
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly stereoselective synthesis of polysubstituted housanes and spiro-oxa-housanes: application and mechanistic insights†
Abdur Rouf Samim Mondal ,
Nakul Abhay Bapat,
Harshita Mishra and
Durga Prasad Hari*
,
Nakul Abhay Bapat,
Harshita Mishra and
Durga Prasad Hari*
Department of Organic Chemistry, Indian Institute of Science, Bangalore, 560012, India. E-mail: dphari@iisc.ac.in
First published on 4th June 2025
Abstract
Ring-strain-enabled transformations have made significant progress, pushed the boundaries of unexplored chemical space, and emerged as a powerful tool for constructing complex molecules selectively and efficiently. Among the strained ring systems, [1.1.1]propellane, bicyclobutane (BCB), and azabicyclobutane (ABB) have garnered substantial attention and found numerous synthetic applications. In contrast, the chemistry of bicyclo[2.1.0]pentane, commonly known as housane, is scantly explored due to the lack of modular synthetic approaches. Herein, we describe a highly stereoselective, catalytic strategy for synthesizing polysubstituted housanes with up to three contiguous all-carbon-quaternary centers. The reaction is very efficient, works under mild conditions, requires visible light and organic dye as a photocatalyst, and exhibits a broad substrate scope. Furthermore, we have also developed a highly diastereoselective Paterno–Buchi reaction for synthesizing spiro-oxa-housanes, which are novel molecular entities in the literature. The post-synthetic diversification of the products via a strain-release driven diastereospecific 1,2-ester migration that allows the rapid synthesis of functionalized bicyclic imides further highlighted the synthetic utility of the current protocol. Combined experimental studies and computational investigations revealed the origin of the reactivity and stereoselectivity.
Introduction
Constructing C(sp3)-rich scaffolds is of great significance in synthetic chemistry as they are invaluable in pharmaceuticals and drug discovery programs, providing improved selectivity, potency, and metabolic stability of drug candidates augmented with minimizing entropic penalty when binding to biological targets.1 In this context, strained ring systems have recently attracted substantial attention since they are ideal sources of C(sp3)-rich rings with rigid and well-defined three-dimensional (3D) spatial arrangements. Moreover, they can undergo a myriad of transformations to generate diverse molecular frameworks owing to their unique bonding and the reactivity enabled by strain release.2 As a result, considerable effort has been directed toward synthesizing and functionalizing strained ring systems. Among these, [1.1.1]propellane, bicyclo[1.1.0]butane (BCB), and azabicyclo[1.1.0]butane (ABB) have been extensively studied and widely applied to generate highly rigid 3D architecture.3 In contrast, bicyclo[2.1.0]pentane, commonly known as housane, has received scant attention. Housane is comprised of two C(sp3)-rich rings-cyclopropane and cyclobutane-with ring-strain energy of 57 kcal mol−1, and it is considered as a potential bioisostere of cyclopentane (Scheme 1A).4 Identifying such rigid counterparts for conformationally flexible cyclopentane is crucial to advancing drug discovery. Furthermore, housane is not only embedded in natural products but also used as a reactive intermediate in the synthesis of complex molecules or natural products (Scheme 1A).5 Nevertheless, the limited synthetic accessibility of housane derivatives thwarted their application in medicinal chemistry discovery programs. Traditionally, housanes are accessed via transannular alkylation of 1,3-functionalized cyclopentane derivatives (Scheme 1B).4c,6 However, this strategy requires multiple steps to prepare the starting material, and accessing different substitution patterns is challenging. Other alternative approaches involve metal-mediated cyclopropanation of cyclobutenes or intramolecular cyclopropanations.7 Recently, the Vicente group reported an elegant strategy through Rh(II)-promoted isomerization of vinylcyclopropenyl carbinols (Scheme 1B).8 While these strategies are highly effective, they require pre-functionalization of the starting material and expensive rhodium catalysts. Furthermore, controlling stereochemistry, especially at quaternary centers, is highly challenging as the number of substituents increases. In this context, developing efficient, sustainable, and catalytic stereoselective strategies for synthesizing polysubstituted housanes with contiguous quaternary carbon centers is highly desirable. | ||
| Scheme 1 (A) Overview of housanes. (B) Known methods for the synthesis of housanes and our reaction design. (C) This work: highly diastereoselective synthesis of polysubstituted housanes and spiro-oxa-housanes and their synthetic application. | ||
Over the last decade, visible light photocatalysis has evolved as a novel paradigm in chemical synthesis, harnessing photons as a traceless energy source, enabling numerous transformations under mild and sustainable conditions for constructing complex scaffolds.9 In this vein, [2 + 2] cycloadditions under visible light have garnered significant interest, not only due to their efficiency but also, their practical applicability in converting simple feedstock materials into structurally complex and unique skeletons.10 A few studies on the cyclopropene–alkene [2 + 2] cycloaddition reactions under photocatalysis have been reported for housane synthesis; however, they suffered from stereoselectivity issues and limited substrate scope.11
We envisioned that cyclopropene 1, a highly strained smallest carbocycle with ring-strain energy of 54.5 kcal mol−1,4b could generate a biradical intermediate I under energy transfer photocatalysis. Subsequent radical addition to an alkene 2 would furnish 1,4-biradical II that could undergo radical–radical recombination to give housane 3. This proposal would provide a straightforward and modular approach for the construction of polysubstituted housane derivatives (Scheme 1B). The major challenges associated with this cycloaddition would be overcoming the diastereoselectivity issue12 and subduing the potential oxidative ring-opening13 and dimerization of cyclopropene or alkene.14
Notwithstanding these challenges, herein we report a highly diastereoselective, catalytic strategy for the synthesis of polysubstituted housanes bearing three contiguous all-carbon-quaternary centers under energy transfer photocatalysis using an organic dye (Scheme 1C). The protocol displays a broad substrate scope and excellent functional group compatibility. We have further developed a diastereoselective Paterno–Buchi reaction to synthesize spiro-oxa-housanes, a novel class of compounds not reported in the literature. The applicability of this method is demonstrated by developing strain-release driven skeletal rearrangement of the obtained housanes via an unusual 1,2-ester migration, enabling a highly diastereospecific approach to densely functionalized bicyclic imide derivatives. DFT calculations combined with experiment-based mechanistic studies were used to rationalize the observed reactivity and the origin of stereoselectivity.
Results and discussion
We commenced our investigation by studying the reaction of cyclopropene 1a with maleimide 2a using [Ru(bpy)3]Cl2 (PC-1) as a photocatalyst under blue light irradiation in CH3CN at 20 °C. However, we didn't observe the formation of the desired housane 3a, and the starting materials were simply reisolated (Fig. 1, column 1). The lack of reactivity is attributed to the high triplet energy of cyclopropene 1a (ET = 47.2 kcal mol−1) over the Ru-catalyst (ET = 46.5 kcal mol−1).9c Next, we screened photocatalysts that have more triplet energies than cyclopropene 1a. To our delight, Ir(ppy)3 (PC-2, ET =![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 58.1 kcal mol−1) gave two diastereomeric products 3a and 3a′ out of four possible products in 25% yield with complete trans selectivity and 3:1 endo/exo selectivity at C1 (Fig. 1, column 2). The yield could be significantly improved to 82% using a higher triplet energy photocatalyst [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (PC-3, ET =61.8 kcal mol−1) (Fig. 1, column 3).
58.1 kcal mol−1) gave two diastereomeric products 3a and 3a′ out of four possible products in 25% yield with complete trans selectivity and 3:1 endo/exo selectivity at C1 (Fig. 1, column 2). The yield could be significantly improved to 82% using a higher triplet energy photocatalyst [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (PC-3, ET =61.8 kcal mol−1) (Fig. 1, column 3).
 | ||
| Fig. 1 Reaction conditions: from column 1 to 8, 1a (0.10 mmol), 2a (2 equiv), solvent (0.1 M), photocatalyst (PC) (2.5 mol%), 440 nm, 20 °C, 20 h. CH3CN was used as a solvent while screening the photocatalysts. NMR yields using dibromomethane as the internal standard, and dr was determined by 1H-NMR from the crude reaction mixture. Column 9 – using 1b. Column 10 – using 1c. Column 11 – without PC. Column 12 – without light. | ||
We also explored metal-free energy transfer photocatalysts and found that 4CzIPN (PC-4, ET = 62.0 kcal mol−1) is very effective and gave 92% yield (Fig. 1, column 4).15 Among the other solvents (DCM, DCE, EtOAc, and PhCF3) tested, DCM was the optimal solvent for this transformation, giving the desired product in 95% yield (Fig. 1, column 5). Controlling the endo/exo selectivity of this reaction would further enrich its synthetic application. We then shifted our focus to improving the selectivity of the reaction. We considered whether steric effects could contribute to improved selectivity. However, the incorporation of a bulky group such as tert-butyl on the ester side didn't improve the selectivity of the reaction (product 3b, Fig. 1, column 9). We were pleased to find that the introduction of a simple methyl group on the ortho-position of the phenyl in cyclopropene 1c improved the endo-selectivity significantly, affording the desired product 3c with complete stereocontrol over five contiguous chiral centers (Fig. 1, column 10). Finally, control experiments confirmed that the reaction did not occur without light or a photocatalyst (Fig. 1, columns 11 and 12).
With the optimized reaction conditions in hand, we examined the scope of the reaction with a variety of cyclopropenes using N-phenylmaleimide 2a (Scheme 2). First, we examined the steric and electronic influences of various substitutions on the aryl group at the C1 position on cyclopropene. Electron-donating and-withdrawing substituents at the ortho position are well tolerated, affording the desired products (3c–3e) in excellent yields and selectivity. ortho-Chloro and bromo-substituted aryl groups underwent the desired transformation successfully and furnished the products 3f and 3g with good yields and selectivity. meta-CF3-substituted aryl group also participated in this reaction, albeit in low yield and moderate selectivity (product 3h). Next, we explored different substitutions at C2 and C3 positions on cyclopropene. We were pleased to find that the reaction worked well with a simple H at the C2 position and gave the desired product 3i with excellent selectivity. Additionally, the scale up (2 mmol) of 3i didn't hamper reaction efficiency. Various substitutions on the aryl group at the C3 position, including chloro, methyl, fluoro, and ethoxy groups, were well tolerated (3j–3m). Ethyl-ester-derived cyclopropene is also a viable substrate in this reaction, giving the product 3n in excellent selectivity. The structure of product 3n was unambiguously determined by X-ray analysis (CCDC 2404132†). Diester containing cyclopropene yielded the housane 3o in moderate yield. The reaction is not limited to esters at the C1 position; we could also use alkyl groups, resulting in the formation of housanes 3p (CCDC 2449293†) and 3q (CCDC 2451397†). Unfortunately, cyclopropenes containing a methyl group at C2 and a phenyl group at C3, as well as non-styrenic cyclopropenes, were found to be unsuitable for this transformation (see ESI† for details on unsuccessful substrates).
 | ||
| Scheme 2 Reaction conditions: 0.3 mmol of 1, 0.6 mmol of maleimide 2a, 3.0 mL of dry DCM, 2.5 mol% of 4CzIPN, 440 nm, 20 °C, 20 h. Yields are of isolated products, and dr was determined by 1H-NMR and 19F-NMR from the crude reaction mixture. a2 mmol scale. | ||
We subsequently investigated the scope of the reaction with N-substituted maleimides (Scheme 3). A broad range of electron-withdrawing groups on the aryl group of N-substituted maleimides, including fluoro, chloro, bromo, cyano, and trifluoromethyl groups, smoothly participated in the reaction to afford the corresponding housanes 3r–3y in good yields with excellent selectivity. A slight drop in the reaction yield was observed when a strongly donating para-methoxy group was introduced (product 3z). Next, we extended our focus to test the viability of N-alkylated maleimides. Various N-alkyl maleimides, including methyl, cyclopropyl, and cyclohexyl, were well tolerated (products 3aa–3ad). Notably, simple maleimide and N-phthalimide were also well tolerated, affording the products 3ae and 3af in 68% and 85% yields with excellent selectivity. To our delight, bis-maleimide participated and gave the desired bis-housane 3ag in excellent yield. Ethyl acrylate could also yield the cycloaddition product 3ah with moderate yield and excellent diastereoselectivity. Furthermore, to improve the selectivity with cyclopropenes having simple phenyl and para-substituted aryls at the C1 position, we employed N-(ortho-tert-butyl phenyl) maleimide. As expected, the products 3ai–3ak were obtained with improved selectivity (∼2.5 fold times) and moderate to good yields. Surprisingly, methyl substituted maleimides, maleic anhydride and other alkenes didn't participate in this reaction (see ESI† for unsuccessful substrates).
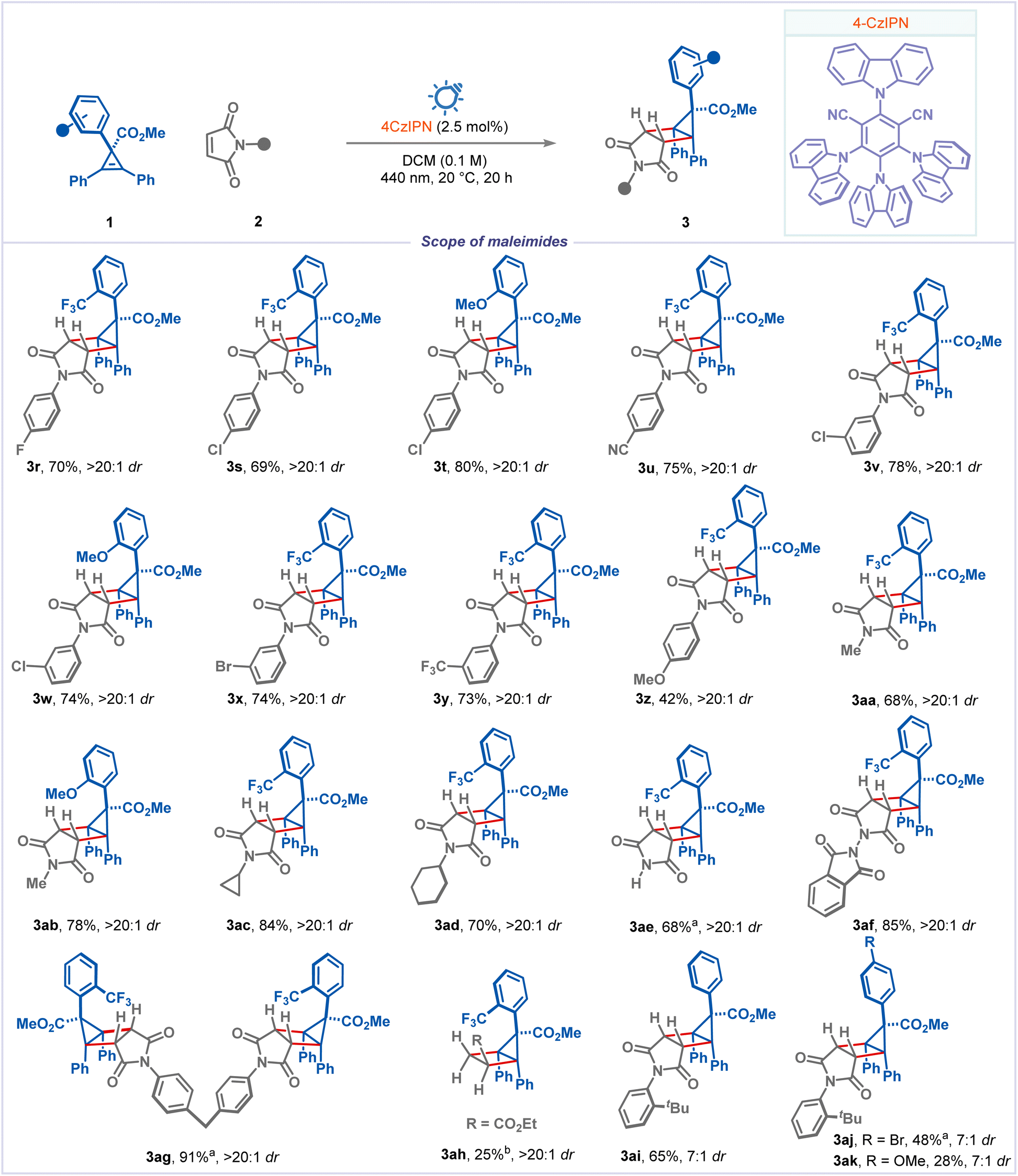 | ||
| Scheme 3 Reaction conditions: 0.3 mmol of 1, 0.6 mmol of maleimide 2, 3.0 mL of dry DCM, 2.5 mol% of 4CzIPN, 440 nm, 20 °C, 20 h. Yields are of isolated products, and dr was determined by 1H-NMR and 19F-NMR from the crude reaction mixture. aNMR yield. bUsing PC-3 ([Ir(dF(CF3)ppy)2(dtbbpy)]PF6). | ||
The synthesis of new spirocyclic scaffolds is an important research area in medicinal chemistry since their incorporation dramatically alters the properties of a molecule, including chemical reactivity, biological activity, metabolic stability, and synthetic tractability.16 Owing to their structural rigidity and well-defined exit vectors, they can mark important features of ring structures beyond their native functionalities, distinguishing them as prominent structural motifs in drug discovery programs.17 Therefore, we next wanted to develop a Paterno–Buchi reaction using cyclopropene for the synthesis of spiro-oxa-housanes, which are novel molecular entities in the literature. To our delight, we obtained the desired spiro-oxa-housane 5a in a 30% yield when we reacted benzoquinone 4a with cyclopropene 1o under visible light irradiation (Scheme 4).18 After briefly investigating various parameters, such as solvents, and reaction time, we found that the yield could be improved to 64% (see ESI† for the complete optimization studies). With the optimized conditions in hand, we investigated the scope of the reaction. Diesters bearing cyclopropenes participated well in this reaction, giving spiro-oxa-housanes 5a and 5b in 52% and 64% yields, respectively. para-Fluoro substituted phenyl at the C3 position of cyclopropenes can also be accommodated in this protocol (product 5c). Dichloro-substituted benzoquinone also underwent the desired cycloaddition, yielding spiro-oxa-housanes 5d–5f in moderate to good yields. Notably, aryl and ester substituents at the C1 position of cyclopropenes can also be tolerated, giving the products as single diastereomers (5g–5i). The structure of product 5h was confirmed by X-ray analysis (CCDC 2425129†). Unfortunately, the reaction didn't work with naphthyl-substituted cyclopropene (at C1), di-tert-butyl benzoquinone and 2-oxo-2-phenylacetate (see ESI† for unsuccessful substrates).
 | ||
| Scheme 4 Reaction conditions: 0.3 mmol of 1, 0.6 mmol of benzoquinone derivative 4, 6.0 mL of dry CH3CN, 440 nm, rt, 2.5 h. Yields are of isolated products and dr was determined by 1H-NMR and 19F-NMR from the crude reaction mixture. aNMR yield. | ||
Synthetic application
In the realm of nitrogen-heterocycles, bicyclic imides and their derivatives are among the most important scaffolds serving as vital structural components within natural products, pharmaceuticals, and bioactive compounds, and, hence, lead to them spanning a wide range of drug classes, including anticancer, GG tase-I inhibitor, antidiabetic agents (Scheme 5A).19 Moreover, owing to its restricted conformation, the bicyclic pyrrolidine motif is considered a potential bioisosteric replacement for piperidine. Petrukhin and co-workers showed that replacing piperidine moiety in A1120 with bicyclic pyrrolidine significantly enhanced its binding towards RBP4 and improved pharmacokinetic and pharmacodynamic properties.20 Nevertheless, further probing such opportunities is currently impeded by the dearth of methods that enable access to such heterobicyclic motifs. The most common method for the construction of bicyclic imides is through an intermolecular [3 + 2] cycloaddition strategy (Scheme 5A).19a Recently, “skeletal remodeling” has garnered significant attention as a powerful tool in organic synthesis to remodel skeletal frameworks and has been widely used as a guiding strategy for assembling unique complex skeletons from simple molecular structures via deconstruction or re-editing the core ring structure through breaking and reforming carbon–carbon or carbon–heteroatom bonds.21 In this regard, radical-mediated rearrangements, especially functional group migration (FGM) strategies, have become powerful tools for making complex scaffolds.22 To demonstrate the potential synthetic utility of our strategy, we envisioned the possibility of strain release-enabled skeletal rearrangement of the obtained housane derivatives by breaking the strained C–C σ bond, followed by 1,2-functional group migration which would directly lead to densely functionalized bicyclic imide derivatives (Scheme 5B). Such 1,2-functional group migrations other than HAT are scarcely explored.4d,5c To our delight, the housane 3e underwent the strained C–C σ bond scission under thermal conditions afforded a bicyclic scaffold 6a in 20% yield with 100% diastereospecificity (ds). Surprisingly, the crystal structure of 6c (CCDC 2409070†) revealed that product 6a emerged from a very rare 1,2-ester migration.23 Noteworthy, the reaction is diastereospecific and obtained product 6c has three contiguous chiral centers. A brief optimization of the reaction conditions by screening temperature, reaction time, and concentration improved the yield to 85% (see ESI† for more details). Gratifyingly, we found that the reaction can be performed in one pot, giving similar yield and selectivity. Subsequently, we investigated the scope of this reaction (Scheme 5C). We were pleased to find that various N-substituted maleimides participated well, affording the corresponding substituted bicyclic scaffolds 6a–6d in good yield with complete diastereospecificity. Simple phenyl and various substitutions on the aryl group at the C1 position of cyclopropene were well tolerated (products 6e–6h). Interestingly, the diester-containing housane yielded the decarboxylated product 6i (see ESI† for mechanism). Furthermore, the bicyclic motifs 6d and 6e could be reduced to give bicyclic pyrrolidine derivatives 6j and 6k. Next, the alcohol functional group in 3q was utilized for the subsequent ring-fragmentation reaction. Treating 3q with stoichiometric p-TsOH gave cyclobutenyl-1,3-dienes 7. The formation of 7 might involve carbocation generation, ring-opening, and deprotonation. Finally, the heating of compound 3q led to an unusual isomerization, resulting in the formation of 3q′ (CCDC 2448186†). The probable mechanism from 3q to 3q′ involves the cleavage of the strained C–C bond in 3q, resulting in a planar 1,3-biradical (I) that allows C–C sigma bond rotation followed by ring-closing (see ESI† for mechanism). | ||
| Scheme 5 (A) Importance and synthesis of bicyclic imide and derivatives. (B) our reaction design. (C) This work: Skeletal remodeling. Reaction conditions: see ESI.† aUsing major diastereomer. Yields are of isolated products and dr was determined by 1H-NMR from the crude reaction mixture. ds = diastereospecificity. | ||
Mechanistic studies
Next, a variety of mechanistic experiments were conducted to shed light on the mechanism of the reaction. First, UV/vis spectra of the individual reaction components revealed that the photocatalyst 4-CzIPN is the only light-absorbing species around the operational wavelength (Scheme 6A). Next, Stern–Volmer quenching studies clearly demonstrated that cyclopropene 1a is an effective quencher (vide infra) of the excited state photocatalyst compared to maleimide 2i and cyclopropene 1v (Scheme 6B). The addition of 2,2,6,6-tetramethylpiperidinyloxy (TEMPO) to the reaction mixture significantly decreased the reaction yield, suggesting radical intermediacy during the reaction (Scheme 6C). To understand the nature of the quenching, we compared the triplet state energies and redox potentials of various photocatalysts and cyclopropene 1a (E1/2 = +1.26V, vs. Ag/AgCl in MeCN) (Scheme 6D and E). This data suggests that the yields of housane 3a and 3a′ correlated with the triplet state energies of photocatalysts, but no trend was observed with their redox potential. Furthermore, direct UV-light irradiation (λmax = 365nm) of the reaction mixture 1a and 2i furnished the housane 3al in 25% yield (see ESI†). These results alluded to the fact that the Dexter-type triplet–triplet EnT process is likely involved in the reaction.24 In addition, when we subjected a non-styrenic cyclopropene 1v to the standard conditions using N-phenyl maleimide 2a, we did not observe the desired housane product, and both the starting materials remained unreacted. This prompted us to gauge the correlation between triplet-state energy of cyclopropenes and reaction efficiency. From Scheme 6F, it is clearly evident that styrenic cyclopropenes 1a and 1o, which have triplet state energies lower than the photocatalyst (ET = 62.0 kcal mol−1), undergo cycloaddition to give the corresponding products 3a and 3o. In contrast, non-styrenic cyclopropene 1v, which has a higher triplet state energy (ET = 64.5 kcal mol−1) than the photocatalyst, did not produce the desired product. These results suggest that the reaction mechanism involves an energy transfer process. Additionally, the reduction potential of 1v (E1/2 = +1.21 V) indicates that the reaction could proceed if it were based on an electron transfer mechanism.
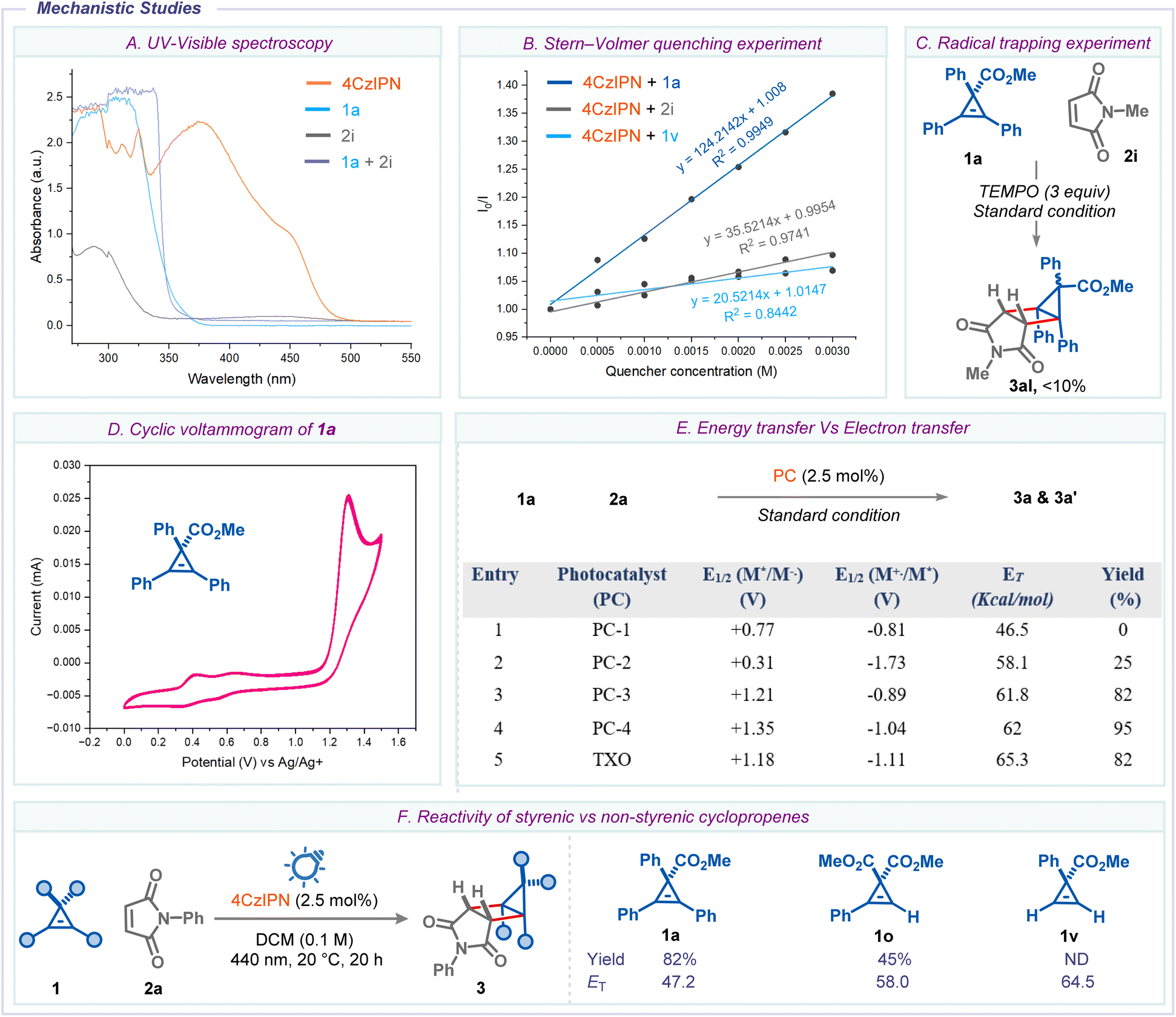 | ||
| Scheme 6 Mechanistic studies (A) Ultraviolet-visible absorption spectra of the reaction components. (B) Stern–Volmer quenching studies. (C) Radical trapping experiment. (D) Cyclic voltammetry measurements of 1a. (E) Energy transfer vs. Electron transfer. (F) Reactivity of cyclopropene. | ||
We subsequently performed density functional theory (DFT) calculations to get insight into the reaction mechanism and the origin of diastereoselectivity. Calculations were performed at the SMD(Dichloromethane)/(U)M06/6-311+g(d,p)//(U)M06/6-31+g(d,p) level of theory (Scheme 7A). The triplet state energies of cyclopropene 1d and maleimide 2i were computed to be 47.5 kcal mol−1 and 55.0 kcal mol−1, respectively, and this data is aligned with our luminescence quenching experiments. The mechanism of the reaction starts with the excitation of cyclopropene 1d by photocatalyst to afford triplet intermediate 31d, which then combines with the ground state of the maleimide 2i and forms two diastereomeric triplet intermediates 3Int3aa and 3Int3aa′ via the transition states 3TS3aa and 3TS3aa′ with an activation barrier of 11.8 kcal mol−1 and 14.4kcal mol−1, respectively (Scheme 7A). The difference in energy of these two transition states (2.6 kcal mol−1) is consistent with the observed high diastereoselectivity of product 3aa. The conversion of 3Int3aa and 3Int3aa′ to their corresponding open-shell singlet state counterparts 1Int3aa and 1Int3aa′ can be achieved through the minimum energy crossing point (MECP). Finally, radical–radical recombination affords the two diastereomeric products 3aa and 3aa′. Next, we focused on rationalizing the low diastereoselectivity that was obtained with our model substrate 1a (see ESI†). Following sensitization of the cyclopropene 1a, initial C–C bond formation requires an activation energy of 9.7 and 9.1 kcal mol−1, respectively, for the formation of two diastereomeric triplet intermediates 3Int3al and 3Int3al′. The small energy difference (0.6 kcal mol−1) between the two diastereomeric transition states 3TS3al and 3TS3al′ is consistent with the observed low diastereoselectivity. Finally, we shed light on the unprecedented 1,2-ester migration over 1,2-aryl migration using DFT studies (Scheme 7B). Transition states leading two possible products 6h (by a 1,2-ester migration) and 6h′ (by a 1,2-aryl migration) were computed. Indeed, we found that the 1,2-ester migration pathway via 1TS6h is favored by about 6 kcal mol−1 compared to the 1,2-aryl migration pathway via 1TS6h′ which explains the observed exclusive formation of 6h.
 | ||
| Scheme 7 DFT studies. (A) Using cyclopropene 1d. (B) 1,2-ester migration of 3al. | ||
Conclusions
In conclusion, we have developed a visible-light-mediated, catalytic, highly stereoselective approach to synthesize polysubstituted housanes with up to three all-carbon-quaternary centers. This reaction operates under mild conditions, uses an organo-catalyst, and tolerates diverse functional groups on cyclopropenes and maleimides. Furthermore, we have extended our chemistry to synthesizing spiro-oxa-housanes, a novel class of compounds not reported in the literature. The synthetic utility of this transformation was highlighted by developing a strain-release driven diastereospecific 1,2-ester migration process that gives rapid access to highly functionalized bicyclic imides and their derivatives. Furthermore, subsequent diversification of housane derivatives provided access to cyclobutenyl-1,3-diene motif. The observed reactivity and stereoselectivity have been rationalized by experiment-based mechanistic studies combined with DFT computation, illuminating cyclopropene's biradical nature and its interaction with maleimide. We believe that this strategy will provide a unique avenue to access and expand the chemical space of structurally intriguing housanes and spiro-oxa-housanes with high molecular complexity.Data availability
General information, experimental procedures, characterization data for all new compounds, NMR spectra as well as mechanistic investigations and computational investigations are in the ESI.† Data for the crystal structure reported in this paper have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under the deposition number CCDC 2404132 (for compound 3n), CCDC 2449293† (for compound 3p), CCDC 2451397† (for compound 3q), CCDC 2425129† (for compound 5h), CCDC 2409070† (for compound 6c), and CCDC 2448186† (for compound 3q′).Author contributions
A. R. S. M. and D. P. H. conceived and designed the project. A. R. S. M. carried out optimization studies, substrate scope, and mechanistic studies. N. A. B. performed DFT studies. H. M. carried out substrate scope. D. P. H. and A. R. S. M. wrote the manuscript with suggestions from N. A. B and H. M. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Science and Engineering Research Board (SERB), Government of India (File CRG/2022/007372), is greatly acknowledged. D. P. H. thanks the Indian Institute of Science (IISc) Bangalore for the infrastructure. A. R. S. M. thanks the University Grant Commission (U. G. C.) for doctoral fellowship. N. A. B. thanks KVPY for the fellowship. H. M. thanks the Indian Institute of Science (IISc) Bangalore for a doctoral fellowship. We thank Shashikant J for solving the X-ray crystal structures.Notes and references
- (a) F. Lovering, Med. Chem. Commun., 2013, 4, 515–519 RSC; (b) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS.
- J. Turkowska, J. Durka and D. Gryko, Chem. Commun., 2020, 56, 5718–5734 RSC.
- (a) P. Bellotti and F. Glorius, J. Am. Chem. Soc., 2023, 145, 20716–20732 Search PubMed; (b) M. M. D. Pramanik, H. Qian, W.-J. Xiao and J.-R. Chen, Org. Chem. Front., 2020, 7, 2531–2537 Search PubMed; (c) J. M. Lopchuk, K. Fjelbye, Y. Kawamata, L. R. Malins, C.-M. Pan, R. Gianatassio, J. Wang, L. Prieto, J. Bradow, T. A. Brandt, M. R. Collins, J. Elleraas, J. Ewanicki, W. Farrell, O. O. Fadeyi, G. M. Gallego, J. J. Mousseau, R. Oliver, N. W. Sach, J. K. Smith, J. E. Spangler, H. Zhu, J. Zhu and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 3209–3226 CrossRef CAS; (d) R. Gianatassio, J. M. Lopchuk, J. Wang, C.-M. Pan, L. R. Malins, L. Prieto, T. A. Brandt, M. R. Collins, G. M. Gallego, N. W. Sach, J. E. Spangler, H. Zhu, J. Zhu and P. S. Baran, Science, 2016, 351, 241–246 CrossRef CAS PubMed; (e) J. L. Tyler and V. K. Aggarwal, Chem.–Eur. J., 2023, 29, e202300008 CrossRef CAS PubMed.
- (a) S.-J. Chang, D. McNally, S. Shary-Tehrany, H. Sister Mary James and R. H. Boyd, J. Am. Chem. Soc., 1970, 92, 3109–3118 CrossRef CAS; (b) P. v. R. Schleyer, J. E. Williams and K. R. Blanchard, J. Am. Chem. Soc., 1970, 92, 2377–2386 CrossRef CAS; (c) V. V. Semeno, V. O. Vasylchenko, B. V. Vashchenko, D. O. Lutsenko, R. T. Iminov, O. B. Volovenko and O. O. Grygorenko, J. Org. Chem., 2020, 85, 2321–2337 Search PubMed; (d) B. K. Carpenter, Org. Biomol. Chem., 2004, 2, 103–109 RSC.
- (a) P. G. Gassman and K. T. Mansfield, J. Am. Chem. Soc., 1968, 90, 1517–1524 CrossRef CAS; (b) M. J. Williams, H. L. Deak and M. L. Snapper, J. Am. Chem. Soc., 2007, 129, 486–487 Search PubMed; (c) Y. S. Park and R. D. Little, J. Org. Chem., 2008, 73, 6807–6815 CrossRef CAS; (d) S. K. Nistanaki, L. A. Boralsky, R. D. Pan and H. M. Nelson, Angew. Chem., Int. Ed., 2019, 58, 1724–1726 Search PubMed; (e) B. Gu, B. Goldfuss, G. Schnakenburg and J. S. Dickschat, Angew. Chem., Int. Ed., 2023, 62, e202313789 CrossRef CAS PubMed; (f) S.-K. Wang, M.-J. Huang and C.-Y. Duh, J. Nat. Prod., 2006, 69, 1411–1416 CrossRef CAS; (g) H. L. Deak, S. S. Stokes and M. L. Snapper, J. Am. Chem. Soc., 2001, 123, 5152–5153 CrossRef CAS PubMed.
- (a) M. Jung and V. N. G. Lindsay, J. Am. Chem. Soc., 2022, 144, 4764–4769 CrossRef CAS PubMed; (b) Y.-C. Chang, C. Salome, T. Fessard and M. K. Brown, Angew. Chem., Int. Ed., 2023, 62, e202314700 Search PubMed; (c) Y. Liu, S. Tranin, Y.-C. Chang, E. B. Piper, T. Fessard, R. Van Hoveln, C. Salome and M. K. Brown, J. Am. Chem. Soc., 2025, 147, 6318–6325 Search PubMed.
- (a) R. Criegee and A. Rimmelin, Chem. Ber., 1957, 90, 414–417 CrossRef CAS; (b) G.-q. Shi and W.-l. Cai, J. Chem. Soc., Perkin Trans. 1, 1996, 2337–2338, 10.1039/P19960002337; (c) J. C. Sharland and H. M. L. Davies, Org. Lett., 2023, 25, 5214–5219 Search PubMed.
- D. Coto, D. Suárez-García, S. Mata, I. Fernández, L. A. López and R. Vicente, Angew. Chem., Int. Ed., 2024, 63, e202409226 Search PubMed.
- (a) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 Search PubMed; (b) S. Dutta, J. E. Erchinger, F. Strieth-Kalthoff, R. Kleinmans and F. Glorius, Chem. Soc. Rev., 2024, 53, 1068–1089 RSC; (c) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 Search PubMed; (d) Y. Jiang, Y. Wei, Q.-Y. Zhou, G.-Q. Sun, X.-P. Fu, N. Levin, Y. Zhang, W.-Q. Liu, N. Song, S. Mohammed, B. G. Davis and M. J. Koh, Nature, 2024, 631, 319–327 Search PubMed; (e) B. Muriel and J. Waser, Angew. Chem., Int. Ed., 2021, 60, 4075–4079 CrossRef CAS.
- (a) S. Poplata, A. Tröster, Y.-Q. Zou and T. Bach, Chem. Rev., 2016, 116, 9748–9815 CrossRef CAS PubMed; (b) J. Iriondo-Alberdi and M. F. Greaney, Eur. J. Org. Chem., 2007, 2007, 4801–4815 Search PubMed; (c) D. Sarkar, N. Bera and S. Ghosh, Eur. J. Org. Chem., 2020, 2020, 1310–1326 Search PubMed; (d) Z. Lu and T. P. Yoon, Angew. Chem., Int. Ed., 2012, 51, 10329–10332 CrossRef CAS; (e) M. Golfmann, L. Glagow, A. Giakoumidakis, C. Golz and J. C. L. Walker, Chem.–Eur. J., 2023, 29, e202202373 Search PubMed.
- (a) A. Padwa, G. D. Kennedy, G. R. Newkome and F. R. Fronczek, J. Am. Chem. Soc., 1983, 105, 137–139 CrossRef CAS; (b) A. Padwa, K. E. Krumpe, L. W. Terry and M. W. Wannamaker, J. Org. Chem., 1989, 54, 1635–1642 CrossRef CAS; (c) D. R. Arnold and R. M. Morchat, Can. J. Chem., 1977, 55, 393–406 CrossRef CAS; (d) We have deposited our work on 18th of December 2024 on Chemrxiv: doi: 10.26434/chemrxiv-2024-ljfs0. Meanwhile, Davies and co-workers reported [2+2] cycloaddition strategy for the synthesis of housanes. However, Our work is the first to achieve high stereocontrol, significant molecular complexity, and a broad substrate scope under visible-light photocatalysis using an organic dye as the photocatalyst. See Davies paper: B. Keen, C. Cong, A. Castanedo, G. H. M. Davies, R. R. Knowles and H. M. L. Davies, Org. Lett., 2025, 27, 1673–1678 Search PubMed.
- (a) P. C. Wong and D. R. Arnold, Can. J. Chem., 1979, 57, 1037–1049 Search PubMed; (b) A. Padwa, G. D. Kennedy and M. W. Wannamaker, J. Org. Chem., 1985, 50, 5334–5341 Search PubMed.
- Q. Ye, H. Ye, D. Cheng, X. Li and X. Xu, Tetrahedron Lett., 2018, 59, 2546–2549 Search PubMed.
- A. Padwa and G. D. Kennedy, J. Org. Chem., 1984, 49, 4344–4352 Search PubMed.
- A. R. Meyer, M. V. Popescu, A. Sau, N. H. Damrauer, R. S. Paton and T. P. Yoon, ACS Catal., 2024, 14, 12310–12317 CrossRef CAS.
- (a) A. A. Kirichok, I. O. Shton, I. M. Pishel, S. A. Zozulya, P. O. Borysko, V. Kubyshkin, O. A. Zaporozhets, A. A. Tolmachev and P. K. Mykhailiuk, Chem.–Eur. J., 2018, 24, 5444–5449 CrossRef CAS PubMed; (b) C. M. Marson, Chem. Soc. Rev., 2011, 40, 5514–5533 Search PubMed.
- (a) E. M. Carreira and T. C. Fessard, Chem. Rev., 2014, 114, 8257–8322 Search PubMed; (b) J. A. Burkhard, B. Wagner, H. Fischer, F. Schuler, K. Müller and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 3524–3527 CrossRef CAS; (c) A. R. Samim Mondal, B. Ghorai and D. P. Hari, Org. Lett., 2023, 25, 4974–4979 Search PubMed.
- (a) P. Franceschi, S. Cuadros, G. Goti and L. Dell'Amico, Angew. Chem., Int. Ed., 2023, 62, e202217210 Search PubMed; (b) A. Sharma, A. Choi, D. Yim, H. Kim and H. Kim, Adv. Synth. Catal., 2024, 366, 2257–2263 CrossRef CAS.
- (a) F. Zhong, G.-Y. Chen, X. Han, W. Yao and Y. Lu, Org. Lett., 2012, 14, 3764–3767 Search PubMed; (b) O. S. Liashuk, I. A. Ryzhov, O. V. Hryshchuk, Y. M. Volovenko and O. O. Grygorenko, Chem.–Eur. J., 2024, 30, e202303504 CrossRef CAS PubMed.
- C. L. Cioffi, N. Dobri, E. E. Freeman, M. P. Conlon, P. Chen, D. G. Stafford, D. M. C. Schwarz, K. C. Golden, L. Zhu, D. B. Kitchen, K. D. Barnes, B. Racz, Q. Qin, E. Michelotti, C. L. Cywin, W. H. Martin, P. G. Pearson, G. Johnson and K. Petrukhin, J. Med. Chem., 2014, 57, 7731–7757 Search PubMed.
- (a) J. Jurczyk, J. Woo, S. F. Kim, B. D. Dherange, R. Sarpong and M. D. Levin, Nat. Synth., 2022, 1, 352–364 CrossRef CAS; (b) J. Jurczyk, M. C. Lux, D. Adpressa, S. F. Kim, Y.-h. Lam, C. S. Yeung and R. Sarpong, Science, 2021, 373, 1004–1012 CrossRef CAS.
- X. Wu and C. Zhu, Acc. Chem. Res., 2020, 53, 1620–1636 CrossRef CAS.
- (a) P. Dowd, M. Shapiro and K. Kang, J. Am. Chem. Soc., 1975, 97, 4754–4757 CrossRef CAS PubMed; (b) P. Dowd, S.-C. Choi, F. Duah and C. Kaufman, Tetrahedron, 1988, 44, 2137–2148 CrossRef CAS; (c) G. Zhao, S. Lim, D. G. Musaev and M.-Y. Ngai, J. Am. Chem. Soc., 2023, 145, 8275–8284 CrossRef CAS; (d) G. Zhao, A. Khosravi, S. Sharma, D. G. Musaev and M.-Y. Ngai, J. Am. Chem. Soc., 2024, 146, 31391–31399 Search PubMed.
- F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2404132, 2449293, 2451397, 2425129, 2409070 and 2448186. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc02288f |
| This journal is © The Royal Society of Chemistry 2025 |