
Organic fluorine involved intramolecular hydrogen bonds in the derivatives of imides: NMR evidence corroborated by DFT based theoretical calculations†
Sandeep Kumar Mishra and
N. Suryaprakash*
NMR Research Centre, Solid State and Structural Chemistry Unit, Indian Institute of Science, Bangalore-560012, India. E-mail: nsp@nrc.iisc.ernet.in; Fax: +91 8023601550; Tel: +91 8022933300
First published on 30th September 2015
Abstract
The rare occurrence of intramolecular hydrogen bonds (HBs) of the type N–H⋯F–C is detected in the derivatives of imides in a low polarity solvent by using multi-dimensional and multinuclear NMR experiments. The observation of 1hJFH, 2hJFN, and 2hJFF, where the spin magnetization is transmitted through space among the interacting NMR active nuclei, provided strong and unambiguous evidence for the existence of intra-molecular HBs. The variation in the chemical shifts of labile protons depending on physical conditions, such as the solvent dilution and the systematic alteration of temperature confirmed the presence of weak interactions through intramolecular HBs in all the investigated fluorine substituted molecules. The self or cross dimerization of molecules is unequivocally discarded by the analysis of the rates of diffusion obtained using pseudo-two dimensional DOSY experiments. The Density Function Theory (DFT) calculations based on the Quantum Theory of Atoms In Molecules (QTAIM) and Non Covalent Interaction (NCI), are in close agreement with the NMR experimental findings.
Introduction
Among the several inter- and intra-molecular interactions which are inherently present in diverse molecules, weak hydrogen bonds (HBs) occupy a special place in chemistry and biology.1–4 The importance of HBs in the self-assembly of molecules5–17 is well documented in the literature. The majority of the reported intramolecular HBs mainly pertain to motifs of the type O⋯H–N and N⋯H–N.18–22 Furthermore it is well known that nearly 30% of the commercially available drug molecules contain at least one fluorine atom. In addition, organofluorine molecules have enormous importance due to their applicability as biomaterials and agrochemicals, in molecular imaging,23,24 crystal engineering25–27 and also in functional materials design.28 The bio-origin of the medicinal and bio-inorganic properties of fluorinated compounds is a consequence of the binding nature of fluorine to enzyme active sites29–33 through intermolecular hydrogen bonded bridges of the type X–H⋯F–C (X = O, N). Nevertheless there are very few reports on the participation of organic fluorine in HBs in the solution state.34–36 Nuclear magnetic resonance (NMR) and X-ray crystallographic studies have been reported on N–H⋯F–C HBs in foldamers and benzanilides.37,38 It is also well known that organic fluorine generally does not get involved in the intramolecular HBs.39–45 A report by Dunitz and co-workers concluded that “organic fluorine hardly ever accepts hydrogen bonds”.46–49 As far as the HBs in the solution state are concerned, the first NMR spectroscopic report involving organic fluorine was reported by detecting the through space coupling (1hJFH)50 mediated by HBs. Subsequently Limbach and his co-workers have made enormous contributions to the development of this field and have explored several examples where not only organic fluorine but also other halogens participated in the intermolecular HBs.51–54 Exclusive NMR studies on the existence of intramolecular HBs of the type X–H⋯F–C (X = O, N), in the solution state, have been reported in benzanilide and benzamide44,45 derivatives. Our group has recently utilized NMR techniques and detected the participation of organic fluorine in intramolecular HBs in the derivatives of hydrazides and bisoxamides.55,56 In a continuation of the ongoing research in our group, in the present work we are reporting the extensive studies carried out on fluorine substituted derivatives of imides. The imides are the diacyl derivatives of ammonia or the primary amines.57 Different derivatives of imides have been proven to be very important in high strength electrically conductive polymers,58–60 synthetic applications,61 medicinal activity,62 as ionic fluids,63 in pharmacology64 and as synthetic precursors.65 The NH linker of an imide provides ample scope to synthesize different derivatives with the desired substitution on the acyl group(s). The procedure for their synthesis is already available in the literature.62,66,67 In the present study we have synthesised the selected derivatives of imides using the microwave assisted method66 of imide synthesis and characterized them through the extensive utility of NMR techniques and ESI-HRMS spectrometry. The chemical structures of the investigated molecules are reported in Scheme 1. These molecules can be classified into two categories; symmetric ones where X = X′ (molecules 1–3) and the asymmetric ones where X ≠ X′ (molecules 4–7). The NMR derived information on the intramolecular HBs has been unequivocally supported by Density Functional Theory (DFT)68,69 based Non Covalent Interaction (NCI),70 and Quantum Theory of Atom In Molecule (QTAIM)71 calculations. | ||
| Scheme 1 The chemical structures of the derivatives of 2-X-N-(2-X′-benzoyl)benzamide. | ||
Results and discussion
The basic information on the HBs can be derived by monitoring the variation in the chemical shift in the NMR spectrum as a function of the physical parameters, such as a systematic variation in the temperature or solvent dilution. The hydrogen bonded proton will have its resonance peak shifted downfield as a consequence of the decrease in the electron density surrounding it. This change in the chemical shift arises due to the variation in the concentration, the alteration in the polarity of the solvent, and the temperature alteration. In order to distinguish the intra- and inter-molecular HBs and to monitor the effect of atmospheric monomeric water on the HBs, solvent titration experiments34–36,72 were performed on the molecules 1–7 in the solvent CDCl3. The plot of the variation of the chemical shift as a function of the solvent concentration is reported in Fig. 1A. From this figure it is clearly evident that there is not any significant change in the chemical shift of the NH proton in the 1H NMR spectra, thereby discarding the possibility of intermolecular HBs, aggregation or dimerization. In addition the chemical shift of the 1H resonance of water was invariant confirming the negligible effect of monomeric water on the intramolecular HBs.73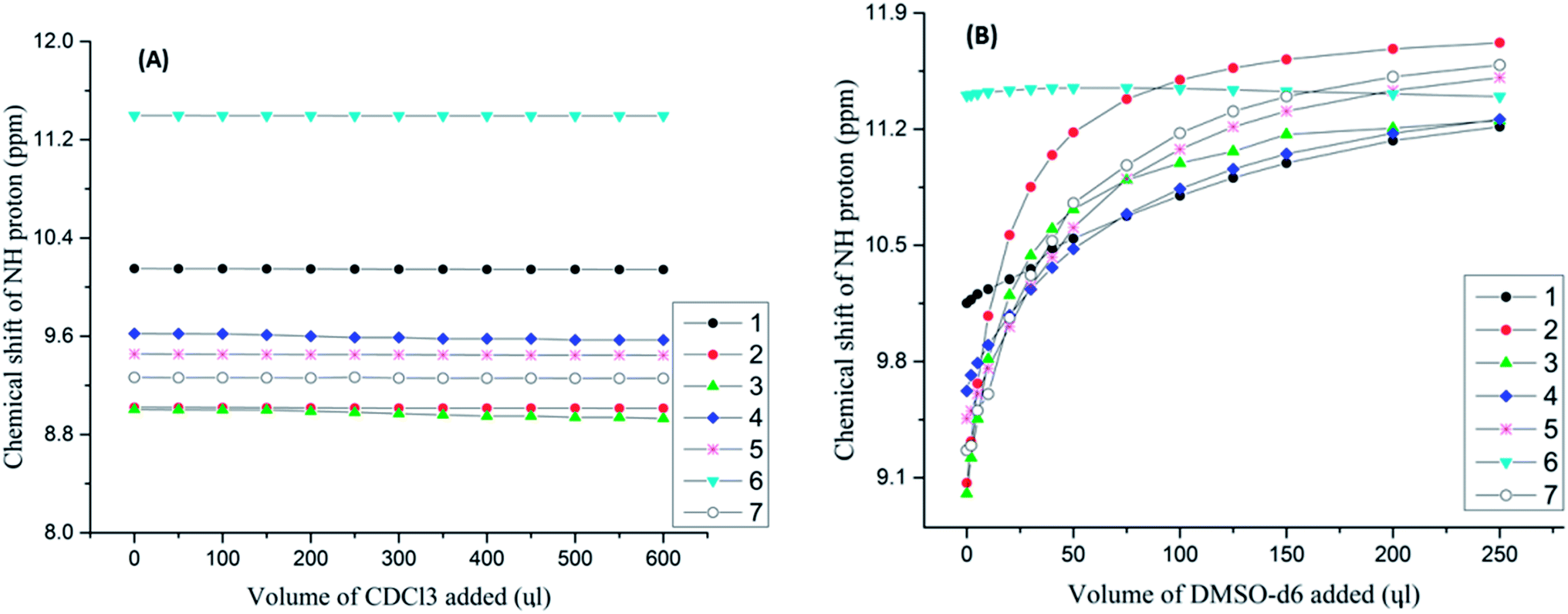 | ||
| Fig. 1 (A and B) The variation in chemical shifts of NH protons as a function of volume of CDCl3 and the volume of DMSO-d6 respectively for the molecules 1–7. The initial concentration taken was 10 mM in the solvent CDCl3. (A) The plot of NH chemical shift with the incremental addition of CDCl3 to an initial volume of 450 μl, at 298 K. (B) The incremental addition of DMSO-d6 to an initial volume of 450 μl in CDCl3, at 298 K. The molecules 1–7 are identified by the symbols given in the inset. | ||
The high polarity solvent dimethyl sulphoxide (DMSO) is a very good HB acceptor and hence it can rupture a variety of inter- and intra-molecular HBs.44,45 To derive qualitative information on the relative strengths of the intramolecular HBs a titration study with the solvent DMSO-d6 was carried out on the molecules 1–7. The variation of the chemical shift of the NH proton as a function of the solvent concentration is reported in Fig. 1B. The excessive deshielding of the NH proton is observed due to the disruption of the intramolecular HBs44,45 due to the strong interaction with DMSO. Interestingly molecule 6 exhibited an upfield shift for the NH peak on the addition of DMSO indicating that, in this particular example, the intramolecular HB formed between the oxygen atom of the methoxy group and the NH proton might be stronger than its interaction with DMSO.55
The strength of the HBs gets increased on lowering the temperature. As a result the deshielding of the NH proton in the 1H NMR spectrum is observed due to the larger displacement of the hydrogen bonded proton towards the HB acceptor, providing evidence of intramolecular HBs.74–76 The chemical shifts of the NH protons as a function of temperature (over the range of 298–220 K) for the molecules 1–7 are compiled in Fig. 2A. The downfield shift of the NH proton resonance on lowering the temperature is observed due to strengthening of the HBs.
 | ||
| Fig. 2 Variation of (A) chemical shifts of NH protons as a function of temperature for the molecules 1–7 and (B) the through space mediated HF coupling constant as a function of temperature for the molecules 1 and 4–7. The molecules are identified by the symbols given in the inset. The initial concentration was 10 mM in the solvent CDCl3. | ||
Another interesting feature observed is the change in the FH coupling value in the 1H NMR spectrum. Such a variation is detected only when the organic fluorine is interacting with the NH proton mediated through a HB. The variation in the coupling constant (through space) as a function of temperature is reported in Fig. 2B, for the molecules 1 and 4–7. This phenomenon provides strong evidence in favour of the HBs.
The variation in the chemical shift values during the titration studies are assimilated in Table 1. The 1H NMR chemical shifts derived using the GIAO77 and CSGT77 DFT methods of NMR simulation were compared with the experimentally determined 1H NMR chemical shifts of the NH protons for the molecules 1–7. It is observed that the CSGT method gives values that are in close agreement with the experimental values. The calculated values using the CSGT method are compiled in Table 1.
| Molecule | HB type (X⋯HN) | Change in chemical shift (ppm) | Change in through-space coupling (Hz) | Theoretical chemical shift of NH proton (ppm) | Experimental chemical shift of NH proton (ppm) | ||
|---|---|---|---|---|---|---|---|
| On adding 600 μl CDCl3 | On adding 250 μl DMSO | Temperature varied from 298 to 220 K | Temperature varied from 298 to 220 K | ||||
| 1 | (F⋯HN) | −0.0052 | 1.0423 | 0.4309 | 3.09 | 9.8 | 10.15 |
| 2 | (Cl⋯HN) | −0.0079 | 2.6999 | 0.2868 | 9.0 | 9.07 | |
| 3 | (H⋯HN) | −0.0739 | 2.2485 | 0.3456 | 9.2 | 9.00 | |
| 4 | (F⋯HN and H⋯HN) | −0.0063 | 1.5813 | 0.46225 | 1.98 | 9.5 | 9.62 |
| 5 | (F⋯HN and Cl⋯HN) | +0.00135 | 2.0087 | 0.3917 | 2.44 | 9.4 | 9.46 |
| 6 | (F⋯HN and MeO⋯HN) | −0.0008 | −0.0954 | 0.4001 | 2.75 | 10.5 | 11.40 |
| 7 | (F⋯HN and CF3⋯HN) | −0.0062 | 2.2942 | 0.3076 | 2.67 | 9.2 | 9.26 |
There is a possibility that molecules of this type might undergo self-dimerization. The absence of any type of dimerization of the investigated molecules was confirmed by pseudo two dimensional 1H DOSY78,79 experiments and ESI-HRMS analysis. Since it is well known that the molecule 2-methoxy-N′-(2-methoxybenzoyl)benzohydrazide labelled as 8 in Fig. 3, does not show any type of self-dimerization or aggregation,55 even in 20 mM solution, the 1H-DOSY NMR experiment was therefore carried out for a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio mixture (20 mM final solution) of molecules 8 and 1 in the solvent CDCl3. The corresponding DOSY spectrum is reported in Fig. 3.
:1 molar ratio mixture (20 mM final solution) of molecules 8 and 1 in the solvent CDCl3. The corresponding DOSY spectrum is reported in Fig. 3.
 | ||
| Fig. 3 500 MHz 1H-DOSY NMR spectrum of a 20 mM solution of the mixture of molecules 8 and 1 at a 1:1 molar ratio in the solvent CDCl3. | ||
From Fig. 3 one can visualize that both molecules have different coefficients of diffusion. Furthermore the rate of diffusion for molecule 8 is slower than that for molecule 1, as expected in the absence of any aggregation or self-dimerization by molecule 1. Thus it conclusively eliminates any possibility of self- or cross-dimerization.
The unambiguous evidence for the existence of HBs can be obtained by detecting the through space couplings between hydrogen bonded NMR active nuclei, where the magnetization transfer takes place through hydrogen bonds. One and two dimensional homo- and hetero-nuclear NMR correlation experiments can be employed for the detection of through space couplings, where the spin polarization is transferred across the hydrogen bonds.80–83 The 1/2 spin and 100% natural abundance of 19F renders it a favourable nucleus for NMR detection. Thus various NMR experiments involving 19F were carried out. The 1H and 1H{19F}45 spectra of molecule 1 in the solvent CDCl3, and the 1H spectrum in the solvent DMSO-d6 are given in Fig. 4. The NH peak of molecule 1 is a triplet with the separation between the adjacent transitions of the triplet corresponding to 13.03 Hz (Fig. 4a). This triplet collapses into a singlet in the 1H{19F}45 experiment confirming the presence of coupling between 1H and 19F (Fig. 4b). Such a large value of coupling being mediated through a covalent bond (5JFH) between 1H and 19F is very unlikely44,45 and the most likely possibility would be mediation through a HB (1hJFH). This is further ascertained by acquiring the spectrum in a high polarity solvent DMSO-d6 which resulted in the collapsing of the triplet to a singlet (Fig. 4c).
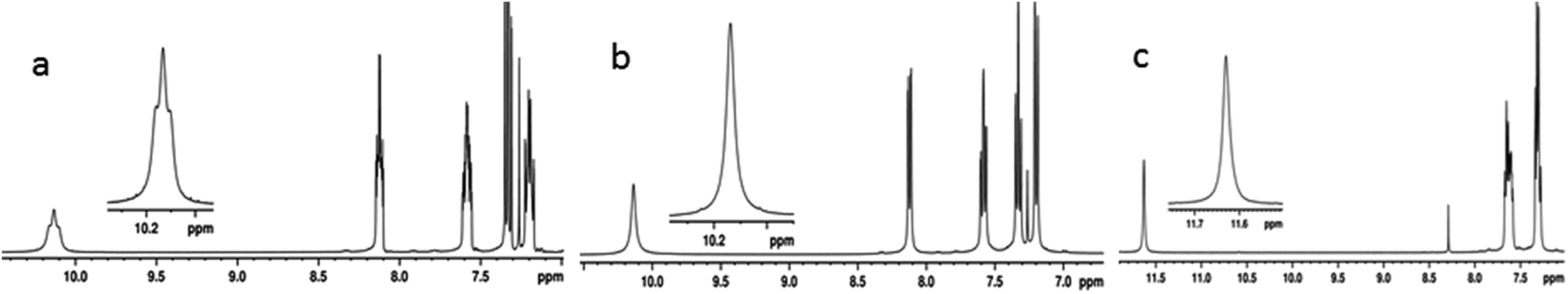 | ||
| Fig. 4 400 MHz 1H NMR spectra of molecule 1; (a) in the solvent CDCl3; (b) 1H{19F} NMR spectrum in the solvent CDCl3; and (c) 1H NMR spectrum in the solvent DMSO-d6. | ||
For obtaining stronger evidence for the involvement of organic fluorine in the intramolecular HBs we have carried out 2D NH-coupled and 1H decoupled 1H–15N HSQC NMR experiments, where 15N is present in its natural abundance. The corresponding spectra for molecule 1 are reported in Fig. 5a and b respectively. The coupled 1H–15N-HSQC spectra are also helpful in assigning the relative signs of couplings mediated through HBs. The measured through space couplings in both the direct and indirect dimensions of the HSQC spectrum are labelled with letters and their values with the relative signs are also reported in the spectrum. The observation of through space couplings of significantly larger strengths, such as, 1hJFH and 2hJFN gives strong and direct evidence for the involvement of organic fluorine in the intramolecular HBs. The NH-coupled 1H–15N HSQC experiment was also carried out in the solvent DMSO-d6 and the corresponding spectrum is reported in Fig. 5c. In the solvent DMSO, except for 1JNH, all the other couplings disappeared. This gives further strong and unambiguous evidence that the measured couplings 1hJFH and 2hJFN in the solvent CDCl3 are mediated through HBs. The 1H–15N HSQC spectra of all the other investigated molecules, along with the measured magnitudes and relative signs of the couplings are reported in the ESI.†
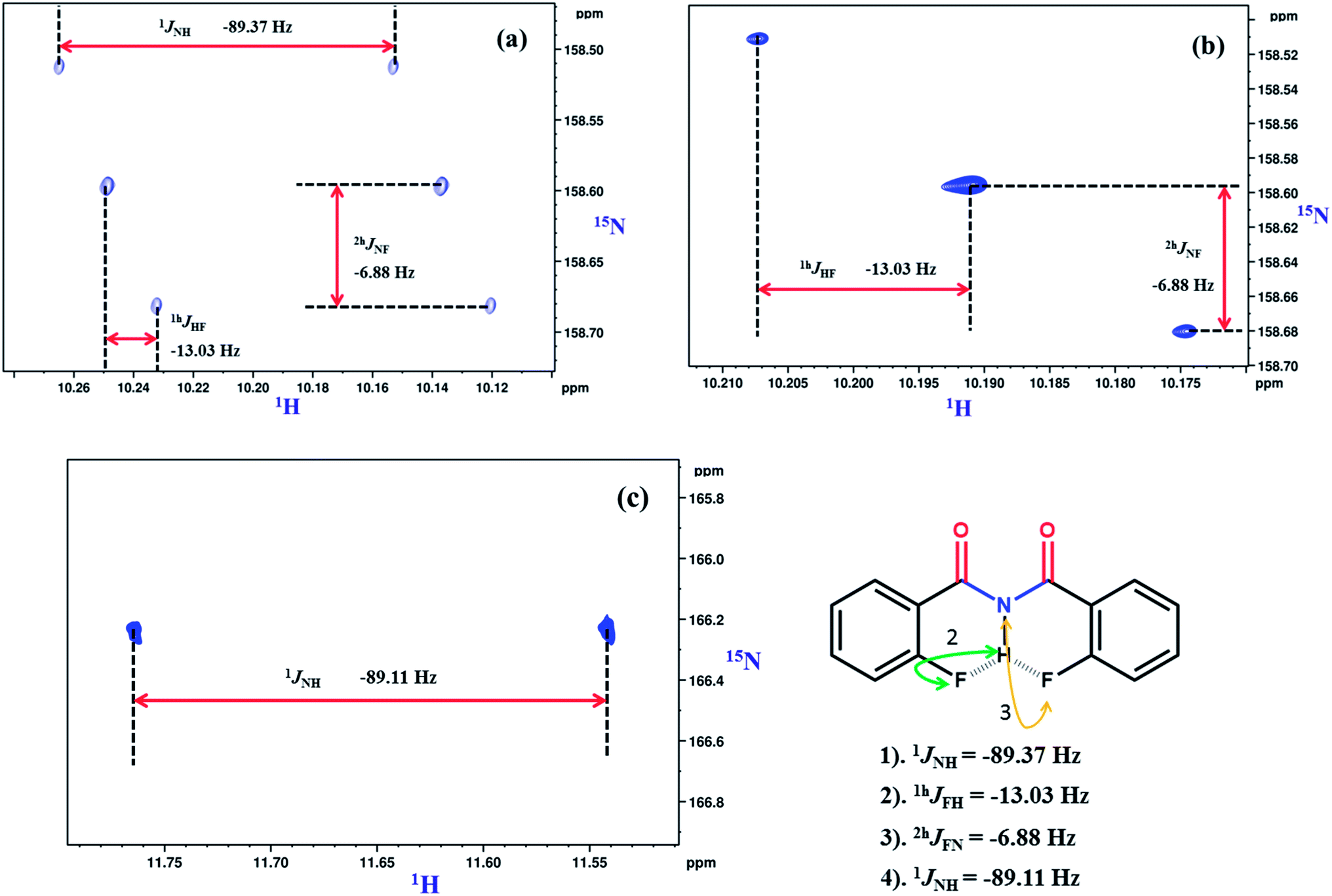 | ||
| Fig. 5 800 MHz spectrum of molecule 1 in the solvent CDCl3 (a) 1H–15N-HSQC (NH-coupled); (b) 1H–15N-HSQC (NH-decoupled); (C) 400 MHz 1H–15N-HSQC spectrum (NH-coupled) in the solvent DMSO-d6. The chemical structures with the magnitudes of scalar and through space couplings are identified by double headed arrows. The measured coupling values and their relative signs derived from the relative slopes of the displacement of cross sections are also given. | ||
From Fig. 5a and b the magnitudes and the relative signs of the couplings could be derived. Based on earlier reports44,45 the relative signs of 1hJFH and 2hJFN are assigned to be negative. All the measured magnitudes of the couplings along with their appropriate signs are reported in Fig. 5.
There is also a great significance in the value of 1JNH in understanding the nature of the HBs. The extent of 1JNH will increase if the HB is predominantly electrostatic84 and decrease if the HB is predominantly a covalent type.85 The 1JNH values of all the investigated molecules obtained from NH coupled 15N–1H HSQC experiments in the solvent CDCl3 are compiled in Table 2. From the visual comparison of the 1JNH couplings of all other molecules with that of an unsubstituted molecule 3, it is evident that the 1JNH couplings of the substituted molecules 1, 2 and 4–7 are substantially smaller than for molecule 3, providing strong and unambiguous evidence that the nature of the HBs in the derivatives of imides is predominantly of the covalent type.
| Molecule | HB type (X⋯HN) | Electron density (ρ(r)) (a.u.) | Laplacian of electron density (∇2ρ(r)) | Energy of HB (EHB) (Kcal mol−1) | Energy difference (cis–trans) ΔEcis–trans (Kcal mol−1) | 1JNH in the solvent CDCl3 |
|---|---|---|---|---|---|---|
| a Shared-shell interaction because the Laplacian of electron density is negative. | ||||||
| 1 | (F⋯HN) | 0.0237 | 0.1006 | −6.4283 | −10.97 | −89.37 |
| 2 | (Cl⋯HN) | 0.0179 | 0.0617 | −3.4298 | −1.88 | −88.07 |
| (Cl⋯Cl) | 0.0109 | 0.0444 | −2.0711 | |||
| 3 | (H⋯HN) | 0.0297 | −0.8246 | a | −86.43 | |
| 4 | (F⋯HN) | 0.0268 | 0.1127 | −7.4767 | −6.33 | −88.22 |
| (H⋯HN) | 0.0145 | 0.0568 | −2.7708 | |||
| 5 | (F⋯HN) | 0.0209 | 0.0870 | −5.5176 | −7.63 | −89.17 |
| (Cl⋯HN) | 0.0217 | 0.0723 | −4.4714 | |||
| (F⋯Cl) | 0.011 | 0.0483 | −2.6671 | |||
| 6 | (F⋯HN) | 0.0184 | 0.0777 | −4.7723 | −10.68 | −89.59 |
| (MeO⋯HN) | 0.0326 | 0.1300 | −8.9233 | |||
| 7 | (F⋯HN) | 0.0256 | 0.1083 | −7.0360 | −3.83 | −88.52 |
| (CF3⋯HN) | 0.0107 | 0.0424 | −2.5233 | |||
The Nuclear Overhauser Effect (NOE) can also be used to correlate the strength of the intramolecular HBs in a molecule. The spatial proximity between the two spins involved in a dipolar interaction is correlated with the change in the intensity of the peak in a two dimensional Nuclear Overhauser Effect spectrum. Thus 2D 1H–19F hetero NOE spectroscopy (HOESY)86–88 experiments were carried out for all the fluorine containing molecules 1 and 4–7, where the through space correlation is established between the NH proton and the F atom. Hexafluorobenzene (C6F6) is used as an internal reference for all the 19F spectra. The 2D 1H–19F HOESY spectrum of molecule 1 is reported in Fig. 6b. The HOESY spectra of the other molecules, 4–7 are reported in the ESI.† The close spatial proximity between F and the NH proton is established by detecting the correlated peak between the NH proton and the F atom in all the molecules. Thus the results derived from the different NMR experiments provided strong evidence for the involvement of organic fluorine in the intramolecular HBs in all the investigated fluorine containing molecules.
 | ||
| Fig. 6 376.5 MHz (a) 19F and (b) 2D 1H–19F HOESY spectra of molecule 1 in the solvent CDCl3. | ||
Theoretical calculations
The molecular weak interactions established by the NMR experimental findings were also further corroborated by DFT68,69 optimized structure based theoretical calculations. To optimize the lowest energy structures for the investigated molecules 1–7 the DFT calculations were performed using the Gaussian09 suite,89 with a B3LYP/6-311+g(d,p) level of theory with the default – chloroform – as the solvation medium. The harmonic vibrational frequency values were utilized to obtain the minimum energy structures. The coordinates of these energy minimised molecular geometries were used to generate the wave function files for the QTAIM, and NCI studies.Non covalent interaction (NCI) calculations
The powerful non covalent interaction (NCI)90 approach is used to detect non covalent interactions in real space which is dependent on the electron density and its derivatives. It provides a strong representation of the steric repulsion, van der Waals interactions and the HBs. There is a very large positive gradient of the reduced density gradient (RDG), and the RDG values will be small, approaching zero in the density tail (i.e., regions far from the molecule, where the electron density exponentially decays to zero). This is the condition for both the covalent and non-covalent bonding regions. There is a strong correlation of electron density (ρ(r)) with the weak interactions in the corresponding regions. The correlations for the HBs are negative and positive for the steric effect, while the van der Waals interaction will always have very small ρ(r) values90 (near to zero). The calculated grid points are plotted for a defined real space function, with sign(λ2(r))ρ(r) as function 1 and the reduced density gradient (RDG) as function 2 using the Multiwfn91 program for the molecules 1–7. The program VMD92 is used for plotting colour filled isosurfaces from these grid points. The plot of sign(λ2(r))ρ(r) vs. RDG, and the coloured isosurfaces for molecule 1 are reported in Fig. 7a and b respectively. | ||
| Fig. 7 (a) The plot of sign(λ2(r))ρ(r) as function 1 vs. the RDG as function 2, and (b) coloured isosurface plot (green colour denotes weak H-bond and red colour stands for steric effect) for molecule 1. The plots for the remaining molecules, 2–7, are given in the ESI.† | ||
The four spikes on the left hand side of Fig. 7a (i.e. sign(λ2(r))*ρ(r) is negative) for molecule 1 denote four types of weak interactions, viz., N–H⋯F, C–H⋯O, F⋯F and a very weak O⋯O. These four HBs can be visualized in Fig. 7b as isosurfaces of green colour. The red coloured isosurface in the plot (Fig. 7b) represents the steric interactions arising from the aromatic phenyl ring and other HB mediated rings. This is seen as five spikes in Fig. 7a on the right hand side (i.e. sign(λ2(r))*ρ(r) is positive). The similar plots and discussions for the remaining molecules 2–7 are reported in the ESI.†
Atoms in molecules (AIM) calculations
After confirming the presence of HBs with NCI plots, the energy of interaction must be known for in depth understanding of the molecular properties influenced by the weak HBs. The topology analysis technique was reported as the “atoms in molecules” (AIM) theory, and also cited as “the quantum theory of atoms in molecules” (QTAIM)71,93–96 dependent on the quantum observables (electron density ρ(r) and the energy densities). The points (except at infinity) where the gradient norm of the function value is zero are called critical points (CPs) in topology analysis. According to the negative Eigenvalues of the Hessian matrix of real space functions71,93–96 CPs can be of four types. Out of these the (3, −1) CP is called a bond critical point (BCP). There is a great significance in the value of the real space function at the BCP. For example the bond strength and bond type respectively are related closely to the value of the electron density (ρ(r)) and the sign of the Laplacian of electron density (∇2ρ(r)) at the BCPs.71,93–96 The magnitudes of ρ(r) and signs of ∇2ρ(r) for the BCPs of the HBs of interest are calculated using AIM calculations and compiled in Table 2. At the corresponding (3, −1) critical points (rcp) the gradients of the electron density (ρ(r)) vanish. The energy of the HB (EHB) is directly related to the potential energy density (V(r)) by the straightforward relationship97 EHB = V(rbcp)/2. The EHB of the X⋯HX type of HB can be calculated by using this relationship. The calculated values of EHB for all the located BCPs of interest are assimilated in Table 2.Conformational study
The molecules of the investigated imides can exhibit conformational isomerism and the number of such possible isomers is dependent on the substitution on the phenyl ring. For the investigated molecules, a maximum of 3–4 conformers are possible, which are shown in Scheme 2. | ||
| Scheme 2 The possible conformers of the investigated imide molecules arising due to ring flip. | ||
Out of the four possible conformers the conformers cis–cis and trans–trans were optimized using the Gaussian09 suite,89 with a B3LYP/6-311+g(d,p) level of theory with the default – chloroform – as the solvation medium and the difference ((cis–cis) – (trans–trans)) in the minimum energy was taken. The energy difference ΔEcis–trans of all the investigated molecules is listed in Table 2. From Table 2 it is evident that the energy of the cis–cis conformers is always lower than the trans–trans, due to the presence of HBs.
The visualization of the BCPs and bond paths of the HBs for molecule 1 is reported in Fig. 8 and for the other investigated molecules 2–7 in the ESI.†
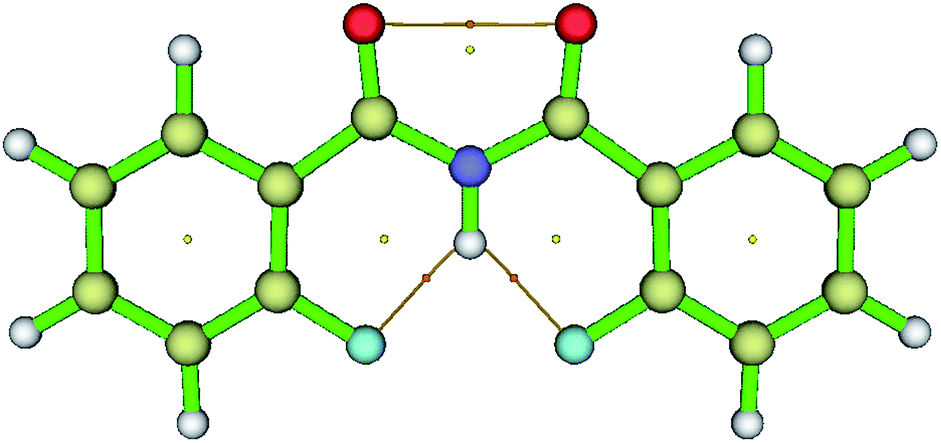 | ||
| Fig. 8 The visualization of BCPs and bond paths of HB for molecule 1 plotted using multiwfn software. Dots represent the CPs and thin bars represents the HB interactions. | ||
The presence of significant interactions at the (3, −1) BCPs is confirmed by the significantly large values of ρ(r) (Table 2). The sign of (∇2ρ(r)) at the BCP has significance in discriminating the shared-shell (covalent bond (−ve)) and closed-shell (ionic, van der Waals interaction, and HB (+ve)). From Table 2 it is confirmed that all BCPs other than for molecule 3 are coming under closed-shell (HB) interactions. The BCP of interest for molecule 3 shows a −ve value of (∇2ρ(r)) indicating that these interactions as expected are shared-shell. If the calculated strengths of the HBs in Table 2 are compared with the chemical shifts of the NH protons in Table 1, it will become evident that the strength of the HBs is directly related to the chemical shift of the NH (H-bonded) proton (a downfield shift with increasing strength or vice versa). Thus under identical experimental conditions we could compare the relative strengths of the HBs by utilizing the NMR chemical shifts.
Relaxed potential energy scan
A relaxed potential energy surface for the internal rotation of the phenyl ring through a single bond was calculated at the B3LYP/6-311G(d,p) level of calculation. The rotation of 360° (−180 to 0 to +180) through a single bond was scanned in 20 steps with an 18° size of rotational segment. The scanned graph of the energy (kcal mol−1) versus the dihedral angle (θ) is reported in Fig. 9 and the animation of the scan is also reported in the ESI.† From the graph it is clear that the energy of the cis conformer is lower than the others, and the energy is at a maximum for the trans conformation. | ||
| Fig. 9 Relaxed potential energy surface for the internal rotation of the phenyl ring through a single bond was performed at the B3LYP/6-311G(d,p) level of calculation. The dihedral angle and the rotation direction are highlighted in the structure of molecule. | ||
Experimental
The NMR spectra of all the investigated molecules were acquired using Bruker AVANCE 400, 500 and 800 MHz NMR spectrometers. TMS (0.0 ppm) is taken as the internal reference for the proton chemical shifts and hexafluorobenzene (−164.9 ppm) is used as the internal reference for all the 19F spectra. All the NMR spectra for the characterization of the synthesized molecules were acquired at 298 K. Deuterated solvents, such as, CDCl3, and DMSO-d6 were purchased from Cambridge Isotopes Limited and used as received. Fresh CDCl3 was used to avoid the possibility of artificial alteration in the interaction, because the strength of the non-covalent interactions is found to be dependent on the solvent and substrate purity. The molecular masses of the synthesized molecules were confirmed using electrospray ionization high resolution mass spectrometry (ESI-HRMS). The standard programs of the Bruker NMR spectrometer library were used to acquire the two dimensional HSQC, HOESY and DOSY spectra.General procedure for synthesis
Different derivatives of imides were synthesized using the corresponding benzamide and benzoyl chlorides.66 All the benzamide and benzoyl chlorides of high purity were purchased and used as received. The AR grade solvents n-hexane (C6H14), chloroform (CHCl3), and n-pentane (C5H12) and HPLC grade methanol (CH3OH) were used in the purification.Synthesis of substituted imides
200 mg of the corresponding benzamide and the benzoyl chloride of interest were taken in a 1:1.2 molar ratio in a silica crucible and mixed using a clean spatula. The resulting mixture was exposed to microwave irradiation at 450 W for 5 min. The reaction was monitored using TLC until completion. For further purification the compound was passed through a column loaded with neutral alumina (Al2O3) using a mixture of the solvents n-hexane and chloroform. The gradient of the solvent was varied from 5% to 30% chloroform with gradual increments. The obtained product was crystalized using methanol (CH3OH) and dichloromethane (CH2Cl2) in a 2:1 ratio.
Conclusions
The combined multinuclear and multidimensional NMR experiments and DFT based theoretical calculations unambiguously establish the presence of intramolecular HBs in all the investigated fluorine derivatives of imides. Any possibility of self or cross dimerization has been discarded by CDCl3 titration (a concentration variation study) and also by the 1H DOSY NMR technique. The 1H–19F HOESY (heteronuclear through space correlation) experiment indirectly provided the information about the possibility of intramolecular HBs. Through space coupling via HBs is detected in 1D 1H and 2D 1H–15N HSQC experiments whose magnitudes varied over the range of 6–15 Hz in different fluorine containing molecules. A very sensitive NCI calculation has been used as a tool for the detection of non-covalent interactions and confirmed the existence of bifurcated intramolecular HBs in the molecules 1–7. The QTAIM calculations were utilized to derive the energy and strengths of the HBs. The calculated energies of the HBs (EHB) for different investigated molecules are found to be in the range of −2.07 to −8.92 kcal mol−1. The HBs are discriminated from the covalent bonds by using the signs of the Laplacian of the electron density. We strongly believe that the present studies lead to a better understanding of the HBs in the derivatives of imides and open up opportunities in designing different new drugs, foldamers and supramolecules of pharmacological, chemical and biological importance.Acknowledgements
SKM would like to thank UGC, New Delhi, for SRF. NS gratefully acknowledges the generous financial support by the Board of Research in Nuclear Sciences, Mumbai (Grant No. 2013/37C/4/BRNS).References
- W.-H. Hu, K.-W. Huang, C.-W. Chiou and S.-W. Kuo, Macromolecules, 2012, 45, 9020–9028 CrossRef CAS.
- M. Saccone, V. Dichiarante, A. Forni, A. Goulet-Hanssens, G. Cavallo, J. Vapaavuori, G. Terraneo, C. J. Barrett, G. Resnati, P. Metrangolo and A. Priimagi, J. Mater. Chem. C, 2015, 3, 759–768 RSC.
- A. J. P. Teunissen, M. M. L. Nieuwenhuizen, F. Rodríguez-Llansola, A. R. A. Palmans and E. W. Meijer, Macromolecules, 2014, 47, 8429–8436 CrossRef CAS.
- T. Xiao, X. Feng, S. Ye, Y. Guan, S.-L. Li, Q. Wang, Y. Ji, D. Zhu, X. Hu, C. Lin, Y. Pan and L. Wang, Macromolecules, 2012, 45, 9585–9594 CrossRef CAS.
- A. Ajayaghosh, S. J. George and A. P. H. J. Schenning, in Top. Curr. Chem., ed. F. Würthner, Springer, Berlin Heidelberg, 2005, vol. 258, 135681, pp. 83–118 Search PubMed.
- E. A. Archer, H. Gong and M. J. Krische, Tetrahedron, 2001, 57, 1139–1159 CrossRef CAS.
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4098 CrossRef CAS PubMed.
- M. M. Conn and J. Rebek, Chem. Rev., 1997, 97, 1647–1668 CrossRef CAS PubMed.
- F. Huang and H. W. Gibson, Prog. Polym. Sci., 2005, 30, 982–1018 CrossRef CAS PubMed.
- D. S. Lawrence, T. Jiang and M. Levett, Chem. Rev., 1995, 95, 2229–2260 CrossRef CAS.
- O. Lukin and F. Vögtle, Angew. Chem., Int. Ed., 2005, 44, 1456–1477 CrossRef CAS PubMed.
- J. S. Nowick, Acc. Chem. Res., 1999, 32, 287–296 CrossRef CAS.
- L. J. Prins, D. N. Reinhoudt and P. Timmerman, Angew. Chem., Int. Ed., 2001, 40, 2382–2426 CrossRef CAS.
- C. Schmuck and W. Wienand, Angew. Chem., Int. Ed., 2001, 40, 4363–4369 CrossRef CAS.
- R. Shenhar and V. M. Rotello, Acc. Chem. Res., 2003, 36, 549–561 CrossRef CAS PubMed.
- R. P. Sijbesma and E. W. Meijer, Chem. Commun., 2003, 5–16, 10.1039/b205873c.
- F. Zeng and S. C. Zimmerman, Chem. Rev., 1997, 97, 1681–1712 CrossRef CAS PubMed.
- B. Gong, Chem.–Eur. J., 2001, 7, 4336–4342 CrossRef CAS.
- I. Huc, Eur. J. Org. Chem., 2004, 2004, 17–29 CrossRef PubMed.
- Z.-T. Li, J.-L. Hou, C. Li and H.-P. Yi, Chem.–Asian J., 2006, 1, 766–778 CrossRef CAS PubMed.
- J. W. Lockman, N. M. Paul and J. R. Parquette, Prog. Polym. Sci., 2005, 30, 423–452 CrossRef CAS PubMed.
- A. R. Sanford, K. Yamato, X. Yang, L. Yuan, Y. Han and B. Gong, Eur. J. Biochem., 2004, 271, 1416–1425 CrossRef CAS PubMed.
- I. Muegge, S. L. Heald and D. Brittelli, J. Med. Chem., 2001, 44, 1841–1846 CrossRef CAS PubMed.
- B. E. Smart, J. Fluorine Chem., 2001, 109, 3–11 CrossRef CAS.
- D. Chopra and T. N. G. Row, CrystEngComm, 2011, 13, 2175–2186 RSC.
- G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952–9967 CrossRef CAS PubMed.
- K. Reichenbacher, H. I. Suss and J. Hulliger, Chem. Soc. Rev., 2005, 34, 22–30 RSC.
- R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496–3508 RSC.
- R. H. Abeles and T. A. Alston, J. Biol. Chem., 1990, 265, 16705–16708 CAS.
- M.-C. Chapeau and P. A. Frey, J. Org. Chem., 1994, 59, 6994–6998 CrossRef CAS.
- T. Kovacs, A. Pabuccuoglu, K. Lesiak and P. F. Torrence, Bioorg. Chem., 1993, 21, 192–208 CrossRef CAS.
- D. O’Hagan and H. S. Rzepa, Chem. Commun., 1997, 645–652, 10.1039/a604140j.
- L. H. Takahashi, R. Radhakrishnan, R. E. Rosenfield, E. F. Meyer and D. A. Trainor, J. Am. Chem. Soc., 1989, 111, 3368–3374 CrossRef CAS.
- I. Alkorta, J. Elguero, H.-H. Limbach, I. G. Shenderovich and T. Winkler, Magn. Reson. Chem., 2009, 47, 585–592 CrossRef CAS PubMed.
- C. Bartolome, P. Espinet and J. M. Martin-Alvarez, Chem. Commun., 2007, 4384–4386, 10.1039/b710304b.
- S. R. Chaudhari, S. Mogurampelly and N. Suryaprakash, J. Phys. Chem. B, 2013, 117, 1123–1129 CrossRef CAS PubMed.
- I. E. Kareev, G. S. Quiñones, I. V. Kuvychko, P. A. Khavrel, I. N. Ioffe, I. V. Goldt, S. F. Lebedkin, K. Seppelt, S. H. Strauss and O. V. Boltalina, J. Am. Chem. Soc., 2005, 127, 11497–11504 CrossRef CAS PubMed.
- G. K. S. Prakash, F. Wang, M. Rahm, J. Shen, C. Ni, R. Haiges and G. A. Olah, Angew. Chem., Int. Ed., 2011, 50, 11761–11764 CrossRef CAS PubMed.
- R. Banerjee, G. R. Desiraju, R. Mondal and J. A. K. Howard, Chem.–Eur. J., 2004, 10, 3373–3383 CrossRef CAS PubMed.
- C. Li, S.-F. Ren, J.-L. Hou, H.-P. Yi, S.-Z. Zhu, X.-K. Jiang and Z.-T. Li, Angew. Chem., Int. Ed., 2005, 44, 5725–5729 CrossRef CAS PubMed.
- Y. Mido and T. Okuno, J. Mol. Struct., 1982, 82, 29–34 CrossRef CAS.
- H. Takemura, N. Kon, M. Yasutake, S. Nakashima, T. Shinmyozu and T. Inazu, Chem.–Eur. J., 2000, 6, 2334–2337 CrossRef CAS.
- X. Zhao, X.-Z. Wang, X.-K. Jiang, Y.-Q. Chen, Z.-T. Li and G.-J. Chen, J. Am. Chem. Soc., 2003, 125, 15128–15139 CrossRef CAS PubMed.
- K. Divya, S. Hebbar and N. Suryaprakash, Chem. Phys. Lett., 2012, 525–526, 129–133 CrossRef PubMed.
- G. N. Manjunatha Reddy, M. V. Vasantha Kumar, T. N. Guru Row and N. Suryaprakash, Phys. Chem. Chem. Phys., 2010, 12, 13232–13237 RSC.
- D. Chopra, Cryst. Growth Des., 2011, 12, 541–546 Search PubMed.
- J. D. Dunitz, ChemBioChem, 2004, 5, 614–621 CrossRef CAS PubMed.
- J. D. Dunitz and R. Taylor, Chem.–Eur. J., 1997, 3, 89–98 CrossRef CAS PubMed.
- J. A. K. Howard, V. J. Hoy, D. O’Hagan and G. T. Smith, Tetrahedron, 1996, 52, 12613–12622 CrossRef CAS.
- I. D. Rae, J. A. Weigold, R. H. Contreras and R. R. Biekofsky, Magn. Reson. Chem., 1993, 31, 836–840 CrossRef CAS PubMed.
- H. Benedict, I. G. Shenderovich, O. L. Malkina, V. G. Malkin, G. S. Denisov, N. S. Golubev and H.-H. Limbach, J. Am. Chem. Soc., 2000, 122, 1979–1988 CrossRef CAS.
- N. S. Golubev, I. G. Shenderovich, S. N. Smirnov, G. S. Denisov and H.-H. Limbach, Chem.–Eur. J., 1999, 5, 492–497 CrossRef CAS.
- I. G. Shenderovich, S. N. Smirnov, G. S. Denisov, V. A. Gindin, N. S. Golubev, A. Dunger, R. Reibke, S. Kirpekar, O. L. Malkina and H.-H. Limbach, Phys. Chem., 1998, 102, 422–428 CrossRef CAS.
- I. G. Shenderovich, P. M. Tolstoy, N. S. Golubev, S. N. Smirnov, G. S. Denisov and H.-H. Limbach, J. Am. Chem. Soc., 2003, 125, 11710–11720 CrossRef CAS PubMed.
- S. K. Mishra and N. Suryaprakash, Phys. Chem. Chem. Phys., 2015, 17, 15226–15235 RSC.
- A. Lakshmipriya, S. R. Chaudhari, A. Shahi, E. Arunan and N. Suryaprakash, Phys. Chem. Chem. Phys., 2015, 17, 7528–7536 RSC.
- IUPAC, Compendium of Chemical Terminology, (the "Gold Book"), ed. A. D. McNaught and A. Wilkinson. Blackwell Scientific Publications, Oxford, 2nd edn, 1997. XML on-line corrected version: http://goldbook.iupac.org, 2006, created by M. Nic, J. Jirat, B. Kosata; updates compiled by A. Jenkins. ISBN 0-9678550-9-8. DOI:10.1351/goldbook.
- W. W. Walter and H. A. Michael, "Polyimides" in Ullmann’s Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2002, DOI:10.1002/14356007.a21_253.
- J. Prinos, Ch. Tselios, D. Bikiaris and C. Panayiotou, Polymer, 1997, 38, 5921–5930 CrossRef CAS.
- L. Luo, Y. Zheng, J. Huang, K. Li, H. Wang, Y. Feng, X. Wang and X. Liu, J. Appl. Polym. Sci., 2015, 132, DOI:10.1002/app.42001.
- Y. Kanaoka, Acc. Chem. Res., 1978, 11, 407–413 CrossRef CAS.
- J. Stec, Q. Huang, M. Pieroni, M. Kaiser, A. Fomovska, E. Mui, W. H. Witola, S. Bettis, R. McLeod, R. Brun and A. P. Kozikowski, J. Med. Chem., 2012, 55, 3088–3100 CrossRef CAS PubMed.
- H. Wang, S. P. Kelley, J. W. Brantley, G. Chatel, J. Shamshina, J. F. B. Pereira, V. Debbeti, A. S. Myerson and R. D. Rogers, ChemPhysChem, 2015, 16, 993–1002 CrossRef CAS PubMed.
- D. Sprecher, M. Maxwell, J. Goodman, B. White, C.-M. Tang, V. Boullay and A.-C. de Gouville, Eur. J. Pharmacol., 2015, 756, 1–7 CrossRef CAS PubMed.
- R. Mhidia, E. Boll, F. Fécourt, M. Ermolenko, N. Ollivier, K. Sasaki, D. Crich, B. Delpech and O. Melnyk, Bioorg. Med. Chem., 2013, 21, 3479–3485 CrossRef CAS PubMed.
- Y. Li, Y. Wang and J. Wang, Russ. J. Org. Chem., 2008, 44, 358–361 CrossRef CAS.
- S. Ahmad, K. M. Ali, J. Charalambous and M. J. Frazer, Inorg. Chim. Acta, 1978, 31, 69–76 CrossRef CAS.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- R. G. Parr and W. Yang, Density-Functional Theory of Atoms and Molecules, Oxford University Press, New York, 1989 Search PubMed.
- J. Contreras-García, W. Yang and E. R. Johnson, J. Phys. Chem. A, 2011, 115, 12983–12990 CrossRef PubMed.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, 1990 Search PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- M. Nakahara and C. Wakai, Chem. Lett., 1992, 21, 809–812 CrossRef.
- S. H. Gellman, B. R. Adams and G. P. Dado, J. Am. Chem. Soc., 1990, 112, 460–461 CrossRef CAS.
- S. H. Gellman, G. P. Dado, G. B. Liang and B. R. Adams, J. Am. Chem. Soc., 1991, 113, 1164–1173 CrossRef CAS.
- F. J. Martinez-Martinez, A. Ariza-Castolo, H. Tlahuext, M. Tlahuextl and R. Contreras, J. Chem. Soc., Perkin Trans. 2, 1993, 1481–1485, 10.1039/P29930001481.
- J. R. Cheeseman, G. W. Trucks, T. A. Keith and M. J. Frisch, J. Chem. Phys., 1996, 104, 5497–5509 CrossRef CAS PubMed.
- K. F. Morris and C. S. Johnson, J. Am. Chem. Soc., 1992, 114, 3139–3141 CrossRef CAS.
- K. F. Morris and C. S. Johnson, J. Am. Chem. Soc., 1993, 115, 4291–4299 CrossRef CAS.
- T. Axenrod, P. S. Pregosin, M. J. Wieder, E. D. Becker, R. B. Bradley and G. W. A. Milne, J. Am. Chem. Soc., 1971, 93, 6536–6541 CrossRef CAS.
- A. J. Dingley, J. E. Masse, R. D. Peterson, M. Barfield, J. Feigon and S. Grzesiek, J. Am. Chem. Soc., 1999, 121, 6019–6027 CrossRef CAS.
- A. Dunger, H.-H. Limbach and K. Weisz, J. Am. Chem. Soc., 2000, 122, 10109–10114 CrossRef CAS.
- S. Grzesiek, F. Cordier, V. Jaravine and M. Barfield, Prog. Nucl. Magn. Reson. Spectrosc., 2004, 45, 275–300 CrossRef CAS PubMed.
- A. V. Afonin, I. A. Ushakov, L. N. Sobenina, Z. V. Stepanova, O. g. V. Petrova and B. A. Trofimov, Magn. Reson. Chem., 2006, 44, 59–65 CrossRef CAS PubMed.
- M. M. King, H. J. C. Yeh and G. O. Dudek, Org. Magn. Reson., 1976, 8, 208–212 CrossRef CAS PubMed.
- P. L. Rinaldi, J. Am. Chem. Soc., 1983, 105, 5167–5168 CrossRef CAS.
- C. Yu and G. C. Levy, J. Am. Chem. Soc., 1983, 105, 6994–6996 CrossRef CAS.
- C. Yu and G. C. Levy, J. Am. Chem. Soc., 1984, 106, 6533–6537 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford, CT, USA, 2009.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
- R. F. W. Bader, S. G. Anderson and A. J. Duke, J. Am. Chem. Soc., 1979, 101, 1389–1395 CrossRef CAS.
- R. F. W. Bader and H. Essén, J. Chem. Phys., 1984, 80, 1943–1960 CrossRef CAS PubMed.
- R. F. W. Bader, T. T. Nguyen-Dang and Y. Tal, Rep. Prog. Phys., 1981, 44, 893–948 CrossRef.
- G. R. Runtz, R. F. W. Bader and R. R. Messer, Can. J. Chem., 1977, 55, 3040–3045 CrossRef CAS.
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra19537c |
| This journal is © The Royal Society of Chemistry 2015 |