
A facile and general synthesis strategy to doped TiO2 nanoaggregates with a mesoporous structure and comparable property†
Gang Cheng*a,
Feifan Xua,
Florian J. Stadlerb and
Rong Chen*a
aSchool of Chemistry and Environmental Engineering, Wuhan Institute of Technology, Xiongchu Avenue, Wuhan 430073, PR China. E-mail: gchenglab@163.com; rchenhku@hotmail.com
bNanshan District Key Lab for Biopolymers and Safety Evaluation, College of Materials Science and Engineering, Shenzhen University, Shenzhen 518060, PR China
First published on 21st July 2015
Abstract
A facile and general route was explored to fabricate porous metal-doped TiO2 nanoaggregates from a titanium glycolate precursor and metal salts, in which the metal salts not only provide the dopant precursor but also favor the formation of a porous structure. The as-prepared Co–TiO2 and Ni–TiO2 nanoaggregates showed higher photocatalytic performance than pure TiO2 nanoparticles, P25, and Zn–TiO2 nanoaggregates, indicating that doping with Co or Ni is an effective approach to improve the photocatalytic efficiency.
Titanium dioxide (TiO2) has been recognized as one of the most promising semiconductor materials for energy and environmental applications due to its environmental friendliness and low cost.1–3 However, a large band gap, low electric conductivity, and the poor photochemical, electrochemical, and electronic properties of TiO2, resulting from the intrinsic crystal structure, largely limit its efficient applications such as photocatalysis, water splitting, dye-sensitized solar cells, and lithium-ion batteries.4,5 Accordingly, many recent works have been conducted to solve the aforementioned intrinsic problems.
Element doping is an established, significant technique to effectively modify the electrical conductivity and optoelectronic properties of materials. Doping TiO2 nanocrystals with a second element can narrow the band gap of TiO2 via electronic coupling effects between doping ions and TiO2 or introduce some impurity energy level just above the valence band or just below the conduction band of TiO2 to extend the visible absorption.6 In addition, the as-produced impurity energy level resulting from including dopants can accelerate electron transport or prolong lifetime of photogenerated hole–electron pairs, leading to an enhanced short circuit current density or a high photocatalytic activity. For instance, Doping TiO2 nanoparticles with Zn7 or Sn8 were found to improve the photocurrent and, thereby, enhance the energy conversion efficiency of dye-sensitized solar cells when used as the photoanode materials. At the same time, various different kinds of dopants, including nonmetals (B, C, N, F, P, S, etc.), transition metals (Fe, Cu, Mn, Co, etc.), noble metals (Au, Ag, Pd, Pt, etc.), and rare earths (La, Ce, Er, Y, Eu, etc.), have been widely tested to design doping materials for TiO2 to improve the photocatalytic capability.6,9–12 Therefore, it is of significance to explore an effective fabrication approach to doped TiO2 materials.
Recently, synthesis of porous TiO2 nanoaggregates has attracted great interest owing to the benefits of the porous structure in enhancing the adsorption capacity of the dyes in dye-sensitized solar cells system, and promoting the diffusion of the reactants and products as well as in facilitating access to the reactive sites on the surface of the photocatalysts in photocatalysis system.13–18 For example, Xiao et al. prepared cerium-doped TiO2 mesoporous nanofibers, which show improved visible light photocatalysis performance for the degradation of rhodamine B.19 However, most of the synthesis methods involved the use of template, the employ of long reaction time and high temperature, and the control of complex process.
Herein, porous TiO2 nanoparticles aggregates with transition metal doping are obtained by a simple, economical, and environmentally benign method from titanium glycolate nanoprecursors and transition metal salts. Compared to previous methods for Co20 or Zn7 or Ni21-doped TiO2, this strategy avoids the process of calcinations treatment with high temperature. In particular, this synthesis provides a general approach, and does not involve any surfactant or toxic materials. By controlling the kind of metal salts, the structure of the products can be tailored from well-crystallized and dispersed TiO2 nanoparticles to porous TiO2 nanoaggregates with Co, Zn, or Ni doping.
Fig. 1a and b show the typical SEM and TEM images of titanium glycolate precursor synthesized according to the previous work.22,23 It was observed that the precursor shows the morphology of aggregated particles with the size of about 100–200 nm. Once putting those precursor powders into the autoclave and performing the hydrothermal treatment at 180 °C for 6 h, as shown in Fig. 1c and d, aggregates consisting of many small TiO2 nanocrystals were obtained. A representative HRTEM image of a single TiO2 nanoparticle shown as the inset in Fig. 1d displays well-resolved lattice fringe, and further confirms that the as-synthesized TiO2 is well-crystallized. The plane spacing of ∼0.35 nm corresponds to the lattice plane of (101) in anatase TiO2.
 | ||
| Fig. 1 SEM (a and c) and TEM (b and d) images of the as-synthesized products: (a and b) titanium glycolate precursor, (c and d) titanium oxide. Inset of (d) is a HRTEM image of a single TiO2 nanocrystal. | ||
The metal doping into the TiO2 aggregates were confirmed by XRD and XPS analysis, Raman spectroscopy, and diffuse reflectance UV-vis spectra measurements. Fig. 2a shows the XRD patterns of pure and doped TiO2 products. The diffraction peaks are assigned well with standard XRD pattern (JCPDS no. 71-1187) of pure tetragonal anatase TiO2, which indicates that the doping of metal does not change the anatase crystal structure of TiO2. It was observed that the (101) diffraction peak in all the samples is the strongest, suggesting the predominant crystal growth along the same plane of anatase TiO2.24 Fig. 2b shows the enlarged (101) and (004) diffraction peaks of pure and doped TiO2 products. It can be seen that there is a small shift to a higher 2θ angle value of the XRD diffraction peaks for doped TiO2 products as compared with those of pure TiO2. This change indicates the lattice deformation of doped TiO2 products.
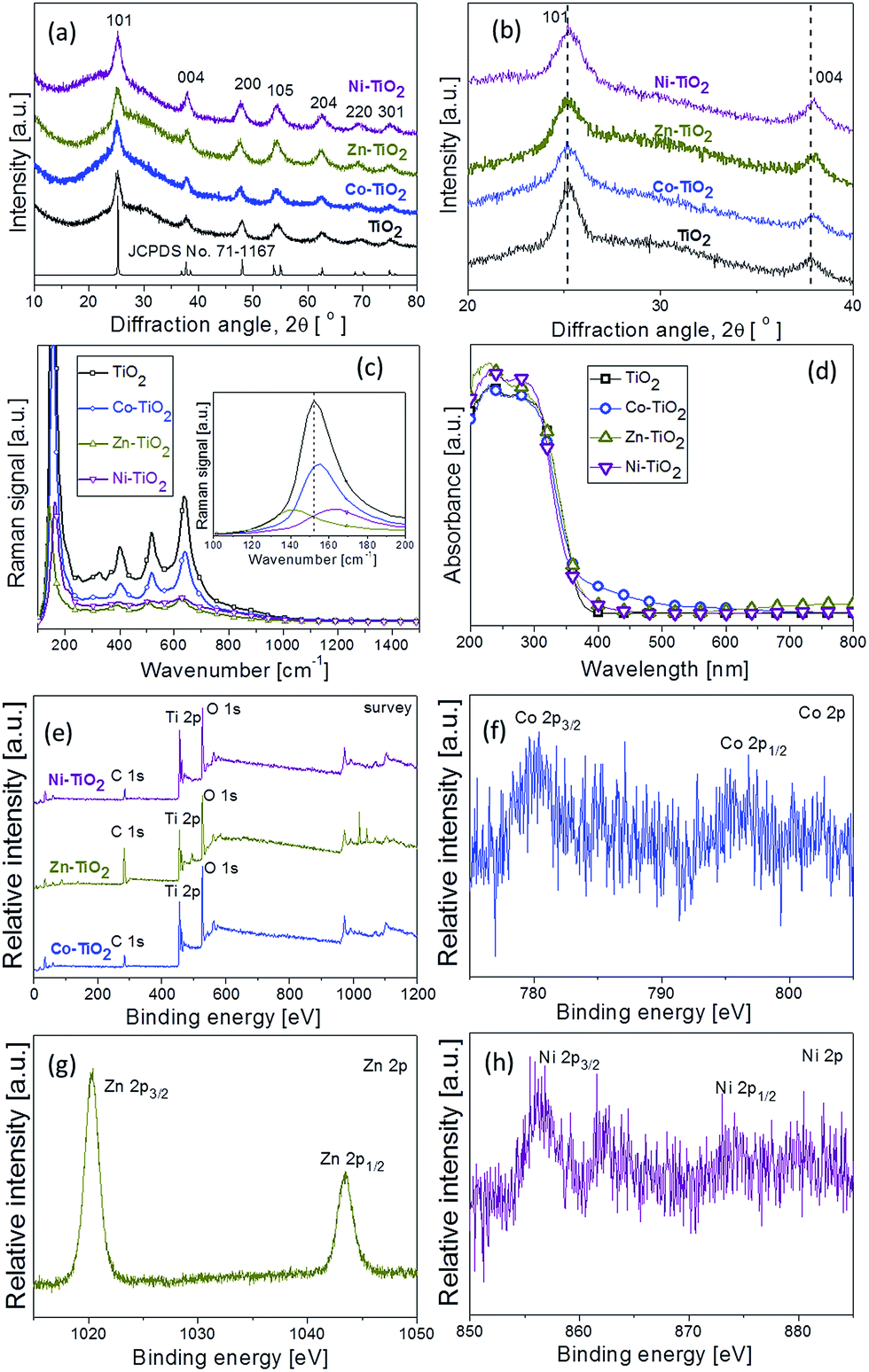 | ||
| Fig. 2 (a) XRD pattern, (b) the enlarged (101) and (004) diffraction peaks, (c) Raman spectra, (d) UV-vis diffuse reflectance spectrum, (e) XPS survey spectra of pure and doped TiO2 products; (f) Co binding-energy peak; (g) Zn binding-energy peak, and (h) Ni-binding-energy peak. | ||
Raman scattering is a very useful technique to analyse the microstructural change of nanocrystalline materials. Fig. 2c shows the typical Raman spectra of pure and doped TiO2 products. In pure TiO2, the prominent Raman modes including Eg(1), B1g(1), A1g + B1g(2), and Eg(2) are observed, assigning well with four characteristic bands at about 152, 400, 516, and 637 cm−1, respectively.25 The typical anatase TiO2 phase is found, which was also consistent with the XRD results. The low-frequency modes at Eg of 152 cm−1 and those at B1g of 400 cm−1 are O–Ti–O bending-type vibrations, and the modes at Eg of 637 cm−1 and those at A1g + B1g of 516 cm−1 are the Ti–O bond stretching type vibrations.26,27 As shown in inset of Fig. 2c, an obvious shift in Raman scattering is observed for the Eg(1)-peak, the strongest mode in the spectrum. Particularly, the Eg(1) Raman mode of Co-doped TiO2 product is slightly shifted to 155 cm−1 from 152 cm−1 for pure TiO2 product, while the one of Zn- and Ni-doped TiO2 product is significantly shifted to 144 and 162 cm−1, respectively, implying the incorporation of Co, Zn, and Ni into the TiO2 lattice.26,28
The corresponding UV-vis diffuse reflectance spectra of the pure TiO2 and doped TiO2 samples are shown in Fig. 2d. Similar results were also observed by Swati et al.29 for Bi3+-doped and Zhang et al.30 for Ce3+-doped TiO2 nanomaterials, respectively. All doped TiO2 samples exhibit an important visible absorption beyond 400 nm, indicating that the metal doping changes the optical absorption ability of TiO2 products. The absorption edges of doped TiO2 samples showed regular and slight red-shifts compared to pure TiO2 products. Fig. S1† shows the plot of (αhν)1/2 versus photon energy (hν) for the as-synthesized pure TiO2 and doped TiO2 samples, where α, n, and h are the absorption coefficient, light frequency, and constant, respectively. The band gap energy (Eg) of Co–TiO2, Zn–TiO2,and Ni–TiO2 estimated from a plot of (αhν)1/2 versus photon energy (hν) was about 2.97, 3.08, and 3.10 eV, respectively, which shows a tendency of narrowing comparing to pure TiO2 (Eg = 3.16 eV). These results indicate that the preparation of doped TiO2 samples from titanium glycolate precursor and metal salts via such a hydrothermal process is successful.
The X-ray photoelectron spectroscopy (XPS) in Fig. 2e shows the intrinsic characteristic and the chemical states of the compositional elements of the pure TiO2 and doped TiO2 samples. All spectra have been calibrated with the adventitious C 1s peak at 284.2 eV. Fig. S2† shows the Ti2p XPS spectra of the pure TiO2 and doped TiO2 samples. The two peaks of the Ti2p spectra for both of Co–TiO2 and Zn–TiO2 show a slight negative shift, while for Ni–TiO2 has a shift to lower binding energy comparing with the spectra for pure TiO2, which is likely attributed to the interaction between titanium atoms, oxygen atoms and metal atoms.31,32 The high-resolution scans of Co 2p, Zn 2p, and Ni 2p are shown in Fig. 2f–h. As shown in Fig. 2f, the binding energies of Co 2p3/2 and Co 2p1/2 peaks are located at 781.2 and 796.5 eV, respectively, indicating Co2+ in anatase phase structure.28,33 There are also two symmetric peaks in Fig. 2g. The first one located at 1020.2 eV is ascribed to Zn 2p3/2 and the other one centered at 1043.4 eV corresponds to Zn 2p1/2,34,35 which suggests the Zn(II) chemical state in Zn–TiO2 products. In Fig. 2h, the binding energies observed at 856.4 and 874.4 eV for pure Ni–TiO2 can be ascribed to Ni(II) 2p3/2 and Ni(II) 2p1/2, respectively, matching the reference values.36 The above XPS analysis revealed that all the doped metals in the TiO2 products are in the +2 oxidation state.
The morphology and composition of doped TiO2 products were characterized by SEM, EDX, TEM, HRTEM, and SAED analysis. Fig. 3a, c, and e show the SEM images of Co–TiO2, Zn–TiO2, Ni–TiO2, respectively. It can be seen that all the doped TiO2 products show significant differences to pure TiO2 product displayed in Fig. 3c. Insteading of well-dispersed nanoparticles, nanoparticle aggregates of Co–TiO2, Zn–TiO2, Ni–TiO2 were obtained. The EDX spectrum was also used to analyze the elemental compositions of the as-synthesized doped TiO2 nanoaggregates. As shown in Fig. 3b, the Ti, O, and Co signals observed indicated that the samples were composed of Ti, O, and Co, which was in good agreement with the XPS result of Co–TiO2. Fig. 3d and f also suggest the as-prepared Zn–TiO2 and Ni–TiO2 consist of Ti, O, Zn and Ti, O, Ni, respectively.
 | ||
| Fig. 3 (a, c, and e) SEM images and (b, d, and f) EDX spectra of doped TiO2 products: (a and b) Co–TiO2; (c and d) Zn–TiO2; (e and f) Ni–TiO2. | ||
The structure of the doped TiO2 nanoaggregates was further investigated by TEM and HRTEM images. As displayed in Fig. 4a, c, and e, the Co–TiO2, Zn–TiO2, Ni–TiO2 products are in the form of spherical structure, comprising of nanoaggregates assembled from many small nanocrystals. The corresponding HRTEM images (Fig. 4b, d, and f) show that all the doped materials are composed of fine nanoparticles, leading to the formation of a mesoporous structure. Further, it can be seen from the clear lattice fringes of a single TiO2 nanoparticle with a d-spacing of ∼0.35 nm, corresponding to the (101) lattice plane, that the TiO2 nanoparticle aggregates are well-crystallized and have a high order of crystallinity. The insets of Fig. 4a, c, and e show the corresponding SAED pattern, taken at a selected area of the TiO2 nanoparticle aggregates, exhibiting several diffraction ring patterns, thus, indicating a polycrystalline structure.
 | ||
| Fig. 4 (a, c, and e) TEM and (b, d, and f) HRTEM images of doped TiO2 products: (a and b) Co–TiO2; (c and d) Zn–TiO2; (e and f) Ni–TiO2. Inset of (a, c, and e) is the corresponding SAED pattern. | ||
The above results demonstrate that the introduction of metal salts not only provides the dopant precursor but also lead to the formation of aggregated structure. In previous work, we have systematically investigated the formation of TiO2@RGO and TiO2@C nanohybrids from titanium glycolate precursor,22,23 in which it was proven that the water could penetrate into the spherical titanium glycolate precursor, resulting in the formation of a porous or nanoparticulate structure. In the present work, it is proposed that the metal salt could adsorb on the surface of the titanium glycolate precursor and to some extent inhibit the penetration of water, leading to the fabrication of such porous structure.
The Brunauer–Emmett–Teller (BET) specific surface areas and porosity of the doped TiO2 nanoaggregates were studied by nitrogen adsorption and desorption measurements. Fig. 5a shows the nitrogen adsorption–desorption isotherms of the as-synthesized doped TiO2 nanoaggregates. It was observed that all the curves display type II adsorption–desorption isotherms with a distinct hysteresis loop in the range of 0.5–1.0p/p0, being typical of porous materials, which is in good agreement with TEM results. The BET specific surface area of porous Co–TiO2, Zn–TiO2, Ni–TiO2 product was found to be 208.9, 247.9, 193.4 m2 g−1, respectively. Fig. 5b displays the pore size distribution of the doped TiO2 nanoaggregates. It was found that the Zn–TiO2 nanoaggregates contained small mesopores with an average diameter of 3.2 nm, determined from the Barret–Joyner–Halenda (BJH) method. The formation of such mesopores may be ascribed to the aggregation of nanoparticles during the crystal growth process. Using the same method, the Co–TiO2 nanoaggregates contained a bimodal distribution of small mesopores with average diameters of 4.6 and 8.8 nm, while the Ni–TiO2 product have bimodally distributed mesopores with diameters of 4.9 and 7.6 nm. The result suggests that the as-prepared TiO2 nanoaggregates doping with different metals have typical mesoporous structure.
 | ||
| Fig. 5 (a) Nitrogen adsorption–desorption isotherms and (b) pore size distribution of doped TiO2 products; (c) change of rhodamine B concentration (Ct/C0) with irradiation time over different photocatalysts and photolysis of rhodamine B from a 500 W Xe lamp with a 420 nm cutoff filter; (d) rhodamine B removal rate upon exposure to different samples; (e) photocurrents of doped TiO2 products under a 500 W Xe lamp light irradiation at a constant potential; (f) PL spectra of doped TiO2 products recorded at room temperature with the excitation wavelength of 200 nm. | ||
Furthermore, the photocatalytic behaviour of the doped TiO2 nanoaggregates for the degradation of rhodamine B dyes under visible-light irradiation was investigated. As a benchmark, the direct photolysis of rhodamine B, the performances of pure TiO2, and of commercial photocatalyst P25 were also investigated. Those experiments were performed in a way that the solution of the photocatalyst and RhB was irradiated for 270 min, after 30 min soaking time to get the establishment of adsorption–desorption equilibrium. As shown in Fig. S3,† the as-synthesized doped TiO2 nanoaggregates display higher adsorption capacity than pure TiO2, which might be due to the unique mesoporous structure and specific surface areas. Fig. 5c depicts the variations of rhodamine B concentration (Ct/C0) with irradiation time over different photocatalysts, where C0 was the initial concentration of rhodamine B and Ct was the rhodamine B concentration at time t. It was clearly observed that the rhodamine B concentration changed slightly with the increase of irradiation time in the absence of photocatalysts, while pure TiO2 nanoparticles, P25, and the doped TiO2 nanoaggregates exhibited different photocatalytic activities for the rhodamine B degradation. Fig. 5d shows rhodamine B removal rate upon different samples, which was found to be much higher for both Co–TiO2 and Ni–TiO2 nanoaggregates than that of pure TiO2 nanoparticles and P25, while Zn–TiO2 nanoaggregates show an inferior performance, indicating that Co- or Ni-doping rather than Zn-doping improve the photocatalytic activity of TiO2 materials.
Photocurrent response and PL emission determination were widely used to investigate separation efficiency and recombination rate of the photo induced electron and holes in the photocatalysts during the photocatalysis reaction,37,38 respectively. As shown in Fig. 5e, a fast and uniform photocurrent response is observed upon both the light-on and light-off in the doped TiO2 nanoaggregates samples. The photocurrent produced by the Ni–TiO2 nanoaggregate sample is much higher than that of the Co–TiO2 and Zn–TiO2 samples. This reflects the higher separation efficiency of photoinduced electrons and holes in the Ni–TiO2 nanoaggregate. Fig. 5f demonstrates that the tested photocatalysts showed the main PL emission peaks at similar position of 456 nm, which can be ascribed to the recombination of electron and holes. The Ni–TiO2 nanoaggregate showed a diminished intensity in comparison to Co–TiO2 and Zn–TiO2, indicating that the introduction of Ni would inhibit the recombination of the carriers generated in TiO2. For pure TiO2, it can be observed that the TiO2 nanoparticles show both high photocurrent and high PL intensity, demonstrating that a highly efficient generation of photoinduced carriers and a fast recombination rate of photogenerated electron and hole. Based on the existing results, it was concluded that the enhancement of photocatalytic activity of Ni–TiO2 nanoaggregate photocatalysts was due to the higher separation efficiency of photogenerated carriers and lower recombination efficiency of photoinduced electron–hole pairs. Further study on the surface structure tailoring and photocatalytic activity optimization of doped TiO2 nanomaterials by different dopant concentrations as well as the related photocatalytic mechanism is still in progress.
Conclusions
In summary, a facile and general synthesis strategy was explored to prepare porous metal-doped TiO2 nanoaggregates from titanium glycolate precursor and metal salts, in which the metal salts not only provided the dopant precursor but also favor for the formation of a mesoporous structure. The composition of the as-synthesized Co–TiO2, Zn–TiO2, Ni–TiO2 products were characterized by XRD, XPS, and EDX analysis. SEM, TEM, HRTEM images, and BET surface areas measurement showed that the as-prepared doped TiO2 products had porous structure, and were composed of many small TiO2 nanocrystals. The UV-vis diffuse reflectance spectra evaluation showed that the involving of metal dopant enhance the visible light absorption of TiO2 materials. The photocatalytic behavior of the doped TiO2 products was investigated by photocatalytic degradation of rhodamine B under visible light irradiation. Both Co–TiO2 and Ni–TiO2 nanoaggregates showed much higher photocatalytic performance than pure TiO2 nanoparticles and P25, while Zn–TiO2 nanoaggregates worsen it, indicating the doping of Co or Ni is an effective approach to improve the photocatalytic efficiency in the present work. It is expected that the as-synthesized doped mesoporous materials have potential application in energy conversion and storage and the synthesis method provides a new approach for the preparation of others doped functional materials.Experimental section
All of the chemical reagents were of analytical grade and were used directly without further purification. Deionized water was used in all experiments.Doped porous TiO2 nanoparticles aggregates were synthesized by reacting titanium glycolate powder precursor with metal salt in aqueous solution. The titaniumglycolate precursor was prepared according to the previously reported procedure.22,23,39 In a typical synthesis, 2 mL tetrabutoxytitanium was added to 50 mL ethylene glycol and magnetically stirred for 2 h at 60 °C in a water bath. After cooling to room temperature, the mixture was immediately poured into a solvent mixture solution which contains 150 mL acetone and 30 mL deionized water under vigorous stirring, and then the above mixture was further stirred for about 1.5 hours. Finally, the white precipitate was collected and washed with deionized water and ethanol five times by centrifugation, and then dried at 60 °C overnight to obtain titanium glycolate powder. For the synthesis of doped TiO2 nanoaggregates, 0.1 g of the as-synthesized titanium glycolate precursor and 1 mmol of one of the following metal salts (CoSO4·7H2O, Zn(CH3COO)2, (NH4)2Ni(SO4)2·6H2O) were sonicated in 30 mL of water until to form a homogeneous colloidal solution. Then the solution was transferred to a 50 mL Teflon-lined autoclave and hydrothermally treated at 180 °C for 6 h. After cooling to room temperature, the precipitates were collected and washed eight times with de-ionized water and ethanol via centrifugation, and finally dried for 12 h at 80 °C to get powder products. The nanoparticles synthesized in the presence of CoSO4·7H2O, Zn(CH3COO)2, and (NH4)2Ni(SO4)2·6H2O in the reaction system are referred to as Co–TiO2, Zn–TiO2, and Ni–TiO2, respectively.
The crystal phase, morphology, and structure of the synthesized precursor powder, pure, and doped TiO2 products were characterized by powder X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), scanning electron microscopy (SEM), transmission electron microscopy (TEM), selected area electron diffraction (SAED), UV-vis diffuse reflectance spectrum (UV-DRS), and Raman spectroscopy. XRD was performed on a high-performance X-ray diffractometer with CuKα radiation (λ = 1.54 Å, PANalytical), at 2θ, from 10° to 90°, and with a scanning rate of 0.03° s−1. XPS was carried out using an AXIS-NOVA CJ109 (Kratos Inc.) in the range of 0–800 eV to evaluate the surface composition of the products. The binding energies were calibrated from the C1s photoelectron peak (284.2 eV). SEM images were taken on a field-emission electron microscope (ZEISS, SUPRA 40VP) operating at an acceleration voltage of 3 kV. TEM images and SAED patterns were recorded on a JEOL 2010 electron microscope at an accelerating voltage of 200 kV. A UV-2550 UV-vis spectrophotometer (Shimadzu) was used to measure the UV-vis diffuse reflectance spectrum of the as-synthesized materials. The Raman spectrum of the as-synthesized product was carried out by a Raman microscope (Renishaw). Nitrogen adsorption was performed with Micromeritics ASAP 2020 (USA) to determine the Brunauer− Emmett−Teller (BET) specific surface area of the as-synthesized products. Photoluminescence spectra (PL) were detected with a HITACHI F4600 fluorescence spectrophotometer.
The photocatalytic activities of the as-synthesized products were evaluated by the degradation of rhodamine B under visible light irradiation of a 500 W Xe lamp with a 420 nm cutoff filter. In a typical test, 0.05 g photocatalyst were added into 100 mL rhodamine B solution with a concentration of 1.0 × 10−5 mol L−1. Prior to irradiation, the suspensions were stirred in the dark for 30 min to reach adsorption–desorption equilibrium. After that, the solution was exposed to visible light irradiation under magnetic stirring. At each irradiation time interval, 3 mL of the suspensions were collected and the slurry samples, including the photocatalyst and rhodamine B solution were centrifuged to remove the photocatalyst particles. The solutions were analyzed by a Shimadzu UV2800 spectrophotometer, and the characteristic absorption of rhodamine B at 554 nm was used to monitor the photocatalytic degradation. All of the measurements were carried out at room temperature.
The electrochemical measurement were conducted on a CHI 660E electrochemical system (Shanghai, China) using a standard three-electrode cell with a working electrode, a platinum wire counter electrode, a standard calomel electrode (SCE) reference electrode and 0.5 M Na2SO4 was used as the electrolyte. The working electrode was prepared according to the following process: 20 mg of as-prepared sample was mixed with 1 mL of DMF and 0.01 mL of Nafion solution (5%, DuPont) to form a homogeneous ink. Then 0.1 ml of the catalyst ink was dip coated on a 10 mm × 10 mm indium–tin oxide (ITO) glass electrode. The as-prepared electrode was dried at 60 °C overnight. Photocurrent responses of the photocatalyst as light on and off were measured at open-circuit potential, with simulated light irradiation provided by a 500 W Xe lamp.
Contributions
GC designed the project, organized the entire research, prepared the materials, and wrote the manuscript. FX did the photocatalysis, photocurrent, and PL measurements. FJS and RC were involved in interpretation of the result and commented on the manuscript. All authors reviewed the manuscript.Acknowledgements
GC would like to thank the Key Program of Hubei Provincial Department of Education (D20151504). RC Would like to thank High-Tech Industry Technology Innovation Team Training Program of Wuhan Science and Technology Bureau (2014070504020243). FJS would like to acknowledge Nanshan District Key Lab for Biopolymers and Safety Evaluation (No. KC2014ZDZJ0001A) and Shenzhen City (JCYJ20140509172719311). The help of Mr Fan Tian for the discussion of the results is also appreciated.Notes and references
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- Z. Sun, J. H. Kim, Y. Zhao, F. Bijarbooneh, V. Malgras, Y. Lee, Y.-M. Kang and S. X. Dou, J. Am. Chem. Soc., 2011, 133, 19314–19317 CrossRef CAS PubMed.
- S. Ameen, M. S. Akhtar, H.-K. Seo and H.-S. Shin, Langmuir, 2014, 30, 12786–12794 CrossRef CAS PubMed.
- L.-L. Long, A.-Y. Zhang, J. Yang, X. Zhang and H.-Q. Yu, ACS Appl. Mater. Interfaces, 2014, 6, 16712–16720 CAS.
- S. Ameen, M. S. Akhtar, H.-K. Seo and H.-S. Shin, CrystEngComm, 2014, 16, 3020–3028 RSC.
- X. Wu, S. Yin, Q. Dong, C. Guo, T. Kimura, J.-i. Matsushita and T. Sato, J. Phys. Chem. C, 2013, 117, 8345–8352 CAS.
- F. Huang, Q. Li, G. J. Thorogood, Y.-B. Cheng and R. A. Caruso, J. Mater. Chem., 2012, 22, 17128–17132 RSC.
- Y. Duan, N. Fu, Q. Liu, Y. Fang, X. Zhou, J. Zhang and Y. Lin, J. Phys. Chem. C, 2012, 116, 8888–8893 CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- R. Asahi, T. Morikawa, H. Irie and T. Ohwaki, Chem. Rev., 2014, 114, 9824–9852 CrossRef CAS PubMed.
- C. Wang, Z. Chen, H. Jin, C. Cao, J. Li and Z. Mi, J. Mater. Chem. A, 2014, 2, 17820–17827 CAS.
- S. Bingham and W. A. Daoud, J. Mater. Chem., 2011, 21, 2041–2050 RSC.
- D. K. Roh, J. A. Seo, W. S. Chi, J. K. Koh and J. H. Kim, J. Mater. Chem., 2012, 22, 11079–11085 RSC.
- H. J. Koo, Y. J. Kim, Y. H. Lee, W. I. Lee, K. Kim and N. G. Park, Adv. Mater., 2008, 20, 195–199 CrossRef CAS PubMed.
- W. Zhou and H. Fu, ChemCatChem, 2013, 5, 885–894 CrossRef CAS PubMed.
- W. Cheng, W. Li, S. Ji, D. Yang, L. Wang, X. Qu, C. Zhang, F. Liang, Q. Wang, J. Li and Z. Yang, J. Mater. Chem. A, 2013, 1, 8023–8028 CAS.
- A. A. Ismail and D. W. Bahnemann, J. Mater. Chem., 2011, 21, 11686–11707 RSC.
- W. Zhou, W. Li, J.-Q. Wang, Y. Qu, Y. Yang, Y. Xie, K. Zhang, L. Wang, H. Fu and D. Zhao, J. Am. Chem. Soc., 2014, 136, 9280–9283 CrossRef CAS PubMed.
- G. Xiao, X. Huang, X. Liao and B. Shi, J. Phys. Chem. C, 2013, 117, 9739–9746 CAS.
- K. Das, S. N. Sharma, M. Kumar and S. K. De, J. Phys. Chem. C, 2009, 113, 14783–14792 CAS.
- L. Zhu, D. Zhang, Y. Wang, C. Feng, J. Zhou, C. Liu and S. Ruan, RSC Adv., 2015, 5, 28105–28110 RSC.
- G. Cheng, M. S. Akhtar, O. B. Yang and F. J. Stadler, ACS Appl. Mater. Interfaces, 2013, 5, 6635–6642 CAS.
- G. Cheng and F. J. Stadler, J. Colloid Interface Sci., 2015, 438, 169–178 CrossRef CAS PubMed.
- N. Roy, Y. Sohn and D. Pradhan, ACS Nano, 2013, 7, 2532–2540 CrossRef CAS PubMed.
- Q. Xiang, J. Yu and M. Jaroniec, Nanoscale, 2011, 3, 3670–3678 RSC.
- H. Zhang, T. Ji, Y. Liu and J. Cai, J. Phys. Chem. C, 2008, 112, 8604–8608 CAS.
- F. Tian, Y. Zhang, J. Zhang and C. Pan, J. Phys. Chem. C, 2012, 116, 7515–7519 CAS.
- N. Roy, Y. Sohn, K. T. Leung and D. Pradhan, J. Phys. Chem. C, 2014, 118, 29499–29506 CAS.
- S. Sood, S. K. Mehta, A. Umar and S. K. Kansal, New J. Chem., 2014, 38, 3127–3136 RSC.
- J. Zhang, W. Peng, Z. Chen, H. Chen and L. Han, J. Phys. Chem. C, 2012, 116, 19182–19190 CAS.
- Y. Hu, Y. Cao, P. Wang, D. Li, W. Chen, Y. He, X. Fu, Y. Shao and Y. Zheng, Appl. Catal., B, 2012, 125, 294–303 CrossRef CAS PubMed.
- Y. Duan, N. Fu, Q. Liu, Y. Fang, X. Zhou, J. Zhang and Y. Lin, J. Phys. Chem. C, 2012, 116, 8888–8893 CAS.
- J. Xu, S. Shi, L. Li, X. Zhang, Y. Wang, X. Chen, J. Wang, L. Lv, F. Zhang and W. Zhong, J. Appl. Phys., 2010, 107, 053910 CrossRef PubMed.
- S. K. Md Saad, A. A. Umar, H. Q. Nguyen, C. F. Dee, M. M. Salleh and M. Oyama, RSC Adv., 2014, 4, 57054–57063 RSC.
- Y. Su, B. Zhu, K. Guan, S. Gao, L. Lv, C. Du, L. Peng, L. Hou and X. Wang, J. Phys. Chem. C, 2012, 116, 18508–18517 CAS.
- Z. Yao, F. Jia, S. Tian, C. Li, Z. Jiang and X. Bai, ACS Appl. Mater. Interfaces, 2010, 2, 2617–2622 CAS.
- H. Li, Y. Sun, B. Cai, S. Gan, D. Han, L. Niu and T. Wu, Appl. Catal., B, 2015, 170–171, 206–214 CrossRef CAS PubMed.
- X. Li, S. Fang, L. Ge, C. Han, P. Qiu and W. Liu, Appl. Catal., B, 2015, 176–177, 62–69 CrossRef CAS PubMed.
- G. Cheng, M. S. Akhtar, O. B. Yang and F. J. Stadler, Electrochim. Acta, 2013, 113, 527–535 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra11099h |
| This journal is © The Royal Society of Chemistry 2015 |