
Immobilizing and de-immobilizing enzymes on mesoporous silica†
Vladimir Zlateski,
Tobias C. Keller,
Javier Pérez-Ramírez and
Robert N. Grass *
*
Institute for Chemical and Bioengineering, ETH Zurich, Zurich, 8093, Switzerland. E-mail: robert.grass@chem.ethz.ch; Fax: +41 44 633 10 83; Tel: +41 44 633 63 34
First published on 8th October 2015
Abstract
Beta glucosidase was immobilised as a model enzyme within mesoporous silica (MCF) at a high loading (80 mg g−1). The enzyme was further entrapped within the material by precipitating additional silica within the channels. This entrapment was performed by the polycondensation of tetraethoxysilane under very mild conditions (pure water). Although unreactive while entrapped, in this state the enzyme was highly stable towards heat treatments of 60–70 °C. Upon release from the matrix by a mild silica dissolution step involving a fluoride comprising buffer, the enzyme regained most of its original activity. With this we developed a novel protein entrapment/release scheme, which is designed along the principles of orthogonal protection group chemistry as the protection/deprotection steps do not affect the integrity of the (bio)molecule. The principle can be adopted to many previously developed mesoporous silica/enzyme biocomposites and will allow the application of enzyme dependent diagnostic devices in applications involving demanding environmental storage requirements.
Introduction
Mild and environmentally friendly reaction conditions in combination with high chemo-, regio- and stereoselectivity as well as high turnover rates compared to synthetic catalysts, contributed to the increased use of enzymes in the last decades, thus fostering the idea of using sustainable methodologies for chemical reactions.1,2 Even though enzymes are extremely efficient as biocatalysts for many chemical reactions, their application is often hampered by the lack of long-term storage stability, considering the fact that many temperature sensitive enzymes need to be continuously stored at −20 °C. Operational stability, namely inactivation caused by mechanical treatment or heat denaturation, and difficulties in recovery and recycling are other every-day problems.3 The effort invested to circumvent these issues led to the development of enzyme immobilization techniques on solid supports (physical adsorption or covalent binding), entrapment and cross-linking, which have proven to enhance enzyme stability and enable re-use.1,4–6Physical adsorption is based purely on hydrogen bonds, electrostatic and hydrophobic interactions between the support surface and the protein of interest. Compared to the other immobilization methods it is the simplest, with which denaturation/deactivation of the enzymes can be avoided and good enzymatic activities can be maintained.7 Adsorption of enzymes on pre-fabricated porous inorganic supports, such as mesoporous silicates (MPS), is currently one of the most attractive enzyme immobilization methods due to the offered simplicity, support stability and large surface area.6,8–11 Despite its high loading, intrinsic problems remain: immobilized enzymes are less active than free enzymes; activity may be further lost12 due to enzymes leaching out from the support and due to spatial constraints, the reaction of immobilized enzymes with large substrates (proteins/DNA/polysaccharides) is very limited.
Previous studies have demonstrated attempts to tackle the leaching and stability problems by fine-tuning the channel to the enzyme size (snug fit).13,14 However, this approach is enzyme specific and results in low protein loadings and substrate diffusion problems.15 Another strategy is based on selective silylation thus reducing the size of the pore openings of the mesoporous supports. In their work, He and co-workers managed to slightly reduce the pore opening diameter of the lipase immobilized MPS by employing 3-(trimethoxysilyl)propyl methacrylate (PMA) and polymerization of the anchored vinyl groups with free PMA.16 Similar work was done by Ma and co-workers, where they employed the same enzyme but a different grafting strategy.17 However, the conditions used in both cases are quite harsh (toluene, 70 °C or 35 °C in the latter case) for most enzymes to survive and be active again. Besides, mechanical and thermal stability of the proteins was not investigated. Both strategies however, have not solved the large substrate limitations.
Following on previous unsuccessful attempts to directly encapsulate proteins in silica in analogy to encapsulating DNA/RNA (see ESI†),18–21 we took advantage of literature knowledge on enzyme immobilization on mesoporous materials. To further overcome the disadvantages that immobilized enzymes have during application, we herein report a novel approach to stably entrap enzymes and release them on demand (Scheme 1).
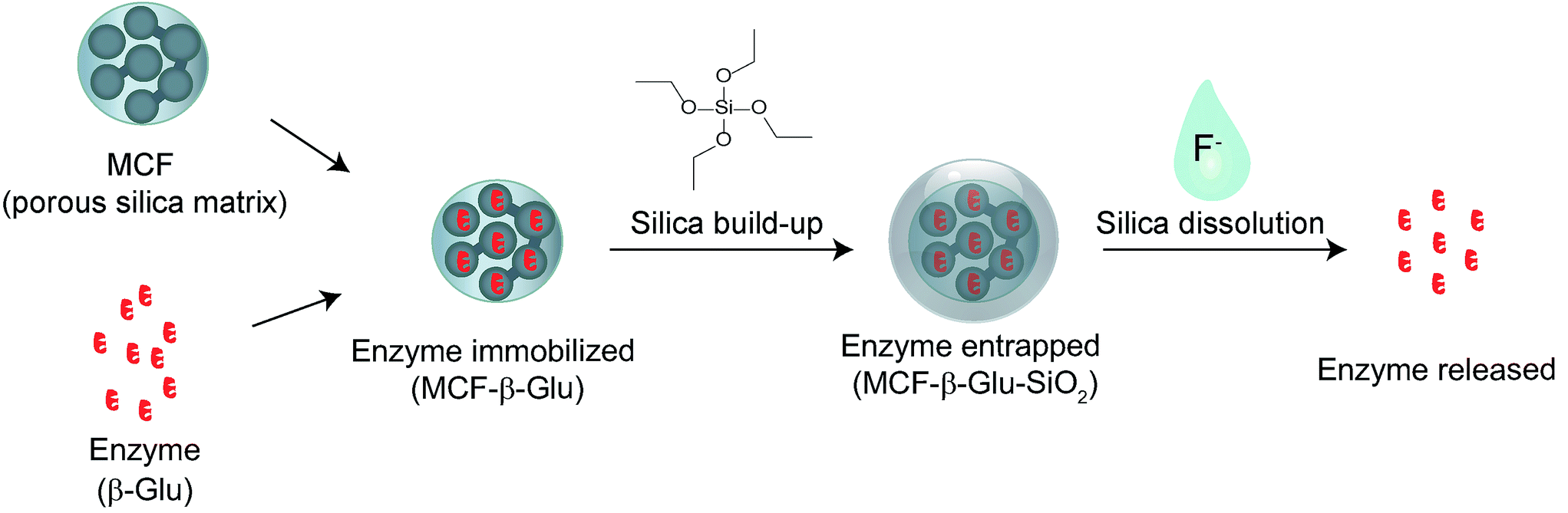 | ||
| Scheme 1 Silica entrapment and fluoride buffer triggered release of enzymes. | ||
Experimental
MCF mesoporous silica synthesis
Mesocellular foam (MCF) was prepared according to a method reported previously with some modifications.22 The mesoporous silica was synthesized from TEOS (tetraethyl orthosilicate) (Sigma-Aldrich), pluronic P123 (poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol)) (Sigma-Aldrich) and mesitylene (Sigma-Aldrich). First, pluronic P123 (4 g) was added to water (120 mL), followed by KCl (6.1 g). The mixture was stirred on a magnetic stirring plate (Heidolph) at 500 rpm at room temperature until it became translucent. Next, mesitylene (3 g) and 12 N HCl (23.6 g, Sigma-Aldrich) were added and the mixture was stirred for 2 h at room temperature. TEOS (8.5 g) was then added to the mixture and all together stirred vigorously for 30 min at room temperature. The solution was transferred to a Teflon-lining steel autoclave and was aged for 24 h at 35 °C in an incubator (Binder GmbH). Later, the solution was subsequently aged for an additional 24 h at 130 °C in a drying oven (T 6030, Heraeus Instruments), filtered and washed with water and ethanol. After all the ethanol was evaporated, the produced powder was calcined in a furnace (Nabertherm) at a heating rate of 60 °C h−1 and held at 500 °C for 6 h.Mercury intrusion experiment
Hg intrusion in the pressure range of 0.01–400 MPa was carried out in a Micromeritics Autopore IV 9510 instrument assuming a contact angle = 140° with a pressure equilibration time of 10 s. Pore size distributions were smoothed using a 2nd order Savitzky–Golay filter over a window of 10 points to eliminate noise from the differentiation.Nitrogen sorption experiment
Nitrogen sorption at −196 °C was carried out in a Micromeritics TriStar II instrument. The MCF was evacuated for 3 h at 300 °C, whereas the enzyme-loaded analogues were outgassed at room temperature. The total surface area (SBET) of the samples was determined by the BET method, and the t-plot method was used to determine the external surface area (Smeso). Pore size distributions were determined by applying the BJH model to the adsorption branch of the isotherm (Table 1).| Sample | dpore (nm) | Vpore (cm3 g−1) | Smeso (m2 g−1) | SBET (m2 g−1) |
|---|---|---|---|---|
| MCF | 23 | 1.95 | 410 | 434 |
| MCF-β-Glu | 21 | 1.70 | 323 | 368 |
| MCF-β-Glu-SiO2 | 13 | 0.48 | 234 | 341 |
Small angle X-ray scattering (SAXS)
The small-angle X-ray scattering (SAXS) curve was recorded on an Empyrean powder diffractometer (PANalytical B.V., The Netherlands), operating in transmission mode with Cu Kα radiation (45 kV, 40 mA). The interlayer spacing was calculated by the Bragg's law (n × λ = 2 × d × sin(θ)), where λ = 0.154 nm.TEM and SEM analysis
For transmission electron microscopy (TEM), the samples were dispersed in ethanol and some droplets of the suspension were deposited on a lacey carbon foil supported on a Cu grid. TEM was performed on a Tecnai F30 (FEI, field emission gun (FEG), SuperTwin lens (point resolution ca. 0.2 nm), operated at 300 kV). For scanning electron microscopy (SEM), the samples were resuspended in i-PrOH, and loaded onto copper/carbon grids. The microscope (FEI Nova NanoSEM) was operated at 30 kV.β-Glucosidase immobilization
MCF (20 mg) was suspended in MQ H2O (0.5 mL) by 30 s ultrasonicating and 15 s vortexing. Separately, β-glucosidase from almonds (20 mg, Sigma-Aldrich) was dissolved in MQ H2O (1 mL) and split into two Eppendorf tubes (0.5 mL each). In one, the MCF suspension was added, whereas in the second MQ H2O (0.5 mL) was added. Both tubes were shaken on an orbital shaker (VXR basic, IKA) for 3 h at room temperature followed by centrifugation at maximum speed for 4 min (CT15E, Hitachi Koki Co., Ltd). The pellet (mesoporous silica plus enzyme) was washed with MQ H2O (1 mL) and finally suspended in the same volume of MQ H2O.β-Glucosidase entrapment in silica
MCF-β-glucosidase (1 mg, corresponding to 50 μL suspension after immobilization) was suspended in MQ H2O (0.45 mL) by 30 s ultrasonicating and 15 s vortexing, followed by TEOS addition (4 μL). The final mixture was left shaking for 5 days at 500 rpm on an orbital shaker, with subsequent additions of TEOS (4 μL) after 24, 48 and 72 h. Afterwards, the sample was washed twice with MQH2O and stored in the fridge until the next activity measurement. In parallel, the two control samples (1) β-glucosidase in water and (2) MCF-β-glucosidase were treated the same way but without any TEOS addition.Enzyme release
The release of the enzyme was triggered by the fluoride buffer, which was added to the entrapped enzymes in order to dissolve the silica support. The buffer was prepared in polyethylene, polypropylene or Teflon containers according to the following protocol: for 10 mL fluoride buffer we dissolved 0.23 g of NH4FHF in 5 mL of H2O and 0.19 g of NH4F in 5 mL of H2O eventually pooling the two solutions together (pH ∼ 4; measure pH with pH paper and not with a pH electrode). This solution is stable at room temperature for at least 2 months. In order to release the enzyme, enough fluoride buffer was added in order to obtain a clear solution. In the case of the fluoride buffer at pH 5, the pH was adjusted by carefully adding NaOH (1 M, Merck). Fluoride comprising waste was collected in a saturated calcium carbonate solution.18Enzymatic activity assays
In the case of the silica-entrapped enzyme we took 10 μL of the reaction mix (as described in the “β-glucosidase entrapment in silica” section) and diluted up to 100 μL with water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10). Then from the dilution we used 48 μL in the assay (as mentioned above). In the cases where fluoride buffer was utilized for either enzyme release or fluoride buffer resistance tests, 25 μL of the fluoride buffer were added to the 10 μL of the enzyme sample, the volume was brought up to 100 μL with water and again 48 μL were taken for the assay.
:10). Then from the dilution we used 48 μL in the assay (as mentioned above). In the cases where fluoride buffer was utilized for either enzyme release or fluoride buffer resistance tests, 25 μL of the fluoride buffer were added to the 10 μL of the enzyme sample, the volume was brought up to 100 μL with water and again 48 μL were taken for the assay.
:1 water/DMSO mixture), CaCl2 (80 μL, 2 M, Fluka) and 100 μL of the sample to make up a final volume of 3 mL. Immediately after the enzyme addition, the mixture was transferred to a plastic disposable cuvette and absorbance was monitored each minute for up to 30 minutes.Enzymatic activity calculation
The specific enzymatic activities (U mg−1) of both free and immobilized β-glucosidase and α-chymotrypsin were calculated with the given formula: specific activity = (ΔA × Vt × Df)/(ε × l × Vs × C) where ΔA = (ΔA test − ΔA blank) per min. At the desired wavelength (390 or 405 nm); Vt = total volume of the reaction mixture; Df = dilution factor; ε = extinction coefficient; l = path length; Vs = volume of the sample; C = protein concentration. The extinction coefficient in the case of the p-nitrophenol (β-glucosidase assay) was calculated under our assay conditions (ε = 13394.43 M−1 cm−1) and in the case of the p-nitroaniline (α-chymotrypsin assay, ε = 12500 M−1 cm−1). The path length is l = 1 cm.
Protein concentration measurement
The amount of protein bound on the mesoporous material was estimated from the C, H, N percentage mass increase after immobilization obtained by elemental microanalysis measurement (Vario Micro Cube, Elementar) and from knowing the elemental content of the enzymes (% N and % C).Thermal stability test
Two samples, β-glucosidase free in water and β-glucosidase entrapped in the silica material, were submitted to thermal stress by incubating them for 1 h at 4 different temperatures: room temperature, 50 °C, 60 °C and 70 °C in a thermomixer (compact, Eppendorf). The samples were let to cool down to and enzymatic activity was measured where the enzyme entrapped in silica was first dissolved with a proper amount of fluoride buffer (as described in the “β-glucosidase assay” enzymatic activity section).Fluoride buffer influence on enzymatic activity
Both β-glucosidase and α-chymotrypsin free in solution were used to check the influence of the fluoride buffer on the enzymatic activity. The same concentrations of free enzyme in solution as used for the immobilization process were used and enzymatic assays were performed by using 10 μL of the enzyme solutions and 25 μL of the following fluoride buffer solutions: (1) fluoride buffer pH 4 (preparation steps shown in the “enzyme release” section of this materials and methods chapter), (2) 1:10 diluted fluoride buffer pH 4 and (3) fluoride buffer pH 5 (pH adjusted to 5 with NaOH). The enzyme solutions were incubated in the presence of the respective fluoride buffer for 2–3 minutes and then transferred to a glass vial for neutralization of the excess F− ions. The enzyme assay solutions were added into the vials and the desired absorbance was subsequently measured.
Results and discussion
For entrapment, we combined the well-known advantages of the mesoporous silicas with the simplicity of the adsorption process in order to obtain high loadings of highly active enzymes immobilized on the MCF carrier. The ultralarge cage-like mesopores of this support are ideal for entrapping enzymes of different dimensions in high loadings.23 Additionally, MCFs possess a three-dimensional, interconnected pore structure which would facilitate substrate diffusion.24,25 Having that as a starting point, we developed a procedure to further entrap enzymes within the porous material, by growing additional silica inside the MCF cells in a protein-friendly environment (water).MCF material with a mesopore size of 23 nm was synthesized as the immobilization support of choice. The mesopores of the calcined MCF were confirmed by measuring nitrogen adsorption–desorption isotherms of the dried sample and plotting the corresponding pore-size distribution curve. (Fig. 1, black squares). N2 sorption analysis evidences the typical type IV isotherm (well defined hysteresis loop) obtained for uniform-size mesoporous materials26 and gives an external surface of 410 m2 g−1 with a total pore volume of 1.95 cm3 g−1 (Fig. 1, Table 1). The large uniform mesopores (dpore = 23 nm), which are in good agreement with the microscopy analysis (Fig. 2(a) and (c)) and the X-ray data (Fig. 3), can be attributed to the organic cosolvent mesitylene addition.22,27,28 Hg intrusion experiment resulted in a pore window size of ∼11 nm and 75% of the mesopore volume determined by nitrogen sorption was proven to be accessible for Hg intrusion (Fig. S1 and S2†). Analysis of the scattering data shows that no indexing of higher order peaks to any plane or space group (e.g. p6mm) is possible and no SBA-15-like structure could be found (Fig. 3).
 | ||
| Fig. 1 N2 sorption isotherms and pore-size distribution curves (inset) obtained from the adsorption branch by the Barret–Joyner–Halenda (BJH) method of: MCF (black squares), MCF-β-Glu (blue circles), and MCF-β-Glu-SiO2 (red triangles). | ||
 | ||
| Fig. 2 TEM and SEM images of the MCF ((a) and (c)) and MCF-β-Glu-SiO2 ((b) and (d)). The texture of the material after silica deposition drastically changed and the obvious mesopore openings in the original material were no longer visible. | ||
 | ||
| Fig. 3 Small-angle X-ray scattering (SAXS) from a MCF with an interlayer spacing of 25.6 nm. No ordered structure is evident. | ||
β-Glucosidase from almonds was chosen as a model enzyme to be immobilized and further entrapped in the silica matrix. Typically, proteins enable binding interactions for adsorption to take place mainly due to the amino and carboxylic acid groups present on their surface. Knowing that the surface of the silicas carries a negative charge at pH values above 2,29 which is much lower than the isoelectric point (pH at which no overall electric charge is carried) values of most proteins, we are able to tune the charge of the protein surface by changing the pH of the solution. However, if the proteins get too much positively charged strong self-repulsion may occur.8,30 Taking advantage of improved adsorption in solutions which have a pH near the pI value of the enzyme, we immobilized β-glucosidase (pI = 7.3) in water at pH 7. We managed to retain activity up to 95% after immobilization with an enzyme loading of ∼80 mg g−1. The pore network is well-preserved after the loading of the enzyme, although the size of the mesopores, and consequently their volumes are reduced (Fig. 1 (blue circles), Table 1).
With the aim to increase the enzyme stability and prevent enzyme leaching, we further entrapped the enzymes within the matrix. For this we utilized silica sol–gel synthesis, a well-known and fundamental reaction that brings about the conversion of silicate precursors (i.e. TEOS) to silica gels.31 In order to minimize any perturbation of the enzyme integrity, we performed the polycondensation reaction by simply mixing the immobilized enzyme with highly diluted tetraethyl orthosilicate (TEOS) in water (opposed to the traditional procedure in alcohol and base catalysis). The entrapment process was monitored by evaluating the activity of the enzyme after various time points. As shown on Fig. 4, this entrapment process was relatively slow and proceeded over several days, which is due to the slow polycondensation reaction of TEOS in the absence of a suitable catalyst.32 After a silica growth process of 5 days, the enzyme activity had dropped by >80% indicating that new silica material had formed within the MCF cells hindering the substrate diffusion to the enzyme. This observation was confirmed by nitrogen sorption and electron microscopy, evidencing that the silica deposition led to a pronounced textural modification (Fig. 2 & Table 1). After the reaction the material evidences both micro- and shallow mesopores. The mesopore volume is reduced by 75%, and the formerly uniform pore-size is transformed into a broader distribution centred at around dpore = 13 nm (Fig. 1, red triangles), indicating that the cells are gradually filled with amorphous silica. Furthermore, the broad hysteresis loop points towards the presence of ink-bottle-like pores, i.e. pores accessible only through a narrow opening, further corroborated by the increased intensity of the forced closure of the isotherm at p/p0 = 0.45.33,34
 | ||
| Fig. 4 Enzymatic activities during the sol–gel synthesis of MCF-β-Glu-SiO2 (stars) and β-Glu free in solution (squares). In order to rule out that the loss of activity is due to enzyme degradation during the 5 day reaction, free enzyme was subjected to the same conditions (mechanical stress and temperature/but no TEOS addition) and showed only a minor loss of activity. | ||
While the above shows that the entrapment of the enzyme within the support was successful, it also displays that as long as the enzyme is entrapped within the inorganic material, it is not very active. As both the support structure (MCF) and the additional material grown within the pores consists entirely of silica, we investigated on a de-encapsulation scheme by dissolving the silica, again without perturbing the enzyme structure. It is well known that silica dissolves rapidly in fluoride comprising buffers (e.g. buffered oxide etch, a pH stabilized ammonium fluoride solution). However, these reagents are very rarely used in biochemistry as they are feared due to their toxicological potential und unclear implications with proteins. Still the application of fluoride solutions is a commonplace in classical protection group chemistry (e.g. potassium fluoride or tetra butylammonium fluoride) and even dentistry. So in order to avoid the handling of dangerous HF, we prepared small volumes of a 4 wt% F− buffered oxide etch solution from ammonium fluoride and ammonium hydrogen fluoride. For reference, dental care products (fluoride gels) contain up to 1.23 wt% F− and 5 mg F− per kg bodyweight is considered the probable toxic dose for humans.18
In order to de-encapsulate the entrapped enzymes, 20 μg of the material was mixed with a small volume of buffered oxide etch (50 μL), resulting in a clear solution. To evaluate the intactness of the active site of the released enzyme the β-glucosidase activity of the resulting solution was measured in an appropriate buffer. As shown on Fig. 5 this procedure resulted in a great boost in the catalytic activity, indicating that the silica support had been dissolved and that the enzyme was released unharmed. The activity of the released enzyme went up to ∼250% of the entrapped one and was comparable to the activity of β-glucosidase free in solution after 5 days storage in the fridge (Fig. 5, dense columns), attributing almost no activity loss to the entrapment/release process itself. It is worth mentioning is that no increase in activity is evident when the immobilized (not entrapped) enzyme is incubated in fluoride buffer (Fig. S3†). As an additional control to this we measured the compatibility of β-glucosidase with fluoride comprising buffers (see Fig. S4†). In addition, another widely used enzyme, α-chymotrypsin, was submitted to the same fluoride buffer compatibility test (see Fig. S4†). A minor activity decrease (10–20%) could be measured for both enzymes, in the three cases where fluoride buffer at pH 4, fluoride buffer at pH 4 diluted (1:10) and fluoride buffer at pH 5 was used for incubation.
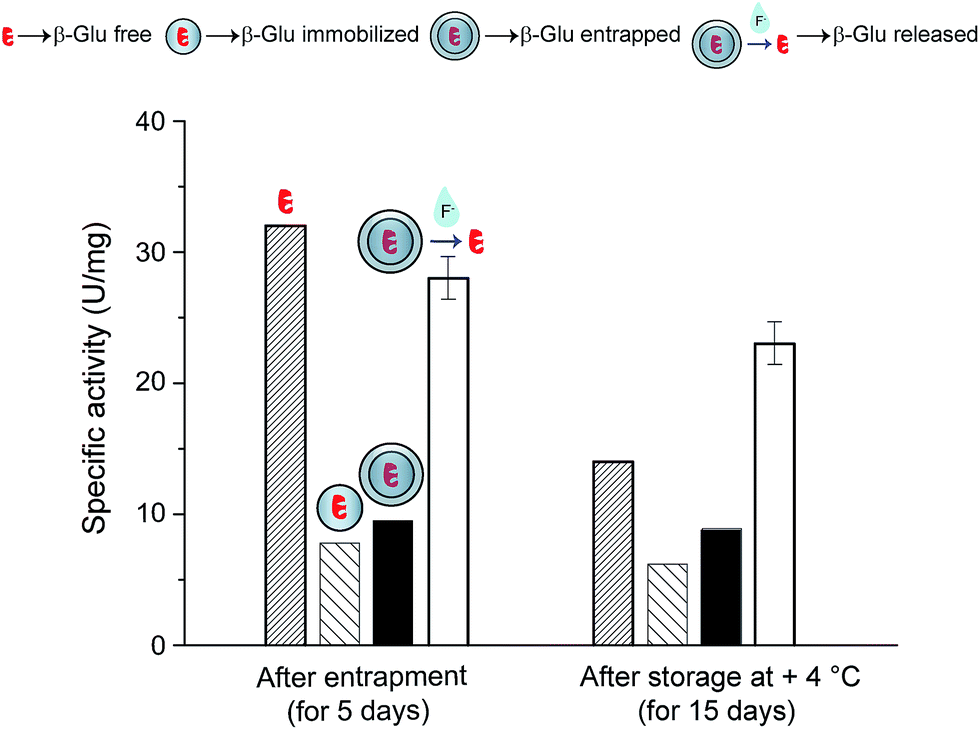 | ||
| Fig. 5 Highly active β-Glu (white columns) was released after MCF-β-Glu-SiO2 (black columns) was treated with fluoride-containing buffer. The activities of the MCF-β-Glu (sparse columns) and β-Glu free in solution (dense diagonal lines) are given for comparison. The storage stability was assessed after 15 days and further shows the advantage of the silica entrapment/release scheme for enzyme storage. | ||
It is worth highlighting the importance of the second comparison on Fig. 5, displaying data after 15 days of wet storage in the fridge. While the free enzyme lost more than half of its activity, no substantial activity loss was observed for the entrapped enzyme after the release.
Besides the high mechanical and storage stabilities already discussed, the encapsulated enzyme shows a high resistance towards heat treatment. MCF-β-Glu-SiO2 and β-Glu free in solution were submitted to 1 hour of incubation at room temperature, 50 °C, 60 °C and 70 °C followed by a subsequent release of the enzyme and enzymatic activity measurement. While β-glucosidase free in solution performed poor (very low and no activity at 60 and 70 °C), β-glucosidase released from its entrapped state resulted in 100% activity recovery at 50 and 60 °C and a high ∼75% retained activity after the 70 °C treatment (Fig. 6). This resistance to heat can be attributed to the protective effect of the silica matrix, which prevents the protein undergoing extensive conformational changes inside the material.
 | ||
| Fig. 6 Enzymatic activities of the β-Glu free in solution (dense stripes columns) and β-Glu released (white columns) after heat treatment; the activities given are relative to the corresponding activities at room temperature. | ||
The entrapment not only allows for improved operational, storage and heat stability of enzymes, but one could also take the advantage of the triggered release to perform enzymatic reactions with large substrates, thus tackling some of the biggest problems faced when working with solid support immobilization techniques. Additional work on this in connection with proteins, DNA and polysaccharides is currently ongoing.
Since there is already extensive work and knowledge on the loading of enzymes to nanoporous silica supports we anticipate that the fluoride de-encapsulation step presented here can be adapted to many other systems and will offer new avenues for immobilized enzymes.
Conclusions
In summary, this work demonstrates the synthesis of a novel enzyme-in-silica material with improved operational and storage stability, that can undergo enzyme release in solution upon a chemical trigger. For this purpose, we utilized the well-known advantages of the mesoporous silicas (high surface area and stability) and the simplicity of enzyme adsorption and further optimized them according to our needs in order to obtain high loadings of active β-glucosidase as a model enzyme. Furthermore, we developed a procedure to additionally silica-entrap the previously immobilized enzymes which led to high mechanical, storage and heat stability of the biomolecules. Last, we utilized a non-harmful way to dissolve the support and trigger an immediate release of the enzyme molecules, giving a possibility to select from both large and small substrates. In this way, one could store enzymes for a long time and release them upon need. In the future, one could pay special attention to sensitive enzymes, which are very delicate to handle and require low storage temperatures. The idea of replacing the freezer with the shelves, eliminating the multiple freeze–thaw cycles and the large number of sensitive enzymes available on the market, shows a great application potential and is certainly worth further detailed experimentation.Acknowledgements
The authors thank ICB/ETH Zurich, ETH research grant ETH-31 13-1 and the EU/ITN network Mag(net)icfun (PITN-GA-2012-290248) for financial support, Dr Frank Krumeich for TEM, Gediminas Mikutis for SEM imaging and Ofer Hirsch for SAXS measurement.Notes and references
- R. A. Sheldon, Adv. Synth. Catal., 2007, 349, 1289–1307 CrossRef CAS PubMed.
- M. Alcalde, M. Ferrer, F. J. Plou and A. Ballesteros, Trends Biotechnol., 2006, 24, 281–287 CrossRef CAS PubMed.
- U. T. Bornscheuer, Angew. Chem., Int. Ed., 2003, 42, 3336–3337 CrossRef CAS PubMed.
- C. Garcia-Galan, Á. Berenguer-Murcia, R. Fernandez-Lafuente and R. C. Rodrigues, Adv. Synth. Catal., 2011, 353, 2885–2904 CrossRef CAS PubMed.
- J. Kim, H. Jia and P. Wang, Biotechnol. Adv., 2006, 24, 296–308 CrossRef CAS PubMed.
- M. Hartmann and D. Jung, J. Mater. Chem., 2010, 20, 844–857 RSC.
- L. Cao, in Carrier-bound Immobilized Enzymes, Wiley, 2006, pp. 1–52 Search PubMed.
- S. Hudson, E. Magner, J. Cooney and B. K. Hodnett, J. Phys. Chem. B, 2005, 109, 19496–19506 CrossRef CAS PubMed.
- S. Hudson, J. Cooney and E. Magner, Angew. Chem., Int. Ed., 2008, 47, 8582–8594 CrossRef CAS PubMed.
- C. Ispas, I. Sokolov and S. Andreescu, Anal. Bioanal. Chem., 2009, 393, 543–554 CrossRef CAS PubMed.
- Z. Zhou and M. Hartmann, Chem. Soc. Rev., 2013, 42, 3894–3912 RSC.
- H. Takahashi, B. Li, T. Sasaki, C. Miyazaki, T. Kajino and S. Inagaki, Chem. Mater., 2000, 12, 3301–3305 CrossRef CAS.
- N. W. Fadnavis, V. Bhaskar, M. L. Kantam and B. M. Choudary, Biotechnol. Prog., 2003, 19, 346–351 CrossRef CAS PubMed.
- J. Aburto, M. Ayala, I. Bustos-Jaimes, C. Montiel, E. Terrés, J. M. Domínguez and E. Torres, Microporous Mesoporous Mater., 2005, 83, 193–200 CrossRef CAS PubMed.
- H. H. P. Yiu, P. A. Wright and N. P. Botting, Microporous Mesoporous Mater., 2001, 44–45, 763–768 CrossRef CAS.
- J. He, Z. Song, H. Ma, L. Yang and C. Guo, J. Mater. Chem., 2006, 16, 4307–4315 RSC.
- H. Ma, J. He, D. G. Evans and X. Duan, J. Mol. Catal. B: Enzym., 2004, 30, 209–217 CrossRef CAS PubMed.
- D. Paunescu, M. Puddu, J. O. B. Soellner, P. R. Stoessel and R. N. Grass, Nat. Protoc., 2013, 8, 2440–2448 CrossRef CAS PubMed.
- M. Puddu, W. J. Stark and R. N. Grass, Adv. Healthcare Mater., 2015, 4, 1332–1338 CrossRef CAS PubMed.
- B. Liu, Y. Y. Cao, Z. H. Huang, Y. Y. Duan and S. N. Che, Adv. Mater., 2015, 27, 479–497 CrossRef CAS PubMed.
- B. Liu, L. Han and S. A. Che, J. Mater. Chem. B, 2013, 1, 2843–2850 RSC.
- S.-I. Matsuura, T. Baba, M. Chiba and T. Tsunoda, RSC Adv., 2014, 4, 25920–25923 RSC.
- J. Fan, C. Yu, F. Gao, J. Lei, B. Tian, L. Wang, Q. Luo, B. Tu, W. Zhou and D. Zhao, Angew. Chem., Int. Ed., 2003, 115, 3254–3258 CrossRef PubMed.
- P. Schmidt-Winkel, W. W. Lukens, D. Zhao, P. Yang, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1999, 121, 254–255 CrossRef CAS.
- Y. Han, S. S. Lee and J. Y. Ying, Chem. Mater., 2006, 18, 643–649 CrossRef CAS.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS.
- T. P. B. Nguyen, J.-W. Lee, W. G. Shim and H. Moon, Microporous Mesoporous Mater., 2008, 110, 560–569 CrossRef CAS PubMed.
- J. S. Lettow, Y. J. Han, P. Schmidt-Winkel, P. Yang, D. Zhao, G. D. Stucky and J. Y. Ying, Langmuir, 2000, 16, 8291–8295 CrossRef CAS.
- R. Ryoo, S. H. Joo and S. Jun, J. Phys. Chem. B, 1999, 103, 7743–7746 CrossRef CAS.
- M. Miyahara, A. Vinu, K. Z. Hossain, T. Nakanishi and K. Ariga, Thin Solid Films, 2006, 499, 13–18 CrossRef CAS PubMed.
- M. F. Bechtold, R. D. Vest and L. Plambeck, J. Am. Chem. Soc., 1968, 90, 4590–4598 CrossRef CAS.
- D. Paunescu, R. Fuhrer and R. N. Grass, Angew. Chem., Int. Ed., 2013, 52, 4269–4272 CrossRef CAS PubMed.
- J. C. Groen, L. A. A. Peffer and J. Pérez-Ramírez, Microporous Mesoporous Mater., 2003, 60, 1–17 CrossRef CAS.
- K. Morishige, M. Tateishi, F. Hirose and K. Aramaki, Langmuir, 2006, 22, 9220–9224 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S4. See DOI: 10.1039/c5ra19568c |
| This journal is © The Royal Society of Chemistry 2015 |