
Synthesis and characterisation of permethylindenyl zirconium complexes and their use in ethylene polymerisation†
Jean-Charles Buffet,
Thomas A. Q. Arnold,
Zoë R. Turner ,
Phakpoom Angpanitcharoen and
Dermot O'Hare*
,
Phakpoom Angpanitcharoen and
Dermot O'Hare*
Chemistry Research Laboratory, Department of Chemistry, University of Oxford, 12 Mansfield Road, OX1 3TA, Oxford, UK. E-mail: dermot.ohare@chem.ox.ac.uk; Tel: +44 (0)1865 272686
First published on 8th October 2015
Abstract
We report the synthesis of two zirconocenes, dimethylsilylbis(hexamethylindenyl) zirconium dichloride, rac-(SBI*)ZrCl2, and nbutyldimethylsilyl(hexamethylindenyl) zirconium trichloride, [(Ind*SiMe2nBu)Zr(μ-Cl)Cl2]2. The complexes were characterised by NMR spectroscopy and X-ray crystallography, and the bonding was evaluated using density functional theory. rac-(SBI*)ZrCl2 demonstrated a very high activity for solution phase polymerisation of ethylene (ca. 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 kgPE−1 molZr−1 h−1 bar−1). Both rac-(SBI*)ZrCl2 and rac-(EBI*)ZrCl2 (EBI* = ethylenebis(hexamethylindenyl)) have been supported on MAO modified silica and AMOST layered double hydroxides (AMO-LDHs), and evaluated as catalysts in the slurry-phase polymerisation of ethylene. The highest catalytic polymerisation activities for rac-(SBI*)ZrCl2 and rac-(EBI*)ZrCl2 on the layered double hydroxides were 9657 and 4325 kgPE−1 molZr−1 h−1 bar−1 respectively, for MAO modified Mg2Al–SO4 LDH. However, rac-(EBI*)ZrCl2 was a three times more active catalyst than rac-(SBI*)ZrCl2 when supported on silica.
500 kgPE−1 molZr−1 h−1 bar−1). Both rac-(SBI*)ZrCl2 and rac-(EBI*)ZrCl2 (EBI* = ethylenebis(hexamethylindenyl)) have been supported on MAO modified silica and AMOST layered double hydroxides (AMO-LDHs), and evaluated as catalysts in the slurry-phase polymerisation of ethylene. The highest catalytic polymerisation activities for rac-(SBI*)ZrCl2 and rac-(EBI*)ZrCl2 on the layered double hydroxides were 9657 and 4325 kgPE−1 molZr−1 h−1 bar−1 respectively, for MAO modified Mg2Al–SO4 LDH. However, rac-(EBI*)ZrCl2 was a three times more active catalyst than rac-(SBI*)ZrCl2 when supported on silica.
Introduction
Well-defined group 4 metallocenes have offered an important alternative technology to the ethylene polymerisation field dominated by Ziegler–Natta and Philips catalysts.1,2 In contrast to the heterogeneous Ziegler–Natta systems, metallocene catalysts produce polyethylene with narrow molecular weight distributions and polydispersity indices close to two. Furthermore, the single-site nature of their active sites enables the polymer properties to be fine-tuned.3Studies of bridged metallocene compounds have shown that slight variations of the ring substituents and bridging groups can massively influence the activity of ethylene polymerisation.4,5 The electronic structure and spatial organisation of the active site of the catalyst, influenced by ligand structure, have profound effects on the microstructure of the polyethylene obtained and hence, the overall polymer properties.6,7
Support materials provide a convenient way to serve as a template for the growing polymer particle.8 Many materials have been tested as polymerisation supports including inorganic solids such as clays,9 ZrO2,10 SiO2,11 and MgCl2.2 Silica (SiO2) is the most commonly used support for immobilising metallocene complexes.11 Modified silicas, such as mesoporous SBA-15,12,13 MCM-41,14,15 and nanostructured silica materials,16,17 have also been investigated. Layered double hydroxides (LDHs) are a class of hydrotalcite-like clays containing positively charged layers with intercalated anions between them.18–24 It is most typical for the metal ions to be divalent and trivalent. The common divalent metal ions include Mg2+ and Ca2+, while the trivalent metal ion is typically Al3+. The intercalated anions show more variety, and while they can include large anionic organic salts, they are more typically inorganics such as CO32−, SO42− or Cl−. Recently, we developed a new generation of LDHs using an aqueous miscible solvent treatment (AMOST) method.19–21 The AMO-LDHs made in this way exhibit surface areas two orders of magnitude in excess of those previously reported (ca. 400 m2 g−1). We have demonstrated via an EXAFS study that they are well-ordered supports for slurry phase polymerisation of ethylene.22,23
We report here the synthesis and characterisation of two new permethylated indenyl zirconium complexes, their reaction with both an MAO-treated layered double hydroxide and silica to form supported catalyst systems, and their use in the slurry and solution polymerisation of ethylene.
Results and discussion
Synthesis of pro-ligands
The synthesis of the dimethylsilylbis(hexamethylindene), (SBI*)H2, ligand was previously reported by O'Hare and co-workers but the compound was not crystallographically characterised.24Single crystals of rac-(SBI*)H2 suitable for an X-ray diffraction study were obtained from a saturated hexane solution at room temperature. The molecular structure of (SBI*)H2 is shown in Fig. 1. Selected bond lengths and angles are given in Table S1.† The stereochemistry of the two chiral centres and the space group (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) show that the crystal corresponds to a racemic mixture, rac-(SBI*)H2.
) show that the crystal corresponds to a racemic mixture, rac-(SBI*)H2.
 | ||
| Fig. 1 Molecular structure of (S,S)-rac-(SBI*)H2. Ellipsoids are drawn at 50% probability level. Hydrogen atoms, except for at the chiral centres, are omitted for clarity. | ||
Reaction of one equivalent of (SBI*)H2 with two equivalents of nbutyllithium in thf for 16 h afforded lithium dimethylsilylbis(hexamethylindenide), (SBI*)Li2, as yellow powder in quantitative yield.24
However, when the reaction was carried out in a pseudo one-pot starting from the hexamethylindene and with a shortened reaction time of 2 h (from 16 h), lithium nbutyldimethylsilyl(hexamethylindenide), LiInd*SiMe2nBu, was afforded as a colourless solid in 83% yield (Scheme 1).
 | ||
| Scheme 1 Synthesis of lithium nbutyldimethylsilyl(hexamethylindenide) (LiInd*SiMe2nBu). | ||
The 1H NMR spectrum of LiInd*SiMe2nBu in pyridine-d5 (Fig. S5†) shows six singlets corresponding to the ring methyl groups at approximately 2.4–3.1 ppm and a singlet at 0.73 ppm, with intensity six, is attributed to the silicon methyl groups. A triplet at 0.92 ppm corresponds to the butyl terminal CH3, while the three multiplets at 1.27, 1.47 and 1.72 ppm represent the methylene groups α, γ and β to the silicon respectively.
Synthesis of complexes
Stoichiometric reaction of (SBI*)Li2 and ZrCl4 in benzene at room temperature led to the formation of rac-(SBI*)ZrCl2 as an orange crystalline material in 34% yield (Scheme 2). No evidence of meso-(SBI*)ZrCl2 was seen in any reaction aliquots analysed by 1H NMR spectroscopy – a marked change from the previous syntheses of (EBI*)ZrCl2 and (SBI*)Fe where 50:50 rac-:meso-isomeric mixtures were obtained.7,24 Reactions in toluene or thf led to yields below 10%.
 | ||
| Scheme 2 Synthetic pathway to rac-(SBI*)ZrCl2. | ||
Single crystals suitable for an X-ray crystallographic study were grown from a benzene solution at room temperature. The molecular structure is represented in Fig. 2. Selected bond lengths and angles are given in Table S3.†
 | ||
| Fig. 2 Molecular structure of rac-(SBI*)ZrCl2. Hydrogen atoms are omitted for clarity. Thermal ellipsoids drawn at 50%. | ||
rac-(SBI*)ZrCl2 crystallises in the monoclinic space group P21/n, with one molecule in the asymmetric unit and four molecules in the unit cell – two of each enantiomer. The first point of note is that the value of torsion angle, TA′‡ (145.15°) is larger than 90° showing that the two indenyl rings are pointed in opposite directions as is expected for a rac-isomer. This value of TA′‡ agrees well with that reported for rac-(SBI)ZrCl2, 145.20°,25 and implies that the constraints of the dimethyl silicon bridge outweigh any steric effects from the permethylation. The tilt angle, α,‡ (58.50°), is slightly lower than that reported for either (SBI)ZrCl2 (61.80°) or (2,4,6-Me3SBI)ZrCl2 (59.18°).26 The value of α is also in good agreement with that of rac-(EBI*)ZrCl2 (57.15°).7 The β‡ value for rac-(SBI*)ZrCl2 (18.89°) slightly exceeds those reported for (2,4,6-Me3SBI)ZrCl2 (17.05°) or (SBI)ZrCl2 (16.45°).27 The average Zr–Cpcent distance (2.251 Å) fits well with other values reported in the literature for ansa-bridged species including (EBI*)ZrCl2 (2.240 Å), (SBI)ZrCl2 (2.241 Å) and (2,4,6-Me3SBI)ZrCl2 (2.231 Å), and also with unbridged examples such as (Ind2*)ZrCl2 (2.257 Å) and (Ind2)ZrCl2 (2.231 Å).8,27
Geometry optimisation calculations were performed at the B3LYP28,30 and BP8631,32 level of DFT with those using the BP86 functional most successfully reproducing the established experimental metrical parameters of rac-(SBI*)ZrCl2; slightly increased rotation and larger tilt angles were observed. (Table 1).
| Experimental | Calculated B3LYP | Calculated BP86 | |
|---|---|---|---|
| Avg. Zr–Cpcent | 2.251 | 2.308 | 2.288 |
| Avg. Zr–Cl | 2.423 | 2.442 | 2.428 |
| C(1)–Si(1)–C(16) | 96.22(7) | 96.31 | 95.85 |
| α | 58.50 | 61.28 | 59.95 |
| TA′ | 145.15 | 148.44 | 146.30 |
Illustrations of the DFT-computed (BP86) HOMO and LUMO are given in Fig. 3. The HOMO primarily consists of ligand p-orbitals; localised double bond character within the benzene rings is implied. In contrast, the LUMO is primarily based on the d0 metal centre (29.0% Zr dxy orbital component).
 | ||
| Fig. 3 Illustration of the DFT-computed (BP86) LUMO (left) and HOMO (right) of rac-(SBI*)ZrCl2. | ||
The 1H NMR spectrum of rac-(SBI*)ZrCl2 in chloroform-d1 shows six singlets between 1.25 and 2.50 ppm. There is one resonance for the two silyl methyl groups which are in the same environment in solution and at low frequency (1.34 ppm).
Stoichiometric reaction of LiInd*SiMe2nBu with ZrCl4 in toluene for two hours led to [(Ind*SiMe2nBu)Zr(μ-Cl)Cl2]2 as a red crystalline solid in 73% yield (Scheme 3).
 | ||
| Scheme 3 Synthesis of nbutyldimethylsilyl(hexamethylindenyl) zirconium trichloride [(Ind*SiMe2nBu)Zr(μ-Cl)Cl2]2. | ||
The crystalline material was found to be suitable for analysis by X-ray diffraction, the molecular structure of [(Ind*SiMe2nBu)Zr(μ-Cl)Cl2]2 is depicted in Fig. 4.
 | ||
| Fig. 4 Molecular structure of [(Ind*SiMe2nBu)Zr(μ-Cl)Cl2]2. Hydrogens omitted for clarity. Thermal ellipsoids drawn at 50%. | ||
[(Ind*SiMe2nBu)Zr(μ-Cl)Cl2]2 crystallises in the triclinic space group P, with half a dimer in the asymmetric unit (Fig. 4). The Zr(1)–Cpcent distance (2.190 Å) is comparable to that in polymeric CpZrCl3 which has Zr–Cpcent distances of 2.196 Å.33 The bond lengths for the terminal chlorides (2.4037(4) and 2.3982(4) Å) are significantly shorter than those of the bridging chlorides (2.5837(4) Å). These agree well with the Zr–Cl distances reported for [CpZrCl3]n of 2.419(3) Å for the terminal chloride and 2.518(3) and 2.728(3) Å for the bridging ligands.
DFT calculations were performed on [(Ind*SiMe2nBu)Zr(μ-Cl)Cl2]2 and highlight the localised double bonds in the benzene rings in the HOMO. The LUMO is again primarily Zr-based with a 27.3% dxy orbital component on each metal centre. (Fig. 5 and Table 2).
 | ||
| Fig. 5 Illustration of the DFT-computed (BP86) LUMO (top) and HOMO (bottom) of [(Ind*SiMe2nBu)Zr(μ-Cl)Cl2]2. | ||
| Experimental | Calculated B3LYP | Calculated BP86 | |
|---|---|---|---|
| Zr–Cpcent | 2.190 | 2.234 | 2.215 |
| Avg. Zr–Clterminal | 2.4011 | 2.420 | 2.418 |
| Avg. Zr–Clbridging | 2.5835 | 2.656 | 2.643 |
| Cl(1)–Zr(1)–Cl(2) | 91.233(17) | 90.74 | 91.59 |
[(Ind*SiMe2nBu)Zr(μ-Cl)Cl2]2 was analysed by 1H NMR spectroscopy in benzene-d6, Fig. S7,† six singlets representing the indenyl methyl groups appear in the range 1.96–2.57 ppm and there are two singlets at 0.55 and 0.65 ppm corresponding to the silyl methyl groups. Multiplets relating to the butyl substituent appear at 0.86, 1.00, 1.26 and 1.29 ppm for the δ, α, γ and β positions respectively.
Synthesis of solid ethylene polymerisation pre-catalysts
We have investigated the use of two different catalyst supports, silica and AMO-LDH, due to their proven excellent activities in the literature.22,23,34,35 Both support materials were calcined before use: SiO2 at 600 °C for two hours under a flow of N2 and AMO-LDH at 150 °C for six hours under high vacuum (2 × 10−2 mbar). The surface of both supports was then treated with 0.5 equivalents of MAO; producing MAO modified silica (SSMAO) and MAO modified AMO-LDH (LDHMAO) with a maximum loading of 15 and 13.5 wt% aluminium (from methylaluminoxane) in the resulting SSMAO and LDHMAO respectively. Immobilisation of the complex was achieved by addition of toluene to a mixture of the complex and MAO-activated support, followed by heating at 60 °C for one hour. After work-up, all the solids were afforded as coloured powders in good yield (>80%).Scanning electron microscopy (SEM) images for the silica immobilisation illustrates an uniform shape and size distribution, Fig. S10.† The particles are not spherical but granular with an average size of approximately 10 μm. They do not change in either shape or dimension on reaction with MAO, nor on the immobilisation of rac-(SBI*)ZrCl2.
SEM-EDX analysis demonstrated the desired elements on the surface (C, 14.3; Al, 2.6; O, 62.9; Si, 19.4 (%)) (Fig. S10†).
13C CPMAS NMR spectroscopy was used to characterise LDHMAO–(SBI*)ZrCl2 (Fig. S8†). The dominant resonance at −9 ppm corresponds to the highly shielded methyl environment of MAO. Resonances between 22 and 34 ppm correspond to the methyl groups around the rings of rac-(SBI*)ZrCl2 while a slightly weaker resonance is also observed at 13 ppm which corresponds to the low frequency silicon methyl groups. The 29Si CPMAS NMR spectrum contains a resonance at −105 ppm corresponding to a single silicon environment found for the SiO2 within SSMAO; the resonance for the silyl methyl group in the ligand was too weak to be identified (Fig. S9†).
Solution phase polymerisation of ethylene
The solution phase polymerisations were all carried out under ethylene at a pressure of two bar, with 50 mL hexanes, 0.2 mg of rac-(SBI*)ZrCl2 and with an Al:Zr ratio (MAO:rac-(SBI*)ZrCl2) of 2000:1. All polymerisation runs were carried out for 30 minutes, or until the stirring ceased entirely. Complete polymerisation data is reported in Tables S7 and S8.† rac-(SBI*)ZrCl2 was used to catalyse the polymerisation of ethylene at a range of temperatures from 40–90 °C using methylaluminoxane (MAO) as the co-catalyst and scavenger (Fig. 6).
 | ||
| Fig. 6 Solution phase ethylene polymerisation activity dependence of rac-(SBI*)ZrCl2 on temperature. MAO (2000:1); 2 bar ethylene; 0.2 mg complex loading; 50 mL hexanes; timed until cessation of stirring. | ||
The activity achieved at 60 °C (22622 kgPE−1 molZr−1 h−1 bar−1) compares well with some of the highest reported values in the literature; rac-(EBI*)ZrCl2 (61800 kgPE−1 molZr−1 h−1 bar−1, 70 °C);7 rac-(SBI)ZrCl2, rac-(2,4,6-Me3SBI)ZrCl2 and Cp2ZrCl2 (18450, 44760 and 30450 kgPE −1molZr −1h −1 bar respectively, 30 °C).36,37 There is a sharp increase in activity seen between 50 and 60 °C (1123 and 22622 kgPE−1 molZr−1 h−1 bar−1 respectively). Similar features were reported by Eskelinen et al. who attributed a sharp rise in activity between 70 and 80 °C to an increase in the solubility of the PE chain (in n-heptane), allowing for better monomer diffusion to the catalytic centre.38 A marked decline in activity is also noted between 70 and 80 °C (18703 and 8130 kgPE−1 molZr−1 h−1 bar−1), suggesting decomposition at elevated temperatures.39 These values are much higher than the recently reported bis(pentamethyl)zirconium dichloride complexes (activity around 400 kgPE−1 molZr−1 h−1 bar−1).40 The weight average molecular weights, Mw, were high (213927 and 261337 kg mol−1 at 60 and 70 °C respectively).
A study was undertaken at 70 °C with rac-(SBI*)ZrCl2 to test the effect that the Al:Zr ratio has on activity (Fig. 7).
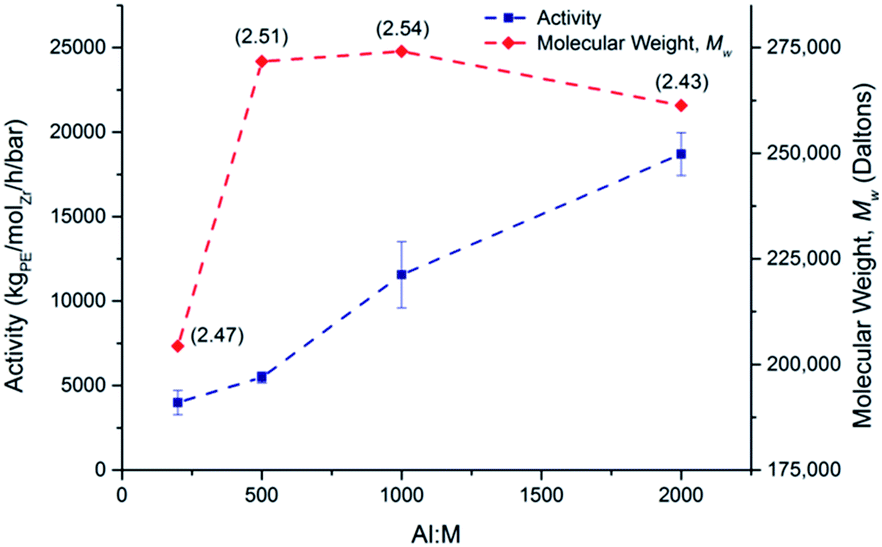 | ||
| Fig. 7 Dependence of solution phase ethylene polymerisation activity and molecular weight, Mw, for rac-(SBI*)ZrCl2 on the Al:Zr co-catalyst (MAO) ratio. PDIs are given in parentheses. 70 °C; 2 bar ethylene; 0.2 mg catalyst loading; 50 mL hexanes; timed until cessation of stirring. | ||
The activity at 2000:1 is more than four times greater than that at 200:1 (18703 and 3996 kgPE−1 molZr−1 h−1 bar−1 respectively). At an Al:Zr ratio of 200:1, Mw was 204374 kg mol−1 rising quickly to 271713 kg mol−1 at a ratio of 500 and plateauing on further increases. This contrasts with reports in the literature that suggest that Mw will decrease on addition of more MAO,41 or at the very least remain constant.42 Attempts were also made, not only to vary the Al:Zr ratio, but to vary the source of aluminium. Polymerisation attempts with 2000 equivalents of AliBu3 (TIBA) and AlMe3 (TMA) failed to produce any polyethylene. A comparison of the solution phase polymerisation activity of rac-(SBI*)ZrCl2 with other zirconocene catalysts is presented in Table 3.
| Activity (kgPE−1 molZr−1 h−1 bar−1) | Mw (g mol−1) | Mw/Mn | Ref. | |
|---|---|---|---|---|
| a 60 °C, 2 bar ethylene, 50 mL hexanes.b 70 °C, 10 bar ethylene, 1.8 L isobutene.c 30 °C, 2 bar ethylene, 5 mL toluene.d 30 °C, 2 bar ethylene, 5 mL toluene. | ||||
| rac-(SBI*)ZrCl2 | 22622 |
213927 | 2.32 | This worka |
| rac-(EBI*)ZrCl2 | 61800 |
215000 | 2.40 | 7b |
| rac-(SBI)ZrCl2 | 18450 |
260000 | 2.30 | 37c |
| rac-(2,4,6-Me3SBI)ZrCl2 | 44760 |
250000 | — | 36c |
| Cp2ZrCl2 | 30450 |
62000 |
2.00 | 37c |
| rac-(Ind#)ZrCl2 | 433 | 313893 | 3.07 | 40d |
Slurry phase polymerisation of ethylene
Slurry phase polymerisations of ethylene were carried out using methylaluminoxane derived silica (SSMAO) and AMO-LDH (LDHMAO) supported permethylindenyl catalysts. The slurry phase polymerisations were carried out similarly to the solution phase polymerisation with 10 mg of pre-catalysts. No leaching occurred during the slurry phase polymerisation. Complete polymerisation data is reported in Tables S9–S11.†We previously reported the solution phase polymerisation of ethylene using rac-(EBI*)ZrCl2 but no slurry polymerisations were carried out.7 Fig. 8 and 9 depict the ethylene polymerisation activity and the molecular weight data for SSMAO-rac-(EBI*)ZrCl2. The activities have an optimum of 2151 kgPE−1 molZr−1 h−1 bar−1 at 60 °C and are comparatively stable (i.e. above 1500 kgPE−1 molZr−1 h−1 bar−1) from 50–80 °C, higher than the reported results for (Ind)2ZrCl2 supported on silica with trimethylalumnium (400 kgPE−1 molZr−1 h−1 bar−1).43
 | ||
| Fig. 8 Dependence of slurry phase ethylene polymerisation activity and Mw for rac-(EBI*)ZrCl2 on temperature. PDIs are given in parentheses. Supported on SSMAO (200:1 loading); TIBA co-catalyst; 2 bar ethylene; 10 mg catalyst; 50 mL hexanes; 1 hour. | ||
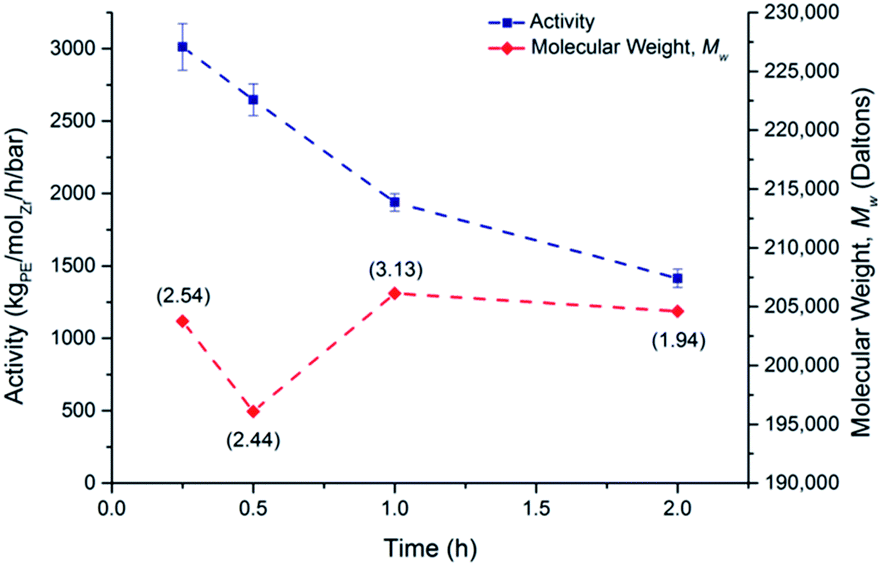 | ||
| Fig. 9 Dependence of slurry phase ethylene polymerisation activity and molecular weight, Mw, for rac-(EBI*)ZrCl2 on length of run. PDIs are given in parentheses. Supported on SSMAO (200:1 loading); TIBA co-catalyst; 70 °C; 2 bar ethylene; 10 mg catalyst; 50 mL hexanes. | ||
The molecular weight observed in the temperature range 50–80 °C is remarkably constant at just over 200000 kg mol−1 before dropping off to 162273 kg mol−1 at 90 °C.
The activities decreased with increasing time of polymerisation (3012 to 1414 kgPE−1 molZr−1 h−1 bar−1 for 15 minutes to two hours), consistent with literature reports.41
Data for the slurry phase polymerisation of ethylene using SSMAO-rac-(SBI*)ZrCl2 are depicted in Fig. 10 and 11.
 | ||
| Fig. 10 Dependence of slurry phase ethylene polymerisation activity and Mw for rac-(SBI*)ZrCl2 on temperature. PDIs are given in parentheses. Supported on SSMAO (200:1 loading); TIBA co-catalyst; 2 bar ethylene; 10 mg catalyst; 50 mL hexanes; 30 minutes. | ||
 | ||
| Fig. 11 Dependence of slurry phase ethylene polymerisation activity and Mw for rac-(SBI*)ZrCl2 on length of run. PDIs are given in parentheses. Supported on SSMAO (200:1 loading); TIBA co-catalyst; 70 °C; 2 bar ethylene; 10 mg catalyst; 50 mL hexanes. | ||
The activities increase from 340 to up to 725 kgPE−1 molZr−1 h−1 bar−1 (40 to 80 °C respectively). However, as expected the molecular weight decreased with increasing temperature (around 261002 to 160049 kg mol−1).
A comparison of two different loadings of rac-(SBI*)ZrCl2 on SSMAO led to the observation of the opposite trend seen for rac-(EBI*)ZrCl2. The activities obtained were lower on decreased loading at 70 °C: 725 and 611 kgPE−1 molZr−1 h−1 bar−1 (200:1 and 300:1 respectively).
These results confirmed the findings in solution phase that the ethylene bridge complex was faster the silyl bridge. However, the molecular weights were higher and the polydispersities lower for silyl based systems.
The permethylindenyl complexes rac-(EBI*)ZrCl2 and rac-(SBI*)ZrCl2 were also supported on an MAO-modified AMOST layered double hydroxides (AMO-LDH)21–23 and their polymerisation was carried out in similar conditions as the silica supported (Fig. 12).
 | ||
| Fig. 12 Dependence of slurry phase ethylene polymerisation activity and Mw for rac-(EBI*)ZrCl2 and rac-(SBI*)ZrCl2 on various support. PDIs are given in parentheses. Supported on LDHMAO (200:1 loading); TIBA co-catalyst; 2 bar ethylene; 10 mg catalyst; 50 mL hexanes; 1 hour. | ||
rac-(SBI*)ZrCl2 demonstrated higher activity than its ethylene bridged analogue (rac-(EBI*)ZrCl2) and also rac-(EBI)ZrCl2 with activities of 9657, 4325 and 1841 kgPE−1 molZr−1 h−1 bar−1 respectively. When the LDH support was varied, the catalyst based on Mg2Al–SO4 led to the highest activity, followed by Mg2Al–CO3 and Mg2Al–Cl with activities of 9657, 2719 and 2373 kgPE−1 molZr−1 h−1 bar−1 respectively. These data corroborate well with our recent studies.21,22 There is a decrease in the polydispersity when using the permethylindenyl in comparison to the parent indenyl complex.
Conclusions
We have reported the synthesis and characterisation of two new permethylindenyl complexes, rac-(SBI*)ZrCl2 and [(Ind*SiMe2nBu)Zr(μ-Cl)Cl2]2.rac-(SBI*)ZrCl2 demonstrated very high activity for the solution phase polymerisation (ca. 22500 kgPE−1 molZr−1 h−1 bar−1). However, the analogous ethylene-bridged compound demonstrated more than double this activity, albeit in a larger scale polymerisation with a higher ethylene pressure.7
MAO-modified AMOST layered double hydroxides support materials led to higher activity with respect to silica supported pre-catalysts. rac-(EBI*)ZrCl2 supported on the AMO-LDH Mg2Al–SO4, Mg2Al–CO3 and Mg2Al–Cl displayed polymerisation activities of 4325, 2719 and 2373 kgPE−1 molZr−1 h−1 bar−1 respectively. rac-(SBI*)ZrCl2 supported on the MAO-modified AMO-LDH Mg2Al–SO4 was the most active solid catalyst system at 9657 kgPE−1 molZr−1 h−1 bar−1.
However, rac-(EBI*)ZrCl2 was three times faster than rac-(SBI*)ZrCl2 when supported on silica.
Experimental details
Synthesis of LiInd*SiMe2nBu
Hexamethylindene (2.00 g, 10 mmol, 1 eq.) was dissolved in thf (50 mL) and nBuLi (2.5 M in hexanes; 4.4 mL, 11 mmol, 1.1 eq.) was added dropwise at room temperature, effecting a change in colour to pale pink. After 2 h, Me2SiCl2 (0.64 g, 0.60 mL, 5 mmol, 0.5 eq.) was added causing a slight decolouration in the solution to pale yellow. After 1 h, nBuLi (2.5 M in hexanes; 4.4 mL, 11 mmol, 1.1 eq.) was added to the reaction mixture and, following stirring at room temperature for 3 h, the reaction was dried under vacuum and LiInd*SiMe2nBu was obtained as an off white solid in an 83% yield (1.33 g, 4.15 mmol). 1H NMR (300 MHz, 298 K, pyridine-d5): δ 0.73 (s, 6H, SiMe2), 0.92 (t, 3H, 3JHH = 7.0 Hz, δ-Bu), 1.27 (m, 2H, α-Bu), 1.48 (m, 2H, γ-Bu), 1.72 (m, 2H, β-Bu), 2.47 (s, 3H, Ar–Me), 2.29 (s, 3H, Ar–Me), 2.82 (s, 3H, Cp–Me), 2.97 (s, 3H, Ar–Me), 3.03 (s, 3H, Cp–Me), 3.05 (s, 3H, Ar–Me). 13C{1H} NMR (75 MHz, 298 K, pyridine-d5): δ 5.96 (SiMe2), 14.84 (δ-Bu), 16.78 (Me), 17.33 (Me), 17.99 (Me), 18.33 (Me), 18.69 (Me), 23.11 (Me), 23.30 (α-Bu), 28.04 (γ-Bu), 28.77 (β-Bu), 90.71 (C–Si), 105.11 (Cp), 119.02 (Ar), 119.48 (Ar), 122.49 (Ar), 122.97 (Ar), 132.30 (Ar), 135.69 (Cp), 138.02 (Ar). 7Li NMR (156 MHz, 298 K, pyridine-d5): δ 1.54.Synthesis of rac-(SBI*)ZrCl2
Benzene (50 mL) was added to a mixture of (SBI*)Li2 (2.00 g, 2.47 mmol, 1 eq.) and ZrCl4 (0.98 g, 2.47 mmol, 1 eq.) and the reaction mixture was stirred at room temperature for 2 h. The resultant red solution was filtered away from the LiCl by-product, concentrated in vacuum by half and left overnight at room temperature to yield orange crystals. The crystals isolated by filtration and dried to afford orange crystalline material in 34% yield (0.89 g, 1.45 mmol). 1H NMR (400 MHz, 298 K, chloroform-d1): δ 1.34 (s, 6H, SiMe2), 1.88 (s, 6H, Cp–Me), 2.18 (s, 6H, Ar–Me), 2.30 (s, 6H, Ar–Me), 2.39 (s, 6H, Cp–Me), 2.45 (s, 12H, Ar–Me). 13C{1H} NMR (100 MHz, 298 K, chloroform-d1): δ 10.94 (SiMe2), 15.93 (Ar–Me), 16.09 (Ar–Me), 16.50 (Ar–Me), 17.52 (Cp–Me), 17.71 (Ar–Me), 22.12 (Cp–Me), 78.02 (Cp–Si), 127.81 (Me), 129.90 (Me), 130.17 (Me), 130.35 (Me), 130.89 (Me), 133.17 (Me), 134.31 (Me), 135.40 (Me). IR (KBr) (cm−1): 2961, 2923, 1543, 1454, 1384, 1260, 1097, 1020, 850, 813, 668. MS (EI): found 615.7972; calculated 615.7908. Major fragmentation peaks noted for M−Me (600.78).Synthesis of [(Ind*SiMe2nBu)Zr(μ-Cl)Cl2]2
To a mixture of LiInd*SiMe2nBu (1.00 g, 3.12 mmol, 1 eq.) and ZrCl4 (0.72 g, 3.12 mmol, 1 eq.) was added toluene (50 mL). This resulted in a bright orange slurry which became vermillion in colour on stirring for a further 2 h. The slurry was allowed to settle and the supernatant was filtered off and left to stand at −35 °C for three days affording [(Ind*SiMe2nBu)Zr(μ-Cl)Cl2]2 as a red crystalline solid suitable for an X-ray diffraction study in 73% yield (1.16 g, 2.28 mmol). 1H NMR (400 MHz, 298 K, benzene-d6): δ 0.55 (s, 3H, Si–Me2), 0.65 (s, 3H, Si–Me2), 0.86 (t, 3JHH = 7.0 Hz, 3H, δ-Bu), 1.01 (m, 2H, α-Bu), 1.25 (m, 2H, β-Bu), 1.29 (m, 2H, γ-Bu), 1.96 (s, 3H, Ar–Me), 2.03 (s, 3H, Ar–Me), 2.31 (s, 3H, Cp–Me), 2.42 (s, 3H, Ar–Me), 2.48 (s, 3H, Cp–Me), 2.57 (s, 3H, Ar–Me). 13C{1H} NMR (100 MHz, benzene-d6): δ 3.37 (Si–Me2), 3.75 (Si–Me2), 13.98 (δ-Bu), 15.92 (Cp–Me), 16.78 (Ar–Me), 17.31 (Ar–Me), 17.31 (Ar–Me), 17.87 (Cp–Me), 19.79 (α-Bu), 21.71 (Ar–Me), 26.82 (γ-Bu), 26.89 (β-Bu), 116.31 (Cp), 126.23 (Cp), 130.46 (Ar), 132.48 (Ar), 133.23 (Ar), 134.05 (Ar), 136.57 (Ar), 137.24 (Ar), 143.54 (Cp). IR (KBr) (cm−1): 3573, 2957, 1457, 1385, 1261, 1217, 1098, 814, 690, 480. HRMS (EI): found 508.0464; calculated 508.0460.Synthesis of solid polymerisation catalysts
The quantity of catalyst immobilised on the surface of the support is given in terms of the Al:Zr ratio of the methylaluminoxane component to the organometallic complex. In the case of SSMAO and LDHMAO, 0.5 eq. MAO is used to activate the surface of each support. The ratios used in this work are 300:1 and 200:1. The molecular weights used for SiO2 and LDH (Mg0.75Al0.25(OH)2(SO4)0.125) are 59.97 and 70.73 kg mol−1 respectively. This gives rise to molecular masses of SSMAO and LDHMAO of 177.94 and 199.46 kg mol−1. SSMAO and LDHMAO are prepared by weighing out silica or LDH and the MAO into a Schlenk tube in the glovebox and adding toluene to the mixture which is then heated at 60 °C for 1 h with regular swirling, until no further effervescence is observed. The solid is allowed to settle, the solution decanted, and the product dried under vacuum with gentle heating (50 °C) to ensure all the solvent is removed. In the glovebox, the activated support and complex was weighed out into a second Schlenk tube. Toluene (50 mL) is added and the reaction mixture is swirled at 60 °C for 1 h. The coloured solid is allowed to settle from the clear, colourless solution which is decanted, and the solid is dried in vacuo (40 °C, 1 × 10−2 mbar).
Solution phase ethylene polymerisation studies
rac-(SBI*)ZrCl2, and the methylaluminoxane were charged into an ampoule in a glovebox and 50 mL of hexanes was added. The reactions were performed under 2 bar of ethylene in a 200 mL ampoule, with 0.2 mg of the complex. The reactions were run for 15–120 minutes at varying temperatures. The resulting polyethylene was immediately filtered under vacuum through a dry sintered glass frit. The polyethylene product was then washed with pentane (2 × 5 mL) and then dried on the frit for at least 1 h. The tests were carried out at least twice for each individual set of polymerisation conditions. The activity values were reported as an average with ±1 SD error.Slurry phase ethylene polymerisation studies
The silica-supported zirconocene catalysts were tested for their ethylene polymerisation activity under slurry conditions in the presence of tri(isobutyl)aluminium (TIBA), an aluminium-based scavenger. The reactions were performed under 2 bar of ethylene in a 200 mL ampoule, with 10 mg of the catalyst suspended in 50 mL of hexanes. The polyethylene work-up was similar to the solution phase polymerisation.X-ray crystallography
Crystals were mounted on MiTeGen MicroMounts using perfluoropolyether oil, then transferred to a goniometer head on the diffractometer and cooled rapidly to 150 K in a stream of cold nitrogen using an Oxford Cryosystems CRYOSTREAM unit. For rac-(SBI*)ZrCl2 and [(Ind*SiMe2nBu)Zr(μ-Cl)Cl2]2, raw frame data were collected at 150 K using a Enraf-Nonius Kappa CCD diffractometer using graphite monochromated Mo Kα radiation (λ = 0.71073 Å),44 reduced using DENZO-SMN45 and corrected for absorption using SORTAV.46 The structures were solved using direct methods (SHELXS)47 or a charge-flipping algorithm (SUPERFLIP)48 and refined by full-matrix least-squares procedures using the Win-GX program suite.49 The data collection for (SBI*)H2 was carried out on an Oxford Diffraction Supernova diffractometer using mirror-monochromated Cu Kα radiation (λ = 1.54178 Å) and data were processed using CrysalisPro50 and refined using full matrix least-squares procedures using CRYSTALS.51,52, a = 9.52710(10) Å, b = 11.12740(10) Å, c = 11.36160(10) Å, α = 78.1654(5)°, β = 87.3403(5)°, γ = 77.9022(5)°, V = 1152.67(2) Å3, Z = 2, λ = 0.71073 Å, T = 150(2) K, μ = 0.88 mm−1, 5248 independent reflections, Rint = 0.013; R1 = 0.030, wR2 = 0.073. CCDC 1419346.Solid-state NMR spectroscopy data. LDHMAO-rac-(SBI*)ZrCl2
13C CPMAS NMR: δ −9.32 (AlOMe), 12.87 (SiMe2), 22.44 (Ar–Me), 24.57 (Ar–Me), 29.54 (Ar–Me), 31.10 (Ar–Me), 74.97 (Cp), 128.39 (Ar). 27Al CPMAS NMR: δ −527, −28, 470. SSMAO-EBI*ZrCl2. 13C CPMAS NMR: δ −9.03 (AlOMe). 27Al CPMAS NMR: δ −309, −113, 3, 182, 336. 29Si CPMAS NMR: δ −106.Acknowledgements
The authors would like to thank SCG Chemicals Ltd, Thailand for financial support, Dr. Nicholas H. Rees (University of Oxford) for the solid state NMR spectroscopy experiments and Chemical Crystallography (University of Oxford) for the use of the diffractometers. Z.R.T gratefully acknowledges Trinity College, Oxford for a Junior Research Fellowship.Notes and references
- L. Resconi, L. Cavallo, A. Fait and F. Piemontesi, Chem. Rev., 2000, 100, 1253 CrossRef CAS PubMed.
- G. G. Hlatky, Chem. Rev., 2000, 100, 1347 CrossRef CAS PubMed.
- (a) W. Kaminsky, A. Funck and H. Hähnsen, Dalton Trans., 2009, 8803 RSC; (b) W. Kaminsky, Macromol. Chem. Phys., 1996, 197, 3907 CrossRef CAS PubMed; (c) H. Sinn, W. Kaminsky, H. J. Vollmer and R. Woldt, Angew. Chem., Int. Ed., 1980, 19, 390 CrossRef PubMed.
- P. C. Möhring and N. J. Coville, J. Organomet. Chem., 1994, 479, 1 CrossRef.
- B. Wang, Coord. Chem. Rev., 2006, 250, 242 CrossRef CAS PubMed.
- S. Ahmadjo, H. Arabi, M. Nekoomanesh, S. M. M. Mortazavi, G. Zohuri, M. Ahmadi and S. Bolandi, Chem. Eng. Technol., 2011, 34, 249 CrossRef CAS PubMed.
- P. Ransom, A. E. Ashley, N. D. Brown, A. L. Thompson and D. O'Hare, Organometallics, 2011, 30, 800 CrossRef CAS.
- R. Duchateau, Chem. Rev., 2002, 102, 3525 CrossRef CAS PubMed.
- P. A. Zapata, C. Belver, R. Quijada, P. Aranda and E. Ruiz-Hitzky, Appl. Catal., A, 2013, 453, 142 CrossRef CAS PubMed.
- T. Pothirat, B. Jongsomjit and P. Praserthdam, Catal. Commun., 2008, 9, 1426 CrossRef CAS PubMed.
- P. Wongwaiwattanakul and B. Jongsomjit, Catal. Commun., 2008, 10, 118 CrossRef CAS PubMed.
- D. Bianchini, F. C. Stedile and J. H. Z. dos Santos, Appl. Catal., A, 2004, 261, 57 CrossRef CAS PubMed.
- E. Casas, R. van Grieken and J. M. Escola, Appl. Catal., A, 2012, 437–438, 44 CrossRef CAS PubMed.
- J. Moreno, R. van Grieken, A. Carrero and B. Paredes, Macromol. Symp., 2011, 302, 198 CrossRef CAS PubMed.
- J. M. Campos, M. R. Ribeiro, J. P. Lourenço and A. Fernandes, J. Mol. Catal. A: Chem., 2007, 277, 93 CrossRef CAS PubMed.
- J. M. Campos, J. P. Lourenço, H. Cramail and M. R. Ribeiro, Prog. Polym. Sci., 2012, 37, 1764 CrossRef CAS PubMed.
- P. A. Zapata, R. Quijada, I. Lieberwirth and H. Palza, Appl. Catal., A, 2011, 407, 181 CrossRef CAS PubMed.
- (a) X. Duan and D. G. Evans, Layered double hydroxides, Springer Verlag, 2006 CrossRef; (b) V. Rives, Layered Double Hydroxides: Present and Future, Nova Science Publishers, 2001 Search PubMed; (c) F. Cavani, F. Trifiro and A. Vaccari, Catal. Today, 1991, 11, 173 CrossRef CAS; (d) Q. Wang and D. O'Hare, Chem. Rev., 2012, 112, 4124 CrossRef CAS PubMed.
- Q. Wang and D. O'Hare, Chem. Commun., 2013, 49, 6301 RSC.
- Q. Wang, J. Undrell, Y. Gao, G. Cai, J.-C. Buffet, C. A. Wilkie and D. O'Hare, Macromolecules, 2013, 46, 6145 CrossRef CAS.
- (a) C. Chen, M. Yang, Q. Wang, J.-C. Buffet and D. O'Hare, J. Mater. Chem. A., 2014, 2, 15102 RSC; (b) M. Yang, O. McDermott, J.-C. Buffet and D. O'Hare, RSC Adv., 2014, 4, 51676 RSC; (c) C. Chen, A. Wangriya, J.-C. Buffet and D. O'Hare, Dalton Trans., 2015, 44, 16392 RSC.
- J.-C. Buffet, N. Wanna, T. A. Q. Arnold, E. K. Gibson, P. P. Wells, Q. Wang, J. Tantirungrotechain and D. O'Hare, Chem. Mater., 2015, 27, 1495 CrossRef CAS.
- J.-C. Buffet, Z. R. Turner, R. T. Cooper and D. O'Hare, Polym. Chem., 2015, 6, 2493 RSC.
- F. M. Alias, S. Barlow, J. S. Tudor, D. O'Hare, R. T. Perry, J. M. Nelson and I. Manners, J. Organomet. Chem., 1997, 528, 47 CrossRef CAS.
- W. A. Herrmann, J. Rohrmann, E. Herdtweck, W. Spaleck and A. Winter, Angew. Chem., Int. Ed., 1989, 28, 1511 CrossRef PubMed.
- A. V. Churakov, J. A. K. Howard and A. Z. Voskoboynikov, J. Organomet. Chem., 2005, 690, 1067 CrossRef PubMed.
- T. Repo, M. Klinga, I. Mutikainen, Y. Su, M. Leskelä, M. Polamo, M. N. Homsi, F. K. H. Kuske, M. Haugg, N. Trabesinger-Rüf and E. G. Weinhold, Acta Chem. Scand., 1996, 50, 1116 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1986, 84, 4524 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS.
- J. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822 CrossRef.
- L. M. Engelhardt, R. I. Papasergio, C. L. Raston and A. H. White, Organometallics, 1984, 3, 18 CrossRef CAS.
- F. Silveira, M. Alves, F. C. Stedile, S. B. Pergher and J. H. Z. dos Santos, J. Mol. Catal. A: Chem., 2010, 315, 213 CrossRef CAS PubMed.
- R. Guimarães, F. C. Stedile and J. H. Z. dos Santos, J. Mol. Catal. A: Chem., 2003, 206, 353 CrossRef.
- W. Kaminsky, J. Chem. Soc., Dalton Trans., 1998, 1413 RSC.
- W. Kaminsky, R. Engehausen, K. Zoumis, W. Spaleck and J. Rohrmann, Makromol. Chem., 1992, 193, 1643 CrossRef CAS PubMed.
- M. Eskelinen and J. V. Seppälä, Eur. Polym. J., 1996, 32, 331 CrossRef CAS.
- J. R. Severn, J. C. Chadwick, R. Duchateau and N. Friederichs, Chem. Rev., 2005, 105, 4073 CrossRef CAS PubMed.
- T. A. Q. Arnold, J.-C. Buffet, Z. R. Turner and D. O'Hare, J. Organomet. Chem., 2015, 792, 55 CrossRef CAS PubMed.
- L. D'Agnillo, J. B. P. Soares and A. Penlidis, Macromol. Chem. Phys., 1998, 199, 955 CrossRef.
- B. Rieger and C. Janiak, Angew. Makromol. Chem., 1994, 215, 35 CrossRef CAS PubMed.
- D.-H. Lee, S.-Y. Shin and D.-H. Lee, Macromol. Symp., 1995, 97, 195 CrossRef CAS PubMed.
- J. Cosier and A. M. Glazer, J. Appl. Crystallogr., 1986, 19, 105 CrossRef CAS.
- Z. Otwinowski and W. Minor, Methods Enzymol., 1997, 276, 307 CAS.
- R. H. Blessing, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 33 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- CrysAlisPRO, Oxford Diffraction/Agilent Technologies UK Ltd, Yarnton, England Search PubMed.
- P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef CAS.
- R. I. Cooper, A. L. Thompson and D. J. Watkin, J. Appl. Crystallogr., 2010, 43, 1100 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: General details, NMR spectroscopy, X-ray crystallography, computational details and definitions of structural parameters. CCDC 1001294, 1001295 and 1419346. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra20465h |
| ‡ Definitions of the structural parameters (TA, RA, α, β) are detailed in the ESI.† |
| This journal is © The Royal Society of Chemistry 2015 |