
An environmentally-friendly enzyme-based nanofibrous membrane for 3,3′,5,5′-tetrabromobisphenol removal†
Ran Xu*,
Rongzhi Tang,
Sijia Liu,
Fengting Li and
Bingru Zhang
State Key Laboratory of Pollution Control and Resource Reuse, College of Environmental Science and Engineering, Tongji University, Shanghai 200092, PR China. E-mail: carol.xuran@tongji.edu.cn; Fax: +86 21 65986313; Tel: +86 21 65980567
First published on 21st July 2015
Abstract
Chitosan (CS)/poly(vinyl alcohol) (PVA) nanofibrous membranes have inherently poor mechanical strength. To improve the mechanical strength of these membranes, nanocrystalline cellulose (NCC) prepared by a simplified method was added to the former system. The results showed that the tensile strength of the membrane with 5% NCC addition was 370% higher than that of the membrane without NCC. Horseradish peroxidase (HRP) was immobilized on the membrane through covalent binding with HRP previously activated with 1,1′-carbonyldiimidazole, and the maximum enzyme loading was approximately 384 mg g−1. The physical, chemical properties of immobilized HRP and its application in 3,3′,5,5′-tetrabromobisphenol (TBBPA) removal were examined. The results showed that HRP immobilized on CS/PVA–NCC membranes showed greater stability and reusability than free HRP and the membrane without NCC. The former also exhibited an effective performance (95.9% removal, 3 h) for TBBPA removal under the optimum conditions (pH 7, 35 °C). The results showed that HRP immobilized on NCC-incorporated CS/PVA membranes could be used to remove brominated flame-retardants, especially TBBPA from wastewater. Thus, these membranes have potential industrial applications.
1. Introduction
3,3′,5,5′-Tetrabromobisphenol A (TBBPA) is a typical representative of brominated flame-retardants, which are widely used in consumer products ranging from fabrics to plastics and electronics.1 The three-dimensional structure of TBBPA is illustrated in Fig. S1.† Previous studies have confirmed that TBBPA is a bioaccumulative, toxic, and persistent compound, which could act as an endocrine disruptor, an immunotoxicity mediator, and a neurotoxicity effector after a long exposure.2–4 Given the extensive global use of TBBPA, it has been detected from a variety of samples, including water and wastewater, indoor air, soil, and even in biological matrices. The measured concentrations reached an alarming amount.5 Therefore, developing an appropriate way to remove TBBPA from contaminated water is necessary.Several methods, such as ozonation, adsorption, anaerobic degradation, as well as oxidation, have been applied to remove TBBPA from water.6–9 TBBPA can be degraded using biological methods with a mean half-life of approximately two months.10 The degradation rate could be higher for nonbiological ones. For example, 96% TBBPA could be removed using multiwalled carbon as adsorbents after 60 min.8 In another case, the removal efficiency of TBBPA was as high as 99.3% through the use of ozonation.11 However, these methods are still greatly limited because of long-cycle length, secondary pollution, high equipment costs, and difficult operations. By contrast, enzyme immobilization on nanofibrous membrane is considered to be a promising technique for the removal of pollutants because of its capability for high efficiency (including both adsorption of nanomaterials and degradation of enzymes), environmental-friendly, and reusability. Our previous studies have found that enzyme immobilized on electrospun nanofibrous membranes (EFMs) have high efficiency on the removal of PPCPs. For example, the removal efficiency for 2,4-dichlorophenol using laccase-CS/PVA nanofibrous membranes could reach as high as 87.6% after 6 h.12 In another case, immobilized HRP showed a great removal efficiency (83.5%) for paracetamol and exhibited excellent reusability.13 Enzyme immobilization was considered to be applicable for TBBPA removal because of its structural similarity with the pollutants above. HRP, which has the inherent advantages of relatively wide ranges of pH, temperature, contaminant concentration, and salinity, was extensively studied as an efficient catalyst for the removal of phenols, bisphenols, anilines, and enzidines.14 Therefore, in the present work, we choose HRP as the model enzyme for the removal of TBBPA.
EFMs generally have the inherent disadvantage of low mechanical strength, which restricts their application in industrial applications. Studies have found that the incorporation of nanoscale particles with robust mechanical properties, such as nanotubes, graphites, nanoclays, and inorganic nanoparticles, would increase the mechanical strength of materials.15 As one of the strongest and stiffest natural materials available, nanocrystalline cellulose (NCC) exhibits remarkable properties, such as high-specific strength, low density, and large surface area, which lead to a distinguished enhancement feature in various matrices.16 Therefore, we attempted to introduce NCC into our matrix to achieve improved mechanical properties.
This study aimed to develop environmental-friendly CS/PVA–NCC electrospun nanofibrous membranes with high mechanical strength. The materials were applied for the first time to the process of HRP immobilization for the removal of TBBPA from aquatic environments. Results showed that the performance of immobilized HRP for the TBBPA removal was distinguished, thus this method is promising for the removal of chlorophenols from water.
2. Materials and methods
2.1. Materials
Low molecular-weight CS (Mw = 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), PVA, TBPPA (97%), guaiacol (98%), coomassie brilliant blue (G250), and 1,1′-carbonyldiimidazole (CDI; 97%), fluorescein isothiocyanate (FITC) isomer were purchased from Sigma-Aldrich. Acetic acid, tetraethyl orthosilicate (TEOS), microcrystalline cellulose (MCC), ethanol, disodium hydrogen phosphate, citric acid, sulfuric acid, and 2,2′-azinobis-(3-ethylbenzthiazoline-6-sulfonate) were obtained from Sinopharm Chemical Reagent Co., Ltd. Horseradish peroxidase (HRP) was obtained from Sinopharm Chemical Reagent Co., Ltd. Deionized water was used in all experiments. All chemicals used were of analytical grade.
000), PVA, TBPPA (97%), guaiacol (98%), coomassie brilliant blue (G250), and 1,1′-carbonyldiimidazole (CDI; 97%), fluorescein isothiocyanate (FITC) isomer were purchased from Sigma-Aldrich. Acetic acid, tetraethyl orthosilicate (TEOS), microcrystalline cellulose (MCC), ethanol, disodium hydrogen phosphate, citric acid, sulfuric acid, and 2,2′-azinobis-(3-ethylbenzthiazoline-6-sulfonate) were obtained from Sinopharm Chemical Reagent Co., Ltd. Horseradish peroxidase (HRP) was obtained from Sinopharm Chemical Reagent Co., Ltd. Deionized water was used in all experiments. All chemicals used were of analytical grade.
NCC was obtained from MCC through acid hydrolysis according to the following procedures.17 Five grams of MCC was mixed with 50 mL of deionized water, the water/MCC-suspension was placed in an ice bath, and then 87 g of concentrated sulfuric acid was added dropwise at 44 °C with gentle stirring for 2 h. Afterwards, 1500 mL of deionized water was quickly added to the mixture to terminate the reaction. The mixture was left to stand for 3 days, during which the supernatant was removed from the sediment and replaced with equal and new deionized water. The suspension obtained was then centrifuged five times, each time involving repeated 5 min centrifuge cycles at 10000 rpm. The final centrifugate was subjected to dialysis with deionized water until the wash water maintained a constant pH. The samples were then sonicated in an ice bath for a certain time and naturally dried into powders.
2.2. Preparation of CS/PVA–NCC EFMs through electrospinning
CS–NCC and PVA–NCC solution were prepared by adding 1 wt% to 8 wt% NCC (dry weight relative to that of dry matrix) to the two solutions containing 3 wt% CS/acetic acid (1 mol L−1) and 10 wt% PVA, and homogenized under vigorous magnetic stirring at room temperature for 4 h, followed by ultrasonication in a water bath for 2 min. Approximately 2 g of TEOS was dissolved in 3 g of 70 wt% acetic acid solution in a round-bottom plastic bottle. Then, 5 g of CS–NCC and 5 g of PVA–NCC were added to the above solution, followed by vigorous stirring for 45 min in a water bath at 60 °C to form a homogeneous CS/PVA sol–gel loaded with NCC.The CS/PVA sol–gel with different NCC loading was added to a plastic syringe with a stainless-steel needle bearing an inner diameter of 0.8 mm. A syringe pump was set to inject the emulsion at a flow rate of 1.2 mL h−1. A copper pin connected to a high-voltage generator was placed in the solution. Electrospinning was conducted at 22 kV with a tip-to-target distance of 20 cm. CS/PVA–NCC EFMs (CPN EFMs) were then collected on a flat glass covered with aluminum foil for 12 h and then dried for 10 h at 40 °C in a vacuum to obtain non-woven fabrics. NCC-free EFMs were also prepared according to our previous study to serve as the control experiment.18
2.3. Characterization of CPN EFMs
Electronic-tensile testing machine UT-2060 from U-CAN company was used to examine the mechanical performance of CPN EFMs according to the determination of tensile properties (GB/T 1030.3-2006). The surface morphology of the CPN EFMs was characterized using scanning electron microscopy (SEM), which was conducted on a field emission XL-30 SEM at 30 kV. The average diameter and diameter distribution were determined by choosing 50 fibers at random SEM images and analyzing them using image analysis software Adobe Photoshop CS6, developed by Adobe Systems Inc. HRP labeled with FITC (HRP-FITC) was used to see whether HRP has been successfully immobilized on to the nanofibers using laser confocal scanning microscopy (LSCM; Leica TCS-SP5, Germany). The HRP-labeling procedure was according to the paper published previously.19 The functional groups of original and enzyme-immobilized nanofibers were determined using a Fourier transform infrared attenuated total reflectance (FTIR/ATR, Bruker-Vector 22) spectrometer that was equipped with a germanium crystal. The background spectra were recorded in air. The residual concentration and activity of HRP was measured through Bradford's method b using an UV-1700 spectrophotometer from Shimadzu.2.4. Immobilization of HRP
CPN EFMs were abundant in hydroxyl groups, which could be activated using CDI to form imidazole carbamate following a reaction with HRP solution. In a typical immobilization experiment, 200 mg of dry composite nanofibers were immersed in 200 mL of anhydrous THF containing 30 mg mL−1 CDI. This mixture was then placed in a 500 mL conical flask, which was fixed on horizontal shaker at 100 rpm and 25 °C for 6 h. Activated fibrous membranes were washed three times with THF to remove excess CDI and reaction by-products. After the final wash, the fibrous membranes were removed from solvent and washed with distilled water several times. Then, 10 mg activated nanofibers was placed in 10 mL of HRP solution (2 mg mL−1 in citric acid–disodium hydrogen phosphate buffer solution) for immobilization at 25 °C under mild shaking (150 rpm). The effects of pH (4.0, 5.0, 6.0, 6.5, 7.0, 7.5, 8.0, and 8.5) and time (0.5, 1, 1.5, 2, 4, 6, 8, 10, and 12 h) on HRP immobilization were analyzed. After enzyme immobilization, the membranes were fetched out from the solution and rinsed with CPBS until no soluble protein was detected in the washings.2.5. Determination of free and immobilized enzymes
A colorimetric assay was used to determine the activity of peroxidase with reference to the method of Nicell.20 In a typical procedure, a suitable amount of free or immobilized HRP sample (supernatant from centrifugation at 4000 rpm) was added to a cuvette containing 3 mL of CPBS (0.1 M, pH 6.0) and 0.05 mL of guaiacol (20 mM), followed by the addition of 1 μL of 30% H2O2 to initiate the reaction. Absorbance at 436 nm was continuously recorded with a UV-1700 spectrometer. One unit of HRP activity was defined as the amount of enzyme that produced 1 μmol tetraguaiacol per min under assay conditions. For data analysis, the activity results were converted to relative activities equal to the percentage of measured activity out of the maximum activity.To determine the effect of immobilization on the activity of HRP, the activity assays were repeated using a series of H2O2 substrates with a concentration ranging from 0.2 mM to 10 mM to achieve corresponding catalytic rates, which were used to calculate Vmax and Km using the Lineweaver–Burk plot.21 Similar assays were also performed with free enzymes as the control experiments.
2.6. Stabilities of the free and immobilized HRP
Thermal stabilities of the catalases were evaluated in terms of different temperature effects on the activity of HRP. The activities of both immobilized and free HRP were measured at pH 6 and different temperatures (4, 20, 30, 40, 50, 60, 70, and 80 °C). The pH stabilities of the catalases were examined by evaluating enzyme activity at 30 °C from pH 3.0 to 10.0 for batch experiments. The operational stability associated with the reusability of immobilized HRP was determined through 10 times of measurements within one day under the optimum conditions. After each reaction, the immobilized HRP was washed with CPBS (pH 6) to remove any residual substrate. Storage stabilities of free and immobilized HRP were determined at 4 °C in CPBS (pH 7) for 30 days. Residual activity was calculated every 3 days.2.7. Removal of TBBPA
Effects of various pH values (4.0, 5.0, 6.0, 7.0, 8.0, 9.0, and 10.0), temperatures (15, 25, 35, 45, and 55 °C), and reaction times (10, 20, 30, 60, 90, 120, and 180 min) on the removal of 3 mg L−1 of TBBPA were studied. CPN EFMs, as well as free and immobilized HRP, were used to treat TBPPA solution. A series of 50 mL conical flasks with tightly closed screw caps were placed on a horizontal shaker at a speed of 150 rpm and were used in all TBBPA treatments. TBBPA concentration in the reaction mixture was analyzed using HPLC (Agilent 1200) as described in the next section. The amount of TBBPA that was degraded using immobilized laccase was calculated with the following eqn (1):| qD = q0 − qS − qA | (1) |
2.8. TBBPA and its analysis with HPLC
An HPLC instrument with two pumps, an auto sampler, and a photodiode array detector was used for the analysis. TBBPA and its oxidation products were extracted from the flasks using a syringe with a filter cap, and the treated solution was analyzed using HPLC. Standard solutions of TBBPA were prepared to obtain a standard curve. The mobile phase was composed of acetonitrile (75%) and 60 mM acetic acid solution (30%), and eluted at 1.0 mL min−1. UV spectra of TBBPA and reaction products were obtained at 207 nm.2.9. Data analysis
One-way ANOVA was applied to measure the statistical significance of the various conditions (pH, temperature) for the HRP removal of TBBPA. Turkey's procedure was used to evaluate differences among carriers, as well as free and immobilized HRP, at a family error rate of 5%. Data were considered as significantly different from one another if p < 0.05. Design Expert 7.0.0 was used throughout the statistical analysis.3. Results and discussion
3.1. Mechanical properties of CPN EFMs
The effect of different NCC loadings ranging from 1% to 8% (w/w) of the final dry weight on the tensile strength of CPN EFMs was studied (Fig. 1a). The reinforced nanofibers added with 1% NCC to 5% NCC accordingly improved tensile-strength values. The optimum amount of NCC incorporated for best tensile strength of CPN EFMs was 4% to 5%, consistent with the previous studies.22,23 A typical example of the stress–strain behavior of CS/PVA loading with 5% (w/w) of NCC electrospun nanofibers and neat CS/PVA EFMs (CP EFMs) is shown in Fig. 1b. The average tensile strength for NCC incorporated nanofibers reached 4.85 MPa compared with 0.85 MPa of neat CS/PVA nanofibers. The 370% tensile-strength increment of 5% NCC-incorporated film compared with non-NCC film can be attributed the reinforcing effect of NCC and was compatible with the former results. This finding could be attributed to the strong intra- and intermolecular forces among NCC, CS, and PVA, as well as the high aspect ratio of NCC itself.24 The formation of the networked structure above the percolation threshold, which was a result of hydrogen bonding, may also contribute to the strong reinforcing effect of NCC.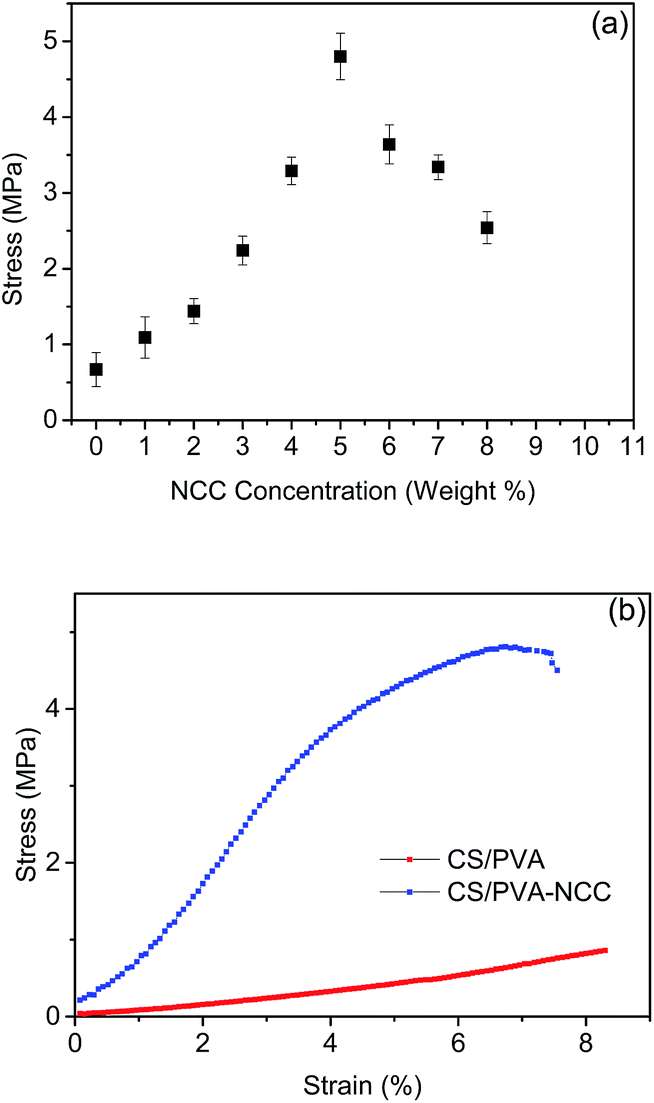 | ||
| Fig. 1 Mechanical properties of nanofiberous membranes: (a) influence of NCC content on the tensile behaviour of the membrane; (b) stress–strain behaviour of CP and CPN EFMs. | ||
3.2. Characterization of CPN EFMs before and after HRP immobilization
CPN EFMs were activated through CDI, following by bio-conjugation with HRP. Specifically, the free hydroxyl groups on EFMs surfaces were activated with CDI, after which the amino groups of the enzymes conducted the condensation reaction with the imidazolyl carbamate groups of the activated EFMs (Fig. S2†). This process is available when attaching HRP on the CPN EFMs, wherein the immobilized HRP exhibited good stability through reutilization.The morphologies of NCC and CPN EFMs with different NCC loadings were characterized using SEM, the images were shown in Fig. S3 and S4,† respectively. As is shown in Fig. S3,† NCC showed a rod-like structure and distributed unevenly, this might be due to the agglomeration of the nanoparticles. We could see from Fig. S4† that nanofibers with NCC loading from 4% to 5% showed best surface morphology. A typical example of 5% NCC-incorporated nanofibers was shown in Fig. S5.† The image demonstrated that the EFMs possessed the feature of being randomly arrayed and bead-free with an average diameter of 138.4 ± 20 nm (Fig. S5a†). The diameter (137 ± 26 nm) and structure of EFMs had no substantial change after binding with HRP. Comparing Fig. S5b with Fig. S5a,† we can find that the surface of the CPN EFMs transformed from smooth to coarse, and many HRP molecules became evenly dispersed on the nanofibers after immobilization. Fig. S5c† showed that the HRP-FITC had been successfully immobilized onto the nanofibers. The strong fluorescence emitted by the nanofibers should be resulted from the combined effect of covalently binding and physical adsorption of HRP-FITC to the nanofibers.
FTIR spectra of CPN EFMs before and after CDI activation were obtained to characterize the EFMs (Fig. S6†). Compared with the original CPN EFMs, a new peak at 1653 cm−1 was found in the spectra of activated CPN EFMs, which was attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations. This change suggested that EFMs had been successfully activated through CDI and could be a favorable carrier for covalent binding.
O stretching vibrations. This change suggested that EFMs had been successfully activated through CDI and could be a favorable carrier for covalent binding.
3.3. Effect of reaction time and pH on HRP activity and enzyme loading on CPN EFMs
The reaction time and pH (p < 0.05) significantly influenced the covalent binding efficiency of the enzyme and EFMs. Fig. 2 showed that the optimum pH for HRP immobilization was 7.5 with a maximum HRP loading of 368 mg g−1, whereas pH lower or higher than 7.5 caused a distinct decrease in the loading efficiency, which might be attributed to changes in the diffusion rate of the enzyme and variations in the microenvironment.25 We could also conclude the effect of time on the immobilization efficiency based on Fig. 2. The immobilization efficiency almost leveled off after 8 h of incubation, which might be because of the stability of imidazole carbamate groups in hydrolysis aqueous buffer that made them react and couple with amines slowly.26 According to Fig. 2, the loading of HRP reached a maximum of 384 mg g−1 after 8 h at pH 7 and 35 °C, which was higher than other supports.27,28 This result may be attributed to the abundant –OH groups on the backbone of the CPN EFMs, which could be activated efficiently through CDI. Thus, sufficient bonding sites are provided for the amino groups of HRP immobilization.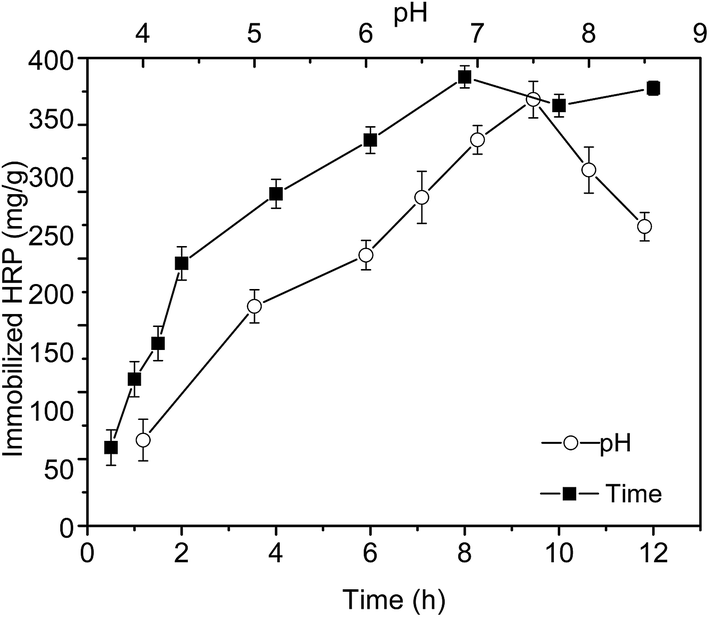 | ||
| Fig. 2 Effects of pH and time on HRP immobilization efficiency. | ||
3.4. Effects of immobilization on kinetic parameters
Kinetic parameters, namely, the Michaelis constant Km and Vmax, were measured using H2O2 as a substrate with Lineweaver–Burk plots. The kinetic parameters for free, HRP-CP EFMs and HRP-CPN EFMs are shown in Table 1.| HRP | Specific activity (U) | Km (μmol mL−1) | Vmax (μmol (mg−1 min−1)) |
|---|---|---|---|
| Free HRP | 237.6 | 2.6 | 670.4 |
| HRP-CP EFMs | 164.7 | 4.1 | 479.2 |
| HRP-CPN EFMs | 192.4 | 3.7 | 529.3 |
According to Table 1, the specific activities of the HRP-CP EFMs and HRP-CPN EFMs was 69.3% and 81% of its free form, respectively. The specific activities of the HRP-CP EFMs and HRP-CPN EFMs were higher than other supports.29,30 These differences can be ascribed to the biocompatible characteristics of the polymers we used, which increased the accessibility between enzyme and carrier. Various methods of immobilization or different sources of HRP can also cause these differences. In contrast with HRP-CP EFMs, the higher specific activities of HRP-CPN EFMs might be resulted from the incorporation of NCC, which increased the biocompatibility of the composite membrane. Compared with free HRP, the immobilized HRP showed a significantly lower Vmax, whereas the Km value was significantly higher. The higher Km of immobilized HRP in this study suggested that the immobilized HRP had a lower affinity for H2O2 than their free form because Km embodies the affinity of an enzyme with its substrate.31 This result may be attributed to the low accessibility of the substrate to the active site of the immobilized enzyme from the increased diffusion limitation. The low possibility to form a substrate–enzyme complex that arised from enzyme-conformational changes may also be an explanation for this phenomenon. As shown in Table 1, the Vmax of the enzymes demonstrate a significant decrease upon immobilization. The reason for this low Vmax could be explained with the steric effects, as well as bulk and diffusional effects.
3.5. Stabilities of free and immobilized HRP
Stabilities are of vital importance for the potential biotechnological applications of the immobilized enzymes. To further investigate whether the incorporation of NCC into CPN EFMs brought any excellent performance for HRP immobilization, comparative stabilities among free HRP, HRP immobilized on CS/PVA, and CPN EFMs (HRP-CP EFMs, HRP-CPN EFMs) in terms of thermal, operational, and storage stability were conducted.Fig. 3 showed the effect of temperature (a) and pH (b) on the catalytic capabilities of the free and immobilized HRPs. Fig. 3a shows that the relative activity of the immobilized HRP changed significantly slower than its free form with the increase of temperature (p < 0.05); the activity of HRP-CPN EFMs declined slower than that of HRP-CP EFMs. Therefore, the thermal stability of three HRP kinds followed the sequence of HRP-CPN EFMs > HRP-CP EFMs > free HRP. The significant improvement of temperature resistance in HRP after immobilization on HRP-CPN EFMs might be a consequence of great mechanical strength resulting from the addition of NCC, as well as a suitable microenvironment from the framework of the carrier.
 | ||
| Fig. 3 Effect of temperature (a) and pH (b) on free and immobilized HRP. | ||
Fig. 3b shows the effects of pH on the relative activity of free and immobilized HRPs. Both free and immobilized HRPs achieved the maximum activity at pH 7, which was similar to that of HRP immobilized on perlite reported using Seyed–Fakhreddin Torabi.32 Within the test pH range, immobilized HRP exhibited higher activity than its free form. A typical example was the case where immobilized HRP retained 75% activity, whereas free HRP retained only 18% at pH 9. Compared with neat CP EFMs, incorporation of NCC increased the mechanical strength and maintained the membrane microstructure, which may explain the high HRP activity.
Compared with immobilized enzymes, free enzymes are sensitive to the surrounding environment and may easily became inactive. The superior performance of storage stability is a great preponderance for immobilized enzymes, which can greatly reduce the cost in their biotechnological and industrial applications. Fig. 4 shows the residual activity of the free and immobilized enzymes. As the storage period increased, the HRP-CPN EFMs showed a higher stability over the other two forms of HRP. The relative activity of free HRP declined sharply as time passed. After 30 days, the immobilized HRP could still retain 72% (CPN EFMs) and 65% (CP EFMs) of its initial activity, whereas the free HRP only retained 9%. This phenomenon might be attributed to the limited conformational changes of the HRP, which helped retain its stability. Accordingly, the support and the methods used in the immobilization provided a long shelf life than their free counterpart and can be a preferable carrier for future applications.
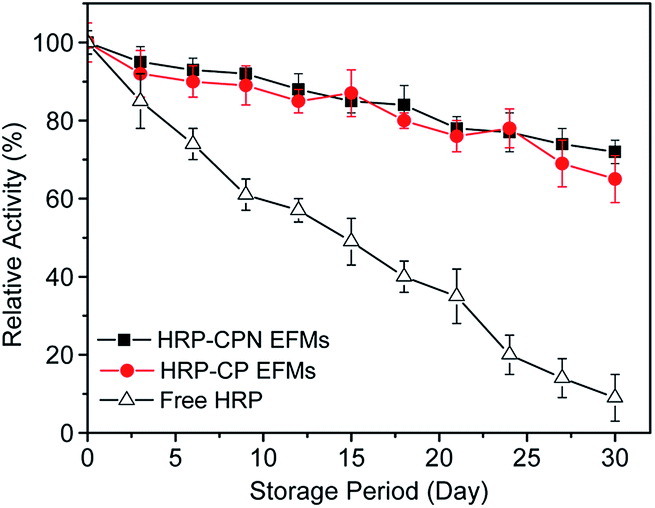 | ||
| Fig. 4 Storage stability of free and immobilized HRP at 4 °C. | ||
3.6. Removal of TBBPA from water by HRP
Fig. 5 shows the influence of pH, temperature, and time on the removal of TBBPA using HRP-CPN EFMs, free HRP, and neat CP EFMs. The removal efficiency of HRP-CPN EFMs remained between 56% and 84% at 35 °C with the pH ranging from 4 to 10 and between 52% and 84% at pH 7, and a temperature range of 15–55 °C.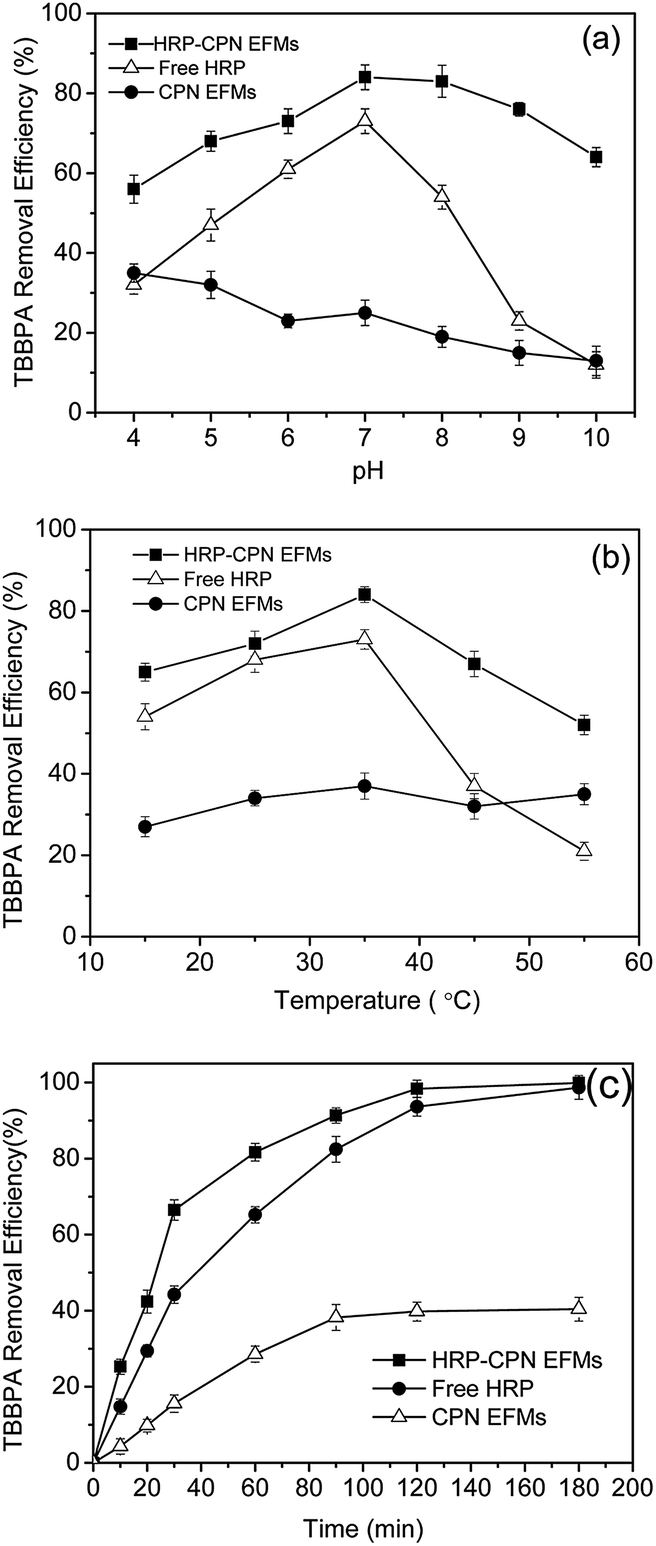 | ||
| Fig. 5 Effect of pH (a), temperature (b) and time (c) on TBBPA removal efficiency of free and immobilized HRP. | ||
Fig. 5a indicates the change of TBBPA removal efficiency at pH values that varied from 4–10. The optimum pH for free enzymes was 7 with a degradation rate of 73%. A high removal efficiency of TBBPA was obtained using HRP-CPN EFMs at pH 4–10, and the maximum degradation rate was 84% at an optimum pH of 7. This result could definitely demonstrate that HRP-CPNEFMs is preferable for the removal of TBBPA from water.
As shown in Fig. 5b, the temperature undoubtedly affected the TBBPA removal efficiency and the optimal temperature for both immobilized and free HRP to remove TBBPA from water was at 35 °C with removal rates of 84% and 73%, respectively. In the temperature range of 35–55 °C, the degradation rate of TBBPA using free HRP decreased sharply as the temperature increased. When temperature was 55 °C, the removal efficiency of TBBPA was only 21% using free HRP, whereas the removal efficiency was 55% using HRP-CPN EFMs. The high thermal stability of immobilized enzymes might be because of the appropriate porous structure and surface characteristics of the membrane, as well as the multipoint complexation of peroxidase with the support.33
As shown in Fig. 5c, the degradation rate of TBBPA using the three forms could be affected with time. When the time was less than 2 h, the removal efficiency increased as the time increased. The degradation rate was almost leveled off after 2 h with a removal rate of 98.34% (HRP-CPN EFMs), 93.66% (free HRP), and 39.8% (neat CP EFMs), which might be because of the decreased concentration of TBBPA, HRP, and H2O2 in the reaction system.34 Furthermore, the generated polymer attacked the active center of the enzyme and combined with the said center, thus the enzyme lost its catalytic activity and lead to the decrease in its reaction rate.35 We can also conclude that the HRP-CPN EFMs were the most effective material among the three forms for the removal of TBBPA from water. For example, the TBBPA removal efficiency using HRP-CPN EFM was 66.5% after 30 min, which was significantly higher than that of free HRP (44.23%) and neat CPN EFM (15.6%).
3.7. Reusability of HRP-CPN EFMs
The main problems for free enzymes are inactivation, easy bleeding and hard to separate.36 The immobilized enzymes were superior to the free HRPs in terms of reusability. The removal efficiency of TBBPA using HRP-CPN and HRP-CP EFMs during different batch operation runs is shown in Fig. 6. The TBBPA removal efficiency using immobilized enzymes decreased with the increase of reuse numbers. The decrease in TBBPA removal efficiency could be explained through the loss and inactivation of the enzyme, as well as the damage of the membrane. After six repeated runs, approximately 60% of TBBPA could be removed through HRP-CPN and HRP-CP EFMs. However, the HRP-CPN EFMs showed better TBBPA removal efficiency than HRP-CP EFMs as the repeated runs increased. This result could be attributed to the improvement of mechanical strength through introducing NCC into the CS/PVA mixed matrix.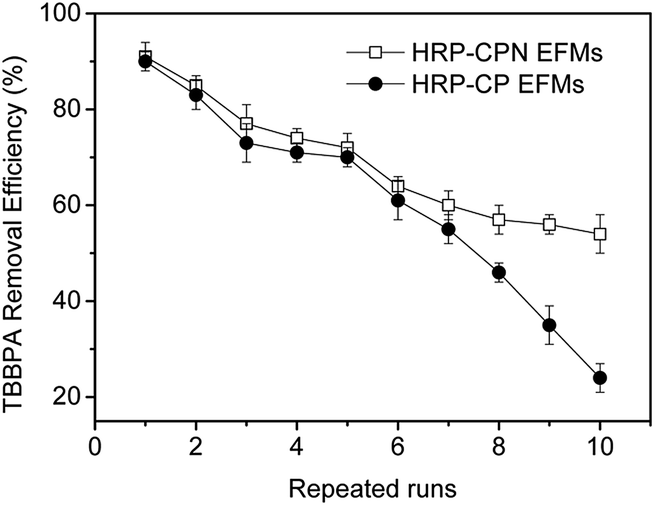 | ||
| Fig. 6 Removal efficiency of TBBPA by HRP-CPN and HRP-CP EFMs during repeated runs. | ||
4. Conclusions
An environmental-friendly nanofibrous membrane was fabricated. Compared with CP EFMs, NCC incorporation increased mechanical strength and enzyme loading. NCC incorporation also effectively maintained the catalytic activity of HRP. Thermal and storage stabilities, as well as pH, were enhanced after immobilization. HRP-CPN EFMs exhibited effective performance (95.9% removal, 3 h) under the optimum conditions. After six repeated runs, removal efficiency remained as high as 60%. Thus, these membranes have potential applications in the field of enzyme immobilization.Acknowledgements
This work was funded by the National Natural Science Foundation of China (51208368). The authors are also grateful for the financial support from the Ministry of Science and Technology, China (Nos. 2010DFA92820 and 2010DFA92800).References
- C. Chignell, S. Han, A. Mouithys-Mickalad, R. Sik, K. Stadler and M. Kadiiska, Toxicol. Appl. Pharmacol., 2008, 230, 17–22 CrossRef CAS PubMed.
- J. Alexander, D. Benford, A. Boobis and Å. Bergman, EFSA J., 2011, 9, 2477 Search PubMed.
- S. Decherf, I. Seugnet, J.-B. Fini, M.-S. Clerget-Froidevaux and B. A. Demeneix, Mol. Cell. Endocrinol., 2010, 323, 172–182 CrossRef CAS PubMed.
- S. Lee, G.-J. Song, K. Kannan and H.-B. Moon, Sci. Total Environ., 2014, 470, 1422–1429 CrossRef PubMed.
- D. Bu, H. Zhuang, X. Zhou and G. Yang, Talanta, 2014, 120, 40–46 CrossRef CAS PubMed.
- M. Fukushima, Y. Ishida, S. Shigematsu, H. Kuramitz and S. Nagao, Chemosphere, 2010, 80, 860–865 CrossRef CAS PubMed.
- M. Shin, B. Duncan, P. Seto, P. Falletta and D.-Y. Lee, Chemosphere, 2010, 78, 1220–1224 CrossRef CAS PubMed.
- I. I. Fasfous, E. S. Radwan and J. N. Dawoud, Appl. Surf. Sci., 2010, 256, 7246–7252 CrossRef CAS PubMed.
- T. Miyamoto, R. Nishimoto, S. Maeno, Q. Zhu and M. Fukushima, J. Mol. Catal. B: Enzym., 2014, 99, 150–155 CrossRef CAS PubMed.
- Z. Ronen and A. Abeliovich, Appl. Environ. Microbiol., 2000, 66, 2372–2377 CrossRef CAS.
- J. Xu, W. Meng, Y. Zhang, L. Li and C. Guo, Appl. Catal., B, 2011, 107, 355–362 CrossRef CAS PubMed.
- R. Xu, Q. Zhou, F. Li and B. Zhang, Chem. Eng. J., 2013, 222, 321–329 CrossRef CAS PubMed.
- R. Xu, Y. Si, F. Li and B. Zhang, Environ. Sci. Pollut. Res., 2014, 1–9 Search PubMed.
- J. Nicell, J. Bewtra, N. Biswas, C. St. Pierre and K. Taylor, Can. J. Civ. Eng., 1993, 20, 725–735 CrossRef.
- F. Hussain, M. Hojjati, M. Okamoto and R. E. Gorga, J. Compos. Mater., 2006, 40, 1511–1575 CrossRef CAS PubMed.
- A. Kaboorani, B. Riedl, P. Blanchet, M. Fellin, O. Hosseinaei and S. Wang, Eur. Polym. J., 2012, 48, 1829–1837 CrossRef CAS PubMed.
- D. Bondeson, A. Mathew and K. Oksman, Cellulose, 2006, 13, 171–180 CrossRef CAS.
- R. Xu, Q. Zhou, F. Li and B. Zhang, Chem. Eng. J., 2013, 222, 321–329 CrossRef CAS PubMed.
- R. Xu, Y. Si, F. Li and B. Zhang, Environ. Sci. Pollut. Res., 2015, 22, 3838–3846 CrossRef CAS PubMed.
- J. A. Nicell and H. Wright, Enzyme Microb. Technol., 1997, 21, 302–310 CrossRef CAS.
- Z. G. Wang, Z. K. Xu, L. S. Wan, J. Wu, C. Innocent and P. Seta, Macromol. Rapid Commun., 2006, 27, 516–521 CrossRef CAS PubMed.
- A. J. Uddin, J. Araki and Y. Gotoh, Biomacromolecules, 2011, 12, 617–624 CrossRef CAS PubMed.
- A. Khan, R. A. Khan, S. Salmieri, C. Le Tien, B. Riedl, J. Bouchard, G. Chauve, V. Tan, M. R. Kamal and M. Lacroix, Carbohydr. Polym., 2012, 90, 1601–1608 CrossRef CAS PubMed.
- M. Pääkkö, M. Ankerfors, H. Kosonen, A. Nykänen, S. Ahola, M. Österberg, J. Ruokolainen, J. Laine, P. T. Larsson and O. Ikkala, Biomacromolecules, 2007, 8, 1934–1941 CrossRef PubMed.
- Z.-G. Wang, L.-S. Wan, Z.-M. Liu, X.-J. Huang and Z.-K. Xu, J. Mol. Catal. B: Enzym., 2009, 56, 189–195 CrossRef CAS PubMed.
- G. T. Hermanson, Bioconjugate techniques, Academic press, 2013 Search PubMed.
- L. Pramparo, F. Stüber, J. Font, A. Fortuny, A. Fabregat and C. Bengoa, J. Hazard. Mater., 2010, 177, 990–1000 CrossRef CAS PubMed.
- M. Monier, D. Ayad, Y. Wei and A. Sarhan, Int. J. Biol. Macromol., 2010, 46, 324–330 CrossRef CAS PubMed.
- Z. G. Wang, B. B. Ke and Z. K. Xu, Biotechnol. Bioeng., 2007, 97, 708–720 CrossRef CAS PubMed.
- D. Wang, G. Sun, B. Xiang and B.-S. Chiou, Eur. Polym. J., 2008, 44, 2032–2039 CrossRef CAS PubMed.
- D. Park, S. Haam, K. Jang, I.-S. Ahn and W.-S. Kim, Process Biochem., 2005, 40, 53–61 CrossRef CAS PubMed.
- S.-F. Torabi, K. Khajeh, S. Ghasempur, N. Ghaemi and S.-O. R. Siadat, J Biotechnol., 2007, 131, 111–120 CrossRef CAS PubMed.
- Z. Karim, R. Adnan and Q. Husain, Int. Biodeterior. Biodegrad., 2012, 72, 10–17 CrossRef CAS PubMed.
- A. Rekuć, J. Bryjak, K. Szymańska and A. B. Jarzębski, Process Biochem., 2009, 44, 191–198 CrossRef PubMed.
- B. Jung and P. Theato, in Bio-synthetic Polymer Conjugates, Springer, 2013, pp. 37–70 Search PubMed.
- F. Quintanilla-Guerrero, M. Duarte-Vázquez, B. García-Almendarez, R. Tinoco, R. Vazquez-Duhalt and C. Regalado, Bioresour. Technol., 2008, 99, 8605–8611 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra09090c |
| This journal is © The Royal Society of Chemistry 2015 |