DOI:
10.1039/C5RA19358C
(Paper)
RSC Adv., 2015,
5, 87139-87150
Synthesis, structure and magnetic characterization of dinuclear copper(II) complexes bridged by bicompartmental phenolate†
Received
20th September 2015
, Accepted 7th October 2015
First published on 8th October 2015
Abstract
The reaction of Cu(II) salts with the bicompartmental 2,6-bis[bis(2-pyridylmethyl)aminomethyl]-4-chlorophenol (LCl-OH) ligand afforded four new dinuclear bridged phenoxido Cu(II) complexes. Three doubly bridged complexes namely [Cu2(μ-LCl-O)(μ-X)](ClO4)2 (1: X = OH−, 3: X = O2P(OC6H5)2−) and 2: [Cu2(μ-LCl-O)(μ-pz)(ClO4)]ClO4 (2) where pz = pyrazolyl anion, and one singly bridged-phenoxido, [Cu2(μ-LCl-O)(dca)2]PF6·2CH3CN (4·2CH3CN) (dca = dicyanamide anion). A complex similar to 4 was also obtained with 2,6-bis[bis(2-pyridylmethyl)aminomethyl]-4-methylphenol (LMe-OH), [Cu2(μ-LMe-O)(dca)2]PF6·2CH3CN (5·2CH3CN) where in both cases dca are acting as terminal monodentate ligands. The complexes were structurally characterized by various spectroscopic techniques (IR, UV-VIS and ESI-MS) and by single crystal crystallography. Magnetic susceptibility measurements at variable temperature revealed strong to very strong antiferromagnetic coupling (AF) in the doubly bridged complexes 1–3 and very weak AF interaction in the dicyanamido compounds 4 and 5. The DFT calculations for the coupling constants, J were in agreement with the experimentally observed behavior. The trend in magnetic properties was attributed to the strength of overlap between the orbitals (dx2−y2/dx2−y2vs. dz2/dx2−y2vs. dz2/dz2) resulting from trigonal bipyramidal (TBP) or square pyramidal (SP) geometries.
Introduction
A variety of compartmental ligands based on phenolic compounds which possess two symmetrical and asymmetrical pendant chelating arms attached to the 2- and 6-positions of the phenol ring have been synthesized.1–11 These ligands are known to accommodate two homo- or hetero-metallic 3d metal ions and hence producing dinuclear metal complexes bridged by the deprotonated phenolic group and in some cases by one or two other groups such as acetate, benzoate or hydroxide ions.4–25 In many of these compounds, the coordination environment around the central metal ions is “coordinatively unsaturated” and/or the metal ion(s) is coordinated to “weakly bound” ligand(s).1,2,4–6,10–13 This property made this class of compounds to serve as good candidates to mimic biological systems and as a consequence they have been extensively employed to elucidate the structural spectroscopic parameters and to mimic the mechanism of metalloenzymes in catecholase oxidases, Mn catalases, metallo-β-lactamases (MβL)7,13,26–30 and particularly in the hydrolytic systems.8,21,31,32 These includes phosphodiester bonds of biomolecules such as DNA, purple acid phosphatases and Zn phosphesterases.8,21,31–33
In addition to the advantages of the compartmental dinuclear metal(II) complexes which derived from phenolic compounds in enhancing our understanding for the role of metal ions in the active sides of metalloenzymes, the compounds could provide interesting magnetic properties as a result of the magnetic coupling between the two paramagnetic metal centers (3d7–9) bridged via the phenoxido group. The close proximity between the bridged metal ions, which is generally within the range of 2.9–4.0 Å provide an excellent pathway for strong antiferromagnetic interaction between the two metallic centers.1,2,4,28,30,34,35 Also, the magnetic coupling between the two metal ions could be enhanced by inserting another bridging ligand which can further propagate the magnetic coupling.4,28,30,34,35
Herein, we report a continuation of our previous studies on dinuclear metal(II) based phenolate ligands.1,2 Our discussion will be limited on symmetrical tetra-methyl pyridyl compounds namely bis[bis(2-pyridylmethyl)aminomethyl]-4-chlorophenol (LCl-OH) and bis[bis(2-pyridylmethyl)aminomethyl]-4-methylphenol (LMe-OH) which are illustrated in Chart 1. We report the synthesis, structure and magnetic characterization of three doubly bridged copper(II)-phenoxido complexes with OH−, pyrazolyl anion (pz−) and diphenylphosphate, as well as two singly bridged complexes with dicyanamide ion N(CN)2− (dca).
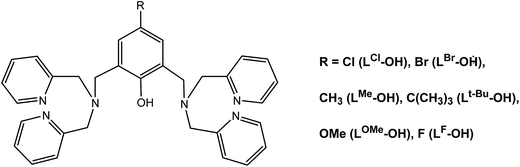 |
| | Chart 1 Structural formula of bis[bis(2-pyridylmethyl)aminomethyl]-4-substitutedphenol (LR-OH). | |
Results and discussion
Synthesis of the complexes
The reaction of a methanolic solution of 2,6-bis[bis(2-pyridylmethyl)aminomethyl]-4-chlorophenol (LCl-OH) with two equivalents of Cu(ClO4)2·6H2O in the presence Na2CO3, pyrazole (Hpz) and diphenyl phosphate afforded the dinuclear Cu(II) complexes [Cu2(μ-LCl-O)(μ2-OH)](ClO4)2 (1), [Cu2(μ-LClO)(μ-pz)(ClO4)]ClO4 (2) and [Cu2(μ-LCl-O)(μ-(O2P(OC6H5)2))](ClO4)2 (3), respectively whereas the corresponding reactions of Cu(NO3)2·3H2O and 2,6-bis[bis(2-pyridylmethyl)aminomethyl]-4-chlorophenol (LCl-OH) or 2,6-bis[bis(2-pyridylmethyl)aminomethyl]-4-methylphenol (LMe-OH) with an aqueous solution of sodium dicyanamide (Nadca) resulted in the formation of [Cu2(μ-LCl-O)(dca)2]PF6·2CH3CN (4·2CH3CN) and [Cu2(μ-LMe-O)(dca)2]PF6·2CH3CN (5·2CH3CN), respectively. The complexes were obtained in moderate to high yield (60–90%) in which the phenolic groups in all complexes and the pyrazine ligand in complex 2 were deprotonated. The isolated complexes were characterized by elemental microanalyses, molar conductivity, IR and UV-VIS spectroscopy, ESI-MS spectrometry, and single crystal X-ray crystallography. The magnetic properties of the complexes were determined at variable temperature. The molar conductivities of the complexes 1–3 (ΛM = 267–298 Ω−1 cm2 mol−1) as measured in CH3CN were consistent with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 electrolyte, whereas values of 161 and 179 Ω−1 cm2 mol−1 determined in complexes 4 and 5 are typical for 1:1 electrolytic behavior. The 1:2 electrolytic nature of complexes 1–3 is clearly obvious from their structural compositions which were also confirmed by the presence of extra bridges of mononegative anions (OH−, pz− or (C6H5O)2PO2−) in the bridged-phenoxido complexes [Cu2(μ-LCl-O)(μ-OH)](ClO4)2 (1), [Cu2(μ-LClO)(μ-pz)(ClO4)]ClO4 (2) and [Cu2(μ-LCl-O)(μ-(O2P(OC6H5)2))](ClO4)2 (3) (see X-ray section).
:2 electrolyte, whereas values of 161 and 179 Ω−1 cm2 mol−1 determined in complexes 4 and 5 are typical for 1:1 electrolytic behavior. The 1:2 electrolytic nature of complexes 1–3 is clearly obvious from their structural compositions which were also confirmed by the presence of extra bridges of mononegative anions (OH−, pz− or (C6H5O)2PO2−) in the bridged-phenoxido complexes [Cu2(μ-LCl-O)(μ-OH)](ClO4)2 (1), [Cu2(μ-LClO)(μ-pz)(ClO4)]ClO4 (2) and [Cu2(μ-LCl-O)(μ-(O2P(OC6H5)2))](ClO4)2 (3) (see X-ray section).
In the complexes 1–3, the two Cu(II) ions are doubly bridged by the phenoxido group and a hydroxido group in 1, pyrazolyl and diphenylphosphate anions in 2 and 3, respectively. Bridged phenoxido complexes similar to 1 have been previously obtained in similar class of ligands.5,28,36 Interestingly, although doubly-bridged mono(diphenylphosphato) complex 3 was isolated here, doubly-bridged bis(diphenylphosphato) complex was reported with Co(II), [Co2(μ-Lt-Bu-O)(μ-(O2P(OC6H5)2))](ClO4)2.37 Attempts made to synthesize the corresponding bis(4-nitrophenylphosphato) (BNP) of 3 were unsuccessful, most likely due to the rapid hydrolysis of BNP under the reaction conditions. Similarly attempts made to synthesize the bridged-azido or bridged-dicyanamido complexes were completely failed and instead, only the bridged-phenoxido complexes [Cu2(LMeO)(N3)2]ClO4 (ref. 2) and [Cu2(μ-LR-O)(dca)2]PF6·2CH3CN (4, R = Cl; 5, R = Me) were produced in which N3− and dca were acting as simple monodentate ligands.
IR spectra of the complexes
The IR spectra of the perchlorate complexes 1–3 displayed the ν(Cl–O) band as broad strong absorption around 1092 cm−1 as in complex 1 or split of the band into two or three bands over the range 1090–1120 cm−1 as observed in complexes 2 and 3, respectively. The broadening or split of the ν(Cl–O) band is attributed to the reduction of the ClO4− ion symmetry from Td to C3ν or C2ν symmetries as a result of the involvement of the counter ClO4− ion in H-bonding with the ligand or its presence in a distorted location. The dicyanamido hexafluorophosphate complexes 4 and 5 displayed strong absorption band around 840 cm−1, due to ν(P–F). The latter two complexes showed two series of bands: a strong absorption in the 2160–2180 cm−1 region corresponding to νs(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) and two weak to medium absorption bands in the 2220–2290 cm−1 region corresponding to νas(CN), and νs + νas(CN)38 and these were in agreement with those observed in other monodentate dicyanamido complexes.39 The frequencies of these peaks were shifted to higher values compared to the corresponding peaks observed in the free dca in its sodium salt (2129, 2232 and 2286 cm−1) indicating its coordination. The bridged hydroxido complex 1, revealed the stretching frequency ν(O–H) band at 3429 cm−1. The complexes also displayed a series of weak to medium intensity bands over the 1610–1440 cm−1 region which are characteristic of the bis(pyridyl) moieties40 and C
N) and two weak to medium absorption bands in the 2220–2290 cm−1 region corresponding to νas(CN), and νs + νas(CN)38 and these were in agreement with those observed in other monodentate dicyanamido complexes.39 The frequencies of these peaks were shifted to higher values compared to the corresponding peaks observed in the free dca in its sodium salt (2129, 2232 and 2286 cm−1) indicating its coordination. The bridged hydroxido complex 1, revealed the stretching frequency ν(O–H) band at 3429 cm−1. The complexes also displayed a series of weak to medium intensity bands over the 1610–1440 cm−1 region which are characteristic of the bis(pyridyl) moieties40 and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C of the phenolate group.
C of the phenolate group.
Electronic spectra of complexes
The acetonitrile spectra of copper complexes under investigation revealed the presence of two broad maxima over the wavelength regions 440–480 and 700–810 nm. The former band in the 440–480 nm region observed in complexes 2–4, most likely corresponds to L → M CT transition between the bridged phenoxido and copper ions. The second observed broad band (710–810 nm region, ε = 140–200 M−1 cm−1) is characteristic for Cu(II) d–d transition in five-coordinate complexes. The long wavelength position of this band suggests a distorted square pyramidal (SP) stereochemistry around the central Cu(II) ions. It is well established that five-coordinate SP Cu(II) complexes are most likely producing a broad band in the visible region which occasionally may or may not be associated with a low-energy shoulder at λ > 800 nm. This band results from dxz, dyz → dx2−y2 transition.41,42 Similar UV-Vis spectral features have been previously reported in related μ-phenoxido dicopper(II) complexes.1,2,28,43 Thus, based on the above criterion in CH3CN solution, the complexes under investigation adopt distorted SP geometry around the central Cu(II) atoms. This assignment in solution was in agreement with those obtained by single crystal X-ray crystallography.
Mass spectra of complexes
ESI-MS spectra of the chlorophenolate complexes 1–4 and the methylphenolate complex 5, recorded in acetonitrile and all are shown in Fig. S1–S5 (ESI†), displayed some general characteristic features which provide qualitative information about their compositions. The mass spectra of the complexes 1, 3 and 4 showed two major peaks at m/z = 835.059 ± 0.001 (this was observed at m/z = 821.020 for complex 2) and 767.071 ± 0.002. The former peak may result from the formation of a species with additional coordination of OH−/H2O/MeCN to the Cu(II) centers such as [Cu2(LClO)(OH)(H2O)(MeCN)3]2+ (calcd m/z = 835.158) in complexes 1, 3 and 4, whereas the corresponding peak observed in complex 2 (m/z = 821.020) could be assigned to [Cu2(LClO)(pz)(H2O)(MeCN)]2+ (calcd m/z = 821.317). The second major peak which was detected in complexes 1–4 at 767.071 ± 0.002 could be attributed to the fragments {[Cu2(LClO)(H2O)]3+ + 2Cl− + H+}2+ (calcd m/z = 767.094) in complexes 1–3 and {[Cu2(LClO)(OH)(H2O)3]2+ + F− + H+}2+ (calcd m/z = 767.031) in complex 4. Similar peaks were observed in complex 5 at m/z = 813.113 and 747.125 corresponding to the species [Cu2(LMeO)(OH)2(MeCN)3]+ (calcd m/z = 813.917) and {[Cu2(LMeO)(OH)(H2O)3]2+ + F−}+ (calcd m/z = 746.798), respectively. The spectra revealed a distinct peak at m/z = 361.035 ± 0.001 for complexes 1–4 and at 351.063 for complex 5 due to doubly charged ions. These were assigned to the fragment [Cu2(LRO)(H2O)(HCN)]3+ (calcd m/z = 361.103 for R = Cl in complexes 1–4 and m/z = 350.894 for R = Me in complex 5). In addition to these peaks, the perchlorate complexes 1–3, and the hexafluorophosphate complexes 4 and 5 displayed an m/z peak at 98.949 (100%) and 144.949 (100%) attributable to the ClO4− (calcd m/z 99.451) and PF6− (calcd m/z 144.642) ions, respectively.
Species with additional coordination such as those observed in the above complexes when MeCN was used as a solvent in measuring the mass spectra, have been recently reported in some dinuclear metal(II) complexes based phenolate.2,44
Crystal structures of the complexes
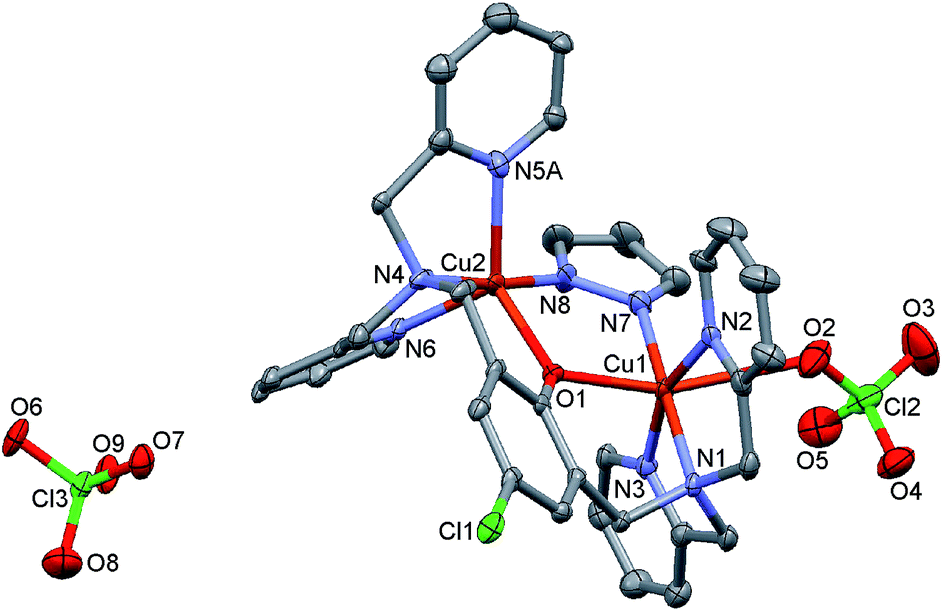
[Cu2(μ-LClO)(μ-OH)](ClO4)2 (1).
The molecular structure of 1 consists of dinuclear [Cu2(LClO)(OH)]2+ complex cations and ClO4− counter ions. A perspective view of the complex cation together with partial atom numbering schemes is given in Fig. 1, and selected bond parameters are summarized in Table S1 (ESI†). Each Cu(II) center within the dinuclear complex cation is penta-coordinated by three N-donor atoms of one bis-pyridylamino group, the bridging O(1) atom of central 4-chlorophenolate moiety and O(10) oxygen atom of bridging hydroxy group. Both CuN3O2 chromophores adopt distorted SP geometry [τ-values: 0.15 and 0.14, for Cu(1) and Cu(2), respectively]45 with N(3) and O(1) atoms in apical sites [Cu(1)–N(3) = 2.205(5), Cu(2)–O(1) = 2.200(4) Å]. The basal Cu–N/O bond distances are in the range from 1.932(3) to 2.044(5) Å. The Cu(1)⋯Cu(2) intra-dimeric distance is 3.0297(13) Å, and the shortest inter-dimer metal–metal separation is 7.166(2) Å. The Cu(1)–O(1)–Cu(2), Cu(1)–O(10)–Cu(2), O(1)–Cu(1)–O(10) and O(1)–Cu(2)–O(10) bond angles are 93.36(15), 103.29(18), 84.49(15) and 78.27(14)°, respectively. Cu(2) forms an additional semi-coordinative bond to O(6A) atom of partially disordered perchlorato anion [Cu(2)–O(6A) (x, 1/2 − y, 1/2 + z) = 2.719(8) Å] (Fig. S6†).
 |
| | Fig. 1 Perspective view and atom numbering scheme of the complex cation, [Cu2(LClO)(μ-OH)]2+ of complex 1. H-atoms are omitted for clarity. | |
[Cu2(μ-LClO)(μ-pz)(ClO4)]ClO4 (2).
The molecular structure of 2 consists of dinuclear [Cu2(LClO)(C3H3N2)(ClO4)]+ complex cations and ClO4− counter ions. A perspective view of the complex cations together with partial atom numbering scheme is given in Fig. 2, and selected bond parameters are summarized in Table S2.† Cu(1) has a 4 + 1 + 1 geometry formed by three N donor atoms of one bis-pyridylamino group, N(7) of a bridging single deprotonated pyrazole, the bridging O(1) atom of central 4-chlorophenolate moiety and O(2) oxygen atom of a terminal perchlorate group. The four short Cu(1)–N bond lengths are in the range from 1.957(5) to 2.020(6) Å. The axial Cu(1)–O(1), Cu(1)–O(2) and O(1)–Cu(1)–O(2) bond parameters are 2.227(4) Å, 2.769(7) Å and 157.87(17)°, respectively. Cu(2) is penta-coordinated by three N donor atoms of second disordered bis-pyridylamino group, N(8) of a bridging single pyrazolyl anion, and the bridging O(1) atom of central 4-chloro-phenolate moiety. The CuN3O2 chromophore adopts a distorted TBP geometry [τ-value: 0.73 (for N5A) or 0.78 (for N5B)].45 The axial sites are occupied by N(4) and N(8) atoms [Cu(2)–N(4) = 2.020(6) Å, Cu(2)–N(8) = 1.994(7) Å, N(4)–Cu(2)–N(8) = 174.1(2)°]. The equatorial Cu(2)–N/O bond distances are in the range from 1.997(10) to 2.186(11) Å. The Cu(1)⋯Cu(2) intra-dimeric distance is 3.4635(10) Å, and the shortest inter-dimer metal–metal separation is 8.0887(11) Å. The Cu(1)–O(1)–Cu(2) bond angles is 108.85(18)°. Packing plot of the compound is shown in Fig. S7.†
 |
| | Fig. 2 Perspective view and atom numbering scheme of the complex [Cu2(LClO)(μ-pz)(ClO4)]ClO4 (2). H-atoms are omitted for clarity. | |
[Cu2(μ-LClO)(μ-O2P(OC6H5)2)](ClO4)2 (3).
The molecular structure of 3 consists of dinuclear [Cu2(LClO)(O2P(OC6H5)2)]2+ complex cations and ClO4− counter ions. A perspective view of the complex cation together with partial atom numbering scheme is given in Fig. 3 and selected bond parameters are summarized in Table S3.† Each Cu(II) center within the dinuclear complex cation is penta-coordinated by three N-donor atoms of one bis-pyridylamino group, the bridging O(1) atom of central 4-chlorophenolate moiety and an oxygen atom of bridging (PO2)(OC6H5)2 group. Both CuN3O2 chromophores adopt distorted SP geometry [τ-values: 0.09 and 0.16, for Cu(1) and Cu(2), respectively]45 with N(2) and N(5) atoms in apical sites [Cu(1)–N(2) = 2.140(4), Cu(2)–N(5) = 2.173(4) Å]. The basal Cu–N/O bond distances are in the range from 1.959(3) to 2.054(4) Å. The Cu(1)⋯Cu(2) intra-dimeric distance is 3.5884(10) Å, and the shortest inter-dimer metal–metal separation is 6.7623(12) Å. The Cu(1)–O(1)–Cu(2), Cu(1)–O(10)–P(1), and Cu(2)–O(11)–P(1) bond angles are 126.90(17), 124.1(2) and 120.1(2)°, respectively. Packing plot of compound 3 is shown in Fig. S8.†
 |
| | Fig. 3 Perspective view and atom numbering scheme of the dinuclear unit, [Cu2(LCl-O)(μ-(PO2(OC6H5)2))]2+ of complex 3. H-atoms are omitted for clarity. | |
[Cu2(μ-LClO)(dca)2]PF6·2MeCN (4·2MeCN) and [Cu2(μ-LMeO)(dca)2]PF6·2MeCN (5·2MeCN).
The molecular structures of these molecules consist of dinuclear complex cations [Cu2(LClO)(dca)2]+ or [Cu2(LMeO)(dca)2]+, PF6− counter ions and MeCN lattice solvent molecules. Perspective views of the crystal structures together with partial atom numbering schemes are depicted in Fig. 4, and selected bond parameters are presented in Tables S4 and S5,† respectively. Each Cu(II) center within a dinuclear complex cation is penta-coordinated by three N donor atoms of one bis-pyridylamino group, a terminal dicyanamide anion in basal sites, and the bridging O(1) atom of central 4-substituted-phenolate moiety, which occupies the axial position of the distorted square pyramids [τ-values: 0.05 and 0.07 for 4 and 0.05 and 0.04 for 5].45 The corresponding axial Cu–O(1) bond distances are 2.190(5), 2.190(5), 2.170(2) and 2.172(2) Å, respectively. The basal Cu–N bond distances are in the range from 1.961(5) to 2.058(6) Å. The Cu(1)–O(1)–Cu(2) bridging bond angles are 136.82(17) and 137.65(8)°, respectively. The intra-dimeric metal⋯metal distances are 4.0727(12) and 4.0492(5) Å, and the shortest inter-dimer metal–metal separations are 8.3646(12) and 8.3684(6) Å, for 4 and 5, respectively (Fig. S9 and S10†). The terminal dicyanamido ligands have the following bond parameters: C–N(nitrile): 1.142(4)–1.174(8) Å, C–N(amide): 1.288(9)–1.342(11) Å, N–C–N: 173.5(3)–175.0(9)°, C–N–C: 117.8(7)–119.9(3)°, Cu–N–C: 160.8(3)–164.7(3)°.
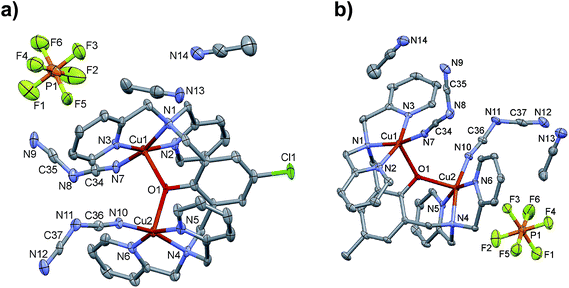 |
| | Fig. 4 Perspective views and atom numbering schemes of the dicyanamido complexes: (a) [Cu2(LClO)(dca)2]PF6·2CH3CN (4·2CH3CN) and (b) [Cu2(LMeO)(dca)2]PF6·2CH3CN (5·2CH3CN). H-atoms are omitted for clarity. | |
Magnetic properties of complexes
The analysis of the magnetic data was based on the spin Hamiltonian for dinuclear system of the form| |  | (1) |
where the isotropic exchange (J) and Zeeman term (g) are included. Then, the molar magnetization can be easily calculated using the following analytical formula46| | 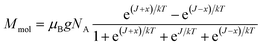 | (2) |
where x = μBgB.
Moreover, the small amount of monomeric paramagnetic impurity (PI) which accounts for increase of molar magnetization (mean susceptibility) at low temperatures was taken into consideration by eqn (3)
| | | Msample = (1 − χPI)Mmol + 2χPIMPI | (3) |
where
MPI was calculated using the Brillouin function. Both temperature and field dependent magnetic data of the studied compounds were included into fitting procedures.
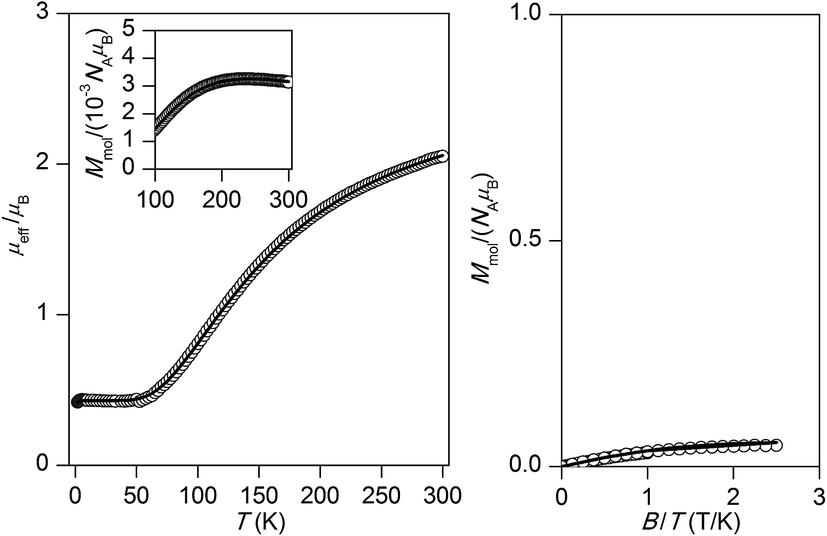
The experimental magnetic data of 1 are depicted in Fig. 5. The theoretical effective magnetic moment μeff for two uncoupled S1 = S2 = 1/2 and g = 2.0 is 2.45 μB or for more typical value of g-factor for copper(II) compounds equaled to 2.2 is μeff/μB = 2.69. The room temperature value of μeff is 2.06 μB and is decreasing on lowering the temperature. The presence of two maxima of Mmolvs. T curve located at Tmax,1 = 52.3 K and Tmax,1 = 213.3 K suggests that most likely the sample was contaminated by unidentified impurity, which was not detected by standard physico-chemical methods. Probably, it should be mentioned that several independently prepared batches of the complex were tested and same result was obtained. However, there is a simple formula derived for dinuclear species which can be used to estimate J-value as46
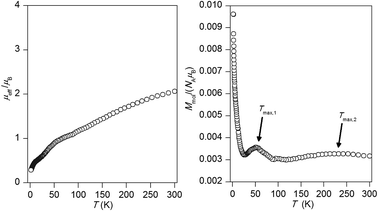 |
| | Fig. 5 The magnetic data for complex 1: the temperature dependence of the effective magnetic moment (left) and molar magnetization measured at B = 1 T (right). The arrows point to maxima found at Tmax,1 = 52.3 K and Tmax,2 = 213 K. | |
Then, the first maximum corresponds to J1 = −58.2 cm−1, while the second one to J2 = −237 cm−1. The J1 spans the interval between DFT calculated values (see next section) JRuiz = −43.1 cm−1 and JYam = −85.5 cm−1, so it can be suggested that J1 matches the predicted magnetism of compound 1, while the J2 may be assigned to the unknown impurity.
Contrary to the case described above, the experimental magnetic data for 2 (Fig. 6) matches well with the expectations where the effective magnetic moment is close to 2.7 μB at 300 K and continuously decreases to 0.24 μB at 1.9 K. The maximum of Mmolvs. T curve is located at Tmax = 55.1 K, which corresponds to J = −61.2 cm−1. Fitting procedure, which was done according to eqn (1)–(3), resulted in J = −61.5 cm−1, g = 2.14, χTIP = 3.9 × 10−9 m3 mol−1, χPI = 0.85%, where χTIP represents the correction to the temperature-independent magnetism (TIP). The strong antiferromagnetic exchange in 3, as compared to 2, is simply evident by its experimental magnetic data (Fig. 7), where Tmax located at 243 K was served as an estimate for J = −270 cm−1. This value is close to that found by the fitting procedure: J = −279 cm−1, g = 2.17, χTIP = 0.0 × 10−9 m3 mol−1, χPI = 2.60%.
 |
| | Fig. 6 The magnetic data for 2. Left: The temperature dependence of the effective magnetic moment and molar magnetization measured at B = 1 T. Right: The isothermal magnetizations measured at T = 2 and 5 K. Open circles – experimental data, solid lines – calculated data using the eqn (1) with J = −61.5 cm−1, g = 2.14, χTIP = 3.9 × 10−9 m3 mol−1, χPI = 0.85%. | |
 |
| | Fig. 7 The magnetic data for 3. Left: The temperature dependence of the effective magnetic moment and molar magnetization measured at B = 1 T. Right: The isothermal magnetizations measured at T = 2 and 5 K. Open circles – experimental data, solid lines – calculated data using the eqn (1), with J = −279 cm−1, g = 2.17, χTIP = 0.0 × 10−9 m3 mol−1, χPI = 2.60%. | |
For the remaining two dicyanamido compounds, [Cu2(μ-LR-O)(dca)2]PF6·2CH3CN (4: R = Cl; 5: R = CH3), DFT calculations predicted almost negligible antiferromagnetic exchange (see next section). Indeed, the effective magnetic moment is almost constant over the whole temperature range (the gradual increase of μeff/μB on heating in the case of 5 can be attributed to small amount of para/ferromagnetic impurity and due to temperature-independent magnetism). There is no maximum of Mmolvs. T curve, which suggests that the value of |J| should be less than 2 cm−1. This agrees well with the fitted values: J = −0.26 cm−1, g = 2.15, χTIP = 1.0 × 10−9 m3 mol−1 for 4 and J = −0.11 cm−1, g = 2.08, χTIP = 1.9 × 10−9 m3 mol−1 for 5 (Fig. 8 and 9).
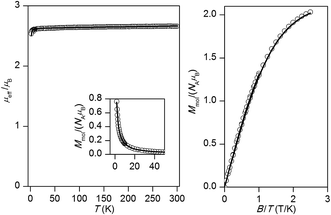 |
| | Fig. 8 The magnetic data for 4. Left: The temperature dependence of the effective magnetic moment and molar magnetization measured at B = 1 T. Right: The isothermal magnetizations measured at T = 2 and 5 K. Open circles – experimental data, solid lines – calculated data using the eqn (1), with J = −0.26 cm−1, g = 2.15, χTIP = 1.0 × 10−9 m3 mol−1. | |
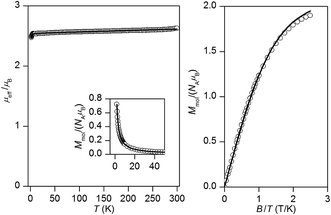 |
| | Fig. 9 The magnetic data for 5. Left: The temperature dependence of the effective magnetic moment and molar magnetization measured at B = 1 T. Right: The isothermal magnetizations measured at T = 2 and 5 K. Open circles – experimental data, solid lines – calculated data using the eqn (1), with J = −0.11 cm−1, g = 2.08, χTIP = 1.9 × 10−9 m3 mol−1. | |
DFT calculations
Our previous study on dinuclear singly bridged-phenoxido metal(II) complexes showed that in the case of copper(II) complex of the type [Cu2(μ-LCl-O)Cl2]PF6·1/2MeOH, there is a very weak antiferromagnetic exchange due to ineffective overlap of non-orthogonal magnetic orbitals.1 In the present study there are additional bridging ligands in compounds 1–3, therefore the DFT calculations were performed to reveal the effect of these extra bridging on the magnetic properties. Thus, we calculated the isotropic exchange parameters J using the ORCA 3.0 software for compounds 1–3 and extended the calculations also for compounds 4 and 5 for comparison purposes. Following our previous study1 we used the B3LYP functional and def2-TZVP(-f) basis set to calculate the energy difference Δ, between high spin (HS) and broken-symmetry (BS) spin states:
This energy difference is then used to calculate J-value for the dinuclear spin Hamiltonian defined as
| | 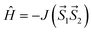 | (6) |
by both Ruiz's approach
| | | JRuiz = 2Δ/[(S1 + S2)(S1 + S2 + 1)] | (7) |
and by Yamaguchi's approach
| | | JYam = 2Δ/[〈S2〉HS − 〈S2〉BS] | (8) |
The results of DFT calculations are summarized in Table 1 and illustrated in Fig. 10. All the J-values were found negative and suggesting the presence of antiferromagnetic coupling ranging from very weak (5) to very strong (3). In case of square-pyramidal geometry (SP) the unpaired electron resides in dx2−y2 orbitals, while the corresponding trigonal-bipyramid (TBP) geometry resulted in magnetic orbital based on dz2 (Cu2 in compound 2). The smallest overlap Sαβ between the non-orthogonal orbitals was found for 5 and the largest for 3, which is in agreement with strength of antiferromagnetic exchange.
Table 1 The DFT-calculated net Mulliken spin densities (ρ), expected values 〈S2〉, overlap Sαβ between the corresponding orbitals and isotropic exchange parameters (J) from high-spin (HS) and broken symmetry spin (BS) states of the dinuclear molecular fragments based on X-ray structures of 1–9 completed with selected structural parameters
| |
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
|
J-values are estimated from maxima of molar susceptibility.
J-values were reported in ref. 28 and 34 and scaled according to eqn (1).
See Chart 1.
The τ-parameters were calculated by assuming pentacoordination of copper atoms, hence neglecting semi-coordination of the perchlorate anion in 1 and 2.
In the case of 2, 3 and 8, 9 the angle was calculated between two bonds Cu–D1 and Cu–D2, where D1 and D2 are donor atoms of bridging ligand X.
In the case of 2, 3 and 8, 9, the dihedral angle was calculated between Cu–D1–D2–Cu atoms, where D1 and D2 are donor atoms of the bridging ligand X.
|
|
ρ
HS(Cu1)/ρHS(Cu2) |
0.63/0.64 |
0.61/0.62 |
0.64/0.63 |
0.61/0.61 |
0.61/0.61 |
0.63/0.63 |
0.63/0.63 |
0.61/0.61 |
0.61/0.61 |
|
ρ
BS(Cu1)/ρBS(Cu2) |
−063/0.63 |
−0.60/0.62 |
−0.63/0.62 |
−0.61/0.61 |
−0.61/0.61 |
−0.63/0.62 |
−0.62/0.62 |
−0.61/0.61 |
−0.60/0.61 |
| 〈S2HS〉 |
2.00 |
2.01 |
2.01 |
2.01 |
2.01 |
2.01 |
2.00 |
2.01 |
2.01 |
| 〈S2BS〉 |
1.00 |
1.00 |
0.99 |
1.01 |
1.01 |
0.99 |
0.97 |
1.00 |
1.01 |
|
S
αβ
|
0.09278 |
0.07118 |
0.13903 |
0.00258 |
0.00272 |
0.13427 |
0.18057 |
0.02449 |
0.00894 |
|
Δ/cm−1 |
−43.150 |
−46.816 |
−194.931 |
−0.031 |
−0.067 |
−142.538 |
−310.304 |
15.762 |
26.894 |
|
J
Ruiz/cm−1 |
−43.1 |
−46.8 |
−194.9 |
−0.03 |
−0.06 |
−142.5 |
−310.3 |
15.8 |
26.9 |
|
J
Yam/cm−1 |
−85.5 |
−93.2 |
−382.3 |
−0.06 |
−0.14 |
−279.9 |
−600.8 |
31.5 |
53.8 |
|
J
mag/cm−1 |
−58.2/−237a |
−61.5 |
−279 |
−0.26 |
−0.09 |
−224b |
−312b |
−80 |
30.8 |
| Type of X-bridgec |
μ-OH |
μ2-C3H3N2 |
μ2-(PO2(OC6H5)2) |
None |
None |
μ-OH |
μ-CH3O |
μ-CH3COO |
μ-CH3COO |
|
τ(Cu1)/τ(Cu2)d |
0.15/0.14 |
0.78/0.21 |
0.09/0.16 |
0.05/0.07 |
0.05/0.04 |
0.60/0.36 |
0.73/0.72 |
0.22/0.22 |
0.12/0.21 |
| Magnetic orbitals |
dx2−y2/dx2−y2 |
dz2/dx2−y2 |
dx2−y2/dx2−y2 |
dx2−y2/dx2−y2 |
dx2−y2/dx2−y2 |
dz2/dx2−y2 |
dz2/dz2 |
dx2−y2/dx2−y2 |
dx2−y2/dx2−y2 |
| ∠(Cu–X–Cu)/°e |
103.29 |
112.01 |
118.04 |
— |
— |
102.10 |
102.40 |
61.08 |
63.43 |
| ∠(Cu–OPh–Cu)/° |
93.7 |
108.88 |
126.91 |
136.82 |
137.65 |
95.66 |
93.45 |
119.50 |
121.28 |
| ∠(Cu–D1–D2–Cu)/°f |
|
31.73 |
60.78 |
|
|
|
|
56.28 |
59.91 |
|
d(Cu–Cu)/10−10 m |
3.030 |
3.464 |
3.588 |
4.073 |
4.049 |
2.966 |
3.002 |
3.548 |
3.603 |
 |
| | Fig. 10 The calculated the isodensity surfaces of the broken symmetry spin states using B3LYP/def2-TZVP(-f) for molecular fragments of 1–4. Positive and negative spin densities are represented by dark blue, and dark red surfaces, respectively. Hydrogen atoms were omitted for clarity. | |
Careful inspection of literature showed that there are only four other examples similar to ours where the magnetic properties were studied for copper(II) dimers containing both LR-OH and X ligands (Chart 1), namely [Cu2(μ-LMeO)(μ-OH)](ClO4)2·THF (6),28 [Cu2(μ-LtBuO)(μ-OCH3)](ClO4)2·H2O (7),34 [Cu2(μ-LMeO)(μ-CH3COO)](PF6)2 (8),4 and [Cu2(μ-LtBuO)(μ-CH3COO)](PF6)2 (9)47 with Jmag = −224, −312, −80, +30.8 cm−1, respectively. The reported Jmag values were scaled according to the spin Hamiltonian definition in eqn (1). Interestingly, compounds 1, 6 and 7 have the same hydroxo/alkoxo-bridging groups (X = OH−/CH3O−) and despite the fact that structural parameters defining the bridges, like Cu–OPh–Cu (93.45–95.66°) and Cu–OX–Cu (102.1–103.29°) angles or Cu⋯Cu distances (2.966–3.030 Å) are almost the same (Table 1), the Jmag values vary significantly. More interesting is also the comparison of the Jmag values for acetato-bridged complexes 8 and 9, which are structurally almost identical (Table 1), their reported magnetic exchanges are either antiferromagnetic or ferromagnetic. Therefore, we utilized the above described DFT procedure to calculate the magnetic exchange parameters also for compounds 6–9 in order to elucidate the observed large variations of the isotropic exchange parameters within this family of coordination compounds. The main results are listed in Table 1 and illustrated in Fig. 10 and 11. Within the structurally similar first group of compounds 1, 6 and 7, it is obvious that the calculated strength of the antiferromagnetic exchange (JRuiz/JYam) increases with increasing the overlap of magnetic orbitals (Sαβ): dx2−y2/dx2−y2 < dz2/dx2−y2 < dz2/dz2. Thus, we may conclude that the main source of variation of magnetic properties in 1, 6 and 7 is due to the different geometry around the central copper ions (SP vs. TB). The large difference in Jmag for compounds 2 (Jmag = −61.5 cm−1) and 3 (Jmag = −279 cm−1) with the bridging ligands X (X = the pyrazolyl anion for 2 and diphenylphosphate anion for 3) can be explained by the fact that in the case of the diphenylphosphato ligand, there is efficient overlap of orbitals (dx2−y2/dx2−y2) through both bridging ligands, whereas in compound 3, the μ-phenoxido ligand is ineffective for overlap of the dz2/dx2−y2 orbitals and thus only the μ-pyrazolato ligand is the one that mediates magnetic exchange (Fig. 10). Next, in singly bridged-phenoxido compounds 4 and 5, the dx2−y2/dx2−y2 orbitals are lying in CuN4 planes perpendicular to the Cu–OPh bond, which leads to their negligible overlap, and hence to a very weak antiferromagnetic exchange similar to the previously observed in [Cu2(μ-LClO)Cl2]PF6·1/2MeOH.1 Finally, we would like to comment on the results of compounds 8 and 9. In these compounds 8, the magnetic orbitals have dx2−y2/dx2−y2 character and their mutual orientation exclude their efficient overlap through the μ-phenoxido ligand and also through acetato ligand (Fig. 11), which results in calculated ferromagnetic exchange, JRuiz/JYam = 15.8/31.5 cm−1 for 8 and JRuiz/JYam = 26.9/53.8 cm−1 for 9 (Table 1). The variation in the calculated J-values in 8 and 9 can be most probably attributed to the small differences in orientation of acetato-ligand with respect to both CuN2O2 planes containing magnetic orbitals with dx2−y2/dx2−y2 character.48 This can be demonstrated by evaluating the angle between plane of the acetato-ligand and CuN2O2 planes, which resulted in values 52.25° and 67.98° for 8 and 60.92° and 66.61° for 9.
 |
| | Fig. 11 The calculated isodensity surfaces of the broken symmetry spin states for molecular fragments of 6 and 7 and high spin states for molecular fragments of 8 and 9 using B3LYP/def2-TZVP(-f). Positive and negative spin densities are represented by dark blue, and dark red surfaces, respectively. Hydrogen atoms were omitted for clarity. | |
Furthermore, the reliability and suitability of the DFT method used here for calculation of J-parameters can be judged by inspecting Fig. 12, where all the calculated J-values are compared to those determined by the experimental magnetic data. We can conclude that except for compound 8, the experimental values Jmag are found within the intervals defined by calculated JRuiz/JYam values, which means that this procedure can reliably determine the nature of the isotropic exchange (antiferromagnetic/ferromagnetic) and also quite accurate in predicting quantitative values of J-parameters for this family of copper(II) dimeric complexes.
 |
| | Fig. 12 Comparison of the experimentally (Jmag) and theoretically (JRuiz and JYam) determined isotropic exchange parameters vs. calculated energy difference Δ, (Δ = EBS − EHS). | |
Experimental
Materials and physical measurements
The compound bis(2-pyridylmethyl)amine (DPA) was purchased from TCI-America. All other chemicals were commercially available and used without further purification. The ligands 2,6-bis[bis(2-pyridylmethyl)aminomethyl]-4-chlorophenol (LCl-OH) and 2,6-bis[bis(2-pyridylmethyl)aminomethyl]-4-methylphenol (LMe-OH) (Chart 1) were prepared and characterized according to the published procedure.1,2 Infrared spectra of all complexes and ligands were recorded on a JASCO FTIR-480 plus spectrometer as KBr pellets, except complex 4 was measured using Nexus 670 spectrometer (ATR) (Thermo Nicolet, USA). Electronic spectra were recorded using an Agilent 8453 HP diode array UV-Vis spectrophotometer. 1H and 13C NMR spectra were obtained at room temperature on a Varian 400 NMR spectrometer operating at 400 MHz (1H) and 100 MHz (13C). 1H and 13C NMR chemical shifts (δ) are reported in ppm and were referenced internally to residual solvent resonances (DMSO-d6: δH = 2.49, δC = 39.4 ppm). ESI-MS spectra were measured on an LC-MS Varian Saturn 2200 spectrometer. The conductivity measurements were performed using a Mettler Toledo Seven Easy conductivity meter and the cell constant was determined by the aid of 1413 μS cm−1 conductivity standard. The molar conductivity of the complexes were determined from ΛM = (1.0 × 103κ)/M, where κ = cell constant and M is the molar concentration of the complex. Magnetic measurements were performed with an MPMS XL7 SQUID magnetometer (Quantum Design, Inc.) (T = 1.9–300 K at B = 1 T; B = 0–5 T at T = 2 and 5 K). The magnetic data were corrected for diamagnetic susceptibilities. Elemental analyses were carried out by the Atlantic Microlaboratory, Norcross, Georgia U.S.A.
Theoretical DFT calculations
The ab initio theoretical calculations were done with the ORCA 3.0 computational package49 using the B3LYP functional50 and polarized triple-ζ quality basis set def2-TZVP(-f) for all the complexes (including all the atoms).51 The single-point energy calculations were done on molecular fragments based on the experimental X-ray geometries: [Cu2(μ-LCl-O)(μ-OH)(ClO4)]+ of 1, [Cu2(μ-LClO)(μ-pz)(ClO4)]+ of 2, [Cu2(μ-LCl-O)(μ-(O2P(OC6H5)2))]2+ of 3, [Cu2(μ-LCl-O)(dca)2]+ of 4·2CH3CN, [Cu2(μ-LMe-O)(dca)2]+ of 5·2CH3CN, [Cu2(μ-LMeO)(μ-OH)]2+ of 6, [Cu2(μ-LtBuO)(μ-CH3O)]2+ of 7, [Cu2(μ-LMeO)(μ-CH3COO)]2+ of 8 and [Cu2(μ-LtBuO)(μ-CH3COO)]2+ of 9. All the calculations utilized the RI approximation with the decontracted auxiliary def2-TZV/J Coulomb fitting basis set and the chain-of-spheres (RIJCOSX) approximation to exact exchange.52 Also, increased integration grids (Grid5 and GridX5 in ORCA convention) and tight SCF convergence criteria were used. The isotropic exchange parameters J were calculated by comparing the energies of high-spin (HS) and broken-symmetry (BS) spin states utilizing both Ruiz's approach53 and Yamaguchi's approach.54 Plots of spin densities were done by means of the VESTA 3 software.55
X-ray crystal structure analysis
The X-ray single-crystal data of compounds 1–5 were collected on a Bruker-AXS APEX CCD diffractometer at 100(2) K. The crystallographic data, conditions retained for the intensity data collection and some features of the structure refinements are listed in Table 2. The intensities were collected with Mo-Kα radiation (λ = 0.71073 Å). Data processing, Lorentz-polarization and absorption corrections were performed using APEX, and the SADABS computer programs.56 The structures were solved by direct methods and refined by full-matrix least-squares methods on F2, using the SHELXTL57 program package. All non-hydrogen atoms were refined anisotropically. The hydrogen atoms were located from difference Fourier maps, assigned with isotropic displacement factors and included in the final refinement cycles by use of HFIX (parent C atom) or DFIX (parent O atom) utility of the SHELXTL program. Molecular plots were performed with the Mercury program.58 In case of 2, split occupancy of 0.508(7) and 0.492(7) were applied to disordered atoms of one pyridyl moiety.
Table 2 Crystallographic data and processing parameters for 1–5 compounds
| Compound |
1
|
2
|
3
|
4·2MeCN |
5·2MeCN |
| Empirical formula |
C32H30Cl3Cu2N6O10 |
C35H33Cl3Cu2N8O9 |
C44H40Cl3Cu2N6O13P |
C40H36ClCu2F6N14OP |
C41H39Cu2F6N14OP |
| Formula mass |
892.07 |
943.14 |
1125.24 |
1036.35 |
1015.93 |
| System |
Monoclinic |
Monoclinic |
Monoclinic |
Orthorhombic |
Orthorhombic |
| Space group |
P21/c |
P21/c |
P21/c |
Pna21 |
Pna21 |
|
a (Å) |
14.958(3) |
16.3363(10) |
9.9409(11) |
16.2623(7) |
16.2326(5) |
|
b (Å) |
10.770(2) |
10.3267(6) |
42.116(5) |
12.0018(5) |
12.0277(4) |
|
c (Å) |
23.043(7) |
23.9249(15) |
10.8358(10) |
21.7768(9) |
21.8320(7) |
|
α (°) |
90 |
90 |
90 |
90 |
90 |
|
β (°) |
103.386(11) |
106.810(2) |
103.104(5) |
90 |
90 |
|
γ (°) |
90 |
90 |
90 |
90 |
90 |
|
V (Å3) |
3611.3(15) |
3863.7(4) |
4418.5(8) |
4250.3(3) |
4262.5(2) |
|
Z
|
4 |
4 |
4 |
4 |
4 |
|
T (K) |
100(2) |
100(2) |
100(2) |
100(2) |
100(2) |
|
μ (mm−1) |
1.465 |
1.373 |
1.256 |
1.181 |
1.115 |
|
D
calc (Mg m−3) |
1.641 |
1.621 |
1.691 |
1.620 |
1.583 |
| Crystal size (mm) |
0.22 × 0.19 × 0.13 |
0.28 × 0.23 × 0.17 |
0.28 × 0.23 × 0.17 |
0.27 × 0.23 × 0.12 |
0.24 × 0.19 × 0.17 |
|
θ max (°) |
25.50 |
26.80 |
25.30 |
26.500 |
29.030 |
| Data collected |
6771 |
40785 |
69771 |
70106 |
55430 |
| Unique refl./Rint |
6771/— |
8253/0.0435 |
7989/0.0994 |
8660/0.0863 |
11115/0.0652 |
| Parameters/restraints |
505/6 |
578/0 |
622/0 |
589/1 |
589/1 |
| Goodness-of-Fit on F2 |
1.172 |
1.204 |
1.348 |
1.115 |
0.920 |
|
R
1/wR2 (all data) |
0.0661/0.1957 |
0.0986/0.2343 |
0.0764/0.2165 |
0.0653/0.1893 |
0.0373/0.0775 |
| Residual extrema (e/Å3) |
1.67/−0.93 |
1.23/−1.27 |
1.46/−0.78 |
0.81/−1.13 |
0.50/−0.48 |
Caution: Salts of perchlorate and their metal complexes are potentially explosive and should be handled with great care and in small quantities.
Syntheses of the complexes
[Cu2(μ-LCl-O)(μ-OH)](ClO4)2 (1).
To a mixture of Cu(ClO)2·6H2O (0.152 g, 0.40 mmol) and 2,6-bis[bis(2-pyridylmethyl)aminomethyl]-4-chlorophenol (0.110 g, 0.20 mmol) dissolved in MeOH (20 mL), an aqueous solution of Na2CO3 (21 mg, 0.20 mmol dissolved in 3 mL H2O) was added. The resulting green solution was heated on a steam-bath for 10 min, filtered while hot through celite and then allowed to stand at room temperature. The precipitate which was obtained was collected by filtration, washed with propan-2-ol and Et2O and then dried at room temperature (overall yield: 105 mg, 59%). Recrystallization of the product from H2O afforded green crystals suitable for X-ray structure determination. Characterization for 1: calcd for C32H31Cl3Cu2N6O10 (MM = 892.07 g mol−1): C, 43.04; H, 3.50; N, 9.41%. Found: C, 42.96; H, 3.46; N, 9.27%. Selected IR bands (cm−1): 3429 (m,b) ν(O–H); 1608 (s), 1573 (w), 1483 (m), 1466 (s), 1440 (m) (pyridyl groups); 1092 (vs., b) νas(Cl–O). UV-VIS spectrum {λmax, nm (ε, M−1 cm−1/Cu atom)} in CH3CN: 805 (137, b). ESI-MS in CH3CN: m/z = 835.056, 767.069, 361.037 (major peaks) and for negative ion: m/z = 98.949 (100%). Molar conductivity, ΛM (CH3CN) = 267 Ω−1 cm2 mol−1.
[Cu2(μ-LClO)(μ-pz)(ClO4)]ClO4 (2).
To a hot solution containing 2,6-bis[bis(2-pyridyl-methyl)aminomethyl]-4-chlorophenol (0.110 g, 0.20 mmol) and Cu(ClO)2·6H2O (0.152 g, 0.40 mmol) dissolved in MeOH (30 mL) pyrazole, Hpz (14 mg, 0.20 mmol) was added. The resulting green solution was heated on a steam-bath for 10 min, filtered while hot through celite and then allowed to stand at room temperature. The greenish-blue precipitate which was obtained after few hours was collected by filtration, washed with propan-2-ol and Et2O and then dried at room temperature (overall yield: 160 mg, 85%). Shiny greenish-blue crystals suitable for X-ray structure determination were obtained from dilute solution. Characterization for 2: calcd for C35H33Cl3Cu2N8O9 (MM = 943.144 g mol−1): C, 44.57; H, 3.53; N, 11.88%. Found: C, 44.38; H, 3.59; N, 11.92%. Selected IR bands (cm−1): 1609 (s), 1485 (w), 1460 (m), 1447 (m), (pyridyl groups); 1120, 1093 (vs.) νas(Cl–O). UV-VIS spectrum {λmax, nm (ε, M−1 cm−1/Cu atom)} in CH3CN: 456 (sh), 799 (194). ESI-MS in CH3CN: m/z = 821.012, 767.069, 361.035 (major peaks) and for negative ion: m/z = 98.950 (100%) [ClO4− = 99.453]. Molar conductivity, ΛM (CH3CN) = 298 Ω−1 cm2 mol−1.
[Cu2(μ-LCl-O)(μ-O2P(OC6H5)2)](ClO4)2 (3).
Diphenyl phosphate (50 mg, 0.20 mmol) which was neutralized with NaOH (0.2 mmol dissolved in 2 mL H2O) was added dropwise to a warm solution containing 2,6-bis[bis(2-pyridylmethyl)aminomethyl]-4-chlorophenol (0.110 g, 0.20 mmol) and Cu(ClO)2·6H2O (0.152 g, 0.40 mmol) in MeOH (20 mL). The resulting green solution was heated on a steam-bath for 10 min, filtered while hot through celite and then allowed to stand at room temperature. The golden single crystals, which separated in the following day, were collected by filtration, washed with propan-2-ol and Et2O and then dried at room temperature (overall yield: 203 mg, 90%). Characterization for 3: calcd for C44H44Cl3Cu2N6O13P (MM = 1129.272 g mol−1): C, 46.97; H, 3.58; N, 7.47%. Found: C, 47.07; H, 3.73; N, 7.58%. Selected IR bands (cm−1): 1611 (m), 1488 (m), 1448 (m) (pyridyl groups); 1121, 1108, 1092 (vs.) νas(Cl–O). UV-VIS spectrum {λmax, nm (ε, M−1 cm−1/Cu atom)} in CH3CN: 440 (542, b). ESI-MS in CH3CN: m/z = 835.060, 767.074, 361.037 (major peaks) and for negative ion: m/z = 98.949 (100%) [ClO4− = 99.453]. Molar conductivity, ΛM (CH3CN) = 294 Ω−1 cm2 mol−1.
[Cu2(μ-LCl-O)(dca)2]PF6 (4·2CH3CN).
To a hot solution containing 2,6-bis[bis(2-pyridylmethyl)aminomethyl]-4-chlorophenol (0.111 g, 0.20 mmol) and Cu(NO3)2·3H2O (97 mg, 0.40 mmol) in MeOH (40 mL) sodium dicyanamide (36 mg, 0.40 mmol) dissolved in H2O (3 mL) was added dropwise and this was followed by the addition of NH4PF6 (100 mg, 0.6 mmol). The resulting greenish-blue solution was heated on a steam-bath for 10 min, filtered while hot through celite and then allowed to stand at room temperature. The crude precipitate which separated in the following day was collected by filtration and recrystallized from acetonitrile to afford shiny greenish-blue single crystals. These were filtered, washed with propan-2-ol and Et2O and then dried at room temperature (overall yield: 122 mg, 59%). Characterization for 4·2CH3CN: calcd for C38H36ClCu2F6N14OP (MM = 1036.341 g mol−1): C, 44.04; H, 3.50; N, 18.92%. Found: C, 44.48; H, 3.53; N, 18.76%. Selected IR bands (cm−1): 2283 (m), 2227 (m), 2161 (vs.); 1610 (m) 1574 (w), 1483 (w), 1445 (m) (pyridyl groups); 837 (s) ν(P–F). UV-VIS spectrum {λmax, nm (ε, M−1 cm−1/Cu atom)} in CH3CN: 457 (117), 656 (155, b). ESI-MS in CH3CN: m/z = 835.060, 767.071, 361.039 (major peaks) and for negative ion: m/z = 144.965 (100%). Molar conductivity, ΛM (CH3CN) = 161 Ω−1 cm2 mol−1.
[Cu2(μ-LMe-O)(dca)2]PF6·2CH3CN (5·2CH3CN).
This complex was prepared using a procedure similar to that described above for 4·2CH3CN except 2,6-bis[bis(2-pyridylmethyl)aminomethyl]-4-methylphenol was used instead of the corresponding 2,6-bis[bis(2-pyridylmethyl)aminomethyl]-4-chlorophenol. Recrystallized of the crude compound from acetonitrile afforded shiny blue well shaped single crystals (overall yield: 144 mg, 71%). Characterization for 5·2CH3CN: calcd for C41H39Cu2F6N14OP (MM = 1015.92 g mol−1): C, 48.47; H, 3.87; N, 19.30%. Found: C, 48.83; H, 4.00; N, 19.45%. Selected IR bands (cm−1): 2291 (m), 2237 (m), 2173 (vs.), 1637 (m), 1611 (m), 1472 (m), 1446 (m) (pyridyl groups); 844 (s) ν(P–F). UV-VIS spectrum {λmax, nm (ε, M−1 cm−1/Cu atom)} in CH3CN: 479 (217), 669 (155, b). ESI-MS in CH3CN: m/z = 813.113, 747.125, 351.063 (major peaks) and for negative ion: m/z = 144.965 (100%) [PF6− = 144.97]. Molar conductivity, ΛM (CH3CN) = 179 Ω−1 cm2 mol−1.
Conclusions
The reaction of copper(II) salts with 2,6-bis[bis(2-pyridylmethyl)aminomethyl]-4-substituted-phenol (LR-OH) affords two categories of dinuclear complexes in which the phenolate ligand is bridging the two Cu(II) atoms via the deprotonated phenol group. The first category is the singly bridged μ-phenoxido complexes of the general formula [Cu2(μ-LR-O)(X)2]+/3+ {R = Cl, X = dca (complex 4); R = X = Cl−;1 R = CH3, X = dca (complex 5), R = CH3, X = OAc−, Cl−, N3−, H2O and CH3CN}.2,4,5,28 This class of compounds mediates very weak antiferromagnetic coupling through the bridged phenoxido group.1,2 The second category of the bicompartmental phenolate ligands is the doubly bridged complexes where in addition to the bridged phenoxido group, an extra bridge exists and this was observed here in this study in complexes [Cu2(μ-LR-O)(μ-X)]+/2+ {1: X = OH−, 2: X = pz− and 3: X = O2P(OC6H5)2−} and in some other related complexes 6–9 {R = CH3/t-Bu, X = OH−, OAc−; R = t-Bu, X = OCH3−}.4,28,34,47 In this case moderate ferromagnetic to very strong antiferromagnetic coupling was observed.
The DFT supported analysis of magnetic properties showed that in the case of the doubly bridged complexes 1–3 and 6–9, the key factor determining the nature and strength of the isotropic exchange is the geometry of copper(II) chromophores (SP–SP, SP–TBP or TBP–TBP), thus mutual orientation of magnetic orbitals based on dx2−y2/dx2−y2, dx2−y2/dz2 or dz2/dz2 orbitals and efficiency of their magnetic orbital overlaps mediated by both μ-LR-Oph and μ-X bridging ligands. Therefore, the strongest antiferromagnetic exchange within the studied compounds was found in complexes 3 (X = O2P(OC6H5)2−) and 7 (X = CH3O), where either dx2−y2/dx2−y2 or dz2/dz2 orbitals resulting from SP–SP or TBP–TBP copper geometries are efficiently overlapping, whereas the strongest ferromagnetic coupling was induced in the acetato-bridged complex 9. Moreover, the herein the DFT method used based on the B3LYP functional and def2-TZVP(-f) basis set seems to predict properly the nature and strength of the magnetic exchange almost for all the studied complexes, thus enabling us to utilize it also for other structurally similar compounds in future. The data in Table 1 revealed that magnetic exchange in the family of doubly hetero-bridged pentacoordinate copper(II) complexes cannot be simply predicted by analyzing the basic structural parameters like ∠(Cu–OPh–Cu) and ∠(Cu–X–Cu) angles but the efficiency of magnetic orbitals overlap is more critical in evaluating the variation of copper chromophores geometries (SP vs. TBP) and their mutual orientation.
Acknowledgements
This research was financially supported by the Department of Chemistry-University of Louisiana at Lafayette. FAM acknowledges support by NAWI Graz. ZT and RH acknowledge support by LO1305.
References
- S. S. Massoud, M. Spell, C. Ledet, T. Junk, R. Herchel, R. C. Fischer, Z. Travnicek and F. A. Mautner, Dalton Trans., 2015, 2110 RSC.
- S. S. Massoud, T. Junk, M. Mikuriya, N. Naka and F. A. Mautner, Inorg. Chem. Commun., 2014, 50, 48 CrossRef CAS.
-
(a) J. K. Bjernemose and C. J. McKenzie, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2003, 59, o1275 CAS;
(b) G. T. Gomes, A. Hazell and C. J. McKenzie, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2000, 56, 382 Search PubMed.
- Y. Nishida, H. Shimo, H. Maehara and S. Kida, J. Chem. Soc., Dalton Trans., 1985, 1945 RSC.
- F. Michel, St. P. Torelli, F. Thomas, C. Duboc, C. Philouze, C. Belle, S. Hamman, E. Saint-Aman and J.-L. Pierre, Angew. Chem., Int. Ed., 2005, 44, 438 CrossRef CAS PubMed.
- K. Selmeczi, C. Michel, A. Milet, I. Gautier-Luneau, C. Philouze, J.-L. Pierre, D. Schnieders, A. Rompel and C. Belle, Chem.–Eur. J., 2007, 13, 9093 CrossRef CAS PubMed.
- Y. Gultneh, Y. T. Tesema, T. B. Yisgedu, R. J. Butcher, G. Wang and G. T. Yee, Inorg. Chem., 2006, 45, 3023 CrossRef CAS PubMed.
- B. Das, H. Daver, M. Pyrkosz-Bulska, E. Persch, S. K. Barman, R. Mukherjee, E. Gumienna-Kontecka, M. Jarenmark, F. Himo and E. Nordlander, J. Inorg. Biochem., 2014, 132, 6 CrossRef CAS PubMed.
- M. Jarenmark, M. Haukka, S. Demeshko, F. Tuczek, L. Zuppiroli, F. Meyer and E. Nordlander, Inorg. Chem., 2011, 50, 3866 CrossRef CAS PubMed.
- P. Comba, L. R. Gahan, V. Mereacre, G. R. Hanson, A. K. Powell, G. Schenk and M. Zajaczkowski-Fischer, Inorg. Chem., 2012, 51, 12195 CrossRef CAS PubMed.
- S. Bosch, P. Comba, L. R. Gahan and G. Schenk, Inorg. Chem., 2014, 53, 9036 CrossRef CAS PubMed.
- S. Svane, F. Kryuchkov, C. J. Lennarston, C. J. McKenzie and F. Kjeldsen, Angew. Chem., Int. Ed., 2012, 51, 3216 CrossRef CAS PubMed.
- S. J. Smith, C. J. Noble, R. C. Palmer, G. R. Hanson, G. Schenk, L. R. Gahan and M. J. Riley, JBIC, J. Biol. Inorg. Chem., 2008, 13, 499 CrossRef CAS PubMed.
- S. Blanchard, G. Blain, E. Riviere, M. Nierlich and G. Blondin, Chem.–Eur. J., 2003, 9, 4260 CrossRef CAS PubMed.
- L. M. Berreau, A. Saha and A. M. Arif, Dalton Trans., 2006, 183 RSC.
- S. Halder, S. Dey, C. Rizzoli and P. Roy, Polyhedron, 2014, 78, 85 CrossRef CAS.
-
(a) A. S. Borovik, M. P. Hendrich, T. R. Holman, E. Munck, V. Papaefthymiou and L. Junior Que, J. Am. Chem. Soc., 1990, 112, 6031 CrossRef CAS;
(b) A. S. Borovik and L. Junior Que, J. Am. Chem. Soc., 1988, 110, 2345 CrossRef CAS.
- T. Manago, S. Hayami, H. Oshio, S. Osaka, H. Hasuyama, R. H. Herber, K. J. Berry and Y. Maeda, J. Chem. Soc., Dalton Trans., 1999, 1001 RSC.
- H. Diril, H.-R. Chang, M. J. Nilges, X. Zhang, J. A. Potenza, H. J. Schugar, S. S. Isied and D. N. Hendrickson, J. Am. Chem. Soc., 1989, 111, 5102 CrossRef CAS.
- R. C. Holz and J. M. Brink, Inorg. Chem., 1994, 33, 4609 CrossRef CAS.
-
(a) K. Matsufuji, H. Shiraishi, Y. Miyasato, T. Shiga, M. Ohba, T. Yokoyama and H. Ōkawa, Bull. Chem. Soc. Jpn., 2005, 78, 851 CrossRef CAS;
(b) D. Saravanakumar, N. K. Sengottuvelan, G. Priyadarshni, M. Kandaswamy and H. Ōkawa, Polyhedron, 2004, 23, 665 CrossRef CAS.
- L. F. Taylor and O. P. Anderson, J. Am. Chem. Soc., 1988, 110, 1986 CrossRef.
- M. Suzuki, M. Mikuriya, S. Murata, A. Uehara and H. Oshio, Bull. Chem. Soc. Jpn., 1987, 60, 4305 CrossRef CAS.
- J. J. Maloney, M. Glogowski, D. F. Rohrbach and F. L. Urbach, Inorg. Chim. Acta, 1987, 127, L33 CrossRef CAS.
-
(a) R. K. Edgal, A. D. Bond and C. J. Mckenzie, Dalton Trans., 2009, 3833 Search PubMed;
(b) R. K. Edgal, F. B. Larsen, A. D. Bond and C. J. Mckenzie, Inorg. Chim. Acta, 2005, 358, 376 CrossRef.
- T. N. Sorrell, D. L. Jameson and C. J. O'Connor, Inorg. Chem., 1984, 23, 190 CrossRef CAS.
- A. Biswas, L. K. Das and A. Ghosh, Polyhedron, 2013, 61, 253 CrossRef CAS.
- S. Torelli, C. Belle, I. Gautier-Luneau, J. L. Pierre, E. Saint-Aman, J. M. Latour, L. le Pape and D. Luneau, Inorg. Chem., 2000, 39, 3526 CrossRef CAS PubMed.
- N. A. Rey, A. Neves, A. J. Bortoluzzi, C. T. Pich and H. Terenzi, Inorg. Chem., 2007, 46, 348 CrossRef CAS PubMed.
- L. J. Daumann, J. A. Larrabee, P. Comba, G. Schenk and L. R. Gahan, Eur. J. Inorg. Chem., 2013, 3082 CrossRef CAS.
- T. P. Camargo, F. F. Maia, C. Chaves, B. de Souza, A. J. Bortoluzzi, N. Castilho, T. Bortolotto, H. Terenzi, E. E. Castellano, W. Haase, Z. Tomkowicz, R. A. Peralta and A. Neves, J. Inorg. Biochem., 2015, 146, 77 CrossRef CAS PubMed.
- A. Neves, M. Lanzanaster, A. J. Bortoluzzi, R. A. Peralla, A. Cassellato, E. E. Castellano, P. Herrald, M. J. Riley and G. Schenk, J. Am. Chem. Soc., 2007, 129, 7486 CrossRef CAS PubMed.
- M. Ghiladi, C. J. Mckenzie, A. Meler, A. K. Powell, J. Ulstrup and S. Wocadlo, J. Chem. Soc., Dalton Trans., 1997, 4011 RSC.
- P. Dalgaard, A. Hazell, C. J. McKenzie, B. Moubaraki and K. S. Murray, Polyhedron, 2000, 19, 1909 CrossRef CAS.
- S. Sarkar, S. Majumder, S. Sasmal, L. Carrella, E. Rentschler and S. Mohanta, Polyhedron, 2013, 50, 270 CrossRef CAS.
- C. Belle, C. Beguin, I. Gautler-Luneau, S. Hamman, C. Phllouze, J. L. Pierre, F. Thomas, S. Torrelli, S. Saint-Aman and M. Bonin, Inorg. Chem., 2002, 41, 479 CrossRef CAS PubMed.
- F. B. Johansson, A. D. Bond, U. G. Nilsen, B. Maubaraki, K. S. Murray, K. J. Berry, J. A. Larrabee and C. J. McKenzie, Inorg. Chem., 2008, 47, 5079 CrossRef CAS PubMed.
- H. Kohler, A. Kolbe and G. Z. Lux, Z. Anorg. Allg. Chem., 1977, 428, 103 CrossRef.
-
(a) S. S. Massoud, A. E. Guilbeau, H. T. Luong, R. Vicente, J. H. Albering, R. C. Fischer and F. A. Mautner, Polyhedron, 2013, 54, 26 CrossRef CAS;
(b) S. S. Massoud, M. C. Lemieux, L. L. le Quan, R. Vicente, J. H. Albering and F. A. Mautner, Inorg. Chim. Acta, 2012, 388, 71 CrossRef CAS;
(c) F. A. Mautner, J. H. Albering, R. Vicente, F. R. Louka, A. A. Gallo and S. S. Massoud, Inorg. Chim. Acta, 2011, 365, 290 CrossRef CAS;
(d) F. A. Mautner, J. B. Soileau, P. K. Bankole, A. A. Gallo and S. S. Massoud, J. Mol. Struct., 2008, 889, 271 CrossRef CAS.
-
(a) S. S. Massoud, R. S. Perkins, F. R. Louka, W. Xu, A. le Roux, Q. Dutercq, R. C. Fischer, F. A. Mautner, M. Handa, Y. Hiraoka, G. L. Kreft, T. Bortolotto and H. Terenzi, Dalton Trans., 2014, 43, 10086 RSC;
(b) F. R. Louka, A. D. Stewart, E. Regel, F. A. Mautner, S. Demeshko, F. Meyer and S. S. Massoud, Inorg. Chem. Commun., 2012, 22, 60 CrossRef CAS;
(c) S. S. Massoud, F. A. Mautner, F. R. Louka, S. Demeshko, S. Dechert and F. Meyer, Inorg. Chim. Acta, 2011, 370, 435 CrossRef CAS.
-
B. J. Hathaway, in Comprehensive Coordination Chemistry, ed. G. Wilkinson, R. D. Gillard and J. A. McCleverty, Pergamon Press, Oxford, England, 1987, vol. 5, p. 533 Search PubMed.
-
(a) S. S. Massoud, F. R. Louka, R. N. David, M. J. Dartez, Q. L. Nguyn, N. J. Labry, R. C. Fischer and F. R. Mautner, Polyhedron, 2015, 90, 258 CrossRef CAS;
(b) F. A. Mautner, M. Mikuriya, Y. Naka, F. R. Louka and S. S. Massoud, Polyhedron, 2015, 85, 110 CrossRef CAS;
(c) S. S. Massoud, L. le Quan, K. Gatterer, J. H. Albering, R. C. Fischer and F. A. Mautner, Polyhedron, 2012, 31, 601 CrossRef CAS;
(d) F. A. Mautner, J. H. Albering, E. V. Harrelson, A. A. Gallo and S. S. Massoud, J. Mol. Struct., 2011, 1006, 570 CrossRef CAS;
(e) M. Schatz, M. Becker, F. Thaler, F. Hampel, S. Schindler, R. R. Jacobson, Z. Tyeklár, N. N. Murthy, P. Ghosh, Q. Chen, J. Zubieta and K. D. Karlin, Inorg. Chem., 2001, 49, 2312 CrossRef.
- T. N. Sorrell, Tetrahedron, 1999, 45, 3 CrossRef.
- l. J. Daumann, P. Comba, J. A. Larrabee, G. Schenk, R. Stranger, G. Cavigliasso and L. R. Gahan, Inorg. Chem., 2013, 52, 2029 CrossRef CAS PubMed.
- A. W. Addison, T. N. Rao, J. Reedijk, J. V. Rijin and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349 RSC.
-
R. Boča, A Handbook of Magnetochemical Formulae, Elsevier, Amsterdam, 2012 Search PubMed.
- S. S. Massoud, T. Junk, R. Herchel, Z. Trávníček, M. Mikuriya, R. C. Fischer and F. A. Mautner, Inorg. Chem. Commun., 2015, 60, 1 CrossRef CAS.
- J. Pasan, F. S. Delgado, Y. Rodriguez-Martin, M. Hernandez-Molina, C. Ruiz-Perez, J. Sanchiz, F. Lloret and M. Julve, Polyhedron, 2003, 22, 2143 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73 CrossRef CAS.
-
(a) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS;
(b) A. D. Becke, J. Chem. Phys., 1993, 98, 1372 CrossRef CAS;
(c) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS;
(d) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623 CrossRef CAS.
-
(a) A. Schaefer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571 CrossRef CAS;
(b) A. Schafer, C. Huber and R. J. Ahlrichs, Chem. Phys., 1994, 100, 5829 Search PubMed;
(c) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98 CrossRef CAS.
-
(a) E. Ruiz, J. Cano, S. Alvarez and P. Alemany, J. Comput. Chem., 1999, 20, 1391 CrossRef CAS;
(b) E. Ruiz, A. Rodríguez-Fortea, J. Cano, S. Alvarez and P. Alemany, J. Comput. Chem., 2003, 24, 982 CrossRef CAS PubMed.
-
(a)
K. Yamaguchi, Y. Takahara and T. Fueno, in Applied Quantum Chemistry, ed. V. H. Smith, Reidel, Dordrecht, 1986, p. 155 Search PubMed;
(b) T. Soda, Y. Kitagawa, T. Onishi, Y. Takano, Y. Shigeta, H. Nagao, Y. Yoshioka and K. Yamaguchi, Chem. Phys. Lett., 2000, 319, 223 CrossRef CAS.
- K. Momma and F. Izumi, VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data, J. Appl. Crystallogr., 2011, 44, 1272 CrossRef CAS.
-
(a)
Bruker, SAINT v. 7.23, Bruker AXS Inc., Madison, Wisconsin, USA, 2005 Search PubMed;
(b)
G. M. Sheldrick, SADABS v. 2, University of Goettingen, Germany, 2001 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- C. F. Macrae, P. R. Edington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, T. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S5 show the mass spectra of the complexes 1–5, respectively whereas Fig. S6–S10 are showing the corresponding packing plots for the crystal structures. Selected bond parameters of complexes 1–5 are summarized in Tables S1–S5, respectively. CCDC 1060469–1060473 contain the crystallographic data in CIF format for 1–5, respectively. For ESI and crystallographic data in CIF file or other electronic format see DOI: 10.1039/c5ra19358c |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Zdeněk
Trávníček
*b,
Roland C.
Fischer
c and
Franz A.
Mautner
*d
b,
Zdeněk
Trávníček
*b,
Roland C.
Fischer
c and
Franz A.
Mautner
*d