
Competitive lithium and sodium intercalation into sodium manganese phospho-olivine NaMnPO4 covered with carbon black
T. Boyadzhievaa,
V. Kolevaa,
E. Zhechevaa,
D. Nihtianovaab,
L. Mihaylovc and
R. Stoyanova*a
aInstitute of General and Inorganic Chemistry, Bulgarian Academy of Sciences, 1113 Sofia, Bulgaria. E-mail: radstoy@svr.igic.bas.bg
bInstitute of Mineralogy and Crystallography, Bulgarian Academy of Sciences, 1113 Sofia, Bulgaria
cFaculty of Chemistry and Pharmacy, Sofia University, 1164 Sofia, Bulgaria
First published on 12th October 2015
Abstract
In this contribution we provide novel data on the reversible lithium and sodium ion intercalation into a sodium-manganese phospho-olivine NaMnPO4, when it is used as a cathode in model lithium-ion cells. The ion-exchange reaction involving the participation of KMnPO4·H2O dittmarite as precursor was chosen for the preparation of NaMnPO4. The NaMnPO4 particles were covered with carbonaceous materials to improve the electrical conductivity and electrolyte wetting. The procedure includes ball-milling of NaMnPO4 with conductive carbon black additives Super C/65, followed by thermal treatment. The mechanically treated samples consist of well crystallized phospho-olivine phase NaMnPO4 free of any anti-site defects and disordered carbon species with graphite like medium-range order. The composite NaMnPO4/C material manifests a reversible capacity between 80–85 mA h g−1 in model lithium cells versus lithium anode. Prior to the electrochemical test, the chemical inertness of NaMnPO4 in the lithium electrolyte is studied by soaking phospho-olivines in the solution of LiPF6 in EC:DMC. The mechanism of the reversible intercalation/deintercalation cycling is investigated using ex situ X-ray powder diffraction, TEM and high-angle annular dark field STEM analysis, infrared spectroscopy and electron paramagnetic resonance spectroscopy (EPR). The study demonstrates, for the first time, that NaMnPO4 is able to intercalate reversibly both Na+ and Li+ ions following the chemical reaction LixNa1−xMnPO4 ↔ Li0.0Na0.5MnPO4 (0.25 ≤ x ≤ 0.45).
Introduction
Lithium-transition metal phosphates with olivine type of structure, LiMPO4 (M = Mn, Fe, Co and Ni), have attracted significant research interest as cathodic materials for high-power lithium ion batteries with potential to power electric vehicles.1,2 Among them, LiFePO4 displays the best electrochemical performance in view of higher rate capability and better cycling stability.1,2 In comparison with LiFePO4, the LiMnPO4 appears to be more promising since it offers a higher potential of Li+ intercalation (4.1 V versus 3.45 V, respectively), as a result of which a higher theoretical energy density can be achieved for LiMnPO4 (701 W h kg−1 versus 586 W h kg−1).3 However, LiMnPO4 displays a limited rate capability due to its lower electronic conductivity (10−10 to 10−14 S cm−1 versus 10−9 S cm−1) and greater lattice distortion due to the Jahn–Teller's instability of Mn3+ ions.4,5 All these factors, together with an anisotropic Li diffusion, determine the critical dependence of the electrochemical properties of LiMnPO4 on the method of synthesis.6 Two main types of synthetic procedures have been developed during the last years.6–8 The first one aims at improving the total electrical conductivity by coating the phospho-olivines with more conductive layers (such as carbon),6,7 the second type of procedure is directed towards designing the morphology of phospho-olivines in order to make the Li diffusion more easy.6,8Recently we have demonstrated that sodium compounds, instead of their Li analogues, can be used directly as cathodes in lithium ion batteries.9–11 This is a consequence of the ability of sodium compounds to participate in ion-exchange reactions between Na+ and Li+ ions. As a result, the in situ generated lithium compounds exhibit different electrochemical properties in comparison with those obtained prior to their use in electrochemical cells.12 The direct use of sodium-containing compounds as electrode materials in lithium ion batteries is beneficial especially in the case of layered oxides on the basis of vanadium, titanium and manganese such as NaxV3O8, Na2/3Mn1−xFexO2, α-Na0.66MnO2.13, sodium titanate.13–16 Concerning polyanion-based compounds, it has been demonstrated that the lithium intercalated jarosite hydroxysulfate Li2+xNaFe3(SO4)2(OH)6 (with 0 < x < 0.5) exhibits reversible electrochemical lithium intercalation/de-intercalation through a solid solution-like process, leading back to the oxidized jarosite LixNaFe3(SO4)2(OH)6 (with 0.3 < x < 0.6), with redox cycling at 2.82 V and a capacity of 110 mA h g−1 at C/20.17 To the best of our knowledge this concept has not yet been examined in the case of manganese-based phospho-olivines.
In comparison with LiMnPO4, NaMnPO4 posses two structure modifications: maricite-type and olivine-type of structure.18,19 Although both structural modifications display the same framework composed of phosphate groups (space group Pnma), the occupancy of M1(4a) and M2(4c) positions by Na+ and Mn2+ ions is different: for the olivine-type of structure, Na+ and Mn2+ occupy preferentially M1 and M2 sites, while the opposite occupancy is observed in the maricite-type of structure. In the olivine structure, Na+-containing octahedra share edges and form zig-zag chains along the b axis.18 This is the preferred direction of more favorable diffusion of alkaline ions through the olivine crystal structure.5,20 The maricite structure is electrochemically inactive since the Na+ ions mobility is blocked.21 However, it is the maricite phase that is the thermodynamically stable modification of NaMnPO4.
Based on ion-exchange reaction, we have already reported a facile low-temperature method for the preparation of both Li- and Na-containing phospho-olivines.22–24 The method is based on the ion exchange reaction by using of KMnPO4·H2O having dittmarite-type of structure as a precursor. The reaction mechanism includes ionic exchange of K+ for Li+ or Na+ in the framework of the dittmarite structure, followed by H2O release and formation of the olivine-type of structure.22,23 The ionic exchange reaction is a consequence of the structural similarity between the dittmarite and olivine structures. Both structures consist of layers composed of corner-sharing metal octahedra bridged through the oxygen atoms of the PO43− groups. The topology of the Mn(2+)-phosphate layer in the ac plane of KMnPO4·H2O matches the topology of the Mn(2+)-phosphate layer in the bc plane of Li/NaMPO4. As a result, the preferred orientation of the precursor dittmarite crystallites remains the same as in the targeted olivine composition.22,23 This method is effective for the preparation of the electrochemically active LiMnPO4 compositions.24
In this contribution, we provide new data on the reversible intercalation of Na+ and Li+ into NaMnPO4 with an olivine-type of structure. The ion-exchange reaction involving the participation of KMnPO4·H2O dittmarite as precursor was chosen for the preparation of NaMnPO4. The NaMnPO4 particles were covered with carbonaceous materials to improve the electrical conductivity and electrolyte wetting. The procedure includes ball-milling followed by thermal treatment. The crystalline structure and morphology of NaMnPO4/C composite was analyzed by means of X-ray powder diffraction, SEM, SAED and HRTEM, IR and Raman spectroscopy. The intercalation properties of NaMnPO4 were tested in model lithium cells versus lithium anode. Prior to the electrochemical test, the chemical inertness of NaMnPO4 in the lithium electrolyte is studied by soaking phospho-olivines in the solution of LiPF6 in EC:DMC. The structural changes during intercalation/deintercalation cycling are monitored by ex situ X-ray powder diffraction, TEM and high angle annular dark field STEM analysis, IR spectroscopy and EPR.
Experimental
Sodium manganese phospho-olivine NaMnPO4 was obtained by an ion-exchange reaction using dittmaritte-type KMnPO4·H2O as precursor at 200 °C. The details are given elsewhere.23 Briefly, the dittmarite precursor KMnPO4·H2O was mixed with NaCH3COO·3H2O at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10. The mixture is heated at 200 °C for 15 hours. After cooling down to room temperature, the product was thoroughly washed with water and ethyl alcohol to remove the unreacted salts, filtered and dried in air for about 5 h. Under these conditions, pure olivine phase of NaMnPO4 was formed. To improve the degree of crystallinity, NaMnPO4 was further annealed at 400 °C for 10 h under argon atmosphere. The composite material was prepared by ball-milling of pristine NaMnPO4 with 15% conductive carbon black additives Super C65 (TIMCAL) for 4 h at a speed of 300 rpm using planetary “Pulverisette 6” mill (Fritsch) with agate balls (ϕ 10 mm). The sample to balls ratio was 10:1. The ball-milled composite NaMnPO4/C was thermally treated under argon at 400 °C for 3 h. This sample will be denoted as NMP/C composite, while pristine NMP will be used for the untreated sample.
:10. The mixture is heated at 200 °C for 15 hours. After cooling down to room temperature, the product was thoroughly washed with water and ethyl alcohol to remove the unreacted salts, filtered and dried in air for about 5 h. Under these conditions, pure olivine phase of NaMnPO4 was formed. To improve the degree of crystallinity, NaMnPO4 was further annealed at 400 °C for 10 h under argon atmosphere. The composite material was prepared by ball-milling of pristine NaMnPO4 with 15% conductive carbon black additives Super C65 (TIMCAL) for 4 h at a speed of 300 rpm using planetary “Pulverisette 6” mill (Fritsch) with agate balls (ϕ 10 mm). The sample to balls ratio was 10:1. The ball-milled composite NaMnPO4/C was thermally treated under argon at 400 °C for 3 h. This sample will be denoted as NMP/C composite, while pristine NMP will be used for the untreated sample.
As a reference sample we used LiMnPO4 obtained by an ion-exchange reaction from the same KMnPO4·H2O precursor. The synthesis procedure was described previously.22,24 In order to eliminate any anti-site defects, LiMnPO4 was annealed at 500 °C. The composite of LiMnPO4 with carbon additives was also prepared following the above mentioned ball-milling process.
The XRD patterns of pristine NMP and NMP/C composite were registered by using a Bruker Advance 8 diffractometer (CuKα radiation), at 0.02° 2θ step of 10 s duration. The structural analysis was made by the Rietveld method with FULLPROF program.25 The diffractometer point zero, the Lorentzian/Gaussian fraction of the pseudo-Voigt peak function, scale factor, the unit cell parameters (a, b and c), the thermal factors for the 4a, 4c and 8d positions and the line half-width parameters were refined. The preferred orientation was estimated by the modified March's function with G1 refinable parameter:26 there is no preferable orientation for G1 = 1 and a preferred orientation with dominant plate-like habit for G1 < 1. The cationic occupancy factors were determined taking into account that the total occupancies of the 4a, and 4c metal sites are equal to unity. The crystallite size of NaMnPO4 compositions was calculated by Scherrer's equation: Dhkl = λ/((β2 − βo2)1/2cosθhkl), where λ(CuKα) = 0.15418 nm, β is the peak width at the half height corrected with instrumental broadening and θhkl is Bragg's angle. The line width was determined by profile analysis using a WinPlotr program.
The morphology of NMP and NMP/C was observed by JEOL JSM-5510 scanning electron microscope. The TEM investigations were performed on a JEOL 2100 transmission electron microscope and a JEOL 2100 XEDS: Oxford Instruments, X-MAXN 80T CCD Camera ORIUS 1000, 11 Mp, GATAN at accelerating voltage of 200 kV. The specimens were prepared by grinding and dispersing the powders in ethanol by ultrasonic treatment for 6 minutes. The suspensions were dripped on standard holey carbon/Cu grids. The analysis was carried out by the Digital Micrograph software.
The IR spectra of pristine NMP and composites NMP/C were recorded on a Fourier transform Nicolet Avatar-320 instrument (resolution < 2 cm−1) using KBr disks. Only the IR spectrum of the electrolyte was measured in Nujol mull. The Raman spectra were obtained by LabRam 300 (Horiba Jobin-Yvon) micro-Raman spectrometer at room temperature using a He–Ne laser operating at 633 nm.
The oxidation state of Mn ions in NMP/C composites was determined by a Bruker EMXplus EPR spectrometer operating in the X-band (9.4 GHz) in the temperature range of 100–400 K.
The electrochemical charge–discharge curves of composites NMP/C were examined by using EL-CELL type two-electrode cells comprising Li|LiPF6 (EC:DMC)|NaMnPO4. The positive electrode, supported onto an aluminium foil, was a mixture containing 80% of the active NaMnPO4/C composition, 7.5% C-NERGY KS 6 L graphite (TIMCAL), 7.5% Super C65 (TIMCAL) and 5% polyvinylidene fluoride (PVDF). The loaded mass of active materials on Al collectors was about of 11 mg. The electrolyte was a 1 M LiPF6 solution in ethylene carbonate and dimethyl carbonate (1:1 by volume) with less than 20 ppm of water (0.2 ml electrolyte solution was used). The lithium electrodes consisted of a clean lithium metal disk with diameter of 18 mm. The cells were mounted in a dry box under argon atmosphere. The intercalation/deintercalation cycling was carried out using an eight-channel Arbin BT2000 system in galvanostatic mode. The charge and discharge rates were expressed as C/h, where h is the hours needed for the insertion of one lithium per formula unit at the applied current intensity. The model lithium cell was cycled between 4.5 and 2.5 V at C/20 and C/50 rates. The CV test was carried out by using a three electrode cell and PAR Potentiostat/Galvanostat, Model 273A, in the potential window of 1.7–4.7 V and at a scan rate of 0.05 mV s−1. The CV curves are recorded after 5 galvanostatic charge/discharge cycles between 2.5 and 4.5 V.
The chemical reactivity of NaMnPO4 with lithium electrolyte was monitored by soaking pristine NMP and composite NMP/C in the solution of LiPF6 salt in EC:DMC for 40 days. All the experiments were carried out in dry box. After the soaking, pristine NMP and composite NMP/C were washed with electrolyte and were dried on pieces of filter paper for 24 h in the dry box. The so treated samples were subjected for XRD and IR spectroscopy measurements. A part of the suspension was also washed with acetone (out of dry box) and the IR spectrum of such a sample was also recorded.
The structural changes of electrode samples during reversible intercalation/deintercalation cycling were analyzed with lithium half-cells stopped at selected potentials. The electrochemical cells were disassembled insight a glove-box, followed by removing and washing of the working electrodes with EC. For the XRD experiments, the electrode samples were covered with parafilm in order to avoid the water contamination. For the TEM experiments, the specimens were dispersed in acetone and the suspensions were dripped on standard holey carbon/Cu grids. For the EPR experiments, the quartz tube was filled with electrodes inside the glove-box.
Results and discussion
Structure and morphology of composite NaMnPO4/C
Fig. 1 compares the XRD patterns of pristine NMP and NMP/C composite. All diffraction patterns were analyzed within the framework of the structural model comprising only one phase with olivine-type of structure. It is worth mentioning that no diffraction peaks due to carbon additives were observed, which is consistent with their amorphous state. The lattice parameters of pristine NMP are identical with that for composite NMP/C: a = 10.5177(3) Å, b = 6.3144(2) Å, c = 4.9873(2) Å, V = 331.227(22) Å3 for pristine NMP and a = 10.5167(4) Å, b = 6.3163(2) Å, c = 4.9885(2) Å, V = 331.378(25) Å3 for NMP/C. On the one hand, these values are in good agreement with those reported previously for NaMnPO4 in the form of single crystal and NaMnPO4 obtained at 200 °C.19,23 On the other, the identical lattice parameters indicate that the olivine structure remains stable after ball-milling procedure. | ||
| Fig. 1 Rietveld refinement of the XRD patterns of pristine NaMnPO4 (a) and NaMnPO4/C composite (b). | ||
Since the olivine modification of NaMnPO4 is thermodynamically unstable, the next issue that should be considered is the possible creation of structural anti-site defects including the exchange of ions between M1 and M2 positions. These kinds of defects have already been observed in LiMnPO4.27,28 The Rietveld analysis evidences that NMP and NMP/C have an ordered olivine-type of structure, where metal 4a and 4c crystallographic sites are entirely occupied by Na and Mn ions, respectively (the refined site occupancy is 1.00 for the two ions). This proves that the ball-milling procedure does not create any defects in the composite material.
The only characteristics that are changed are the XRD peak intensities. The Rietveld's refinement can be interpreted by a preferred orientation of olivine crystallites along the [100] direction: Rb = 0.039, goodness of fit GoF = 1.8 and G1 = 0.76 for pristine sample and Rb = 0.048, GoF = 1.5 and G1 = 0.82 for NaMnPO4/C. The observation of preferred orientation coincides with our previous studies on the formation of NaMnPO4 with controlled surface morphology by using template-directed dittmarite precursors.23 The important finding here is the comparison of the degree of crystallite orientation: there is a trend of increase in the G1-parameter together with decrease in GoF for NMP/C. This means that ball-milling procedure reduces the degree of preferred crystallite orientation in the NaMnPO4/C composite. The line-width increases only slightly after ball-milling: the line broadening corresponds to a slight reduction in crystallites sizes from 60 nm in pristine NaMnPO4 to 55 nm in NaMnPO4/C composite.
The stability of the NaMnPO4 olivine structure is further supported by the IR spectra of the pristine and ball-milled samples (Fig. 2). Both NMP and NMP/C samples display the typical IR bands for the PO43− groups in the olivine-type of structure: five bands at 943 sh/969/1060/1078/1129 cm−1 due to the PO4 stretching vibrations (ν1 and ν3 modes) and the bands at 630 sh/618/580/545 cm−1 originate from the PO4 bending vibrations (ν4 modes) (Fig. 2). The two IR spectra are identical within the experimental resolution (up to 2 cm−1), thus indicating that the local structure of the phosphate groups in NaMnPO4/C olivine phase remains unchanged after the ball-milling.
 | ||
| Fig. 2 IR spectra of pristine NMP (a) and NMP/C composite (b). | ||
The presence of carbon additives in the NMP/C composite is clearly detected by Raman spectroscopy. Fig. 3 represents the Raman spectra of carbon black additives before and after milling with NaMnPO4. The Raman spectra are dominated by two intensive bands at 1326 and 1590 cm−1. These two bands are defined as D (disorder) and G (graphite) peaks and they are fingerprints of disordered carbon with graphite-like medium-range order.29–31 The G band is due to the in-plane stretching motion of pair of sp2 carbon atoms in aromatic and olefinic bonding.29–32 The D band is attributed to disorder-allowed phonon modes which become Raman active as a result of the disrupted symmetry of the graphite sheets (scattering from defects).32 In the case of our samples, the positions of the D and G bands as well as their FWHM (full width at half maximum) values are not changed after the ball-milling process: 133/131 cm−1 for the D bands and 78/82 cm−1 for the G bands. On the other hand, the integrated intensity ratio ID/IG (calculated by D- and G-band areas) shows a tendency to decrease after ball-milling of mixture of carbon black additives with NaMnPO4: 2.2 versus 1.8, respectively. The Raman spectroscopy gives evidence that the carbon is in a disordered state and this amorphous state is not modified by the ball-milling process. In addition, the amorphous state of carbon additives is consistent with the lack of XRD peaks due to carbon skeleton (Fig. 1).
 | ||
| Fig. 3 Raman spectra of NMP (a), NMP/C (b) and carbon additives (c). | ||
In addition to the G and D bands, the Raman spectrum of NMP/C exhibits two low-intensity bands at 950 cm−1 and at 648 cm−1. These bands can be attributed to symmetric stretching and asymmetric bending modes (ν1 and ν4) of the PO4 group. The ν1 band is much weaker than that in pristine NMP, which can be related to a screening effect of the carbon on the surface of the NaMnPO4 particles.
The morphology of phospho-olivine is an important feature that determines their electrochemical properties. Fig. 4 compares the SEM images of NMP and NMP/C. The morphology of pristine NMP (Fig. 4a) consists of rod-like aggregates with a length of about 0.5–1.0 μm and a width of about 0.10–0.15 μm. The ball-milling process, however, changes considerably the morphology of the composite NMP/C. It is visible that spherical aggregates instead of rod-like ones prevail in the SEM images (Fig. 4b and c). This means that during the ball-milling the rods are being broken into small spherical aggregates with sizes below 100 nm (Fig. 4c).
 | ||
| Fig. 4 SEM images of pristine NMP (a) and NMP/C composite (b). For NMP/C, a higher magnification is used (c). | ||
The TEM images show that NMP/C consists of well crystallized particles with sizes of about 50 nm (Fig. 5). Some of the particles are stacked into rod-like aggregates with smaller dimensions: i.e. 500/200 nm. As in the case of the SEM images, the rod-like aggregates appear to become smaller after ball-milling. The SAED pattern viewed along [013] reveals that nanoparticles are composed of single olivine-type of phase. The HRTEM image (Fig. 5) exhibits lattice fringes from (100) plane, the interplanar space of 1.05 nm coincides with that estimated by the XRD analysis (10.5156 Å). The TEM results confirm once again the stability of the olivine-type of structure during ball-milling procedure.
 | ||
| Fig. 5 Bright field micrographs and SAED of composite NMP/C (left, first row); HRTEM image (right, first row); BF-STEM images and corresponding composition map of CKα1–2, MnKα1, PKα1, NaKα1–2 and OKα1. | ||
The important finding is that the olivine particles are covered with carbon additives; the coating thickness varies between 2 and 10 nm. In addition, some separate carbon particles with sizes of about 20 nm are also visible. The TEM investigation reveals that the ball-milling procedure followed by annealing at 400 °C is an effective way to ensure a good inter-phase contact between the olivine particles and the carbon additives.
The distributions of Na, Mn, P, O and C elements, determined using high angle annular dark field STEM images, are compared in Fig. 5. It is clear that nano-particles have composition NaMnPO4, which is homogeneously covered with carbon. The occurrence of separate individual carbon particles is also observed in the image of carbon element distribution.
Chemical inertness of NaMnPO4 phospho-olivine in the lithium electrolyte
Prior to the electrochemical tests it is necessary to understand the chemical inertness of NaMnPO4 phase in the lithium electrolyte. It has been accepted that LiPF6 is a highly reactive reagent due to its thermal instability.33 LiPF6 has been shown to decompose into LiF and PF5 even at room temperature.33 The reaction product PF5 is a strong Lewis acid and it easily reacts with the surface of electrode materials leading to the formation of the solid-electrolyte inter-phase, containing a mixture of organic and inorganic compounds. Based on ex situ micro-Raman spectroscopy, it has been found out that the delithiated LixMnPO4 exhibits chemical instability in the LiPF6-based electrolyte during a charge–discharge cycle, as a result of which pyrophosphate, Li4P2O7 is being formed.34 The degradation of LiMnPO4 is significantly reduced when a phosphate surface is uniformly covered with carbon.34 In addition, several chemical species, such as polycarbonates and carbonates, have been shown to be deposited on the surface of LiMnPO4 during electrochemical cycling especially above 4.0 V vs. Li+/Li.35–37 Taking into account the possible interaction between phospho-olivines and lithium electrolyte, we studied the chemical reactivity of NaMnPO4 with lithium electrolyte by soaking both NMP and NMP/C in the solution of LiPF6 salt in EC/DMC solvent for 40 days. The soaked samples were then characterized by XRD and IR spectroscopic techniques.Fig. 6 compares the XRD patterns of pristine NMP and composite NMP/C before and after soaking them in the electrolyte. The XRD patterns of the soaked samples exhibit high level of background, but it is important that only diffraction peaks due to phospho-olivine phase become visible. Moreover, no shifts and changes in the intensities of the olivine diffraction peaks are observed. These characteristic features are valid for both the pristine and the carbon treated phospho-olivines. The constancy in the peak positions and intensities proves the chemical inertness of both pristine and carbon-treated NaMnPO4 in the lithium electrolyte. This means that ion-exchange reactions of Na+ from NaMnPO4 for Li+ from LiPF6 salt are not taking place during the prolonged soaking.
 | ||
| Fig. 6 XRD patterns of: pristine NMP (a); pristine NMP after soaking in the lithium electrolyte (b); NMP/C composite (c), and NMP/C after soaking in the electrolyte (d). | ||
The lack of side reactions is also confirmed by the IR spectra of the soaked samples. Since pristine and composite samples have one and the same behavior in the electrolyte, Fig. 7 represents only the IR spectrum of NMP/C. To facilitate the assignment of IR bands, the same figure gives also the IR spectrum of fresh electrolyte (i.e. LiPF6 solution in EC/DMC). The IR spectrum of soaked NMP/C is dominated by the vibrational bands due to LiPF6, EC and DMC. In order to eliminate these bands, the soaked NMP/C sample is further washed with acetone. The comparison of IR spectra shows that only the bands due to the phosphate groups are resolved, their positions and intensities match well those of the pristine NMP/C. This observation gives evidence for the chemical inertness of NaMnPO4 after storage in lithium electrolyte LiPF6/EC/DMC. It is worth mentioning that no traces of pyrophosphates or polycarbonates, which are the usual products of side reactions, are detected by IR spectroscopy.
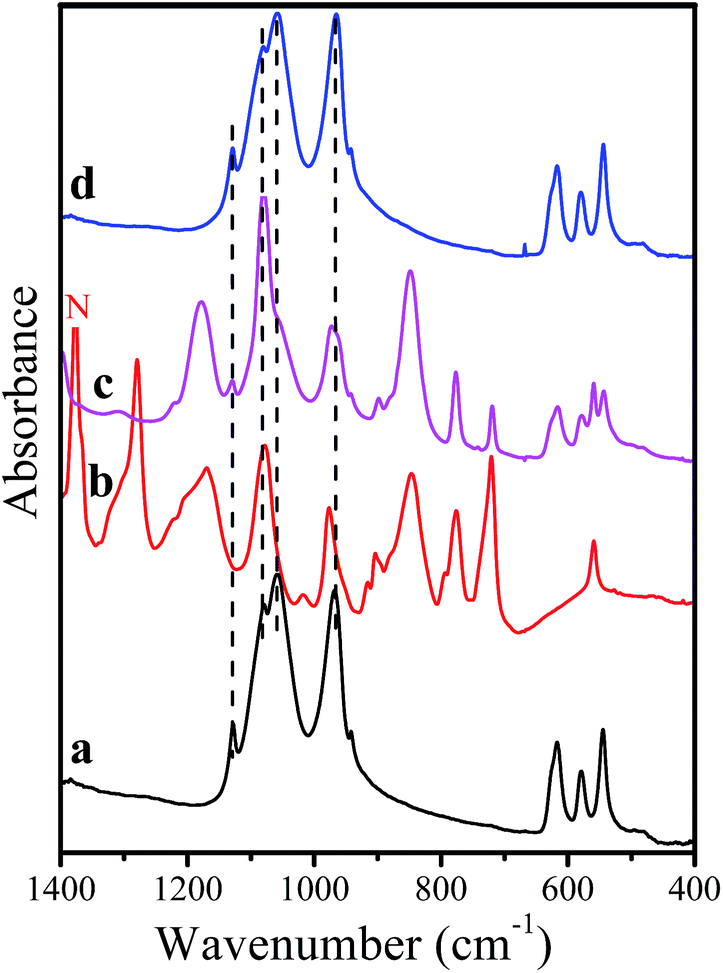 | ||
| Fig. 7 IR spectra of NMP/C composite (a), fresh electrolyte LiPF6 in EC/DMC (b), NMP/C after soaking in the lithium electrolyte (c); NMP/C after soaking in the lithium electrolyte and washed with acetone (d). “N” denotes the band from Nujol oil. | ||
Electrochemical properties of composites NaMnPO4/C
Fig. 8 compares the first charge/discharge curves of NaMnPO4. For the sake of comparison, the same figure gives also the charge/discharge curve of LiMnPO4 analogue covered with carbon following the same treatment procedure. All cells start with a charging mode, where Li+ and Na+ deintercalation from phospho-olivines is expected to occur. For LiMnPO4, the main oxidation and reduction peaks corresponding to the Mn2+/Mn3+ redox couple appear at 4.3 and 3.9 V, respectively. The capacity delivered during the first charging and consecutive discharging reaches 165 and 150 mA h g−1. These values are close to the theoretical one (i.e. 168 mA h g−1), which indicates a lack of polarization for the cell based on LiMnPO4 electrode. The good electrochemical performance can be related to a close contact between carbon additives and LiMnPO4. It is well known that carbon additives play a crucial role in improving the electrochemical performance of LiMnPO4: on the one hand, the carbon additives have a positive impact on the electrical conductivity and chemical stability of phospho-olivines and, on the other hand, they ensure a good wetting of NaMnPO4 with electrolyte solution. The electrochemical performance of LiMnPO4, used by us as a reference, points out that ball-milling procedure followed by thermal treatment at 500 °C is a successful method to overcome the drawbacks typical of LiMnPO4-based electrode material. | ||
| Fig. 8 First charging/discharging curves for NaMnPO4 and LiMnPO4 used as electrodes in model lithium cells. The charge and discharge capacity (black and red symbols) for NaMnPO4 versus the number of cycles. | ||
In comparison with LiMnPO4, its sodium analogue NaMnPO4 delivered a capacity that is slightly lower. During the first charging, the potential corresponding to the Na+ ion deintercalation depends on the charging rate: at a slow rate, the potential tends to that typical of Mn2+/Mn3+ in the LiMnPO4 analogue. On the contrary, the reverse discharging process is proceeding at lower potential than that for LiMnPO4: 3.75 versus 3.95 V. It is worth mentioning that the discharging rate does not affect the discharge potential. This allows assigning the two plateaus at 4.25 (charge) and 3.75 V (discharge) to the Mn2+/Mn3+ couple too. The stability of capacity during cycling is shown in Fig. 8. After the first three cycles, both charge and discharge capacities become close during cycling, the columbic efficiency being more than 98%. The reversible capacity reaches 80–85 mA h g−1, which corresponds to 0.5 mol of intercalated alkaline ions. These results are the first experimental evidence that NaMnPO4 is electrochemically active in the model lithium cell.
As a support of the electrochemical activity of NaMnPO4 in a lithium cell, Fig. 9 gives the CV curve of NaMnPO4/C in a broad potential range from 1.7 to 4.7 V. The CV curve displays one well-resolved peak at 4.38 V in the anodic scan and two peaks at 3.94 and 3.10 V in the cathodic scan. Below 2.5 V, additional less intensive peaks are resolved. Above 4.5 V, the strong increase in the current implies for a possible electrode–electrolyte interaction. Taking into account the CV data, the galvanostatic cycling was limited between 2.5 and 4.5 V. The anodic and cathodic peaks at 4.38 and 3.94 V coincide with that typical for LiMnPO4: for carbon-coated LiMnPO4 with a particle size of 30–70 nm the cathodic and anodic peaks are at 4.093 and 4.307 V, respectively, while for LiMnPO4 with a bigger particle size the peaks are at 3.930 and 4.544 V, respectively.38 In our case of carbon coated NaMnPO4 with a particle size below 100 nm the peak positions are between above limiting cases of LiMnPO4. This result reveals unambiguously that lithium intercalation takes place into NaMnPO4 thanks to the oxidation and reduction of manganese ions (i.e. Mn2+/Mn3+ ionic pair). However, the cathodic peak at 3.10 V is not associated with lithium intercalation. Thus, the main question is: what is the origin of the intercalation reaction of NaMnPO4 in a lithium cell?
 | ||
| Fig. 9 CV curve of NaMnPO4/C in a lithium cell after 5 galvanostatic cycles between 2.5 and 4.5 V. | ||
In order to understand the nature of the electrochemical activity of NaMnPO4 in lithium cells, ex situ XRD, IR, TEM and EPR experiments have been undertaken. Fig. 10 represents the ex situ XRD pattern of an electrode NMP/C after 5 cycles between 4.5 and 2.5 V at C/50. (The cell is stopped at 2.5 V.) For comparison, the XRD pattern of an electrode NMP/C prior to the electrochemical cycling is also given. The non-cycled electrode displays the well-resolved diffraction peaks corresponding to the olivine phase only (Fig. 10a and c). After 5 cycles, the background level increases significantly, but the relatively weak diffraction peaks due to olivine phase are still well resolved (Fig. 10b). On the basis of (200), (210), (020), (301), (311) and (121) diffraction peaks, it is possible to estimate the lattice parameters: a = 10.452 Å, b = 6.266 Å, c = 4.994 Å, V = 327.06 Å3. The estimated lattice parameters are slightly lower than those of pristine NaMnPO4 (a = 10.5177(3) Å, b = 6.3144(2) Å, c = 4.9873(2) Å, V = 331.227(22) Å3), but they are higher in comparison to the lithium ones LiMnPO4 obtained by ion-exchange reaction22 (a = 10.488(1) Å, b = 6.1006(8) Å, c = 4.7518(8) Å, V = 302.901 Å3). The comparison indicates that phospho-olivine phase with sodium content slightly lower than 1 mol participates in a reversible intercalation/deintercalation cycling when NaMnPO4 is used as a cathode in model lithium cell. This means that Na+ intercalation proceeds together with Li+ intercalation during the lithium cell discharging. The occurrence of sodium-rich phospho-olivine phase in discharged lithium cell is an opposite behavior compared to that of sodium transition metal oxides with a layered structure NaxNi0.5Mn0.5O2.12 We have demonstrated that Li+ intercalation proceeds easily in NaxNi0.5Mn0.5O2 during lithium cell discharging, as a result of which mixed lithium–sodium oxides with Li-to-Na ratio higher than 6 are being formed.12
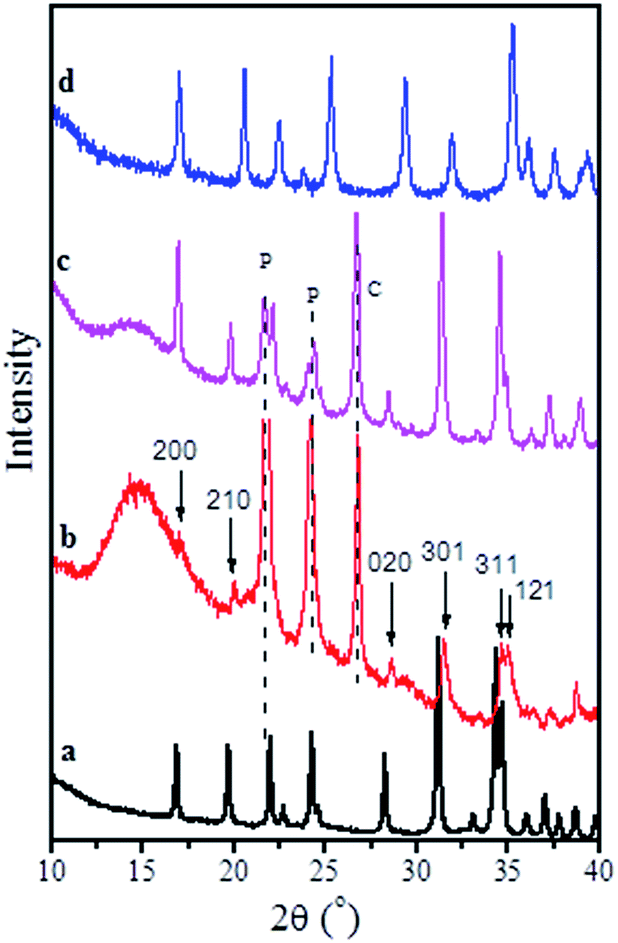 | ||
| Fig. 10 XRD patterns of NMP/C composite (a), NMP/C after 5 charge–discharge cycles between 4.5 and 2.5 V at C/50 (b), NMP/C electrode prior to cycling (c) and LiMnPO4 (d). Symbols “P” and “C” denote the diffraction peaks due to parafilm and graphite. | ||
The TEM analysis allows gaining insight into the structure stability of phospho-olivines during cell operation (Fig. 11). After the intercalation/deintercalation cycling, the morphology of NMP/C is preserved. There are well-crystallized particles with dimensions close to those before the cycling experiment. In addition, the rod-like aggregates are also distinguishable in the TEM image of cycled electrode. The lattice parameters determined by SAED are comparative with those of NaMnPO4. Taking into account the accuracy in the determination of lattice parameters by TEM (less than 1.5%), it is not possible to detect precisely the small deviations of the lattice parameters as it was observed by XRD experiments. However, HRTEM reveals that the degree of crystallinity of phospho-olivine phase is also preserved after the intercalation/deintercalation cycling.
 | ||
| Fig. 11 Ex situ bright field micrographs and SAED of NMP/C electrode cycled 5 times between 2.5 and 4.5 V at C/50 in a model lithium cell (left, first row); HRTEM image (right, first row); BF-STEM images and corresponding composition map of CKα1–2, MnKα1, PKα1, NaKα1–2 and OKα1. | ||
The distributions of Na, Mn, P and O elements in particles with nanometric dimensions are given in Fig. 11. In these particles, all Na, Mn, P and O are homogeneously distributed. It is worth mentioning that, in addition to Na, Mn, P and O, the peak due to F element is also resolved (not shown). This indicates presence of electrolyte on the NMP/C electrode surface. The important finding from STEM is related with the ratio of Na-to-Mn. The Na-to-Mn ratio in NMP/C is 1.04, which matches well with the chemical composition of phospho-olivine NaMnPO4. After the intercalation/deintercalation cycling, there is a decrease in the Na-to-Mn ratio, which varies for the different particles from 0.55 to 0.75. This decrease in the sodium content is in agreement with XRD data, according to which the phospho-olivine phase has a slightly shrunk unit cell. Combining the results from TEM and XRD analysis of the cycled electrodes, it appears that the olivine-phase participates in the reversible intercalation/deintercalation cycling without changing its structure and degree of crystallinity, accompanied by a loss of Na of about 25–45%.
The IR spectroscopy gives evidence for lithium intercalation in NaMnPO4. Fig. 12 compares the IR spectra of two electrode compositions: NMP/C before the cycling experiment and NMP/C after 5 cycles between 2.5 and 4.5 V. The IR spectra of its LiMnPO4 analogue, PVDF and lithium electrolyte are also shown. Two specific features can be distinguished. First, the comparative analysis shows that the IR spectra of electrode compositions before and after cycling are identical in the range, where PO4 vibrations appear. It is noticeable that, in this range, the IR spectra of both sodium and lithium phospho-olivines are also quite close (especially with respect to band positions). Second, the IR spectra of NaMnPO4 and LiMnPO4 analogues can be clearly distinguished in the range 450–490 cm−1, where Li–O translations appear. Close inspection of the IR spectrum of NMP/C after the intercalation/deintercalation cycling indicates the appearance of two weak bands at 496 and 453 cm−1, which have not been detected with NMP/C before the cycling. These bands coincide with those typical of Li–O translations observed for LiMnPO4. It should be taken into account that in this IR range both electrolyte and PVDF compound do not show any strong IR bands. Therefore, the two weak bands at 496 and 453 cm−1 are attributed to Li–O translations. The appearance of these bands in the IR spectrum of cycled electrode is a spectroscopic evidence for the intercalation of Li+ in NaMnPO4 after the cycling. The spectroscopic evidence is in a good agreement with CV data, where the lithium intercalation takes place at 4.38 and 3.94 V (Fig. 9). This allows attributing the second cathodic peak at 3.10 V to sodium intercalation reaction into NaxMnPO4 (Fig. 9). It is worth mentioning that Li+ and Na+ intercalation proceeds at two separate potentials, while their deintercalation processes are not distinguished in the anodic scan of the CV curve (Fig. 9). This is an interesting result, which deserves further investigation. Combining TEM, IR spectroscopic and CV data, the most probable chemical reaction is the reversible exchange of 0.5 mol of alkali ions between Li(0.25–0.45)Na(0.75–0.55)MnPO4 ↔ Li0.0Na0.5MnPO4. The formation of mixed lithium–sodium manganese phospho-olivine in discharged state is an unexpected result, which can serve as guidance to design novel polyanion-based electrode materials.
 | ||
| Fig. 12 IR spectra of NMP/C composite (a), NMP/C electrode prior to electrochemical reaction (b), NMP/C electrode after 5 charging–discharging cycles between 4.5 and 2.5 V at C/50 (c), LiMnPO4 reference (d). For the sake of comparison, PVDF and electrolyte LiPF6/EC/DMC are also given. | ||
To elucidate the nature of the electrochemical activity of NaMnPO4, we used EPR spectroscopy in order to monitor the changes in the oxidation state of manganese ions. Fig. 13 gives the EPR spectra of NMP/C electrodes before and after the cycling experiments. The EPR spectrum of non-cycled NMP/C consists of a single Lorentzian line. The g-factor is constant in the temperature range of 100–300 K, whereas the EPR line width (ΔHpp) increases considerably upon cooling (Fig. 14). The temperature dependence of the EPR signal intensity obeys the Curie–Weiss law between 180 and 300 K, where Weiss constant is −118 ± 6 K. These parameters can be assigned to Mn2+ ions coupled by strong exchange interactions. It is worth mentioning that the magnetic structure of NaMnPO4 has not been studied yet. However, we can compare the EPR data of NaMnPO4 with those for LiMnPO4 analogue, whose magnetic structure is well documented. LiMnPO4 displays antiferromagnetic long-range order below 34 K. The magnetic structure of LiMnPO4 includes in-plane super-exchange interactions between the Mn2+ ions via oxygen (bc-plane) and interlayer coupling through the phosphate groups along the [100] direction.39 The magnetic interactions between Mn2+ are responsible for the appearance of a single Lorentzian line in the EPR spectrum of LiMnPO4 at temperatures above that of the magnetic order.40–42 The comparison between EPR spectra of both sodium and lithium phases shows that their parameters are quite similar: g = 2.004 and ΔHpp = 20 mT for NaMnPO4 in comparison with g = 1.99 and ΔHpp = 18 mT for LiMnPO4.22 This fact confirms that exchange-coupled Mn2+ ions contribute to the origin of the EPR spectrum of NaMnPO4. In addition, the EPR parameters of NaMnPO4 in the fabricated electrode NMP/C before cycling and in the composite NaMnPO4/C in the form of powder are close (Fig. 14).
 | ||
| Fig. 13 EPR spectra at 290 and 120 K of NMP/C electrode before the electrochemical reaction (a and a′); NMP/C electrode after 5 cycles between 2.5 and 4.5 V (b and b′). The black curves are the experimental EPR spectra, while the red and blue curves correspond to the deconvoluted signal 1 and signal 2. | ||
 | ||
| Fig. 14 Temperature dependence of the g-factor (a) and EPR line width (b) for NMP/C powder (square symbols), NMP/C electrode before the electrochemical reaction (triangular symbols) and NMP/C electrode after 5 cycles between 2.5 and 4.5 V at C/50 (open and full circles for Si1 and Si2). | ||
After the cycling, the EPR spectrum undergoes a strong change. There are two overlapping signals (Fig. 13). The first signal (Si1) has a g-factor and line width similar to those before the cycling (Fig. 14). The Weiss constant value, determined from the temperature dependence of the signal intensity, is also similar: −134 ± 19 K. The similar EPR parameters reveal unambiguously that the first signal originates from the Mn2+ ions, localized in the unreacted NaMnPO4 phase. The second signal (Si2) is broader and it possesses higher g-factor in comparison with that of the first signal. In addition, both the g-factor and the line width increase upon cooling down. Although the g-value of Si2 falls within the typical range for Mn2+ ions, the temperature dependence of the g-factor reveals that the second signal is due to a complex spin system rather than to exchange-coupled Mn2+ ions. This can be explained, if we suppose that highly oxidized Mn ions appear together with Mn2+ ions and all these manganese ions contribute to the second broad signal. Taking into account the charging–discharging curves (Fig. 8), the redox couple Mn2+/Mn3+ is responsible for the alkaline ion intercalation in the phospho-olivine matrix. In contrast to Mn2+ ions, the Mn3+ ions are unlikely to be detectable by EPR in the X-band region (i.e. at 9.4 GHz) due to their strong spin-lattice relaxation and large zero-field splitting parameters. However, their effect on the EPR response of Mn2+ ions is easily observable. The same picture has been observed for several layered and spinel oxides (such as lithium manganese spinels, delithiated lithium nickel manganese oxides, sodium deficient manganese oxides), where Mn3+ and Mn4+ coexist.9,43 (It should be reminded that both Mn2+ and Mn4+ are to be observed easily by X-band EPR even at room temperature.) Therefore, the second signal is assigned to Mn2+ ions, whose parameters are perturbed by Mn3+ ions. This is consistent with the temperature dependence of the signal intensity: between 200 and 300 K, the signal intensity is changing following the Curie–Weiss law with a Weiss constant of −365 K. On the one hand; the EPR signal is related to irreversibility of the electrochemical reaction, as a result of which Mn3+ ions appear in addition to Mn2+ ones. On the other hand, this is a direct spectroscopic evidence for oxidation and reduction of manganese ions during the intercalation/deintercalation cycling.
Conclusions
Composite materials NaMnPO4/C are obtained by ball-milling of pristine NaMnPO4 with conductive carbon black additives Super C/65. The ball-milling leads to a fragmentation of the rod-like aggregates of NaMnPO4 into smaller species, accompanied by their dense covering with carbonaceous materials. The covering layer thickness varies between 2 and 10 nm. The carbonaceous skeleton is composed of disordered carbon structure with graphite-like medium-range order, which remains intact during ball-milling process. The phospho-olivine structure is also stable in regard to mechanical milling.The composite NaMnPO4/C material displays relatively good electrochemical activity, when used as a cathode in model lithium ion cells. Prior to the electrochemical test, the composite NaMnPO4/C does not react with the lithium electrolyte containing 1 M LiPF6 solution in ethylene carbonate and in dimethyl carbonate. During the first charging, almost all of Na+ ions are being extracted from NaMnPO4 at voltage plateau of 4.25 V, which coincides well with that typical of LiMnPO4 analogue. During the consecutive discharging, there is a competition between Li+ and Na+ intercalation into NaMnPO4, which is manifested by a sloping galvanostatic voltage curve. The CV curves reveal that Li+ and Na+ intercalation proceeds at two separate potentials, while their deintercalation processes are not distinguished in the anodic scan. The reversible intercalation/deintercalation cycling proceeds with an exchange of 0.5 mol alkaline ions, the most probable chemical reaction being Li(0.25–0.45)Na(0.75–0.55)MnPO4 ↔ Li0.0Na0.5MnPO4. The reversible intercalation of Li+ and Na+ takes place owing to the redox Mn2+/Mn3+ couple.
As far as we know, the reversible Li+ and Na+ intercalation into sodium manganese phospho-olivines is reported experimentally for the first time. The electrochemical performance of NaMnPO4 in lithium ion cells is not yet competitive compared to the well-known LiMnPO4 analogue. However, by optimizing the electrolyte solutions, it would be possible to stimulate the single ion intercalation instead of double ion intercalation. By both facilitating lithium intercalation and depressing sodium intercalation, one can expect to transform NaMnPO4 into LiMnPO4 during the first cycles. Thus, the in situ generated LiMnPO4 will further operate and, most probably, it will be characterized with improved electrochemical performance in comparison with conventional LiMnPO4 due to size-dependent effects. In this context, these first studies demonstrate clearly a significance of the sodium manganese phospho-olivine as an electrode material in alkaline ion batteries.
Acknowledgements
Authors are grateful to the financial support from European Social Fund (Grant BG051PO001-3.3.06-0050). The authors would like to thank Dr Tsvetina Dobrovolska (Institute of Physical Chemistry, Bulgarian Academy of Sciences) and Dr Mladen Mladenov (Institute of Electrochemistry and Energy Systems, Bulgarian Academy of Sciences) for CV experiments.References
- Z. Gong and Y. Yang, Energy Environ. Sci., 2011, 4, 3223 CAS
.
- C. Masquelier and L. Croguennec, Chem. Rev., 2013, 113, 6552 CrossRef CAS PubMed
- D. Choi, D. Wang, I.-T. Bae, J. Xiao, Z. Nie, W. Wang, V. V. Viswanathan, Y. J. Lee, J.-G. Zhang, G. L. Graff, Z. Yang and J. Liu, Nano Lett., 2010, 10, 2799 CrossRef CAS PubMed
- C. Delacourt, L. Laffont, R. Bouchet, C. Wurm, J.-B. Leriche, M. Morcrette, J.-M. Tarascon and C. J. Masquelier, J. Electrochem. Soc., 2005, 152, A913 CrossRef CAS PubMed
- A. Morgan, A. van der Ven and G. Ceder, Electrochem. Solid-State Lett., 2004, 7, A30 CrossRef PubMed
- J. Wang and X. Sun, Energy Environ. Sci., 2012, 5, 5163 CAS
- L. Dimesso, C. Förster, W. Jaegermann, J. P. Khanderi, H. Tempel, A. Popp, J. Engstler, J. J. Schneider, A. Sarapulova, D. Mikhailova, L. A. Schmitt, S. Oswald and H. Ehrenberg, Chem. Soc. Rev., 2012, 41, 5068 RSC
- C. Delacourt, P. Poizot, M. Morcrette, J.-M. Tarascon and C. Masquelier, Chem. Mater., 2004, 16, 93 CrossRef CAS
- M. Yoncheva, R. Stoyanova, E. Zhecheva, E. Kuzmanova, M. Sendova-Vassileva, D. Nihtianova, D. Carlier, M. Guignard and C. Delmas, J. Mater. Chem., 2013, 22, 23418 RSC
- M. Kalapsazova, R. Stoyanova and E. Zhecheva, J. Solid State Electrochem., 2014, 18, 2343 CrossRef CAS
- M. Kalapsazova, R. Stoyanova, E. Zhecheva, G. Tyuliev and D. Nihtianova, J. Mater. Chem. A, 2014, 2, 19383 CAS
- M. Kalapsazova, G. F. Ortiz, J. L. Tirado, O. Dolotko, E. Zhecheva, D. Nihtianova, L. Mihaylov and R. Stoyanova, ChemPlusChem DOI:10.1002/cplu.201500215.
- Y. Tang, D. Sun, H. Wang, X. Huang, H. Zhang, S. Liu and Y. Liu, RSC Adv., 2014, 4, 8328–8334 RSC
- H. Wang, S. Liu, Y. Ren, W. Wang and A. Tang, Energy Environ. Sci., 2012, 5, 6173 CAS
- S. Bach, J. P. Pereira-Ramos and P. Willmann, Electrochim. Acta, 2006, 52, 504 CrossRef CAS PubMed
- M. Shirpour, J. Cabana and M. Doeff, Energy Environ. Sci., 2013, 6, 2538 CAS
- M. Gnanavel, V. Pralong, O. I. Lebedev, V. Caignaert, P. Bazin and B. Raveau, Chem. Mater., 2014, 26, 4521 CrossRef CAS
- P. B. Moore, Am. Mineral., 1972, 57, 1333 CAS
- J. Moring and E. Kostiner, J. Solid State Chem., 1986, 61, 379 CrossRef CAS
- C. A. J. Fisher and M. Saiful Islam, J. Mater. Chem., 2008, 18, 1209 RSC
- S. P. Ong, V. L. Chevrier, G. Hautier, A. Jain, C. Moore, S. Kim, X. Ma and G. Ceder, Energy Environ. Sci., 2011, 4, 3680 CAS
- V. Koleva, E. Zhecheva and R. Stoyanova, Dalton Trans., 2011, 40, 7385 RSC
- V. Koleva, T. Boyadzhieva, E. Zhecheva, D. Nihtianova, S. Simova, G. Tyuliev and R. Stoyanova, CrystEngComm, 2013, 15, 9080 RSC
- V. Koleva, R. Stoyanova, E. Zhecheva and D. Nihtianova, CrystEngComm, 2014, 16, 7515 RSC
- J. Rodrıguez-Carvajal, Commission on Powder Diffraction Newsletter, 2001, vol. 26, p. 12 Search PubMed
- J. Rodriguez-Carvajal, Abstracts of the Satellite Meeting on Powder Diffraction of the XV Congress of the IUCr, Toulouse, 1990 Search PubMed
- H. Fang, Z. Pan, L. Li, Y. Yang, G. Yan, G. Li and S. Wei, Electrochem. Commun., 2008, 10, 1071 CrossRef CAS PubMed
- K. T. Lee, T. N. Ramesh, F. Nan, G. Botton and L. F. Nazar, Chem. Mater., 2011, 23, 3593 CrossRef CAS
- Y. Liu, C. Pan and J. Wang, J. Mater. Sci., 2004, 39, 1091 CrossRef CAS
- F. Tuinstra and J. L. Koenig, J. Chem. Phys., 1970, 53, 1126 CrossRef CAS PubMed
- J. Robertson, J. Non-Cryst. Solids, 2002, 299–302, 798 CrossRef CAS
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095 CrossRef CAS
- K. Xu, Chem. Rev., 2004, 104, 4303 CrossRef CAS
- N. S. Norberg and R. Kostecki, J. Electrochem. Soc., 2012, 159, A1431 CrossRef CAS PubMed
- K. Edström, T. Gustafsson and J. O. Thomas, Electrochim. Acta, 2004, 50, 397 CrossRef PubMed
- J. W. Lee, M. S. Park, B. Anass, J. H. Park, M. S. Paik and S. G. Doo, Electrochim. Acta, 2010, 55, 4162 CrossRef CAS PubMed
- D. Choi, J. Xiao, Y. J. Choi, J. S. Hardy, M. Vijayakumar, M. S. Bhuvaneswari, J. Liu, W. Xu, W. Wang, Z. Yang, G. L. Graff and J.-G. Zhang, Energy Environ. Sci., 2011, 4, 4560 CAS
- K. Wang, Y. Wang, C. Wang and Y. Xia, Electrochim. Acta, 2014, 146, 8 CrossRef CAS PubMed
- J. Li, W. Tian, Y. Chen, J. L. Zarestky, J. W. Lynn and D. Vaknin, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 144410 CrossRef
- D. Arčon, A. Zorko, P. Cevc, R. Dominko, M. Bele, J. Jamnik, Z. Jagličić and I. Golosovsky, J. Phys. Chem. Solids, 2004, 65, 1773 CrossRef PubMed
- N. Wizent, G. Behr, F. Lipps, I. Hellmann, R. Klingeler, V. Kataev, W. Löser, N. Sato and B. Büchner, J. Cryst. Growth, 2009, 311, 1273 CrossRef CAS PubMed
- D. Arčon, A. Zorko, R. Dominko and Z. Jagličić, J. Phys.: Condens. Matter, 2004, 16, 5531 CrossRef
-
(a) R. Stoyanova, E. Zhecheva and S. Vassilev, J. Solid State Chem., 2006, 179, 378 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2015 |