
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible surface reconstruction of metal–organic frameworks for durable oxygen evolution reaction†
Shulin
Li‡
abc,
Zhaoxin
Zhou‡
b,
Jiahui
Li
a,
Yang
Xiao
a,
Ye
Yuan
a,
He
Zhu
 b,
Fengchao
Cui
*a,
Xiaofei
Jing
*a and
Guangshan
Zhu
*a
b,
Fengchao
Cui
*a,
Xiaofei
Jing
*a and
Guangshan
Zhu
*a
aKey Laboratory of Polyoxometalate and Reticular Material Chemistry of Ministry of Education, Northeast Normal University, Changchun 130024, China. E-mail: jingxf100@nenu.edu.cn; zhugs@nenu.edu.cn
bSchool of Science and Engineering, The Chinese University of Hong Kong, Shenzhen 518172, China
cSchool of Chemistry, Chongqing Normal University, Chongqing 400000, China
First published on 11th June 2025
Abstract
Metal–organic frameworks (MOFs) are regarded as prospective electrocatalysts for the oxygen evolution reaction (OER). Nevertheless, controllably reversible reconstruction of MOFs, yielding highly active catalytic sites for durable OER, has not been extensively studied. Herein, Ni-BPM (BPM = 4,4′-dihydroxybiphenyl-3,3′-dicarboxylic acid) equipped with open metal sites was selected as a potential electrocatalyst, and orientated MOF electrodes were fabricated via a sacrificial lattice-matched-template method. Surface reconstruction of Ni-BPM to active γ-NiOOH was detected during the OER, and reconstructed Ni-BPM can also be repaired in the reduction process, resulting in durable OER properties: continuous operation at 100 mA cm−2 for 130 h followed by another 70 h at 500 mA cm−2, surpassing those of most single Ni-based catalysts. The electronic configuration transformation of Ni sites at the interface of Ni-BPM and γ-NiOOH is confirmed using in situ Raman and X-ray absorption spectra together with density functional theory (DFT) calculations. This work has investigated the reversible structural transformation of MOFs during the OER and thereby would help establish a theoretical foundation for the development of durable MOF electrocatalysts.
1. Introduction
The global environmental crisis and energy shortage have driven the attention away from fossil fuels towards the development of high-performance, cost-effective, and eco-friendly systems for energy storage and conversion.1–4 The oxygen evolution reaction (OER) has broad applications that involve cathodic reactions, including water electrolysis, carbon dioxide reduction, ammonia synthesis, and electrocatalytic hydrogenation.5–8 However, sluggish OER kinetics result in significant overpotential that adversely impacts the energy efficiency of these devices.9–11 Therefore, developing cost-effective OER electrocatalysts with high stability is crucial for advancing the development of energy technologies.Ni-based MOFs12–14 are promising OER electrocatalysts benefiting from their highly crystalline nature, abundant active sites, and adjustable skeleton structures.15–17 The majority of MOFs are irreversibly subject to reconstruction into oxyhydroxides during the OER process (Scheme 1a), namely MOOH, due to the impact of the electrochemical environment and their structures and morphologies.18–21 This phenomenon is considered the primary factor contributing to the augmented OER activity, while the activity of synthetic MOOH is lower than that formed from MOF structural transformation.22–24 Wu et al.20 employed an organic acid etching strategy to generate MFc-MOF (M = Ni, Co, Fe, Zn) nanosheet arrays, and in situ Raman spectroscopy and Fourier transform infrared (FTIR) spectroscopy studies showed that the NiFc-MOF surface underwent dynamic structural reconfiguration during OER catalysis, generating NiFeOOH, which was conducive to rapid OER. The surface reconstruction of the catalyst, i.e. the oxidation state change of interfacial metal sites, directly determines the pH-dependent OER activity.25 This is especially significant since alkaline environments provide lower flat-band potentials, thereby enabling the completion of redox transformations of the metal sites at lower potentials, which is favorable for surface reconstruction.
 | ||
| Scheme 1 Illustration of the irreversible reconstruction (a) and reversible reconstruction (b) of MOFs during the reaction. | ||
However, there is a paucity of research regarding the maintenance of elevated OER activity after the self-reconstruction process. Reversibly reconfigurable MOFs have been shown to resist the possible structural damage caused by the reconstruction process during the electrochemical process, thus ensuring durable OER activity.26–28 This implies that the reconstructed MOF surfaces suggest the capacity for complete or partial repair to the original MOF structure (Scheme 1b). In contrast, irreversibly reconfigured MOFs may experience a loss of catalytic sites due to structural damage during the OER process, resulting in a decline in catalytic efficiency and, consequently, a reduction in long-term catalytic stability.29,30 Therefore, it is imperative to develop catalysts with higher activity and stability for practical applications by understanding and controlling the reconstruction behavior of MOFs.
Herein, Ni-BPM (BPM = 4,4′-dihydroxybiphenyl-3,3′-dicarboxylic acid) equipped with open metal sites was identified as an electrocatalyst to investigate the correlation between the reversible reconstruction and elevated OER activity and stability, due to the resemblance in atom distances with the target reconstructed γ-NiOOH active species (Table S1 & Fig. S1†). Orientated Ni-BPM/NF electrodes via a sacrificial lattice-matched-template method (Fig. 1a) were prepared for the OER. Ni-BPM/NF showed outstanding OER activity, reaching 10 mV cm−2 with 218 mV and enabling 5000 CV cycles, surpassing most single Ni-based electrocatalysts. In situ Raman spectra confirmed the reversible Ni-BPM reconstruction during the OER. DFT calculations showed that the γ-NiOOH/Ni-BPM heterostructure during the reconstruction process could tune the electronic configuration of Ni sites, remarkably optimizing the adsorption free energy of O*, thus heightening the OER kinetics. Besides, bimetallic MOF electrodes were also successfully prepared by introducing Fe/Co atoms into Ni-BPM and exhibited ultra-low overpotentials of 198 and 209 mV at 10 mA cm−2, respectively.
 | ||
| Fig. 1 (a) Schematic illustration of Ni-BPM/NF-x (x = 8, 16, 24, 40, and 52 h) electrode preparation. SEM (b and c), TEM and HRTEM (d) and TEM-EDX-mapping (e) images of Ni-BPM/NF-24 h. | ||
2. Results and discussion
2.1 Material synthesis and structural characterization
From the data of Ni–Ni distance and Ni–O bond length in Ni-BPM and NiOOH (Table S1†), Ni-BPM reversible reconstruction to active γ-NiOOH was expected during the OER due to their distance similarities. Ni-BPM electrodes were prepared through a sacrificial lattice-matched-template31 pathway using Ni layered double hydroxide (Ni-LDH) as a template, as the Ni-BPM lattice matched well with that of Ni-LDH (Fig. S2†). As illustrated in Fig. 1a, Ni-LDH nanowires grew on NF directly by hydrothermal synthesis which was denoted as Ni-LDH/NF. Then, Ni-LDH/NF, employed as a sacrificial template, was immersed in a saturated BPM solution and reacted for a certain time to form Ni-BPM/NF electrodes via solvent-thermal synthesis.We adjusted the reaction time to investigate its effect on the morphology of obtained Ni-BPM/NF-x (x = 8, 16, 24, 40, 52 h) electrodes. Meanwhile, we prepared Ni-BPM/NF-D for comparison purposes, where Ni-BPM was directly grown on NF without the Ni-LDH template. The structures of these electrodes were first analyzed by X-ray diffraction (XRD). As displayed in Fig. S3,† the peak located at 11.7° in Ni-LDH/NF was assigned to the (003) crystal plane, the same as that of NiFe-LDH (PDF#40-0215), confirming that Ni-LDH exhibited an isostructural configuration to the NiFe-LDH structure. The XRD patterns of as-prepared Ni-BPM/NF-x and Ni-BPM/NF-D were consistent with that of the simulated one, implying the successful formation of Ni-BPM on the substrate surface. As the reaction time increased, the Ni-BPM peak intensity gradually increased while the characteristic peak of Ni-LDH disappeared. This suggested that Ni-LDH dissolved and the size and crystallinity of Ni-BPM increased gradually.
The morphology of the as-prepared electrodes was characterized via scanning electron microscopy (SEM), transmission electron microscopy (TEM), and high-resolution TEM (HRTEM). As illustrated in Fig. S4, S5a & e,† after 8 h of coordination reaction, the thickness of the Ni-BPM/NF-8 h film reduced from 1.2 to 0.6 µm. Then the electrode underwent a morphological change after 16 h, resulting in a nanorod array morphology with a thickness of 2.1 µm (Fig. S5b & e†). As the reaction proceeded for 24 h, the nanorod arrays became significantly denser, with a concomitant increase in thickness to 3.6 µm (Fig. 1b and c). With the reaction time increasing to 40 and 52 h, it was observed that the nanorods became curved and disordered in arrangement due to overgrowth (Fig. S5c & d†). Meanwhile, a constant thickness was obtained at 40 h (7.1 µm) and 52 h (7.2 µm) samples (Fig. S5e†). In contrast, Ni-BPM/NF-D fabricated by direct synthesis was loosely packed, confirming the importance of Ni-LDH as a sacrificial template for the growth of dense nano-arrayed MOFs (Fig. S6†). The orientated Ni-BPM/NF-24 h sample was selected for the test considering MOF overgrowth observed after 24 h. Fig. 1d shows the nanorod morphology of Ni-BPM/NF-24 h. The HRTEM image of Ni-BPM/NF-24 h exhibited a crystal plane spacing value of 0.227 nm indexing to the (0 1 3) facet of Ni-BPM. In addition, TEM energy-dispersive X-ray spectroscopy (EDX) elemental mapping demonstrated that C, O and Ni elements were uniformly distributed across the nanorods in Ni-BPM/NF-24 h (Fig. 1e). Furthermore, the impact of ligand concentration (0.0425, 0.0375, and 0.0325 mmol) and reaction temperature (60, 80, and 100 °C) on the growth process of MOF electrodes was investigated. As demonstrated in Fig. S7 and S8,† elevated concentrations and temperatures can induce excessive growth in MOF nanostructures, while insufficient conditions may result in incomplete growth and a less dense arrangement. In brief, MOF electrodes with nano-arrayed morphology were successfully fabricated via a lattice-matched-template method, which will produce enhanced pore connectivity, increased accessible active sites, and stronger capillary effects. These properties will improve intrinsic electron transfer and extrinsic mass transfer, facilitating the OER process.
2.2 Evaluation of electrocatalytic performance
To elucidate the OER-activated process of Ni-BPM/NF-24 h, the CV measurement was initially conducted from 1.1 to 1.7 V vs. RHE at a scan rate of 50 mV s−1 without iR compensation (Fig. 2a). Notably, the OER current density gradually increased along with the CV cycles at 1.7 V vs. RHE, until it reached a stable state after 50 CV cycles. A recognized redox couple from 1.35 to 1.45 V was observed, which was consistent with the electrochemical redox of Ni2+ to Ni3+/4+ species.32 Besides, the increasing anode current along with the CV test could suggest the continuous phase transformation from Ni2+ into Ni3+/4+, thereby confirming that the electrode underwent activation during CV tests.33 To gain further insight into OER processes, a quasi-steady-state CV scan was conducted over the potential range of 1.1 to 1.65 V vs. RHE at a scan rate of 5 mV −1 with a 95% iR correction applied after different CV cycles (1st, 5th, 15th, 35th, and 50th) at a scan rate of 100 mV s−1. Since the oxidation peak corresponding to Ni2+ overlapped in the anode scan region, the characteristic OER overpotential was estimated to originate from the cathode scan.34 The results demonstrated that Ni-BPM/NF-24 h exhibited the highest steady-state test performance in the 35th cycle, and a slight decline was observed in the 50th cycle (Fig. S9†). These findings again indicated that Ni-BPM/NF-24 h was undergoing a gradual reconstruction towards a stabilized state during the OER.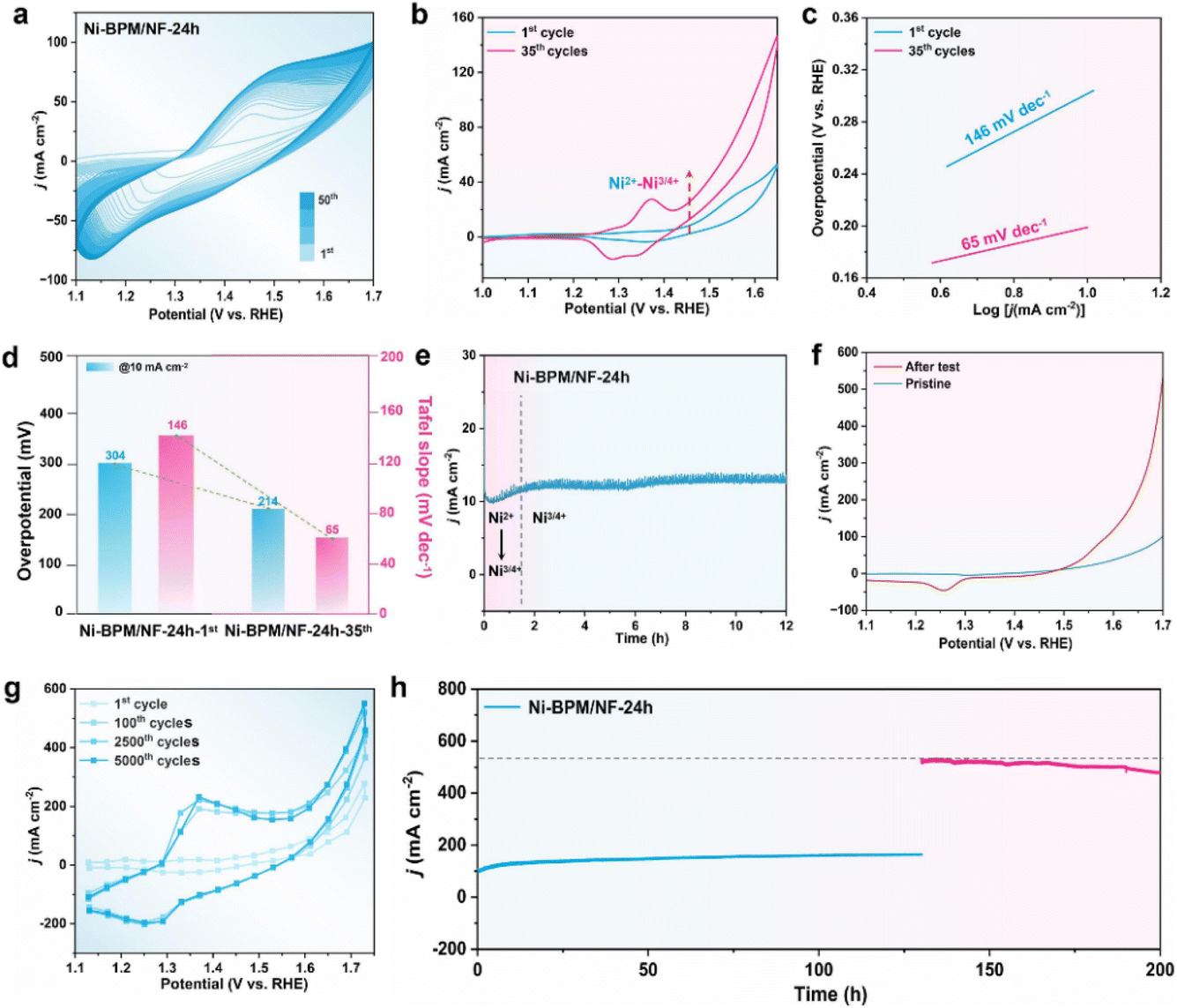 | ||
| Fig. 2 Electrochemical performance of Ni-BPM/NF-24 h. (a) 50 CV cycles in the potential range of 1.1–1.7 V vs. RHE at a scan rate of 50 mV s−1. (b) The quasi-steady-state CV curves recorded at a scan rate of 5 mV s−1 following various CV cycles at a scan rate of 100 mV s−1, between 1 and 1.65 V vs. RHE without iR correction applied. (c) Corresponding Tafel slopes with the increase of CV cycles. (d) The overpotentials required at 10 mA cm−2 and Tafel slopes. (e) Chronopotentiometry curve for 24 h at 10 mA cm−2 in 1 M KOH. (f) LSV curves before and after the chronopotentiometry test. (g) Different CV cycles in the potential range of 1.1–1.75 V vs. RHE at a scan rate of 100 mV s−1. (h) Chronopotentiometry curves for 130 h at 100 mA cm−2 (blue curve), followed by another 70 h at 500 mA cm−2 (pink curve) in 1 M KOH. | ||
Subsequently, the steady-state CV curves of 1st and 35th cycles were selected for further comparison of OER performance. Fig. 2b–d illustrate that the steady-state curve of the 35th cycle necessitated 218 mV to attain a current density of 10 mA cm−2, while the Tafel slope was 66 mV dec−1, surpassing that of the majority of MOF-based electrocatalysts (Table S2†). Notably, the increasing area of the Ni2+/Ni3+/4+ oxidation peak indicated more accessible active species (NiOOH) generated during the OER.35 Furthermore, combined with the in situ Raman test, it is known that the self-reconstruction degree deepens, which was conducive to the improvement of performance (Fig. S10†). To investigate the OER activation process of Ni-BPM/NF-24 h at constant current, chronoamperometry and LSV tests were performed. The current density first enhanced and then remained stable after 1.5 h of subjecting it to the continuous electro-oxidic Ni2+ to Ni3+/4+, and the OER activity was significantly improved in comparison with the initial one (Fig. 2e and f). These findings indicated that the CV-induced structure generated by the self-reconstruction process was conducive to enhanced OER activity.
To gain insight into the long-term stability of Ni-BPM/NF-24 h (stored in a sealed vial for two years), CV tests with 5000 cycles at a sweep rate of 100 mV s−1 and a chronoamperometry test were conducted at high current densities. As depicted in Fig. 2g, the Ni-BPM/NF-24 h 100th cycle curve evinced a pronounced augmentation in the Ni3+/4+ oxidation peak and a substantial enhancement in current density compared with the 1st cycle. Both the current density and oxidation peak gradually increased from the 1st to 100th cycles and reached a plateau at 2500 cycles. After testing for 5000 cycles, the current density was essentially unchanged from the 2500 cycle test. These results demonstrated that Ni-BPM may undergo repair due to the application of a reduction potential leading to high cycling stability. Furthermore, the sample was subjected to a chronoamperometry test at current densities of 100 and 500 mA cm−2. As depicted in Fig. 2h, the current density exhibited an initial increase, followed by a period of stabilization that persisted for a minimum of 130 h of continuous electrolysis at a constant voltage of 1.8 V vs. RHE. Subsequently, when the current density was increased to nearly 500 mA cm−2, Ni-BPM/NF-24 h exhibited only 10% decay of current density after 70 h at a constant voltage of 2.1 V vs. RHE (Fig. 2h), suggesting the superior durability of Ni-BPM/NF-24 h at high current density.
Furthermore, both Ni-BPM/NF-D and Ni-LDH/NF electrodes were selected for comparative testing. Ni-BPM/NF-D exhibited a current density of 10 mA cm−2 at 297 mV, accompanied by a Tafel slope of 69 mV dec−1 which signified a good OER activity in comparison to Ni-LDH/NF (318 mV and 71 mV dec−1) (Fig. S11a & b†). Electrochemical impedance spectroscopy (EIS) was performed to better study catalytic kinetics during the OER. As illustrated in Fig. S11c,† the semicircular diameter of Ni-BPM/NF-24 h (4 Ω) was smaller than those of Ni-BPM/NF-D (4.8 Ω) and Ni-LDH/NF (5.9 Ω). This finding suggested that activated Ni-BPM/NF-24 h displayed the lowest charge transfer resistances (Rct). Besides, the value of Cdl evaluated from CV curves revealed that Ni-BPM/NF-24 h (17.3 mF cm−2) has higher Cdl than Ni-BPM/NF-D (11.4 mF cm−2) and Ni-LDH/NF (7.3 mF cm−2) (Fig. S11d & S12†). The above results suggested that increased electrochemical active area and abundant potential active sites were obtained due to nano-arrayed structures of Ni-BPM/NF-24 h.
2.3 Relationship between MOF reconstruction and performance
In situ Raman spectra were recorded to study the reversible reconstruction process of Ni-BPM surface during the OER. The sample was subjected to a 20-turn CV test at a scan rate of 100 mV s−1 prior to the first test. As shown in Fig. 3, at an applied potential of 1.1 and 1.2 V vs. RHE, the Raman signals of Ni–O, C–H, C–O and C–C bonds at 630, 863,1137 and 1428/1610 cm−1, respectively, are ascribable to the BPM ligand in Ni-BPM.22 As the applied potential increased, two characteristic bands at 470–480 and 545–555 cm−1 appeared which were assigned to the Eg bending vibration (δ(Ni3+–O)) and A1g stretching vibration (ν(Ni3+–O)) mode of γ-NiOOH, respectively.36 And the characteristic peak of Ni-BPM progressively diminished, proving the surface reconstruction of Ni-BPM to NiOOH. The ratio of δ(Ni3+–O) and ν(Ni3+–O) was 2.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, corresponded to γ-NiOOH rather than β-NiOOH.37γ-NiOOH was considered more active than β-NiOOH,33,38,39 which was different from most previously reported results and may produce improved OER activity. This was explained by the similarity between the distance of adjacent Ni atoms and Ni–O bond length of Ni-BPM and those in γ-NiOOH (Table S1†). Surprisingly, when the potential was reversed to 1.1 V vs. RHE, the characteristic peaks of Ni-BPM were observed again, while the intensity of characteristic peaks of γ-NiOOH obviously decreased, suggesting reversible surface reconstruction of Ni-BPM.
:1, corresponded to γ-NiOOH rather than β-NiOOH.37γ-NiOOH was considered more active than β-NiOOH,33,38,39 which was different from most previously reported results and may produce improved OER activity. This was explained by the similarity between the distance of adjacent Ni atoms and Ni–O bond length of Ni-BPM and those in γ-NiOOH (Table S1†). Surprisingly, when the potential was reversed to 1.1 V vs. RHE, the characteristic peaks of Ni-BPM were observed again, while the intensity of characteristic peaks of γ-NiOOH obviously decreased, suggesting reversible surface reconstruction of Ni-BPM.
 | ||
| Fig. 3 In situ Raman spectra of Ni-BPM/NF-24 h during the OER process and the proposed reversible surface reconstruction of Ni-BPM. | ||
The reversible surface reconstruction process was proposed as follows:
In the initial phase, the applied potential in an alkaline medium resulted in the rupture of the Ni–O bond in the BPM ligand molecule or H2O, thereby exposing the uncoordinated Ni centers while simultaneously coordinating hydroxide ions to Ni atoms. With increasing voltage, the Ni–O bond underwent rapid oxidation, forming γ-NiOOH species. The dissociated ligand is hypothesized to remain on the electrode surface through hydrogen bonds or weak coordination bonds. Then, at the applied decreased potential, γ-NiOOH underwent reduction along with free ligand recoordination. Consequently, most γ-NiOOH regenerated to Ni-BPM.
Density functional theory (DFT) calculations were used to gain insight into the improved OER catalytic performance of Ni-BPM. A heterostructure with γ-NiOOH as the upper surface and Ni-BPM as the bottom layer was built to simulate the active structure of Ni-BPM/NF-24 h (Fig. 4a). Based on the four-step adsorbate evolution (AEM) mechanism in alkaline media, the geometrical structures of oxygen-containing intermediates (*OH, *O and *OOH) adsorbed on the interfacial Ni sites of γ-NiOOH/Ni-BPM (Fig. 4a) and the Ni sites of Ni-BPM (Fig. S13†) were optimized, respectively. From the calculated Gibbs free energy depicted in Fig. 4b, the interfacial Ni sites provided a lower overpotential of 0.96 eV compared to the Ni sites of pristine Ni-BPM (1.12 eV) for the rate-determining OER step, where *O formation occurred from *OH. The results indicated that the reconstruction can effectively reduce the energy barrier of Ni-BPM, thus producing enhanced electrocatalytic activity.
 | ||
| Fig. 4 (a) Illustration of intermediates formation during the OER based on the AEM mechanism; (b) free energy diagram of intermediate formation on Ni sites of Ni-BPM and γ-NiOOH/Ni-BPM; (c) DOS plots of γ-NiOOH/Ni-BPM and Ni-BPM. (d) Distribution of interfacial charge. Yellow and blue represent the electron accumulation and dissipation areas, respectively. (e) PDOS plots of Ni 3d of γ-NiOOH/Ni-BPM and Ni-BPM. | ||
To further elucidate the intrinsic OER mechanism underlying the observed potential-dependent OER activity in Ni-BPM, the differential charge density within γ-NiOOH/Ni-BPM was calculated to gain a deeper understanding of its interfacial charge behavior. As depicted in Fig. 4c, the γ-NiOOH/Ni-BPM density of states (DOS) was closer to the Fermi level than that of Ni-BPM, suggesting that γ-NiOOH/Ni-BPM has higher conductivity and enhanced electron-transfer ability compared to individual Ni-BPM.36 The partial density of states (PDOS) of γ-NiOOH/Ni-BPM revealed large orbital overlap between Ni 3d and O 2p (Fig. S14†), indicating strong electronic coupling ability. The charge redistribution at the interfacial Ni sites in the γ-NiOOH/Ni-BPM heterostructure showed more charge accumulation (Fig. 4d), indicating electron transfer from Ni-BPM to γ-NiOOH. A negative shift in the d-band center was observed (Fig. 4e), which would result in a higher occupation degree of the anti-bonding orbital.19 These results suggested that modulating the electronic configuration of Ni sites through surface self-reconstruction during the OER could optimize the adsorption free energy of oxygen-containing intermediates, thereby aiding the OER process.
Post-catalytic study was conducted on Ni-BPM/NF-24 h with the objective of further elucidating the relationship between enhanced OER performance and reversible surface reconstruction of MOFs. The optical photographs revealed that the original MOF electrodes altered to black color following the CV test with varying cycles and then underwent color changes and exhibited their original colors with a slight darkening after CV tests and drying at 60 °C for 2 h (Fig. S15†), suggesting reversible structure reconstruction. XRD patterns of Ni-BPM/NF-24 h after cycle tests showed no discernible change for the (−1 2 0) peak but only intensity decrease of the (0 1 0) peak (Fig. S16†), confirming that the Ni-BPM honeycomb lattice remained intact. SEM images of Ni-BPM/NF-24 h after cycle tests were consistent with the original one (Fig. S17†), and clear lattice fringes of (−1 3 1) planes of Ni-BPM and (2 1 0) planes of γ-NiOOH were observed in HRTEM images after cycle tests (Fig. S18†) indicating structural reconstruction.
The local electronic structure and atomic coordination environment of Ni in pristine Ni-BPM/NF-24 h and after OER CV 35 cycle tests (denoted as Ni-BPM/NF-R) were verified by using X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS). As depicted in XANES spectra (Fig. 5a), the absorption edge of Ni K-edge shifted toward a higher energy side after the OER, indicating that Ni lost electrons and underwent oxidation, consistent with the XPS results.40 In the Fourier-transformed (FT)-EXAFS spectrum, the peak at 1.50 Å was attributed to the Ni–O bond and peak at 2.1 Å was for the distance between two neighboring Ni atoms (Fig. 5b), and the fitting parameter curves of EXAFS spectra are listed in Table S3.† After CV tests, the coordination number of Ni–O increased since the Ni-BPM surface was transformed into γ-NiOOH during the OER. The WT analysis delivered both R-space and k-space information, giving two scatterings at 3.3 and 7.95 Å−1 corresponding to Ni–O and Ni–Ni coordination, respectively (Fig. 5c).
 | ||
Fig. 5 (a) Ni K-edge XANES spectra, (b) the corresponding Fourier transforms of EXAFS spectra of Ni-BPM/NF-24 h and reference samples and (c) WT images. (d) Ni 2p XPS spectra and (e) O 1s XPS spectra of Ni-BPM/NF-24 h and after the 15th and 30th cycles, respectively. The peak corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C from the C 1s high-resolution spectrum was at 284.8 eV and was used for charge calibration.41 C from the C 1s high-resolution spectrum was at 284.8 eV and was used for charge calibration.41 | ||
To gain further insight into the valence state of Ni-BPM/NF-24 h both before and after CV cycle tests and to elucidate the relationship between cycle numbers and the reconstruction degree, X-ray photoelectron spectroscopy (XPS) was conducted. As illustrated in Fig. 5d, the 2p3/2 and 2p1/2 peaks composed of Ni2+ shifted to higher binding energies both after 15 cycles (0.62 eV) and 35 cycles (1.22 eV) tests, respectively, implying an increased proportion of high-valent Ni.19 Likewise, comparable patterns were observed in the O 1s data (Fig. 5e). The O-2 peaks shifted to lower binding energies after 15 cycles (0.37 eV) and 35 cycles (0.87 eV), respectively.20 These findings again confirmed that the Ni-BPM/NF-24 h surface was in situ transformed into γ-NiOOH/Ni-BPM during the OER and the reconstruction degree was found to be positively correlated with cycle numbers.
To further enhance OER performance, Fe/Co atoms were introduced into the Ni-BPM lattice, resulting in bimetallic nano-arrayed MOF electrodes. Successful preparation of all the corresponding electrodes was confirmed through XRD patterns (Fig. S19†), SEM images (Fig. S20†) and ICP analysis (Table S4†). Subsequently, the OER performance of NiFe-BPM/NF and NiCo-BPM/NF was evaluated under 1 M KOH conditions, while an IrO2 catalyst on an NF support was used for comparison. Owing to the strong synergistic effect between Ni and Fe active sites, the ultra-high catalytic activity of NiFe-BPM/NF over NiCo-BPM/NF and Ni-BPM/NF was obtained. As shown in Fig. S21a,† NiFe-BPM/NF only needs 198 mV to deliver a current density of 10 mA cm−2, lower than that of NiCo-BPM/NF-24 h (210 mV), Ni-BPM/NF-24 h (218 mV) and IrO2/NF (365 mV) respectively. At a higher current density of 100 mA cm−2, NiFe-BPM/NF still exhibited the best performance with an overpotential of 287 mV. In addition, NiFe-BPM/NF showed the smallest Tafel slope of 43 mV dec−1 in comparison with NiCo-BPM/NF-24 h (96 mV dec−1), Ni-BPM/NF-24 h (66 mV dec−1) and IrO2/NF (98 mV dec−1), respectively, confirming its faster OER kinetics (Fig. S21b†). The above results confirmed that the construction of bimetallic synergism would efficiently improve Ni-MOF OER activity.
3. Conclusions
In summary, reversible MOF surface reconstruction to active γ-NiOOH during the OER was realized by selecting Ni-BPM as a potential electrocatalyst. By using a sacrificial lattice-matched-template approach, the Ni-BPM/NF-24 h electrode was directionally prepared and showed a continuous increase in catalytic activity until reaching a stable state with an overpotential of 218 mV at 10 mA cm−2. This phenomenon can be attributed to Ni-BPM self-reconstruction during activation, which formed a γ-NiOOH/Ni-BPM heterostructure facilitating the OER process. In situ Raman spectra verified the reversible reconstruction of Ni-BPM, and DFT calculations revealed the altered electronic configuration of Ni sites in the γ-NiOOH/MOF heterostructure, further confirmed by XANES analysis. The reversible surface reconstruction mitigated any potential performance loss due to structural damage that may occur during electrocatalysis and produced durable stability. Ni-BPM/NF-24 h demonstrated the ability to maintain stability in the IT test for at least 200 hours, including 170 h at 100 mA cm−2 followed by another 70 h at 500 mA cm−2. In addition, the introduction of Fe/Co atoms into Ni-BPM resulted in further enhanced OER activities due to bimetallic synergies. This research lays a foundation for the development of reversibly reconstructed MOF electrocatalysts.4. Experimental section
4.1 Materials
4,4′-Dihydroxybiphenyl-3,3′-dicarboxylic acid (H2dobpdc) (Jilin Chinese Academy of Sciences – Yanshen Technology Co. Ltd), nickel foam (NF) (Jilin Tianxuan Economic and Trade Co. Ltd), urea, NiCl2·6H2O (Sigma-Aldrich, 99%), CoCl2·6H2O (Sigma-Aldrich, 99%) and FeCl2·4H2O (Energy-Chemical, 99%) were used as received without further purification. N,N-Dimethylformamide (DMF) and ethanol were purchased from Beijing Chemical Works. All other reagents and solvents were purchased from local commercial suppliers.4.2 Preparation of Ni-LDH/NF and Ni-BPM/NF-D electrodes
Typically, NF was first washed with ethanol and deionized water by ultrasonication to remove the surface organics and then rinsed with 3 M HCl solution to remove the oxide layer and organic residues. Finally, NF was washed with ethanol several times and dried at 80 °C for 30 min. A mixture of NiCl2·6H2O (275 mg, 1.2 mmol), urea (380 mg, 6.23 mmol) and deionized water (10 mL) was added to a 50 mL Teflon-lined autoclave. Then, the cleaned NF (0.8 cm × 1 cm) was placed in the Teflon-lined autoclave and the system was heated at 120 °C for 12 h. After cooling down to room temperature, the resulting Ni-LDH/NF was washed with deionized water and dried at 80 °C for 2 h.NF (1.5 × 2 cm2) was immersed into a 20 mL glass vial containing NiCl2·6H2O (27.5 mg, 0.12 mmol), H2dobpdc (10.3 mg, 0.0375 mmol), DMF (2 mL) ethanol (2 mL) and deionized water (2 mL). The solution was then heated at 80 °C for 24 h, after which the film electrode was taken and thoroughly washed with ethanol several times under ultrasonication. Finally, the Ni-BPM/NF-D electrode was dried at 80 °C overnight.
4.3 Preparation of Ni-BPM/NF-x (x = 8, 16, 24, 40, 52 h) electrodes
Ni-LDH/NF was immersed into a 20 mL glass vial containing H2dobpdc (10.3 mg, 0.0375 mmol), DMF (2 mL), ethanol (2 mL) and deionized water (2 mL) heated at 80 °C for 8, 16, 24, 40 and 52 h, respectively. After cooling down to room temperature, the film electrode was taken and thoroughly washed with ethanol several times under ultrasonication and finally dried at 80 °C overnight.4.4 Preparation of NiM (M = Co, Fe)-BPM/NF electrodes
A similar experimental procedure was employed for fabrication of NiCo-BPM/LDH/NF and NiFe-BPM/LDH/NF with the metal sources of NiCl2·6H2O (0.6 mmol) and CoCl2·6H2O (0.6 mmol) for NiCo-BPM/LDH/NF and NiCl2·6H2O (0.6 mmol) and FeCl2 (0.6 mmol) for NiFe-BPM/LDH/NF, respectively.4.5 Preparation of IrO2/NF electrodes
5 mg IrO2 was dispersed in a mixture of 0.98 mL EtOH and 0.02 mL Nafion solution (5 wt%) to prepare an IrO2 suspension. 0.1 mL of the suspension was dropped on the pretreated NF supports with a size of 1.0 × 0.8 cm2 and then dried at room temperature.4.6 Physical characterization
Powder X-ray diffraction (PXRD) measurements were conducted using a Rigaku SmartLab X-ray diffractometer with Cu-Kα radiation (40 kV, 30 mA, λ = 1.5418 Å) and a scanning step of 0.01°. SEM characterization was performed on an XL-30 ESEM FEG scanning electron microscope FEI COMPANYTM. The TEM was recorded using a JEOL JEM 2100F and the energy spectrum was obtained using an FEI Tecnai G2 F20. More detailed structure information was obtained with a high-resolution transmission electron microscope. ICP anaysis was performed on a PE300D. In situ Raman spectroscopy was performed using a Gaoss Union C031-3 Raman electrolytic cell and spectra were recorded on a Renishaw with a wavelength of 633 nm. X-ray absorption fine structure (XAFS) was measured at the BL11B beamlines at the Shanghai Synchrotron Radiation Facility (SSRF) (Shanghai, China). XPS measurements were performed on a Thermo ESCALAB 250.4.7 Electrochemical characterization
All electrochemical measurements were conducted using a standard three-electrode cell using a CHI 760E electrochemical workstation (Shanghai Chenhua Instrument Co. Ltd) in 1 M KOH (pH = 13.8). All film electrodes with a size of 0.8 cm × 1 cm were directly used as the working electrode, a platinum plate (1 cm × 1 cm) was used as the counter electrode, and Ag/AgCl (3.5 M KCl) was used as the reference electrode. For comparison, IrO2 powder (5 mg) was dispersed in 960 µL ethanol solution including 40 µL 5 wt% Nafion with sonication for 30 min to obtain a uniform catalyst ink. Then, the ink was loaded on clean NF. Before all the tests, the electrolyte was purged with N2 for at least 30 min to remove dissolved oxygen from water. All the measured potentials were converted to RHEs: ERHE = EAg/AgCl + 0.059 × pH + 0.205. EIS was carried out at 1.56 V vs. RHE over a frequency range from 100 kHz to 0.1 Hz with a 5 mV amplitude. CV curves were recorded in the non-faradaic region with an increasing scan rate of 25, 50, 75, 100 and 125 mV s−1, respectively. The chronoamperometry measurement was conducted for 43200 s at an overpotential of 10 mA cm−2.
Data availability
All data that support the findings of this study are included within the article.Author contributions
Shulin Li: conceptualization, methodology, writing the original draft, data curation, visualization. Zhaoxin Zhou: methodology, formal analysis, editing. Jiahui Li and Yang Xiao: data curation. Ye Yuan and He Zhu: investigation. Fengchao Cui: theoretical calculation. Xiaofei Jing: conceptualization, formal analysis, supervision, writing– review & editing. Guangshan Zhu: conceptualization, validation, funding acquisition, resources.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 22475036 and 22131004) and the “111” project (B18012).References
- J. Wang, R. Rao, L. Giordano, Y. Katayama, Y. Yu and Y. Shao-Horn, Science, 2017, 358, 751–756 CrossRef.
- S. Li, Z. Zhou, G. Liu, Q. Zhang, Y. Gao, H. Zhu and S. Zhu, Chem. Eng. J., 2024, 493, 152820 CrossRef CAS.
- Z. Zhang, X. Ma, Y. Li, N. Ma, M. Wang, W. Liu, J. Peng, Y. Liu and Y. Li, J. Am. Chem. Soc., 2024, 146, 8425–8434 CrossRef CAS PubMed.
- X. Peng, L. Chen, Y. Liu, C. Liu, H. Huang, J. Fan, P. Xiong and J. Zhu, Chem. Synth., 2023, 3, 47 CAS.
- J. Han, Q. Liu, Y. Yang and H. Wu, Chem. Sci., 2025, 16, 3788 RSC.
- J. Zhou, Y. Dou, X. Wu, A. Zhou, L. Shu and J. Li, Small, 2020, 16, e1906564 CrossRef PubMed.
- Y. Zhang and H. Liang, Chem. Synth., 2024, 4, 39 CAS.
- L. Jin, H. Xu, K. Wang, Y. Liu, X. Qian, H. Chen and G. He, Chem. Sci., 2025, 16, 329 RSC.
- H. Zhong, X. Wang, G. Sun, Y. Tang, S. Tan, Q. He, J. Zhang, T. Xiong, C. Diao, Z. Yu, S. Xi, W. Lee and J. Xue, Energy Environ. Sci., 2023, 16, 641–652 RSC.
- X. Mu, M. Yu, X. Liu, Y. Liao, F. Chen, H. Pan, Z. Chen, S. Liu, D. Wang and S. Mu, ACS Energy Lett., 2024, 9, 5763–5770 CrossRef CAS.
- X. Wang, H. Zhong, S. Xi, W. Lee and J. Xue, Adv. Mater., 2022, 34, 2107956 CrossRef CAS.
- N. Burtch, H. Jasuja and K. Walton, Chem. Rev., 2014, 114, 10575–10612 CrossRef CAS.
- Y. Wang, B. Liu, X. Shen, H. Arandiyan, T. Zhao and Y. Li, Adv. Energy Mater., 2021, 11, 2003759 CrossRef CAS.
- B. Zhang, Y. Zheng, T. Ma, C. Yang, Y. Peng, Z. Zhou, M. Zhou, S. Li, Y. Wang and C. Cheng, Adv. Mater., 2021, 33, e2006042 CrossRef PubMed.
- S. Li, T. Wang, D. Tang, Y. Yang, Y. Tian, F. Cui, J. Sun, X. Jing, D. Sholl and G. Zhu, Adv. Sci., 2022, 9, 202203712 Search PubMed.
- S. Yuan, J. Peng, B. Cai, Z. Huang, A. Garcia-Esparza, D. Sokaras, Y. Zhang, L. Giordano, K. Akkiraju, Y. Zhu, R. Hubner, X. Zou, Y. Roman-Leshkov and Y. Shao-Horn, Nat. Mater., 2022, 21, 673–680 CrossRef CAS PubMed.
- S. Li, D. Tang and X. Jing, Chem. Synth., 2024, 4, 70 CAS.
- Q. Qi, C. Zhang and J. Hu, Coord. Chem. Rev., 2025, 522, 216235 CrossRef CAS.
- Y. Jiang, T. Chen, J. Chen, Y. Liu, X. Yuan, J. Yan, Q. Sun, Z. Xu, D. Zhang, X. Wang, C. Meng, X. Guo, L. Ren, L. Liu and R. Lin, Adv. Mater., 2024, 36, 202306910 Search PubMed.
- X. Wang, W. Zhou, S. Zhai, X. Chen, Z. Peng, Z. Liu, W.-Q. Deng and H. Wu, Angew. Chem., Int. Ed., 2024, 63, e202400323 CrossRef CAS PubMed.
- Z. Yan, H. Liu, Z. Hao, M. Yu, X. Chen and J. Chen, Chem. Sci., 2020, 11, 10614 RSC.
- L. Zhang, J. Wang, K. Jiang, Z. Xiao, Y. Gao, S. Lin and B. Chen, Angew. Chem., Int. Ed., 2022, 61, e202214794 CrossRef CAS PubMed.
- S. Zhang, Z. Huang, T. Isimjan, D. Cai and X. Yang, Appl. Catal., B, 2024, 343, 123448 CrossRef CAS.
- W. Zheng and L. Lee, ACS Energy Lett., 2021, 6, 2838–2843 CrossRef CAS.
- J. Huang, A. Clark, N. Hales, K. Crossley, J. Guehl, R. Skoupy, T. Schmidt and E. Fabbri, Nat. Chem., 2025, s41557 Search PubMed.
- S. Zhao, C. Tan, C.-T. He, P. An, F. Xie, S. Jiang, Y. Zhu, K.-H. Wu, B. Zhang, H. Li, J. Zhang, Y. Chen, S. Liu, J. Dong and Z. Tang, Nat. Energy, 2020, 5, 881–890 CrossRef CAS.
- N. Heidary, M. Morency, D. Chartrand, K. Ly, R. Iftimie and N. Kornienko, J. Am. Chem. Soc., 2020, 142, 12382–12393 CrossRef CAS PubMed.
- Z. Kou, X. Li, L. Zhang, W. Zang, X. Gao and J. Wang, Small Sci., 2021, 1, 2100011 CrossRef CAS PubMed.
- M. Liu, Y.-Q. Zhang, X. Wang, F. Lang, N. Li and X.-H. Bu, Sci. China:Chem., 2023, 66, 2754–2779 CrossRef CAS.
- Y.-J. Tang and Y.-Q. Lan, Sci. China:Chem., 2023, 66, 943–965 CrossRef CAS.
- Y. Wang, L. Yan, K. Dastafkan, C. Zhao, X. Zhao, Y. Xue, J. Huo, S. Li and Q. Zhai, Adv. Mater., 2022, 34, 202109927 Search PubMed.
- C.-P. Wang, Y. Feng, H. Sun, Y. Wang, J. Yin, Z. Yao, X.-H. Bu and J. Zhu, ACS Catal., 2021, 11, 7132–7143 CrossRef CAS.
- L. Dai, C. Fang, F. Yao, X. Zhang, X. Xu, S. Han, J. Deng, J. Zhu and J. Sun, Appl. Surf. Sci., 2023, 623, 156991 CrossRef CAS.
- J. Duan, S. Chen and C. Zhao, Nat. Commun., 2017, 8, 15341 CrossRef CAS.
- H. Sun, L. Chen, Y. Lian, W. Yang, L. Lin, Y. Chen, J. Xu, D. Wang, X. Yang, M. Rummerli, J. Guo, J. Zhong, Z. Deng, Y. Jiao, Y. Peng and S. Qiao, Adv. Mater., 2020, 32, e2006784 CrossRef.
- X. Ma, D. Zheng, S. Hou, S. Mukherjee, R. Khare, G. Gao, Q. Ai, B. Garlyyev, W. Li, M. Koch, J. Mink, Y. Shao-Horn, J. Warnan, A. Bandarenka and R. Fischer, ACS Catal., 2023, 13, 7587–7596 CrossRef CAS.
- M. Louie and A. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 Search PubMed.
- D. Bediako, B. Lassalle-Kaiser, Y. Surendranath, J. Yano, V. Yachandra and D. Nocera, J. Am. Chem. Soc., 2012, 134, 6801–6809 CrossRef CAS PubMed.
- W. Xu, Y. Tao, H. Zhang, J. Zhu, W. Shao, J. Sun, Y. Xia, Y. Ha, H. Yang, T. Cheng and X. Sun, Small, 2024, 20, 2407328 CrossRef CAS PubMed.
- C. Zhang, Q. Qi, Y. Mei, J. Hu, M. Sun, Y. Zhang, B. Huang, L. Zhang and S. Yang, Adv. Mater., 2023, 35, e2208904 CrossRef PubMed.
- K. Siegbahn, ESCA: atomic molecular and solid state structure studied by means of electron spectroscopy, 1967 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and Fig. S1–S18 and Tables S1–S4 as noted in the text. See DOI: https://doi.org/10.1039/d5sc02536b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |