
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHow to design active sites for tailoring the C1 and C2 products of CO2 photoreduction
Peijin
Du†
a,
Suning
Zhang†
a,
Jinyu
Ding†
 a,
Dongpo
He
a,
Qinyuan
Hu
a,
Xingchen
Jiao
*a and
Yi
Xie
*b
a,
Dongpo
He
a,
Qinyuan
Hu
a,
Xingchen
Jiao
*a and
Yi
Xie
*b
aKey Laboratory of Synthetic and Biological Colloids, Ministry of Education, School of Chemical and Material Engineering, Jiangnan University, Wuxi 214122, China. E-mail: xcjiao@jiangnan.edu.cn
bState Key Laboratory of Precision and Intelligent Chemistry, University of Science and Technology of China, Hefei, Anhui 230026, China. E-mail: yxie@ustc.edu.cn
First published on 21st July 2025
Abstract
Photocatalytic CO2 reduction generally involves three fundamental stages: light absorption, charge carrier separation, and the redox reactions occurring on the surface. Adjusting these processes to optimize the activity and selectivity of CO2 photoreduction towards C1 and C2 products has proven to be an effective strategy. In this review, we summarize the design of active sites through various structural modifications, including defect structure, metal doping structure, heterojunction structure, alloy structure, loading structure, and composite structure, to tailor the product selectivity and activity of CO2 photoreduction. Additionally, we provide a concise overview and outlook on strategies for stabilizing active sites, constructing bio-inorganic systems, and elucidating catalytic mechanisms.
 From left to right: Jinyu Ding, Suning Zhang and Peijin Du | Peijin Du is currently studying for her Master's degree at Jiangnan University. Her current interests include the synthesis and characterization of two-dimensional solid materials, and their applications in photocatalytic CO2/plastic conversion. Suning Zhang is currently pursuing a Master's degree at Jiangnan University. Her current research interests include the preparation and assembly of low-dimensional nanomaterials, as well as photocatalytic CO2 reduction. Jinyu Ding received her Master's degree at Jiangnan University in 2025. Her current interests include the synthesis and characterization of two-dimensional metal oxides, and their applications in photo/electro-catalytic CO2/plastic conversion into valuable fuels. |
 From left to right: Dongpo He, Qinyuan Hu | Dongpo He received his Master's degree at Dalian Polytechnic University in 2023. He is currently pursuing his PhD in the School of Chemistry and Materials Science at Jiangnan University. He mainly focuses on catalytic processes for energy and environmental applications of low-dimensional solid materials, such as the catalytic conversion of waste plastics and the photoreduction of CO2. Qinyuan Hu received his BS degree at Jiangnan University in 2022. He is currently pursuing his PhD degree at Jiangnan University. His current interests include the synthesis and characterization of two-dimensional metal oxides and sulfides, and their applications in catalytic CO2 and plastic conversion. |
 Xingchen Jiao | Xingchen Jiao received his BS degree from the Department of Chemistry at Hefei University of Technology (2014) and PhD degree in Inorganic Chemistry from the University of Science and Technology of China (2019). After that he worked as a postdoctoral fellow at the University of Science and Technology of China. In 2022, he joined Jiangnan University as a professor. His current interests include the synthesis and characterization of nanostructures, as well as their applications in CO2 photo-/electro-conversion into hydrocarbon fuels. |
 Yi Xie | Yi Xie received her BS degree from Xiamen University (1988) and a PhD from the University of Science and Technology of China (USTC, 1996). She is now a full professor of the Department of Chemistry, USTC. She was appointed as the Cheung Kong Scholar Professor of inorganic chemistry in 2000 and elected as a member of the Chinese Academy of Sciences in 2013. Her research interests include cutting-edge research in four major frontiers: solid state materials chemistry, nanotechnology, energy science and theoretical physics. |
Broader contextDue to the burning of fossil fuels and industrial activities, atmospheric carbon dioxide concentrations have risen to critical levels, significantly impacting global climate change. Solar-driven conversion of CO2 into C1 or C2 products (such as methane, methanol or ethylene) represents a promising sustainable technology that mimics natural photosynthesis, transforming sunlight, water, and CO2 into energy-rich molecules. This process involves three key steps: the absorption of light, the separation of charge carriers, and the redox reactions occurring on the surface. Currently, photocatalysts face challenges including narrow light absorption ranges and low charge carrier separation efficiencies. Therefore, optimizing these steps by designing active sites is crucial for enhancing performance. Combining this technology with renewable energy systems further enhances its feasibility and establishes photocatalytic CO2 reduction as a cornerstone for the future circular carbon economy and sustainable energy solutions. |
1 Introduction
The Paris Agreement underscores the imperative to reduce global carbon dioxide (CO2) emissions by 50% by the year 2030 and to achieve complete carbon neutrality by 2050.1,2 The increment in atmospheric CO2 concentrations has posed significant challenges, including climate change and air pollution, representing major issues of the 21st century. The diminution of fossil fuel reserves and the exacerbation of atmospheric conditions due to excessive CO2 emissions have propelled the scientific community to innovate alternative energy solutions to meet the escalating energy needs of the present and the future. Current methodologies for the capture and utilization of CO2, in conjunction with emerging innovative technologies, are instrumental in facilitating the attainment of these ambitious environmental objectives.3 The photocatalytic reduction of CO2 and water into valuable chemicals and fuels, emulating the natural process of photosynthesis, is heralded as one of the most auspicious strategies for reducing atmospheric CO2 levels.4–7 This approach not only contributes to the mitigation of climate change but also facilitates the development of sustainable energy sources, thereby addressing critical environmental challenges. Harnessing solar energy, utilizing water (H2O) as a reductant, and employing a photocatalyst, this innovative process is commonly referred to as artificial photosynthesis.8,9 It diverges from natural photosynthesis, which occurs within the chloroplasts of plant leaves, converting the same reactants into oxygen (O2) and glucose, by differing in both the system architecture and the end products generated. At present, the viability of solar-to-fuel conversion technology as a rival to the natural photosynthetic mechanism in terms of efficiency remains an open question. The efficiency of CO2 photoreduction is predominantly influenced by the selection of the catalyst,10 making it crucial to design and fabricate catalysts with superior efficiency to enhance the overall performance of the process. As such, there is a pressing need to prioritize the development of catalysts that can significantly improve the efficiency of CO2 photoreduction.In recent studies, CO2 photoreduction into a variety of compounds, including carbon monoxide (CO), methane (CH4), formic acid (HCOOH), methanol (CH3OH), formaldehyde (HCHO), and ethanol (C2H5OH)—spanning both C1 and C2 categories—has emerged as a vibrant field of study.11–14 This area has captivated scientific interest for several decades and necessitates deeper exploration. The targeted photoreduction of CO2 to diverse C1 products, such as CO, CH4, CH3OH, and HCOOH, is of particular interest. Typically, CO and CH4 serve as gaseous fuels with lower calorific values, while CH3OH and HCOOH are utilized as liquid fuels.15,16 C2 and higher hydrocarbons/alcohols are utilized as fuels with high calorific values and as raw materials for chemical synthesis, making the conversion of CO2 into C1 and C2 or higher hydrocarbons crucial for meeting future energy and chemical needs. The directed photoreaction of CO2 to produce C1 or C2 products holds considerable appeal and importance, particularly from both academic and industrial perspectives. In the context of a photocatalytic CO2 reduction process, it generally involves three fundamental stages: the absorption of light, the separation of charge carriers, and the redox reactions occurring on the surface.17–20 The complexity of multiple protonation steps during the surface redox reactions presents significant hurdles in attaining the desired specificity, as it can result in the generation of a diverse array of products. To this end, it is indeed possible to modulate these three processes to fine-tune the activity and selectivity of CO2 photoreduction towards C1 and C2 products.
Owing to the significant advancements in CO2 photoreduction in recent years, a multitude of review articles have been published, reflecting the growing interest and progress in this field.21–23 Some of them concentrate on the criteria for selecting photocatalysts, while others delve into the underlying principles. Additionally, there are reviews that explore various strategies aimed at enhancing the performance of these photocatalytic systems.24,25 In a distinctive departure from existing literature, we are pleased to offer a contemporary review that focuses on the design of active sites to customize the C1 and C2 products of CO2 photoreduction by strategically modulating the aforementioned three processes (Scheme 1). First, we focus on strategies to improve the light absorption capacity of photocatalysts, including defect engineering and metal doping engineering, both of which have shown significant potential in boosting CO2 photoreduction performance. Secondly, we emphasize how heterojunction engineering and alloy engineering influence the charge carrier dynamics of photocatalysts. These approaches effectively suppress electron–hole recombination and enhance charge separation efficiency, thereby facilitating the photocatalytic reduction of CO2. Thirdly, we analyze the adsorption mode of hydrogen-containing intermediates and the role of active sites in regulating the protonation process, which critically impact the selective formation of C1 or C2 products. Lastly, comprehensive insights are presented on the rational design of photocatalyst active sites for the photoreduction of CO2 into C1 or C2 products, along with an in-depth analysis of the challenges and practical difficulties encountered in real-world applications. This review comprehensively examines current strategies for active site engineering, elucidates critical limitations, and offers perspectives for addressing these challenges. This review serves to provide valuable guidance and practical assistance for the rational design of active sites in CO2 photoreduction catalysts, aiming to advance the field of sustainable carbon utilization.
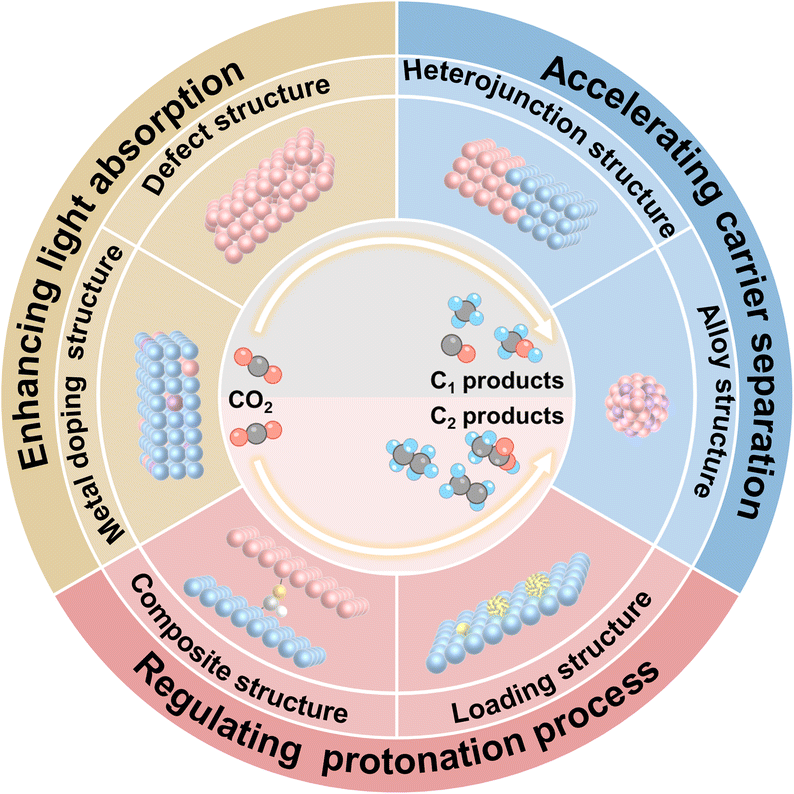 | ||
| Scheme 1 Schematic illustrations of various strategies for designing active sites to tailor the C1 and C2 products of the CO2 photoreduction process. | ||
2 Design strategies for regulating the active sites of photocatalysts
It is well known that the photoreduction of CO2 primarily involves three key processes: light absorption, charge carrier separation, and redox reactions occurring on the surface (Fig. 1). Among these, light absorption has received significant attention, particularly in the visible light region. However, infrared light, which accounts for approximately 50% of sunlight, remains underexplored and underutilized. Thus, expanding the light absorption range is an effective strategy for enhancing photocatalyst performance. Furthermore, improving the efficiency of charge carrier separation is crucial for optimizing charge transfer and increasing the yield of CO2 photoreduction. Lastly, the regulation of the protonation process in surface redox reactions is essential for controlling the transformation of intermediates into target products, thereby achieving the desired outcomes. | ||
| Fig. 1 Schematic diagram of the process of CO2 photoreduction, including light absorption, charge carrier separation, and redox reactions occurring on the surface. | ||
2.1 Enhancing light absorption
 | ||
| Fig. 2 (a) Energy band diagram of solar driven full decomposition of CO2 and H2O into hydrocarbons and O2. (b) ESR spectra and (c) optical absorption spectra of WO3 nanosheets. (d) CO and O2 formation rates over WO3 nanosheets. Reproduced with permission from ref. 28. Copyright 2018, Elsevier. (e) Optical absorption spectra for BiOBr nanosheets. (f) EPR spectra and (g) CO yield of oxygen-deficient BiOBr nanosheets, BiOBr nanosheets, and bulk BiOBr. Reproduced with permission from ref. 31. Copyright 2024, Wiley-VCH. | ||
 | ||
| Fig. 3 (a) The formation of the localized surface plasmon resonance phenomenon on metal nanoparticles. Reproduced with permission from ref. 37. Copyright 2025, Elsevier. (b) UV-vis diffuse reflectance spectra, (c) bandgaps and (d) yields of BiOCl and 1% M/BiOCl (M = Pt, Pd, and Au) samples. Reproduced with permission from ref. 38. Copyright 2024, Wiley-VCH. (e) UV-vis absorption spectra and (f) energy level diagram for Au/TZO, Au/ZrO2 and TZO. (g) The yield of photocatalytic CO2 reduction by using Au/TZO as the catalyst. Reproduced with permission from ref. 39. Copyright 2025, Wiley-VCH. | ||
2.2 Accelerating charge carrier separation
 | ||
| Fig. 4 (a) In2O3/Nb2O5 S-heterojunction formation and charge transfer mechanism. (b) The CO yield and selectivity of different catalysts. (c–e) Kinetic decay curves of the transient absorption spectra of different catalysts. Reproduced with permission from ref. 45. Copyright 2024, Springer Nature. (f) Scheme of a heterojunction with highly connected and matching crystal faces. (g) Product evolution rates of different catalysts. (h) Normalized transient absorption kinetics for CuS–BiWO6. Reproduced with permission from ref. 46. Copyright 2024, Wiley-VCH. | ||
 | ||
| Fig. 5 (a) Schematic diagram of the reaction mechanism of photoreduction of CO2 by the NixCoy–CCN catalyst. (b) XPS spectra of N 1s, (c) PL spectra and (d) time-resolved transient PL decay spectra for NixCoy–CCN. (e) Product evolution rates of NixCoy–CCN. Reproduced with permission from ref. 50. Copyright 2024, Wiley-VCH. (f) The cycling test over CuxAg50−x/UiO66–NH2. (g) PL spectra; (h) time-resolved transient PL decay spectra and (i) Fs-TA spectra of CuxAg50−x/UiO66–NH2. Reproduced with permission from ref. 51. Copyright 2024, Royal Society of Chemistry. | ||
2.3 Tailoring the protonation process
 | ||
| Fig. 6 (a) Schematic diagram of interlayer dual active site fixation intermediates promoting photoreduction of CO2 to CH3OH. (b) Rates of CO2 photoreduction with different catalysts. Quasi in situ XPS spectra of (c) Ni 2p and (d) In 3d for the CoNi2S4–In2O3 composites of nanosheets. (e) In situ FTIR spectra and (f) free energy diagrams for the CO2 photoreduction process on the CoNi2S4–In2O3 composite of the nanosheet slab. Reproduced with permission from ref. 54. Copyright 2024, American Chemical Society. | ||
 | ||
| Fig. 7 (a) Schematic representation of CO2 photoreduction in bimetallic sites. (b) TEM image of Pd–Nb2O5 nanosheets, (c) product evolution rates for CO2 photoreduction over various catalysts. (d) Calculated Bader charge value for a Pd–Nb2O5 nanosheet slab. (e) Free energy diagrams of CO2 photoreduction to CH3COOH over the Pd–Nb2O5 nanosheets. Reproduced with permission from ref. 60. Copyright 2024, Wiley-VCH. (f) TEM of Pd–ZnO nanosheets. Quasi in situ XPS spectra of (g) Pd 3d and (h) Zn 2p for the Pd–ZnO nanosheets during CO2 photoreduction. Reproduced with permission from ref. 61. Copyright 2024, Wiley-VCH. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond (Fig. 8e). Furthermore, the in situ DRIFT spectrum (Fig. 8f) indicated that this process promotes the formation of HCO3− intermediates. Under illumination, when HCO3− was proximal to the OCOH intermediate at hydroxyl sites, spontaneous reactions occur, fostering C–C coupling (Fig. 8g). While this study highlights the synergistic effects between surface vacancies and hydroxyl groups, the research on CO2 photoreduction remains limited in depth. Therefore, the development of a stable and sustainable synergistic photocatalyst integrating vacancy and hydroxyl functionalities holds promising prospects in promoting C–C coupling. However, the challenge of stably adsorbing hydroxyl groups to function as active sites remains to be addressed.
O bond (Fig. 8e). Furthermore, the in situ DRIFT spectrum (Fig. 8f) indicated that this process promotes the formation of HCO3− intermediates. Under illumination, when HCO3− was proximal to the OCOH intermediate at hydroxyl sites, spontaneous reactions occur, fostering C–C coupling (Fig. 8g). While this study highlights the synergistic effects between surface vacancies and hydroxyl groups, the research on CO2 photoreduction remains limited in depth. Therefore, the development of a stable and sustainable synergistic photocatalyst integrating vacancy and hydroxyl functionalities holds promising prospects in promoting C–C coupling. However, the challenge of stably adsorbing hydroxyl groups to function as active sites remains to be addressed.
 | ||
| Fig. 8 (a) Schematic representation of CO2 photoreduction in bimetallic sites. (b) O 1s XPS spectra of an H–WO3 catalyst. (c) UV-vis-NIR absorption spectrum of catalysts. (d) CH3COOH yield increasing with irradiation time. (e) Adsorption of CO2 on the anoxic WO3–0.33H2O surface. (f) In situ DRIFT spectrum of the H–WO3. (g) Schematic diagram of possible reaction pathways in CH3COOH production. Reproduced with permission from ref. 68. Copyright 2018, American Chemical Society. | ||
3 Conclusions and outlook
Comprehensive insights were presented on the rational design of photocatalyst active sites for promoting the photoreduction of CO2 into C1 or C2 products. This review systematically explores strategies from three key perspectives: enhancing light absorption, accelerating charge carrier separation, and regulating the protonation process. The discussed approaches include defect structure, metal doping structure, heterojunction structure, alloy structure, loading structure, and composite structure. In detail, we outlined how defects in WO3 nanosheets and BiOBr nanosheets can promote the generation of intermediate bands and boost the performance of the photoreduction of CO2 to CO. We also emphasized the incorporation of Au, Pt, and Pd in BiOCl, which contributed to broadening the spectral range through their LSPR effect. Secondly, we reviewed how the In2O3/Nb2O5 heterojunction and NiCo alloy-modified CCN accelerated electron transfer and promoted charge carrier separation to enhance the photoreduction performance of CO2. Additionally, we explored the conversion of CO2 to CH3OH by constructing a Ni–C–O–In adsorption structure via the CoNi2S4–In2O3 composite structure and detailed how the composite structure affects the intermediate's adsorption structure. Furthermore, we summarized that loading Pd in the form of clusters or particles on nanosheets can construct charge asymmetric pair sites, thus reducing the C–C coupling energy barrier. Despite their proven effectiveness in regulating the CO2 photoreduction process, there remain areas requiring improvement and innovation within photocatalytic CO2 reduction. Based on this, we propose the following recommendations for advancing the photoreduction of CO2 into high-value products (Fig. 9). | ||
| Fig. 9 Outlook on the design of active sites for CO2 photoreduction. | ||
(a) The photoreduction of CO2 involves a multi-electron reaction process, where precise control of electron transfer to a specific step remains challenging, often resulting in poor product selectivity. To overcome these challenges, the photocatalyst must provide a favorable coordination environment that modulates the adsorption strength of intermediate products. Surface modification methods, such as adjusting the coordination number of active sites, functional group modification of the charge carrier, and introducing corresponding co-catalysts, can enhance product selectivity. These modifications alter the local electronic structure and thermodynamically reduce the adsorption energy of intermediates, thereby regulating reaction pathways and improving product selectivity. Therefore, optimizing the surface coordination environment is a powerful approach to enhancing product selectivity.
(b) The practical application of photocatalytic CO2 reduction is significantly hindered by insufficient catalyst stability, primarily attributed to deactivation mechanisms. Such issues include metal center agglomeration in single-atom photocatalysts during cyclic operations and rapid poisoning of Pd-based catalysts due to excessive *CO intermediate adsorption, both representing critical challenges in catalyst design. Future research should prioritize developing advanced stabilization strategies, particularly those enhancing metal–support interactions to mitigate agglomeration and preserve atomic dispersion of active sites. Furthermore, rational catalyst engineering through co-doping architectures could modulate intermediate adsorption configurations and prevent surface passivation while maintaining catalytic fidelity throughout photoreduction processes.
(c) The integration of solar catalysis with natural enzymatic processes offers a favorable model for efficient solar-to-chemical energy conversion. Biological enzymes exhibit exceptional photostability during light-driven reactions, a feature that current photocatalytic CO2 reduction systems seek to emulate through enzyme encapsulation within engineered matrices. In the future, by developing structures that mimic the active sites of enzymes in natural photosynthesis and combining them with existing inorganic materials, a sophisticated synthesis of photocatalysts that imitate the enzyme sites can be achieved, so as to enhance the catalytic performance of photoreduction of CO2.
(d) A thorough elucidating of the structural evolution of the active sites of photocatalysts during the photoreduction process holds paramount scientific significance. It not only provides fundamental insights into the internal processes such as adsorption and activation of the active sites during the reaction process, but also establishes a strategic framework for the rational design and performance optimization of photocatalysts. It is of critical importance to explore the reaction mechanism by employing advanced in situ characterization methodologies. While in situ XPS62 and in situ infrared spectroscopy69,70 are commonly used, other techniques like in situ mass spectrometry and in situ NMR spectroscopy71 offer potential for deeper insights into intermediate evolution processes, thereby strengthening the theoretical foundation of the research.
In the future, efforts will be focused on elucidating the selective regulatory mechanism of the CO2 photoreduction reaction. We aim to reveal the true reaction pathways responsible for producing C1 or C2 products at different sites. With a particular emphasis on the generation of high-value-added hydrocarbons such as C2, our research will address the current limitation in the field where C1 products predominantly dominate.
Data availability
The data supporting this article have been included within the article. No new data were generated or analysed as part of this review.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the National Key R&D Program of China (2022YFA1502904), the National Natural Science Foundation of China (22275178) and the Fundamental Research Funds for the Central Universities (JUSRP123013, JUSRP123015).References
- K. de Kleijne, S. Hanssen, L. van Dinteren, M. A. J. Huijbregts, R. van Zelm and H. de Coninck, One Earth, 2022, 5, 168–185 CrossRef.
- J. Rogelj, M. den Elzen, N. Höhne, T. Fransen, H. Fekete, H. Winkler, R. S. Chaeffer, F. Ha, K. Riahi and M. Meinshausen, Nature, 2016, 534, 631–639 CrossRef CAS.
- G. G. Zhang, G. S. Li, T. Heil, S. Zafeiratos, F. L. Lai, A. Savateev, M. Antonietti and X. C. Wang, Angew. Chem., Int. Ed., 2019, 58, 3433–3437 CrossRef CAS PubMed.
- Y. Tachibana, L. Vayssieres and J. R. Durrant, Nat. Photonics, 2012, 6, 511–518 CrossRef CAS.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890–1898 CrossRef CAS.
- Y. Hao and A. Steinfeld, Sci. Bull., 2017, 62, 1099–1101 CrossRef CAS.
- T. Zhang and W. B. Lin, Chem. Soc. Rev., 2014, 43, 5982–5993 RSC.
- J. Jia, H. Wang, Z. L. Lu, P. G. O'Brien, M. Ghoussoub, P. Duchesne, Z. Q. Zheng, P. C. Li, Q. Qiao, L. Wang, A. Gu, F. M. Ali, Y. C. Dong, Q. Wang, K. K. Ghuman, T. Wood, C. X. Qian, Y. Shao, C. Y. Qiu, M. M. Ye, Y. M. Zhu, Z. H. Lu, P. Zhang, A. S. Helmy, C. V. Singh, N. P. Kherani, D. D. Perovic and G. A. Ozin, Adv. Sci., 2017, 4, 1700252 CrossRef PubMed.
- L. Hurtado, R. Natividad and H. García, Catal. Commun., 2016, 84, 30–35 CrossRef.
- R. Das, S. Chakraborty and S. C. Peter, ACS Energy Lett., 2021, 6, 3270–3274 CrossRef.
- Q. Y. Hu, M. Q. Li, J. C. Zhu, Z. X. Zhang, D. P. He, K. Zheng, Y. Wu, M. H. Fan, S. Zhu, W. S. Yan, J. Hu, J. F. Zhu, Q. X. Chen, X. C. Jiao and Y. Xie, Nano Lett., 2024, 24, 4610–4617 CrossRef PubMed.
- Y. Wang, S. B. Wang and X. W. Lou, Angew. Chem., Int. Ed., 2019, 58, 17236–17240 CrossRef PubMed.
- Y. Wu, Q. X. Chen, J. C. Zhu, K. Zheng, M. Y. Wu, M. H. Fan, W. S. Yan, J. Hu, J. F. Zhu, Y. Pan, X. C. Jiao, Y. F. Sun and Y. Xie, Angew. Chem., Int. Ed., 2023, 62, e202301075 CrossRef.
- S. Chakraborty, R. Das, M. Riyaz, K. Das, A. K. Singh, D. Bagchi, C. P. Vinod and S. C. Peter, Angew. Chem., Int. Ed., 2023, 62, e202216613 CrossRef PubMed.
- A. Corma and H. Garcia, J. Catal., 2013, 308, 168–175 CrossRef.
- J. Y. Liu, B. Garg and Y. C. Ling, Green Chem., 2011, 13, 2029–2031 RSC.
- X. C. Jiao, K. Zheng, L. Liang, X. D. Li, Y. F. Sun and Y. Xie, Chem. Soc. Rev., 2020, 49, 6592–6604 RSC.
- M. Q. Li, Z. Q. Han, Q. Y. Hu, W. Y. Fan, Q. Hu, D. P. He, Q. X. Chen, X. C. Jiao and Y. Xie, Chem. Soc. Rev., 2024, 53, 9964–9975 RSC.
- S. W. Wang, J. S. Wang, Y. Wang, X. Y. Sui, S. H. Wu, W. X. Dai, Z. Z. Zhang, Z. X. Ding and J. L. Long, ACS Catal., 2024, 14, 10760–10788 CrossRef CAS.
- Y. Wu, Q. Hu, Q. Chen, X. Jiao and Y. Xie, Acc. Chem. Res., 2023, 56, 2500–2513 CrossRef CAS PubMed.
- J. Albero, Y. Peng and H. García, ACS Catal., 2020, 10, 5734–5749 CrossRef CAS.
- G. R. Dey, Petroleum, 2024, 10, 373–398 CrossRef.
- A. Behera, A. K. Kar and R. Srivastava, Mater. Horiz., 2022, 9, 607–639 RSC.
- S. W. Cheng, Z. H. Sun, K. H. Lim, T. Z. H. Gani, T. X. Zhang, Y. S. Wang, H. Yin, K. L. Liu, H. W. Guo, T. Du, L. Y. Liu, G. K. Li, Z. Y. Yin and S. Kawi, Adv. Energy Mater., 2022, 12, 2270079 CrossRef.
- H. B. He, Y. Q. Ren, Y. H. Zhu, R. X. Peng, S. N. Lan, J. C. Zhou, B. Y. Yang, Y. T. Si and N. X. Li, ACS Catal., 2025, 15, 10480–10520 CrossRef CAS.
- C. Han, B. K. Kundu, Y. J. Liang and Y. J. Sun, Adv. Mater., 2024, 36, e2307759 CrossRef PubMed.
- S. Yang, W. J. Byun, F. Zhao, D. Chen, J. Mao, W. Zhang, J. Peng, C. Liu, Y. Pan, J. Hu, J. Zhu, X. Zheng, H. Fu, M. Yuan, H. Chen, R. Li, M. Zhou, W. Che, J.-B. Baek, J. S. Lee and J. Xu, Adv. Mater., 2024, 36, 2312616 CrossRef CAS.
- L. Liang, X. D. Li, Y. F. Sun, Y. L. Tan, X. C. Jiao, H. X. Ju, Z. M. Qi, J. F. Zhu and Y. Xie, Joule, 2018, 2, 1004–1016 CrossRef CAS.
- J. C. Wu, J. C. Zhu, W. Y. Fan, D. P. He, Q. Y. Hu, S. Zhu, W. S. Yan, J. Hu, J. F. Zhu, Q. X. Chen, X. C. Jiao and Y. Xie, Nano Lett., 2024, 24, 696–702 CrossRef CAS PubMed.
- Y. Wu, D. P. He, L. Li, Z. Q. Wang, W. S. Yan, J. F. Zhu, Y. Pan, Q. X. Chen, X. C. Jiao and Y. Xie, Chem. Commun., 2023, 59, 11700–11703 RSC.
- J. Wu, X. D. Li, W. Shi, P. Q. Ling, Y. F. Sun, X. C. Jiao, S. Gao, L. Liang, J. Q. Xu, W. S. Yan, C. M. Wang and Y. Xie, Angew. Chem., Int. Ed., 2018, 57, 8719–8723 CrossRef CAS.
- T. Y. Huang, J. Y. Han, Z. Q. Li, Y. X. Hong, X. F. Gu, Y. F. Wu, Y. J. Zhang and S. Q. Liu, Angew. Chem., Int. Ed., 2025, 64, e202500269 CrossRef CAS.
- X. Y. Jiang, J. D. Huang, Z. H. Bi, W. J. Ni, G. Gurzadyan, Y. A. Zhu and Z. Y. Zhang, Adv. Mater., 2022, 34, 2109330 CrossRef CAS PubMed.
- M. Tahir, B. Tahir and N. A. S. Amin, Appl. Catal., B, 2017, 204, 548–560 CrossRef CAS.
- L. Z. Shi, H. M. Liu, S. B. Ning and J. H. Ye, Catal. Sci. Technol., 2022, 12, 6155–6162 RSC.
- C. Boerigter, R. Campana, M. Morabito and S. Linic, Nat. Commun., 2016, 7, 10545 CrossRef CAS PubMed.
- R. T. Guo, C. Xia, Z. X. Bi, Z. R. Zhang and W. G. Pan, Fuel Process. Technol., 2023, 241, 107617 CrossRef CAS.
- Q. Liu, C. B. Bai, C. X. Zhu, W. J. Guo, G. F. Li, S. Guo, D. Kripalani, K. Zhou and R. Chen, Adv. Sci., 2024, 11, 2400934 CrossRef CAS.
- N. Y. Huang, B. Li, D. J. Wu, Z. Y. Chen, B. Shao, D. Chen, Y. T. Zheng, W. J. Wang, C. Z. Yang, M. Gu, L. Li and Q. Xu, Angew. Chem., Int. Ed., 2024, 63, e202319177 CrossRef CAS.
- W. D. Hou, H. Z. Guo, M. H. Wu and L. Wang, ACS Nano, 2023, 17, 20560–20569 CrossRef PubMed.
- W. Gui, S. Jiang, L. Wang, C. Liu, Z. Huang, L. Wang and J. Yang, Adv. Funct. Mater., 2025, 2505919 CrossRef.
- Z. L. Liu, Y. F. Mu, X. R. Li, Y. X. Feng, M. Zhang and T. B. Lu, Appl. Catal., B, 2025, 366, 125012 CrossRef.
- D. Liu, L. Jiang, D. Chen, Z. Hao, B. Deng, Y. Sun, X. Liu, B. Jia, L. Chen and H. Liu, ACS Catal., 2024, 14, 5326–5343 CrossRef.
- R. Sun, Z. Zhu, N. Tian, Y. Zhang and H. Huang, Angew. Chem., Int. Ed., 2024, 63, e202408862 CrossRef PubMed.
- X. Y. Deng, J. J. Zhang, K. Z. Qi, G. J. Liang, F. Y. Xu and J. G. Yu, Nat. Commun., 2024, 15, 4807 CrossRef.
- J. Tian, Y. Zhang, Z. Shi, Z. Liu, Z. Zhao, J. Li, N. Li and H. Huang, Angew. Chem., Int. Ed., 2025, 64, e202418496 CrossRef.
- X. Wang, H. H. Liao, W. Tan, W. Song, X. Li, J. W. Ji, X. Q. Wei, C. Wu, C. X. Yin, Q. Tong, B. Peng, S. C. Sun, H. Q. Wan and L. Dong, ACS Appl. Mater. Interfaces, 2024, 16, 22089–22101 CrossRef PubMed.
- B.-H. Yan, B.-R. Xu, Y. Jin, H. Xiao, S.-C. Luo, R.-H. Duan, H. Li, X.-Q. Yan, B. Lin and G.-D. Yang, cMat, 2024, 1, e28 CrossRef.
- H. W. Huang, J. W. Zhao, H. L. Guo, B. Weng, H. W. Zhang, R. A. Saha, M. L. Zhang, F. L. Lai, Y. F. Zhou, R. Z. Juan, P. C. Chen, S. B. Wang, J. A. Steele, F. L. Zhong, T. X. Liu, J. Hofkens, Y. M. Zheng, J. L. Long and M. B. J. Roeffaers, Adv. Mater., 2024, 36, 2313209 CrossRef CAS.
- X. F. Kang, Z. Z. He, F. Wang, Y. Liu and L. J. Guo, Adv. Funct. Mater., 2025, 35, 2419802 CrossRef CAS.
- L. P. Jiang, D. Q. Chen, Z. K. Hao, D. X. Cao, R. Q. Liu, J. Y. Cheng, L. M. Chen, X. Liu, B. Y. Jia and D. D. Liu, Energy Environ. Sci., 2024, 17, 8228–8242 RSC.
- Y. Chai, Y. H. Kong, M. Lin, W. Lin, J. N. Shen, J. L. Long, R. S. Yuan, W. X. Dai, X. X. Wang and Z. Z. Zhang, Nat. Commun., 2023, 14, 6168 CrossRef CAS PubMed.
- X. Zhu, Y. Cao, Y. Song, J. Yang, X. She, Z. Mo, Y. She, Q. Yu, X. Zhu, J. Yuan, H. Li and H. Xu, Small, 2021, 17, 2103796 CrossRef CAS.
- J. C. Wu, F. Huang, Q. Y. Hu, D. P. He, W. X. Liu, X. D. Li, W. S. Yan, J. Hu, J. F. Zhu, S. Zhu, Q. X. Chen, X. C. Jiao and Y. Xie, J. Am. Chem. Soc., 2024, 146, 26478–26484 CrossRef CAS PubMed.
- S. K. Xue, C. G. Wei, M. Shen, X. C. Liang, J. L. Wang, C. Yang, W. D. Xing, S. B. Wang, W. Lin, Z. Y. Yu, Y. D. Hou, J. C. Yu and X. C. Wang, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2319751121 CrossRef CAS.
- W. W. Shao, X. D. Li, J. C. Zhu, X. L. Zu, L. Liang, J. Hu, Y. Pan, J. F. Zhu, W. S. Yan, Y. F. Sun and Y. Xie, Nano Res., 2022, 15, 1882–1891 CrossRef CAS.
- L. Cheng, X. Y. Yue, L. X. Wang, D. N. Zhang, P. Zhang, J. J. Fan and Q. J. Xiang, Adv. Mater., 2021, 33, e2105135 CrossRef.
- H. N. Shi, H. Z. Wang, Y. C. Zhou, J. H. Li, P. L. Zhai, X. Y. Li, G. G. Gurzadyan, J. G. Hou, H. Yang and X. W. Guo, Angew. Chem., Int. Ed., 2022, 61, e202208904 CrossRef CAS PubMed.
- C. Chen, C. Ye, X. Zhao, Y. Zhang, R. Li, Q. Zhang, H. Zhang and Y. Wu, Nat. Commun., 2024, 15, 7825 CrossRef CAS.
- J. Y. Ding, P. J. Du, P. P. Li, W. X. Liu, J. Q. Xu, W. S. Yan, Y. Pan, J. Hu, J. F. Zhu, Q. X. Chen, X. C. Jiao and Y. Xie, Angew. Chem., Int. Ed., 2025, 64, e202414453 CrossRef CAS PubMed.
- P. Du, J. Ding, C. Liu, P. Li, W. Liu, W. Yan, Y. Pan, J. Hu, J. Zhu, X. Li, Q. Chen and X. Jiao, Angew. Chem., Int. Ed., 2025, 64, e202421353 CrossRef CAS.
- G. Wang, Z. Chen, T. Wang, D. S. Wang and J. J. Mao, Angew. Chem., Int. Ed., 2022, 61, e202210789 CrossRef CAS.
- Z. Guo, H. Yang, X. Huang, Y. Ning, H. Luo, J. Xie, J. He, Y. Liu and T.-C. Lau, Angew. Chem., Int. Ed., 2025, e202423666 CAS.
- C. Chen, C. Ye, X. Zhao, Y. Zhang, R. Li, Q. Zhang, H. Zhang and Y. Wu, Nat. Commun., 2024, 15, 7825 CrossRef CAS.
- J. Ding, P. Du, J. Zhu, Q. Hu, D. He, Y. Wu, W. Liu, S. Zhu, W. Yan, J. Hu, J. Zhu, Q. Chen, X. Jiao and Y. Xie, Angew. Chem., Int. Ed., 2024, 63, e202400828 CrossRef CAS PubMed.
- W. Gao, L. Shi, W. Hou, C. Ding, Q. Liu, R. Long, H. Chi, Y. Zhang, X. Xu, X. Ma, Z. Tang, Y. Yang, X. Wang, Q. Shen, Y. Xiong, J. Wang, Z. Zou and Y. Zhou, Angew. Chem., Int. Ed., 2024, 63, e202317852 CrossRef CAS PubMed.
- L. Zhang, T. Liu, T. Liu, S. Hussain, Q. Li and J. Yang, Chem. Eng. J., 2023, 463, 142358 CrossRef CAS.
- S. M. Sun, M. Watanabe, J. Wu, Q. An and T. Ishihara, J. Am. Chem. Soc., 2018, 140, 6474–6482 CrossRef CAS.
- Y. Wu, Q. X. Chen, J. C. Zhu, K. Zheng, M. Y. Wu, M. H. Fan, W. S. Yan, J. Hu, J. F. Zhu, Y. Pan, X. C. Jiao, Y. F. Sun and Y. Xie, Angew. Chem., Int. Ed., 2023, 62, e202301075 CrossRef CAS.
- H. Huang, J. Zhao, B. Weng, F. Lai, M. Zhang, J. Hofkens, M. B. J. Roeffaers, J. A. Steele and J. Long, Angew. Chem., Int. Ed., 2022, 61, e202204563 CrossRef CAS.
- K. Zheng, S. Y. Liu, J. C. Zhu, Z. Q. Dai, C. Y. Liu, B. W. Li, Y. B. Zheng, X. Y. Chen, L. Zhai, Y. Wu, W. X. Liu, M. H. Fan, J. Hu, Y. Pan, J. F. Zhu, F. F. Sun, Y. F. Sun and Y. Xie, Angew. Chem., Int. Ed., 2025, e202508259 Search PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |