with the acceptor units imparted helicity to the tetrazole cages. These corners could be characterised as either a right-handed (Δ) or a left-handed (Λ) helix. As there were four corners in cages 40 and 41, multiple diastereomeric and/or isomeric cages could be formed (Fig. 13). The crystal structure showed the formation of both (ΛΛΛΛ) or (ΔΔΔΔ) cages, making the system overall achiral through racemisation.103
 | ||
| Fig. 13 Representation of a right-handed (Δ) configuration and a left-handed (Λ) configuration present in one edge of the tetrahedron 40, formed through the coordination of metal centre (39) and tetrazole ligands (38). | ||
Further, 40 had a hydrophobic cavity with open windows. Thus, it was employed as a host to perform reactions inside its cavity. 40 successfully catalysed Michael's addition reactions of water-insoluble nitro-olefins with 1,3-dimethyl barbituric acid (Fig. 14).103
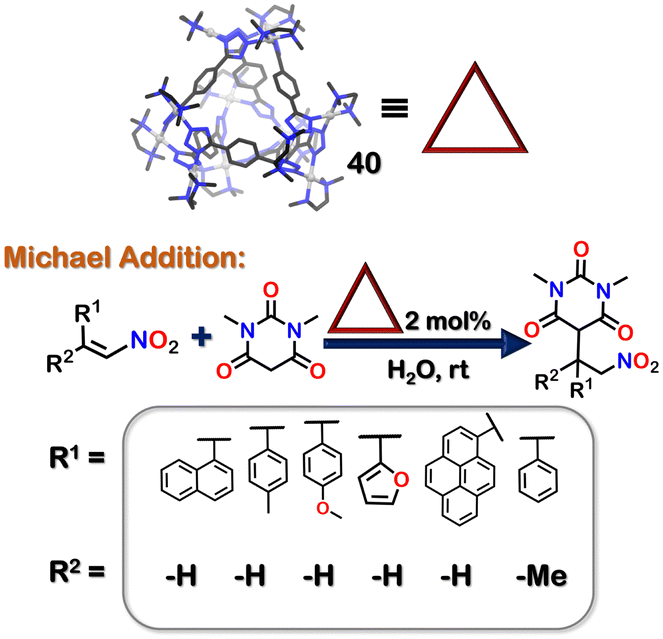 | ||
| Fig. 14 Michael addition reaction of insoluble nitro olefins facilitated by the tetrahedron cage 40. | ||
Changing the metal centre and the orientation of the ligand can also lead to the formation of different architectures. Lee and coworkers used half-sandwich iridium (Ir) complexes and three tetrazole ligands to synthesise three novel architectures (Fig. 15(a)). The combination of [Cp*IrCl2]2 (41), di-tetrazole ligand (42), and Ag(OTf) (43) in a molar ratio of 1:1:3 resulted in the formation of a tetranuclear architecture (44). The tetranuclear rectangular architecture (44) had four Ir centres coordinated to two ligands (42), and a Cl atom bridged between two Ir centres. The tetrazole motif displayed two distinct bidentate modes (mode V and mode VI) (Fig. 15(b)). Interestingly, ligand 42 was an isomer of ligand 37, which was previously used to make tetrahedral cages. This showed how changing the ligand structure and metal centre can lead to the formation of diverse architectures.104
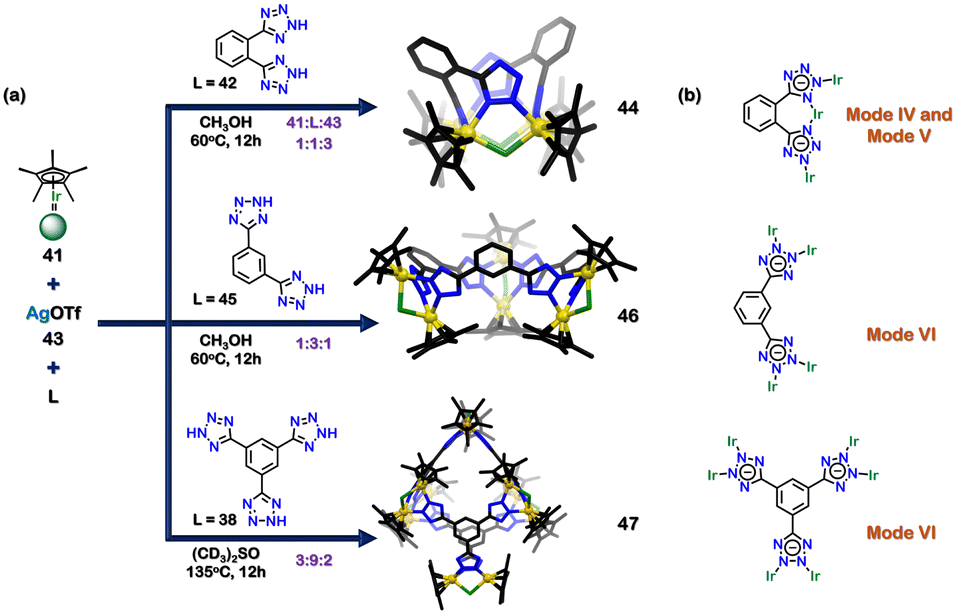 | ||
| Fig. 15 (a) Schematic representation for the formation of tetranuclear rectangular architecture (44), hexanuclear supramolecule architecture (46) and dodecanuclear supramolecular architecture (47). Crystal structures of 44, 46 and 47 were adopted from ref. 104 [color code: blue, N; black, C; golden, Ir (H atoms and anions omitted for clarity)] (b) Representation of binding modes of 42, 45 and 38. | ||
The combination of 41, 43 and isomeric di-tetrazole ligand (45) in a ratio of 1:3:1 resulted in the synthesis of a truncated trigonal pyramidal hexanuclear supramolecule architecture (46). All the tetrazole motifs indicated a bidentate nature with VI binding mode (Fig. 2 and 15(b)). The composition of the hexanuclear complex consisted of six Ir centres and three ligands (45); and a Cl atom bridged between two Ir centres. The isomeric ligands 42 and 45 resulted in the construction of 2D complexes. To develop a 3D architecture, a tridentate ligand (38) was introduced. The mixture of 41, 38, and 43 in a molar ratio of 3:2:9 resulted in a 3D dodecanuclear octahedral cage (47). This 3D architecture had a truncated octahedral geometry with twelve half-sandwich Ir units and four ligands (48); a Cl atom bridged between two Ir centres. The tetrazole ligand displayed a bidentate nature (via VI binding mode) (Fig. 15(b)).104
Interestingly, the previous example used the same ligand (38) with cis-blocked Pd-metal to form a tetrahedral cage.103 Changing the metal acceptor from Pd to Ir caused the binding modes to change, forming an octahedral cage instead of a tetrahedral one.104
Previous reports showed the formation of achiral or racemic cages based on tetrazole moieties. However, tetrazole ligands can also easily fabricate chiral self-assembled architecture. A series of enantiopure homochiral tetrahedral cages by employing chiral enantiopure square planar Pd(II) acceptors ((R,R)/(S,S)(N1,N1,N2,N2-tetramethylcyclohexane-1,2-diamine)Pd(NO3)2) with di-tetrazole linkers were reported. The combination of 1,4-di(1H-tetrazol-5-yl) benzene (37) with (R,R)-Pd(II) acceptor (48) in a 1:2 molar ratio formed homochiral cage 49 with (ΛΛΛΛ) configuration (Fig. 16(a)). When enantiomeric acceptor (S,S) (48) was used, it formed the other enantiomer with (ΔΔΔΔ) configuration. The tetrazole linker displays bidentate coordination via mode V (Fig. 16(b)). The self-assembly of other bi-tetrazole linkers 2,6-di(1H-tetrazol-5-yl)naphthalene (22) and 4,4′-di(1H-tetrazol-5-yl)-1,1′-biphenyl (50) with (R,R) and (S,S) (48) resulted in the formation of enantiopure water-soluble tetrahedral cages with either (ΛΛΛΛ) or (ΔΔΔΔ) configuration respectively.105
 | ||
| Fig. 16 (a) Schematic representation for the formation of homochiral tetrahedral cage 49. Crystal structure of 49 is adopted from ref. 105 [color code: blue, N; black, C; grey, Pd (H atoms and anions omitted for clarity)]. (b) Representation of the binding mode of 37. | ||
The nanocages should interact with chiral guests in a specific manner to form diastereomeric host–guest complexes. The tetrahedral cage formed by the self-assembly of 4,4′-di(1H-tetrazol-5-yl)-1,1′-biphenyl (50) had the largest hydrophobic aperture and was used for selective chiral recognition of the ethyl derivative of (R/S) BINOL (R/S-EtB). The (ΛΛΛΛ) configuration showed selectivity towards R-EtB, whereas the (ΔΔΔΔ) configuration showed selectivity towards S-EtB (Fig. 17).105
 | ||
| Fig. 17 Illustration to display the selective encapsulation of R-EtB over S-EtB by the homochiral tetrahedron cage with (ΛΛΛΛ) configuration. | ||
Throughout this review, multiple examples of the diverse binding modes of tetrazole to metals have been demonstrated. These bindings are not covalent; thus, switching between these binding modes is possible based on external stimuli. Howlader et al. demonstrated that changing the solvent polarity could interchange different molecular architectures. This change caused a dynamic shift between a square-shaped macrocycle and a tetrahedral cage. The binding mode of the tetrazole ligand also changed from mode-VI (in macrocycle) to mode-V (in cage). The self-assembly of di-tetrazole linker 50 [4,4′-bis(1H-tetrazol-5-yl)-1,1′-biphenyl] with acceptor (39) in 1:2 molar ratio in dimethyl sulfoxide (DMSO) and followed by crystallisation, resulted in the formation of a 2D octa-nuclear molecular square (macrocycle) (51). All the tetrazole linkers were coordinated in a bidentate manner (via mode-VI) (Fig. 18). However, the macrocycle (51) was not stable because of the dynamic nature of the metal–ligand bond and the availability of various coordinating sites of tetrazole. Macrocycle 51 transformed into a 3D-tetrahedral cage (52), where the di-tetrazole linker transformed its binding mode to V, maintaining a bidentate nature (Fig. 18). This also displays the weaker affinity of binding mode-VI compared to mode-V. The combination of 50 and 39, along with KPF6 in a 1:2:4 molar ratio in less polar acetonitrile solvent, resulted in the formation of a distinct assembly 52 (Fig. 18). The tetrahedral cage 52 also showed transformation to 51 upon introducing a more polar solvent, DMSO. A similar reaction using a triphenyl amine-based tri-tetrazole linker 53 with square planar Pd(II) acceptor 39 in DMSO resulted in the formation of only a 3D-tetrahedral cage 54, which did not show transformation to macrocycle (Fig. 19). This interconvertible behaviour was unique to the ligand 52 and reluctant to the ligand 53.106
 | ||
| Fig. 18 Schematic representation for the formation of molecular square (51) and 3D-tetrahedral cage (52) and the dynamic reaction between 51 and 52, along with the representation of the dual binding modes adopted by 50. | ||
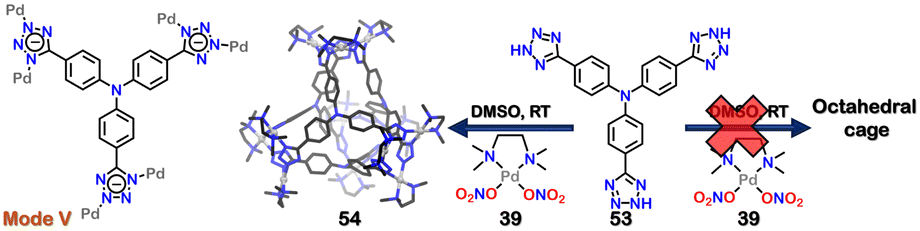 | ||
| Fig. 19 Schematic representation of distinct 3D-tetrahedral cage (54) along with the representation of binding mode of 53. Crystal structure of 54 was adopted from ref. 106 [color codes: blue, N; black, C; grey, Pd (H atoms and anions omitted for clarity)]. | ||
Until now, examples of the self-assembly of tetrazole-based ligands with transition metals of 3d and 4d series have been demonstrated. Generally, the coordination number of these metal ions is restricted to four and six with square planar and octahedral geometries, respectively. The metal ions of the f-block series can be explored to extend the coordination number. The main challenge in designing discrete 4f-series organic polyhedra lies in their complex coordination behaviour, and this absence of precise control could hinder the formation of discrete assemblies. Qing Fu Sun and co-workers reported a series of water-stable anionic lanthanide organic polyhedra (LOPs) by self-assembling three different bis-tetrazolate ligands and Ln (III) metals. The combination of tetrazolate linker 55 with Eu(OTf)3 in a 3:2 molar ratio in DMSO with 12 equivalents of tetraethylammonium hydroxide gave a mixture of Eu6(55)9 (56) and Eu4(55)6 (Fig. 20(a)). This showed that under the given conditions both the tetranuclear and hexanuclear assemblies were formed and could not be separated. A similar reaction was carried out with Tb(OTf)3 and Gd(OTf)3, which also formed a similar mixture of the final assemblies. However, changing the base to KOH resulted in the discrete formation of 56. The crystal structure showed that 56 had a trigonal bipyramidal architecture and the tetrazole moiety acted in a monodentate fashion (via mode II, Fig. 2). The combination of second tetrazole linker 57 with Eu(OTf)3 in a 3:2 molar ratio resulted in the formation Eu8(57)12 cubic structure (58) (Fig. 20(a)). Similar structures were formed with Tb(OTf)3 and Gd(OTf)3 metal acceptors. The combination of third linker 59 with other Ln(OTf)3 also formed similar Ln8(59)12 cubic architectures. The combination of 59 along with EuCl3·6H2O and KOH resulted in the formation of Eu10(59)15 architecture, which could be confirmed by mass analysis. The crystallisation, however, led to the transformation of the Eu10(59)15 structure to a cubic Eu8(59)12 structure (60) (Fig. 20(a)). These Ln(III) based assemblies were stable in aqueous medium due to the electrostatic and chelating interaction between lanthanide metals and ligands. The ligands 57 and 59 also exhibited mode II binding to the metal centre in a monodentate form (Fig. 20(b)). The quantum yield for the cubic structures was obtained for Tb8(57)12 (Φ = 11.2% in water) and Eu8(59)12 (Φ = 76.8% in DMSO). Moreover, the cubic Gd8(59)12 architecture was further employed as an MR (magnetic resonance) imaging contrast agent. Heterometallic cubic assemblies with Eu(III) and Gd(III) could also be created, which showed the dual functionality of luminescent imaging as well as MR imaging.107
 | ||
| Fig. 20 (a) Schematic representation for the formation of Eu6(55)9 trigonal pyramidal architecture (56), Eu8(57)12 cubic structure (58) and Eu8(59)12 architecture (60). Crystal structures of 56, 58 and 60 are adopted from ref. 107 [color codes: blue, N; black, C; red, O; light green, Eu (H atoms and anions omitted for clarity.)] (b) Representation of binding modes of 55, 57 and 59. | ||
Discrete architectures could also be constructed using coordination clusters instead of single metal ions at the vertices of the architectures. The incorporation of coordination clusters as the acceptor units form a new class of coordination-cluster cages. Recently, a series of thiacalixarene-based (Co/Fe) coordination clusters were reported to study oxygen evolution reaction (OER). The solvothermal reaction of metal precursors (CoCl2·6H2O and/or FeCl3·6H2O) along with p-tert-butylsulfonylcalix[4]arene (61), and 1,3-bis(2H-tetrazol-5-yl)benzene (45) in a 4:1:2 resulted in the formation of Co-based coordination cluster (62), Fe-based coordination cluster (63), and heterometallic Co–Fe coordination cluster (64) (Fig. 21(a)). All the clusters were arranged in a box-like fashion where the tetrazole moiety exhibited a bidentate binding (via mode VI, Fig. 21(b)). Three bimetallic complexes {CoFe}16-A, {CoFe}16-B (64), {CoFe}16-C with different molar ratios were synthesised. The {CoFe}16-B (64) cluster was optimised among the three bimetallic clusters to show maximum OER. The synergistic effect in the bimetallic Fe–Co (64) cluster helped to study OER with enhanced activity in comparison to 63 and 62.108 Such calixarene metal conjugates can also be used for the construction of polymeric systems which can show stimuli dependent chain(polymer) to cage(discrete) transformation.109
 | ||
| Fig. 21 (a) Schematic representation for the formation of Co-based coordination cluster (62), Fe-based coordination cluster (63) and heterometallic Co–Fe coordination cluster (64). Crystal structures of 62, 63 and 64 are reproduced from ref. 108 [color code: blue, N; black, C; red, O; yellow, S; dark green, Fe; violet, Co (H atoms and anions omitted for clarity.)] (b) Representation of binding modes of 45. | ||
In the previous examples, tetrazole was used as a ligand, forming coordination bonds with various metals, but such moiety can also be used to functionalize existing metal–organic cages/frameworks. Using the ligand 2-(1H-tetrazol-1-yl)terephthalic acid (65), synthesis of multiple tetrazole-functionalized MOFs (UiO-66-tetrazole, CAU-1-tetrazole, and MIL-101-tetrazole) have been reported. In these MOFs, the tetrazole unit was free and uncoordinated. Synthesis of a porous metal–organic cage (66) by reacting ligand 65 with zirconocene dichloride in a 2:5 molar ratio in DMA (Fig. 22) was also reported.
 | ||
| Fig. 22 Schematic representation of the preparation of 65, along with efficient separation of gases by 66. Reproduced with permission from ref. 110. Copyright 2021 German Chemical Society. | ||
66 had an adsorption capacity of 207.1 cm3 g−1 and Brunauer–Emmett–Teller (BET) surface area for N2 gas was calculated to be around 636.7 m2 g−1. Compared to an amine-functionalized cage (67), the tetrazole-functionalised cage showed higher adsorption capability for acidic gases (CO2, ethylene, and acetylene). The uptake efficiency was based on the order of acidity of the gases. As acetylene (C2H2) is more acidic than ethylene (C2H4) and CO2, 66 showed maximum uptake of acetylene and could selectively uptake acetylene (C2H2) from the binary mixture of C2H2/C2H4 and C2H2/CO2. DFT calculation and GCMC simulations elucidated that 66 interacted with the acetylene molecules through hydrogen bonding (via Ntetrazole⋯H–C) and the cavity provided sufficient space for multiple dipole–dipole interactions between uptaken acetylene molecules through Cδ+⋯Hδ−, increasing the stability of the host–guest complex. Low heat of adsorption for acetylene, high acetylene uptake capability, and moderate separation selectivity made 66 a suitable candidate for acetylene separation.110
3.1 Tetrazole-based cage composites
The lanthanoids are well-established for their distinctive magnetic and optical properties. Due to the highly shielded 4f electrons, lanthanide ions exhibit unique characteristics like sharp emission peaks, long lifetimes, and ligand-independent and environment-independent emission bands. Lanthanide compounds have numerous applications in sensing, bio-imaging, luminescent display, and catalysis. However, most reported discrete LOPs and mononuclear lanthanide compounds suffer from inferior water stability, hindering their utilisation in various applications. This section focuses on incorporating lanthanide-based polyhedra (with tetrazole motif) in polymeric networks or porous materials to increase their stability and effectiveness. The utility of these cage composites in different applications has also been briefly discussed.Sun and co-workers reported the construction of three cage-composites, namely Eu8L12@MSNs (68) (MSN = mesoporous silica nanoparticles), Eu8L12@MSNs-NH2 (69) and Eu8L12@MSNs-biotin (70) by employing the “ship-in-a-bottle” strategy. 68 was functionalised to 69 by applying 3-aminopropyl-triethoxysilane (APS). The attachment of biotin on the surface of 68 resulted in the formation of 70 (Fig. 23). The hybrid materials 68, 69, and 70 displayed relatively high photoluminescence quantum yields of 38.70%, 40.08%, and 44.04%, respectively. Additionally, 68, 69 and 70 exhibited better stability than pristine LOP (Eu8L12). Moreover, the biotin-modified hybrid material 70 revealed excellent cell targeting capability even at a low concentration.111
 | ||
| Fig. 23 Schematic representation of the formation of hybrid 70via the employment of a “ship-in-a-bottle” synthetic strategy. Reproduced with permission from ref. 111. Copyright 2022 Royal Society of Chemistry. | ||
In the second report, different tetrazole-based polyhedra [Gadolinium-based polyhedra (Gd8(59)12)] were employed and encapsulated in MSN (mesoporous silica nanoparticles) by utilising the “ship-in-a-bottle” strategy. The formation of Gd8(59)12@MSN (71) was ensured by the reaction of 59 with Gd(OTf)3 in cyclohexane-suspended MSN in a 3:10 molar ratio in DMSO solvent (Fig. 24(a)). The solid-state optical Diffuse Reflectance Spectrum (DRS) ascertained 71 as a potential n-type semiconductor due to an optical band gap of 3.40 eV, more prominent than commercially available TiO2 and other semiconductors. The hybrid material 71 could degrade organic dye methyl orange (MO) under UV irradiation.
 | ||
| Fig. 24 (a) Schematic representation of the formation of cage-composite 71via the employment of a “ship-in-a-bottle” synthetic strategy. Reproduced with permission from ref. 112. Copyright 2023 Asian Chemistry Editorial Society. (b) Proposed mechanism for the degradation of MO (Methyl Orange) by 71. | ||
It degraded 88.94% of MO (a common macro-pollutant in water) in 2 hours. A plausible mechanism for the photocatalytic degradation of pollutant MO showed the effective generation of electron–hole pairs upon irradiation of UV light (Fig. 24(b)). The reactive holes enabled the formation of hydroxyl radicals, and electron pairs facilitated the formation of superoxide radical anion, which synergistically degraded MO to carbon dioxide and water. Further, 71 demonstrated an affinity for degrading different cationic dyes like MB (methylene blue), RhB (rhodamine-B) and neutral NR (Nile red) under irradiation.112
4. Conclusion & outlook
The discipline of coordination-driven self-assembly has significantly expanded in recent decades, owing primarily to its capacity for facile synthesis of intricate architectures surpassing those attainable through traditional covalent chemistry. Notably, the exceptional versatility of tetrazole in adopting nine distinct coordination modes makes it a compelling building block. The initial segment of this review offers a brief overview of tetrazole-based polymeric assemblies, including coordination polymers and their subclass metal–organic frameworks, which have applications in diverse fields such as sensing, gas adsorption and separation, and catalysis.Subsequently, tetrazole-based discrete assemblies that are soluble in organic and aqueous solvents have been discussed. Precise control over stoichiometry, ligand selection, and metal coordination has facilitated the construction of distinct cages, yielding novel two-dimensional and three-dimensional architectures. These cages have demonstrated utility in catalytic reactions such as Micheal addition and oxygen evolution and have been utilised to design homochiral cages featuring chiral cavities for enantioselective separation. Additionally, discrete cages exhibited gas-separation capabilities, with tetrazole linkers contributing to gas uptake mechanisms. Tetrazole-based LOPs have also emerged as MR contrast agents and luminescent labels. The integration of tetrazole-based lanthanide organic polyhedra (LOPs) into mesoporous silica nanoparticles (MSNs) and their applications in cell imaging and photocatalytic degradation have been highlighted as well.
However, despite the manifold examples of tetrazole-based polymeric assemblies, more investigation into tetrazole linkers for discrete architectures still needs to be done. Thus, many questions on tetrazole-based discrete self-assembly remain unanswered: can tetrazole ligands utilise other binding modes for constructing discrete architecture besides bidentate ones? Can unusual non-platonic architectures or mechanically interlocked structures be fabricated using tetrazole ligands? Can tetrazole or tetrazolium ligands be used to create purely organic cages?
These questions warrant further exploration into the dynamic nature of tetrazole binding modes as the field remains in its infancy. In summation, this review endeavours to consolidate the multifaceted role of the tetrazole motif, advocating for the advancement of tetrazole linker design by incorporating additional functionalities and introducing new applications.
Author contributions
MA and SD have contributed equally to the formulation of the manuscript. DC contributed extensively towards editing the writing and contents of the article, along with a fruitful discussion on tetrazoles. PSM supervised the manuscript and helped in writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the contributions of the co-workers and colleagues whose names are listed as co-authors in the papers we have cited from our group. S. D. acknowledges UGC for fellowship. D. C. acknowledges PMRF for fellowship. P. S. M. acknowledges SERB (India) for research grant as J. C. Bose fellowship.References
- D. Pochan and O. Scherman, Chem. Rev., 2021, 121, 13699 CrossRef CAS PubMed.
- C. J. Pedersen, Angew. Chem., Int. Ed. Engl., 1988, 27, 1021 CrossRef.
- D. J. Cram, Angew. Chem., Int. Ed. Engl., 1988, 27, 1009 CrossRef.
- J.-M. Lehn, Angew. Chem., Int. Ed. Engl., 1988, 27, 89 CrossRef.
- G. Gupta, Y. Sun, A. Das, P. J. Stang and C. Yeon Lee, Coord. Chem. Rev., 2022, 452, 214308 CrossRef CAS PubMed.
- J. Zhao, Z. Zhou, G. Li, P. J. Stang and X. Yan, Natl. Sci. Rev., 2021, 8, nwab045 CrossRef CAS PubMed.
- Z. Yang, F. Esteve, C. Antheaume and J.-M. Lehn, Chem. Sci., 2023, 14, 6631 RSC.
- M. Zenka, J. Preinl, E. Pertermann, A. Lützen and K. Tiefenbacher, Eur. J. Inorg. Chem., 2023, e202300110 CrossRef CAS.
- D.-Y. Zhang, Y. Sang, T. K. Das, Z. Guan, N. Zhong, C.-G. Duan, W. Wang, J. Fransson, R. Naaman and H.-B. Yang, J. Am. Chem. Soc., 2023, 145, 26791 CrossRef CAS PubMed.
- H. Liang, Y. Yang, L. Shao, W. Zhu, X. Liu, B. Hua and F. Huang, J. Am. Chem. Soc., 2023, 145, 2870 CrossRef CAS PubMed.
- M. Li, Y. Liu, L. Shao, B. Hua, M. Wang, H. Liang, N. M. Khashab, J. L. Sessler and F. Huang, J. Am. Chem. Soc., 2023, 145, 667 CrossRef CAS PubMed.
- Y.-X. Hu, X. Hao, D. Wang, Z.-C. Zhang, H. Sun, X.-D. Xu, X. Xie, X. Shi, H. Peng, H.-B. Yang and L. Xu, Angew. Chem., Int. Ed., 2024, 63, e202315061 CrossRef CAS PubMed.
- D. Chakraborty, R. Modak, P. Howlader and P. S. Mukherjee, Chem. Commun., 2021, 57, 3995 RSC.
- G. Yu, T. R. Cook, Y. Li, X. Yan, D. Wu, L. Shao, J. Shen, G. Tang, F. Huang, X. Chen and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 13720 CrossRef CAS PubMed.
- M. G. Sullivan, H. K. Welgama, M. R. Crawley, A. E. Friedman and T. R. Cook, Chem. Mater., 2024, 36, 567 CrossRef CAS.
- R. A. Tromans, T. S. Carter, L. Chabanne, M. P. Crump, H. Li, J. V. Matlock, M. G. Orchard and A. P. Davis, Nat. Chem., 2019, 11, 52 CrossRef CAS PubMed.
- W. Liu, Y. Tan, L. O. Jones, B. Song, Q.-H. Guo, L. Zhang, Y. Qiu, Y. Feng, X.-Y. Chen, G. C. Schatz and J. F. Stoddart, J. Am. Chem. Soc., 2021, 143, 15688 CrossRef CAS PubMed.
- R. Wyler, J. de Mendoza and J. Rebek Jr., Angew. Chem., Int. Ed. Engl., 1993, 32, 1699 CrossRef.
- J. Rebek, Chem. Soc. Rev., 1996, 25, 255 RSC.
- B. Odell, M. V. Reddington, A. M. Z. Slawin, N. Spencer, J. F. Stoddart and D. J. Williams, Angew. Chem., Int. Ed. Engl., 1988, 27, 1547 CrossRef.
- H.-Y. Gong, B. M. Rambo, E. Karnas, V. M. Lynch and J. L. Sessler, Nat. Chem., 2010, 2, 406 CrossRef CAS PubMed.
- D. Chakraborty and P. S. Mukherjee, Chem. Commun., 2022, 58, 5558 RSC.
- E. G. Percástegui, T. K. Ronson and J. R. Nitschke, Chem. Rev., 2020, 120, 13480 CrossRef PubMed.
- R. Banerjee, D. Chakraborty and P. S. Mukherjee, J. Am. Chem. Soc., 2023, 145, 7692 CrossRef CAS PubMed.
- Z. Zhang, L. Ma, F. Fang, Y. Hou, C. Lu, C. Mu, Y. Zhang, H. Liu, K. Gao, M. Wang, Z. Zhang, X. Li and M. Zhang, JACS Au, 2022, 2, 1479 CrossRef CAS PubMed.
- J. Tessarolo, E. Benchimol, A. Jouaiti, M. W. Hosseini and G. H. Clever, Chem. Commun., 2023, 59, 3467 RSC.
- R.-J. Li, A. Tarzia, V. Posligua, K. E. Jelfs, N. Sanchez, A. Marcus, A. Baksi, G. H. Clever, F. Fadaei-Tirani and K. Severin, Chem. Sci., 2022, 13, 11912 RSC.
- J.-M. Lehn, Science, 2002, 295, 2400 CrossRef CAS PubMed.
- C. O. Dietrich-Buchecker and J. P. Sauvage, Chem. Rev., 1987, 87, 795 CrossRef CAS.
- M.-M. Gan, F. Wang, X. Li, L.-Y. Sun, G. Yuan and Y.-F. Han, Inorg. Chem., 2023, 62, 2599 CrossRef CAS PubMed.
- Y.-W. Zhao and X.-M. Zhang, J. Mater. Chem. C, 2020, 8, 4453 RSC.
- C. T. McTernan, T. K. Ronson and J. R. Nitschke, J. Am. Chem. Soc., 2021, 143, 664 CrossRef CAS PubMed.
- N. Hoyas Pérez and J. E. M. Lewis, Org. Biomol. Chem., 2020, 18, 6757 RSC.
- D. Chakraborty, R. Saha, J. K. Clegg and P. S. Mukherjee, Chem. Sci., 2022, 13, 11764 RSC.
- G. Gil-Ramírez, D. A. Leigh and A. J. Stephens, Angew. Chem., Int. Ed., 2015, 54, 6110 CrossRef PubMed.
- J. E. Beves, J. J. Danon, D. A. Leigh, J.-F. Lemonnier and I. J. Vitorica-Yrezabal, Angew. Chem., Int. Ed., 2015, 54, 7555 CrossRef CAS PubMed.
- D. A. Leigh, F. Schaufelberger, L. Pirvu, J. H. Stenlid, D. P. August and J. Segard, Nature, 2020, 584, 562 CrossRef CAS PubMed.
- H.-N. Zhang and G.-X. Jin, Angew. Chem., Int. Ed., 2023, 62, e202313605 CrossRef CAS PubMed.
- M. C. Jiménez, C. Dietrich-Buchecker and J.-P. Sauvage, Angew. Chem., Int. Ed., 2000, 39, 3284 CrossRef.
- S. Zhang, A. Rodríguez-Rubio, A. Saady, G. J. Tizzard and S. M. Goldup, Chem, 2023, 9, 1195 CAS.
- L. Zhang, Y. Qiu, W.-G. Liu, H. Chen, D. Shen, B. Song, K. Cai, H. Wu, Y. Jiao, Y. Feng, J. S. W. Seale, C. Pezzato, J. Tian, Y. Tan, X.-Y. Chen, Q.-H. Guo, C. L. Stern, D. Philp, R. D. Astumian, W. A. Goddard and J. F. Stoddart, Nature, 2023, 613, 280 CrossRef CAS PubMed.
- R. Saha, B. Mondal and P. S. Mukherjee, Chem. Rev., 2022, 122, 12244 CrossRef CAS PubMed.
- S. R. Batten, B. Chen and J. J. Vittal, ChemPlusChem, 2016, 81, 669 CrossRef CAS PubMed.
- O. Yaghi and H. Li, J. Am. Chem. Soc., 1995, 117, 10401 CrossRef CAS.
- V. J. Pastore and T. R. Cook, Chem. Mater., 2020, 32, 3680 CrossRef CAS.
- E. G. Percástegui, J. Mosquera, T. K. Ronson, A. J. Plajer, M. Kieffer and J. R. Nitschke, Chem. Sci., 2019, 10, 2006 RSC.
- Q. Xu, X. Wang, S. Huang, Y. Hu, S. J. Teat, N. S. Settineri, H. Chen, L. J. Wayment, Y. Jin, S. Sharma and W. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202304279 CrossRef CAS PubMed.
- B. Moosa, L. O. Alimi, W. Lin, A. Fakim, P. M. Bhatt, M. Eddaoudi and N. M. Khashab, Angew. Chem., Int. Ed., 2023, 62, e202311555 CrossRef CAS PubMed.
- H.-Y. Lin, Y.-T. Wang, X. Shi, H.-B. Yang and L. Xu, Chem. Soc. Rev., 2023, 52, 1129 RSC.
- T. Hong, Z. Zhang, Y. Sun, J.-J. Tao, J.-D. Tang, C. Xie, M. Wang, F. Chen, S.-S. Xie, S. Li and P. J. Stang, J. Am. Chem. Soc., 2020, 142, 10244 CrossRef CAS PubMed.
- C. Ngai, H.-T. Wu, B. da Camara, C. G. Williams, L. J. Mueller, R. R. Julian and R. J. Hooley, Angew. Chem., Int. Ed., 2022, 61, e202117011 CrossRef CAS PubMed.
- S. Pullen, S. Löffler, A. Platzek, J. J. Holstein and G. H. Clever, Dalton Trans., 2020, 49, 9404 RSC.
- R. Banerjee, D. Chakraborty, W.-T. Jhang, Y.-T. Chan and P. S. Mukherjee, Angew. Chem., Int. Ed., 2023, 62, e202305338 CrossRef CAS PubMed.
- S. M. Bierschenk, J. Y. Pan, N. S. Settineri, U. Warzok, R. G. Bergman, K. N. Raymond and F. D. Toste, J. Am. Chem. Soc., 2022, 144, 11425 CrossRef CAS PubMed.
- D. Chakraborty, S. Ali, P. Choudhury, N. Hickey and P. S. Mukherjee, J. Am. Chem. Soc., 2023, 145, 26973 CrossRef CAS PubMed.
- V. Martí-Centelles, A. L. Lawrence and P. J. Lusby, J. Am. Chem. Soc., 2018, 140, 2862 CrossRef PubMed.
- E. O. Bobylev, J. Ruijter, D. A. Poole III, S. Mathew, B. de Bruin and J. N. H. Reek, Angew. Chem., Int. Ed., 2023, 62, e202218162 CrossRef CAS PubMed.
- L. Catti, R. Sumida and M. Yoshizawa, Coord. Chem. Rev., 2022, 460, 214460 CrossRef CAS.
- M. Shuto, R. Sumida, M. Yuasa, T. Sawada and M. Yoshizawa, JACS Au, 2023, 3, 2905 CrossRef CAS PubMed.
- D. Zhang, T. K. Ronson, R. Lavendomme and J. R. Nitschke, J. Am. Chem. Soc., 2019, 141, 18949 CrossRef CAS PubMed.
- A. Ghosh, J. Pruchyathamkorn, C. Fuertes Espinosa and J. R. Nitschke, J. Am. Chem. Soc., 2024, 146, 2568 CrossRef CAS PubMed.
- Y. Ding, L. O. Alimi, J. Du, B. Hua, A. Dey, P. Yu and N. M. Khashab, Chem. Sci., 2022, 13, 3244 RSC.
- M. Ueda, N. Kishida, L. Catti and M. Yoshizawa, Chem. Sci., 2022, 13, 8642 RSC.
- Y. Hashimoto, Y. Katagiri, Y. Tanaka and M. Yoshizawa, Chem. Sci., 2023, 14, 14211 RSC.
- S. Zhang, L. Ma, W. Ma, L. Chen, K. Gao, S. Yu, M. Zhang, L. Zhang and G. He, Angew. Chem., Int. Ed., 2022, 61, e202209054 CrossRef CAS PubMed.
- P.-P. Jia, Y.-X. Hu, Z.-Y. Peng, B. Song, Z.-Y. Zeng, Q.-H. Ling, X. Zhao, L. Xu and H.-B. Yang, Inorg. Chem., 2023, 62, 1950 CrossRef CAS PubMed.
- J. Gemen, M. J. Białek, M. Kazes, L. J. W. Shimon, M. Feller, S. N. Semenov, Y. Diskin-Posner, D. Oron and R. Klajn, Chem, 2022, 8, 2362 CAS.
- A. Brzechwa-Chodzyńska, G. Markiewicz, P. Cecot, J. Harrowfield and A. R. Stefankiewicz, Chem. Commun., 2023, 59, 6247 RSC.
- A. B. Sainaba, R. Saha, M. Venkateswarulu, E. Zangrando and P. S. Mukherjee, Inorg. Chem., 2024, 63, 508 CrossRef CAS PubMed.
- R. Tabuchi, H. Takezawa and M. Fujita, Angew. Chem., Int. Ed., 2022, 61, e202208866 CrossRef CAS PubMed.
- S. Maji, J. Samanta and R. Natarajan, Chem. – Eur. J., 2024, 30, e202303596 CrossRef CAS PubMed.
- P. Bhandari, R. Modak, S. Bhattacharyya, E. Zangrando and P. S. Mukherjee, JACS Au, 2021, 1, 2242 CrossRef CAS PubMed.
- A. B. Grommet, L. M. Lee and R. Klajn, Acc. Chem. Res., 2020, 53, 2600 CrossRef CAS PubMed.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810 CrossRef CAS PubMed.
- U. Bhatt, Mod. Heterocycl. Chem., 2011, 1401 CAS.
- C. G. Neochoritis, T. Zhao and A. Dömling, Chem. Rev., 2019, 119, 1970 CrossRef CAS PubMed.
- H. Zhao, Z.-R. Qu, H.-Y. Ye and R.-G. Xiong, Chem. Soc. Rev., 2008, 37, 84 RSC.
- S. R. Batten, N. R. Champness, X.-M. Chen, J. Garcia-Martinez, S. Kitagawa, L. Öhrström, M. O’Keeffe, M. P. Suh and J. Reedijk, Pure Appl. Chem., 2013, 85, 1715 CrossRef CAS.
- J. López-Molino and P. Amo-Ochoa, ChemPlusChem, 2020, 85, 1564 CrossRef PubMed.
- X.-M. Kang, M.-H. Tang, G.-L. Yang and B. Zhao, Coord. Chem. Rev., 2020, 422, 213424 CrossRef CAS.
- J.-P. Zhang, Y.-B. Zhang, J.-B. Lin and X.-M. Chen, Chem. Rev., 2012, 112, 1001 CrossRef CAS PubMed.
- D.-C. Zhong, W.-G. Lu, L. Jiang, X.-L. Feng and T.-B. Lu, Cryst. Growth Des., 2010, 10, 739 CrossRef CAS.
- H.-F. Wu, J.-G. Xu, J. Lu, F.-K. Zheng, S.-H. Wang and G.-C. Guo, ACS Omega, 2019, 4, 15107 CrossRef CAS PubMed.
- S. Abedi and A. Morsali, RSC Adv., 2015, 5, 51828 RSC.
- R.-R. Dai, C.-W. Ding, J.-Y. Zhou, R.-J. Wei, X.-Z. Wang, X.-P. Zhou and D. Li, Inorg. Chem., 2021, 60, 565 CrossRef CAS PubMed.
- B. Abeykoon, J.-M. Grenèche, E. Jeanneau, D. Chernyshov, C. Goutaudier, A. Demessence, T. Devic and A. Fateeva, Dalton Trans., 2017, 46, 517 RSC.
- A. Buragohain, M. Yousufuddin, M. Sarma and S. Biswas, Cryst. Growth Des., 2016, 16, 842 CrossRef CAS.
- P. Gu, H. Wu, T. Jing, Y. Li, Z. Wang, S. Ye, W. Lai, M. Ferbinteanu, S. Wang and W. Huang, Inorg. Chem., 2021, 60, 13359 CrossRef CAS PubMed.
- T.-W. Tseng, T.-T. Luo, S.-Y. Chen, C.-C. Su, K.-M. Chi and K.-L. Lu, Cryst. Growth Des., 2013, 13, 510 CrossRef CAS.
- G.-S. Yang, M.-N. Li, S.-L. Li, Y.-Q. Lan, W.-W. He, X.-L. Wang, J.-S. Qin and Z.-M. Su, J. Mater. Chem., 2012, 22, 17947 RSC.
- L. Hou, L.-N. Jia, W.-J. Shi, Y.-Y. Wang, B. Liu and Q.-Z. Shi, Dalton Trans., 2013, 42, 3653 RSC.
- Z. Lu, F. Meng, L. Du, W. Jiang, H. Cao, J. Duan, H. Huang and H. He, Inorg. Chem., 2018, 57, 14018 CrossRef CAS PubMed.
- P. Cui, Y.-G. Ma, H.-H. Li, B. Zhao, J.-R. Li, P. Cheng, P. B. Balbuena and H.-C. Zhou, J. Am. Chem. Soc., 2012, 134, 18892 CrossRef CAS PubMed.
- P. Pachfule and R. Banerjee, Cryst. Growth Des., 2011, 11, 5176 CrossRef CAS.
- G. Orcajo, G. Calleja, J. A. Botas, L. Wojtas, M. H. Alkordi and M. Sánchez-Sánchez, Cryst. Growth Des., 2014, 14, 739 CrossRef CAS.
- H. Molero, B. F. Bartlett and W. T. Tysoe, J. Catal., 1999, 181, 49 CrossRef CAS.
- T.-L. Hu, H. Wang, B. Li, R. Krishna, H. Wu, W. Zhou, Y. Zhao, Y. Han, X. Wang, W. Zhu, Z. Yao, S. Xiang and B. Chen, Nat. Commun., 2015, 6, 7328 CrossRef CAS PubMed.
- J.-Y. Xian, X.-X. Xie, Z.-Y. Huang, Y.-L. Liu, H.-Y. Song, Z.-Q. Chen, Y.-C. Ou and S.-R. Zheng, Cryst. Growth Des., 2023, 23, 1448 CrossRef CAS.
- A. Valverde-González, M. C. Borrallo-Aniceto, U. Díaz, E. M. Maya, F. Gándara, F. Sánchez and M. Iglesias, J. CO2 Util., 2023, 67, 102298 CrossRef.
- S. Jeong, X. Song, S. Jeong, M. Oh, X. Liu, D. Kim, D. Moon and M. S. Lah, Inorg. Chem., 2011, 50, 12133 CrossRef CAS PubMed.
- J. Liu, F. Wang, Q.-R. Ding and J. Zhang, Inorg. Chem., 2016, 55, 12520 CrossRef CAS PubMed.
- Y. J. Park, J. Y. Ryu, H. Begum, M. H. Lee, P. J. Stang and J. Lee, J. Am. Chem. Soc., 2015, 137, 5863 CrossRef CAS PubMed.
- P. Howlader and P. S. Mukherjee, Chem. Sci., 2016, 7, 5893 RSC.
- S. G. Lee, J. Y. Ryu, P. J. Stang and J. Lee, Inorg. Chem., 2018, 57, 8054 CrossRef CAS PubMed.
- P. Howlader, E. Zangrando and P. S. Mukherjee, J. Am. Chem. Soc., 2020, 142, 9070 CrossRef CAS PubMed.
- P. Howlader, P. Bhandari, D. Chakraborty, J. K. Clegg and P. S. Mukherjee, Inorg. Chem., 2020, 59, 15454 CrossRef CAS PubMed.
- Z. Wang, L. He, B. Liu, L.-P. Zhou, L.-X. Cai, S.-J. Hu, X.-Z. Li, Z. Li, T. Chen, X. Li and Q.-F. Sun, J. Am. Chem. Soc., 2020, 142, 16409 CrossRef CAS PubMed.
- X. Hang, X. Wang, M. Wang, M. Chen and Y. Bi, Inorg. Chem. Front., 2023, 10, 926 RSC.
- X. Zhu, S. Wang, H. Han, X. Hang, W. Xie and W. Liao, Cryst. Growth Des., 2018, 18, 225 CrossRef CAS.
- W. Fan, S. B. Peh, Z. Zhang, H. Yuan, Z. Yang, Y. Wang, K. Chai, D. Sun and D. Zhao, Angew. Chem., Int. Ed., 2021, 60, 17338 CrossRef CAS PubMed.
- X.-S. Lin, Y. Yu, L.-P. Zhou, L. He, T. Chen and Q.-F. Sun, Dalton Trans., 2022, 51, 4836 RSC.
- X.-S. Lin, W.-H. Liao, L.-P. Zhou and Q.-F. Sun, Chem. – Asian J., 2023, 18, e202201249 CrossRef CAS PubMed.
Footnote |
| † Soumya Dey and Medha Aggarwal contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |