
Promoting photoreduction selectivity via synergetic utilization between vacancy and nanofiber structure over flexible Zr/TiO2−x nanofiber films†
Shan
Jiang‡
a,
Haoze
Li‡
a,
Wenke
Gui
a,
Yingbing
Zhang
a,
Chenchen
Zhang
b,
Lei
Zhang
 c,
Jianping
Yang
a and
Li
Wang
*a
c,
Jianping
Yang
a and
Li
Wang
*a
aInstitute of Functional Materials, State Key Laboratory for Modification of Chemical Fibers and Polymer Materials, College of Materials Science and Engineering, Donghua University, Shanghai 201620, P. R. China. E-mail: lw66@dhu.edu.cn
bKey Laboratory of Synthetic and Biological Colloids, Ministry of Education, School of Chemical and Material Engineering, Jiangnan University, Wuxi, Jiangsu 214122, China
cCentre for Catalysis and Clean Energy, Gold, Coast Campus, Griffith University, Gold Coast, QLD 4222, Australia
First published on 11th March 2024
Abstract
Photocatalytic conversion of CO2 into value-added hydrocarbon fuels is a promising approach to alleviate the energy crisis caused by the overuse of fossil fuels. Here, a flexible Zr/TiO2−x nanofiber photocatalyst with abundant oxygen-vacancies (OVs) has been fabricated and employed in the CO2 photocatalytic reduction process. The selectivity for the photocatalytic reduction of CO2 to CH4 over H400-Zr/TiO2−x nanofiber films could reach up to 87.6% which is rather high compared with currently reported photocatalytic systems. In-depth experiments demonstrate that the high product selectivity of CH4 originated from the synergetic effect between vacancy and nanofiber structure in H400-Zr/TiO2−x. Density functional theory (DFT) simulation reveals that the existence of a vacancy in H400-Zr/TiO2−x facilitates a reduction in the surface free energy barrier from the intermediate CO* to CHO* during the production of CH4, which is further confirmed by the observed obvious CHO* signal in in situ FTIR spectra. Additionally, the characteristic of one-dimensional long-range orientation and large surface area of nanofiber structure of H400-Zr/TiO2−x is beneficial to providing more catalytic active sites which help to promote the CO2 photoreduction property. This work paves the way for the efficient design of photocatalytic systems towards high conversion of CO2 to CH4.
 Li Wang | Li Wang was appointed as an Associated Professor in the College of Materials Science and Engineering at Donghua University (China). She received her PhD at the University of Wollongong in 2018, and then held research fellow and postdoctoral positions at Monash University and Ludwig-Maximilians-Universität München. Her research interests include the design of functional materials towards energy and environmental applications. |
Introduction
In the past few decades, the continuously excessive burning of fossil fuels has resulted in massive CO2 emissions, causing serious environmental problems and energy crises.1–4 Photocatalytic reduction of CO2 into value-added products represents a crucial pathway to reduce the CO2 level in the atmosphere and thus alleviate the environmental and energy issues.5–8 CH4 is considered to be the most promising hydrocarbon product during the photocatalytic CO2 reduction reaction (CO2RR) as its generation process could consume CO2 and its burning process could produce only H2O and CO2 which helps to achieve a carbon cycle.9–11 However, the current production of CH4 with high selectivity still remains a big challenge due to the side reactions of water splitting and by-products which have similar reduction potentials with CH4.12,13 Hence, developing novel photocatalysts to promote the selectivity of CH4 in the photocatalytic reduction of CO2 is of great importance for the establishment of a carbon-neutral society.An ideal photocatalyst is expected to have a suitable bandgap which can not only enable sufficient harvesting of solar light but can also provide a good match with the chemical generation potential of specific products in the CO2RR.14,15 TiO2 has the advantages of non-toxicity, low cost, earth abundance, high photocatalytic reaction rate and good stability. More importantly, Ti4+ with d-band centers relatively close to the Fermi level promoted CO2 absorption/activation, resulting in CO2 photoreduction to products such as CH4 or CO, and its band energy levels are suitable as its conduction band (CB) position is more negative than the reduction potential of CO2 to CH4 and the valence band (VB) position is corrected compared with the oxidation potential of H2O to O2.16–18 Hence, TiO2 is a promising candidate in realizing high efficiency and selectivity for the photoreduction of CO2 to CH4. Many research works have been carried out over TiO2 for the CO2 photoreduction reaction. For example, Wang et al. adopted metalloporphyrins to support commercial TiO2 (P25) to prepare TCPP-Cu/TiO2 for the CO2 photoreduction process, and the selectivity for CH4 is about 29.8%.19 Wang et al. prepared a ternary Z-scheme Ag–Cu2O/TiO2 catalyst using a one-step reduction method, and the 0.5Ag–0.5Cu2O/TiO2 catalyst has the highest CO2 photoreduction performance with a CH4 selectivity of 30%.20 Despite these achievements, the obtained selectivity for the photoreduction of CO2 to CH4 is still far from satisfactory. Recent theoretical and experimental research has demonstrated that unsaturated metals are extremely efficient in transferring electrons to adsorbed reactants.21,22 OVs are considered to possess abundant localized electrons, which facilitate the adsorption/activation of inert gas molecules (such as CO2) and the formation of intermediates.23–28 Thus, constructing OVs is an efficient strategy to improve the CO2 photoreduction selectivity of TiO2. Jing et al. introduced OVs in iron tetraphenylporphyrin (FeTPP) modified TiO2 to reinforce the efficiency and selectivity of the CO2 photoreduction process, and the obtained selectivity for CO2 to CH4 could achieve 56.1%.29 The nanofiber structure possesses the characteristics of long-range orderliness and large surface area which facilitate the provision of more catalytic active sites and promote the catalytic reactions.30–34 Ding et al. prepared a mesoporous black Nb2O5 nanofiber catalyst and found that the selectivity of CH4 could be increased to 64.8%.35 Even though the introduction of OVs and the construction of a nanofiber structure could be employed to improve the product selectivity of the CO2 photoreduction reaction, whether these two aspects have a synergetic effect in promoting the product selectivity of the CO2 photoreduction process over TiO2 is ambiguous.
In this work, we prepared flexible H400-Zr/TiO2−x nanofiber films (NFs) and employed them in the CO2RR. It is found that H400-Zr/TiO2−x NFs have abundant OVs which broaden the bandgap and increase the adsorption wavelength range. The product selectivity of CH4 in the CO2RR over H400-Zr/TiO2−x NFs is about 87.6% which is rather higher than currently reported works. In-depth investigation demonstrates that a synergetic effect exists between OVs and the nanofiber structure of H400-Zr/TiO2−x NFs in promoting the product selectivity of CH4 during the CO2RR process. The OVs are proved to possess a lower surface free energy barrier for the transformation from the intermediate CO* to CHO* during the photocatalytic CO2RR to CH4 process via DFT simulation, which is further confirmed by the in situ FTIR characterization and is beneficial for the improvement of CH4 selectivity. This work provides guidance for the future design of efficient photocatalysts towards high product selectivity of the CO2RR.
Experimental
Materials
Polyvinylpyrrolidone (PVP) (Mw = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300000) was purchased from Shanghai Macklin Biochemical Co., Ltd. Titanium(IV) tetraisopropanolate (TTIP) (98%) and zirconium acetate (Zr(Ac)4) were purchased from Shanghai Adamas Reagent Co., Ltd. Acetic acid (99.5%) and absolute ethanol (99.7%) were all purchased from Hushi Chemical Co., Ltd, China. All the initial chemicals were used without further purification.
300000) was purchased from Shanghai Macklin Biochemical Co., Ltd. Titanium(IV) tetraisopropanolate (TTIP) (98%) and zirconium acetate (Zr(Ac)4) were purchased from Shanghai Adamas Reagent Co., Ltd. Acetic acid (99.5%) and absolute ethanol (99.7%) were all purchased from Hushi Chemical Co., Ltd, China. All the initial chemicals were used without further purification.
Preparation of flexible Zr/TiO2 NFs
Firstly, 0.5 g PVP (Mw = 1300000) was dissolved in a mixed solution containing 4 mL acetic acid and 10 mL ethanol under vigorous stirring at room temperature for 12 h. Next, 3 g titanium tetraisopropanolate (TTIP) and 0.38 g Zr(Ac)4 were slowly added into the above solution and then stirred in an ice-water bath for 1 h to produce a homogeneous solution. The electrospinning process was performed by using a HZ-TJB-02 spinning apparatus with a stable flow rate of 1.5 mL h−1 at an applied voltage of 17 kV and with a distance of 15 cm between the nozzle. The precursor fibrous membranes were collected on silicone paper covering the roller collector with a rotating speed of 200 rpm. Finally, the precursor nanofibers were collected and calcined in a furnace at 600 °C for 60 min with a heating rate of 2 °C min−1 in air. The same method was used as above to prepare pure TiO2 nanofibers (no Zr(Ac)4 added).
Preparation of flexible Zr/TiO2−x NFs
H400-Zr/TiO2−x NFs were made by placing Zr/TiO2 NFs in a porcelain boat and then calcined in a tube furnace at 400 °C for 3 h with a heating rate of 2 °C min−1 under a H2 atmosphere. H300-Zr/TiO2−x NFs, H500-Zr/TiO2−x NFs and H600-Zr/TiO2−x NFs were prepared by changing the temperature in the hydrogen atmosphere to 300 °C, 400 °C and 600 °C, respectively.Characterization of materials
The surface morphology of Zr/TiO2−x NFs was characterized by SEM (TESCAN/MAIA3). Transmission electron microscopy (TEM) and element mapping measurements were conducted on FEI Talos F200S to check the structure and element distribution of Zr/TiO2−x NFs. Raman was tested by the Dilor LabRam-1B microscope Raman spectrometer with an excitation wavelength of 532 nm. The XRD patterns of the as-prepared catalysts were obtained using a Rigaku D/MAX-2550 PC diffractometer (Tokyo, Japan) equipped with CuKα radiation (40 mA, 40 kV) in the 2θ rang of 10° to 90° at a scan rate of 5° min−1. The surface areas and pore size distribution of Zr/TiO2−x NFs were tested using a Brunauer–Emmett–Teller analyzer (BET, ASAP 2460, Micromeritics Co. USA). The XPS spectra were acquired by Escalab 250Xi.Photocatalytic CO2 reduction
In a closed evacuated system, the CO2/H2O solution was used to carry out CO2 photoreduction experiments. The nanofibers (20 mg) were placed in a quartz reactor equipped with a cooling system, and vacuuming and filling with CO2 were carried out to remove impurity gases. Then, 40 kPa pure CO2 gas and 10 mL distilled water were injected into the closed reactor with a syringe. A 300 W Xe lamp (>420 nm) was used as the excitation source. The gaseous products of CO and CH4 in the reactor effluent were analyzed by using a gas chromatograph (FuliGC9790II) equipped with flame ionization detector (FID).DFT calculations
DFT calculations were performed in the Vienna ab initio simulation package (VASP). The generalized gradient approximation (GGA) with Perdew–Becke–Ernzerhof (PBE) was used to optimize the structures, all-electron plane-wave basis sets with an energy cutoff of 600 eV. A (2 × 2 × 1) Monkhorst–Pack mesh was used for the Brillouin-zone integrations to be sampled. The convergence tolerance of energy and the force of each atom were set as 1 × 10−4 eV and 0.05 eV Å−1. The energy barrier of the various reactions was calculated by subtracting the energy of the initial state from the energy of the transition state.36| ΔG = Etotal − E0 − TΔS |
Results and discussion
Synthesis and characterizations of Zr/TiO2−x NFs
Fig. 1a shows the synthesis procedure of flexible OVs-rich Zr/TiO2−x NFs. Firstly, the spinning solution was prepared with ethanol and acetic acid (C2H4O2) as solvents, polyvinyl pyrrolidone (PVP) as the polymer template, and titanium tetraisopropanolate (TTIP) together with Zr(Ac)4 as the Ti source and Zr source. Then the smooth precursor nanofibers were successfully prepared by electrospinning technology. Here, C2H4O2 acted as a chelating agent to inhibit the rapid hydrolysis of TTIP since it was easily hydrolyzed in ethanol, and PVP could be connected to TTIP via hydrogen bonds to form a stable sol. Next, the precursor nanofibers were calcinated at 600 °C in air to remove carbon from PVP, and the surface of the nanofibers could quickly be changed from smooth to rough during this process. Finally, the OVs were introduced to Zr/TiO2−x NFs by hydrogenation at different temperatures.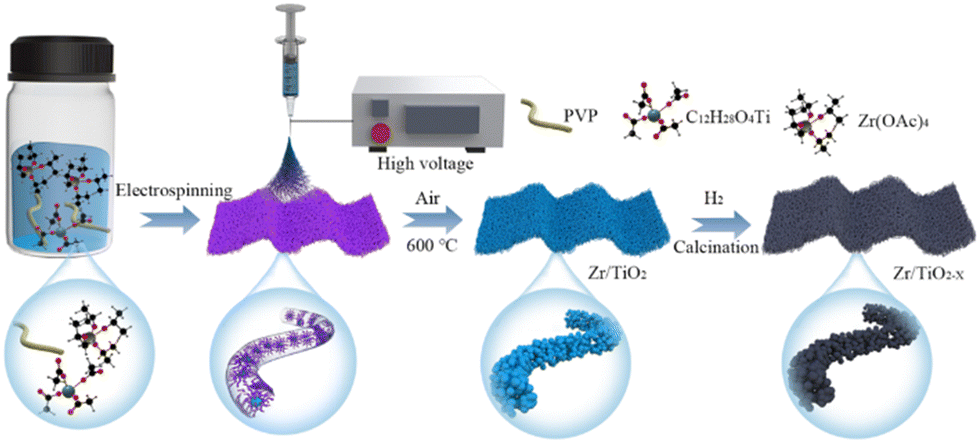 | ||
| Fig. 1 The fabrication process of Zr/TiO2−x NFs by using electrospinning and calcination. | ||
Fig. 2a shows the XRD patterns of Zr–TiO2 NFs and H400-Zr/TiO2−x NFs. It was found that the X-ray diffraction characteristic peaks for these two materials matches well with the standard PDF (JCPDS file no. 21-1276) card of the TiO2 anatase phase, reflecting that the Zr–TiO2 NFs and H400-Zr/TiO2−x NFs are both anatase TiO2. Additionally, the XRD patterns of the Zr–TiO2 NFs and H400-Zr/TiO2−x NFs have negligible difference before and after hydrogenation, and the XRD patterns of Zr/TiO2−x NFs calcinated at different temperatures are also indexed to the TiO2 anatase phase (Fig. S1, ESI†), revealing that hydrogenation process does not affect the composition of TiO2. Scanning electron microscopy (SEM) was used to explore the morphology of H400-Zr/TiO2−x NFs. As displayed in Fig. 2b, a pronounced fiber structure without obvious cross-linking can be observed and the diameter of the nanofibers is around 207 nm (Fig. 2c). Transmission electron microscopy (TEM) was adopted to further analyze the finer structure of H400-Zr/TiO2−x NFs and the results are demonstrated in Fig. 2d and e. It can be observed that the nanofibers were composed of many nanosized grains. The high-resolution TEM image in Fig. 2f demonstrates that the distance of the lattice fringe was 0.35 nm which corresponds to the (101) plane of the TiO2 anatase phase.37 The elemental mapping images by TEM in Fig. 2g illustrate that Ti, O and Zr were uniformly dispersed over the whole nanofiber, indicating that the Zr4+ ions were uniformly dispersed throughout the fibers with no aggregation. The mapping plot of Zr actual doping amount was about 9.65%, which is close to the theoretical doping amount of 10% as shown in Fig. S2 (ESI†).
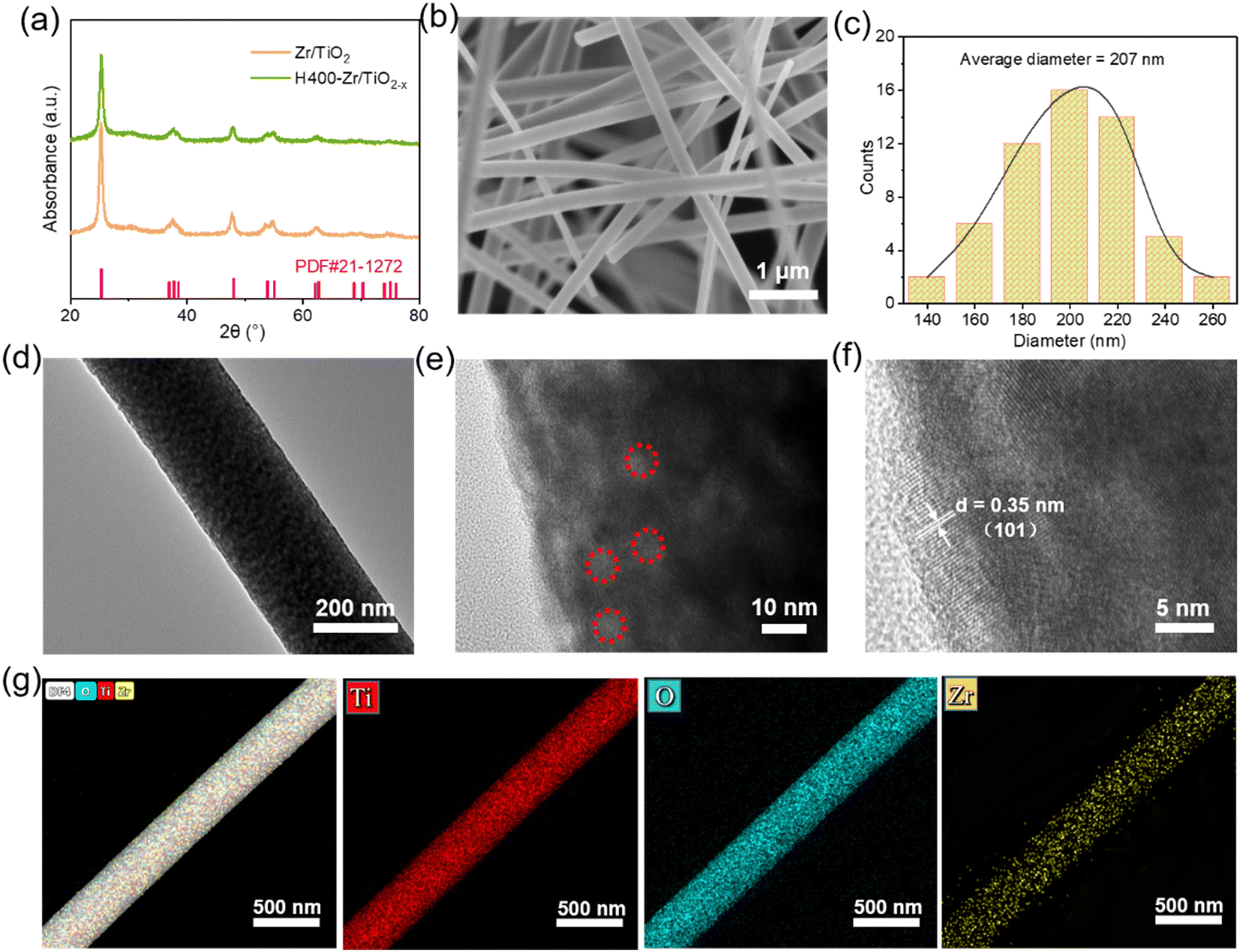 | ||
| Fig. 2 (a) XRD patterns of Zr/TiO2 NFs and H400-Zr/TiO2−x NFs. (b) SEM images of H400-Zr/TiO2−x NFs. (c) The diameter distribution of H400-Zr/TiO2−x NFs counted from (b). (d) and (e) TEM images of H400-Zr/TiO2−x NFs with different magnification. The OVs are marked in a red circle in (e). (f) High-resolution TEM images of H400-Zr/TiO2−x NFs with the lattice distance marked. (g) Elemental mapping of H400-Zr/TiO2−x NFs. | ||
Various characterization technologies were used to explore the changes on the structural and physical properties of Zr/TiO2 NFs caused by the introduction of OVs. Fig. 3a shows the electron paramagnetic resonance (EPR) spectra of Zr/TiO2 and H400-Zr/TiO2−x NFs. It is clear that H400-Zr/TiO2−x NFs present an obvious signal at around g = 2.2 which corresponds to the unpaired electrons and thus confirms the existence of OVs in the as-prepared NFs.38 X-ray photoelectron spectroscopy (XPS) was adopted to further confirm the presence of OVs. As demonstrated in Fig. 3b, compared with Zr/TiO2 NFs, the two deconvolved peaks of O 1s for H400-Zr/TiO2−x NFs moved in the direction of higher binding energy and the Ti 2p peaks of H400-Zr/TiO2−x NFs were redshifted (Fig. S3a, ESI†), which were attributed to the structural relaxation caused by the existence of OVs.39 In Fig. 3c, the Raman peak of H400-Zr/TiO2−x NFs at around 510 cm−1 was found to be slightly redshifted, which is ascribed to the increment of electronic conductivity of TiO2via the introduction of OVs.40
 | ||
| Fig. 3 (a) EPR spectra, (b) O 1s XPS spectra and (c) Raman patterns of Zr/TiO2 NFs and H400-Zr/TiO2−x NFs. (d) and (e) N2 adsorption–desorption isotherms and corresponding specific surface area (SSA) of Zr/TiO2 NFs and H400-Zr/TiO2−x NFs. (f) UV-visible diffuse reflectance spectra of Zr/TiO2 NFs and H400-Zr/TiO2−x NFs. | ||
The surface area and the pore types of the two nanofibers were checked by N2 adsorption–desorption measurements (Fig. 3d). It can be observed that all the samples can be identified as type IV characteristic isotherms which possess capillary condensation, and the specific surface area of Zr/TiO2 NFs and H400-Zr/TiO2−x NFs calculated using the adsorption isotherm were about 67.3 m2 g−1 and 158.6 m2 g−1, respectively (Fig. 3e). The rather large specific surface area of H400-Zr/TiO2−x NFs could provide more active sites on the surface, and could also promote the adsorption of CO2 molecules, which thus help to promote the photocatalytic efficiency. The pore size distribution curves in Fig. S4b (ESI†) show that H400-Zr/TiO2−x NFs mainly possess mesopores with a uniform pore size distribution and the diameter of the pore is centered at around 3.138 nm while the pore size distribution of Zr/TiO2 NFs is relatively dispersed.41 UV-vis diffuse reflectance spectra were measured to study the optical absorption properties of the different samples. As displayed in Fig. 3f, the absorption band edge of Zr/TiO2 NFs and H400-Zr/TiO2−x NFs is about 400 nm which corresponds to the band gap of about 3.1 eV (Fig. S5a, ESI†). The band gaps of Zr/TiO2−x materials calcinated at different temperatures were also slightly reduced (Fig. S5b, ESI†), reflecting that the hydrogenation process has little influence on the band gap of TiO2. Compared with Zr/TiO2 NFs, H400-Zr/TiO2−x NFs present an additional continuous and exponentially decaying absorption tail extended to 800 nm, which is ascribed to the absorption of defect states induced by OVs.42,43 Photoluminescence (PL) spectroscopy was adopted to further investigate the separation and transfer dynamics of photogenerated carriers. It is clear that H400-Zr/TiO2−x NFs exhibit highly suppressed PL intensity as compared to Zr/TiO2 NFs (Fig. S6a, ESI†), suggesting less radiative recombination of photogenerated charge carriers in H400-Zr/TiO2−x NFs.44 As a result, more charge carriers can participate in the photocatalytic reactions. Fig. S6b (ESI†) shows the electrochemical impedance plots (EIS) of Zr/TiO2 NFs and H400-Zr/TiO2−x NFs, the arc radius of H400-Zr/TiO2−x NFs is smaller than that of Zr/TiO2 NFs, implying that the resistance for the charge transfer process is reduced after vacancy modification. To gain a deeper insight into the charge separation and transfer efficiency, photocurrent curves were performed to evaluate the photoelectric properties of these different materials, and the H400-Zr/TiO2−x NFs show largely enhanced photocurrent density, which can be ascribed to the highly efficient charge transfer (Fig. S7a, ESI†).45 Charge carrier dynamics was further investigated using the transient photocurrent response (Fig. S7b and c, ESI†). Generally, the following exponential functions can be used to describe the rise process of the photocurrent response.46
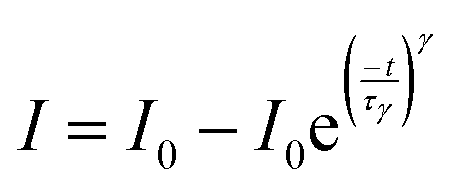
CO2 photocatalytic performance and mechanisms
The CO2 photoreduction performance was characterized to determine the influence of OVs and nanofiber structure on the photocatalytic properties. Fig. 4a and b demonstrate the evolution of CO and CH4 dependent on irradiation time over various as-prepared photocatalysts, and the evolution rates are summarized in Fig. S9a (ESI†). Among these as-prepared photocatalytic systems, Zr/TiO2 NFs present the highest CO yields while H400-Zr/TiO2−x NFs present the highest CH4 yield, indicating that OVs have an effect on the product selectivity for the CO2RR. Fig. S10 (ESI†) shows the change of the O2 characteristic peak in gas chromatography during the photocatalytic CO2 reduction process, which verifies that the catalyst promotes the oxidation of H2O to O2. E(H2O/O2) = 0.82 eV48 was also within the band gap range of the H400-Zr/TiO2−x NFs, which was consistent with theory and practice. The H2O molecules adsorbed on the surface of TiO2 could be decomposed into H+ and OH−, OH− further forms O2, and H+ participates in the reaction of CO2 to form CH4.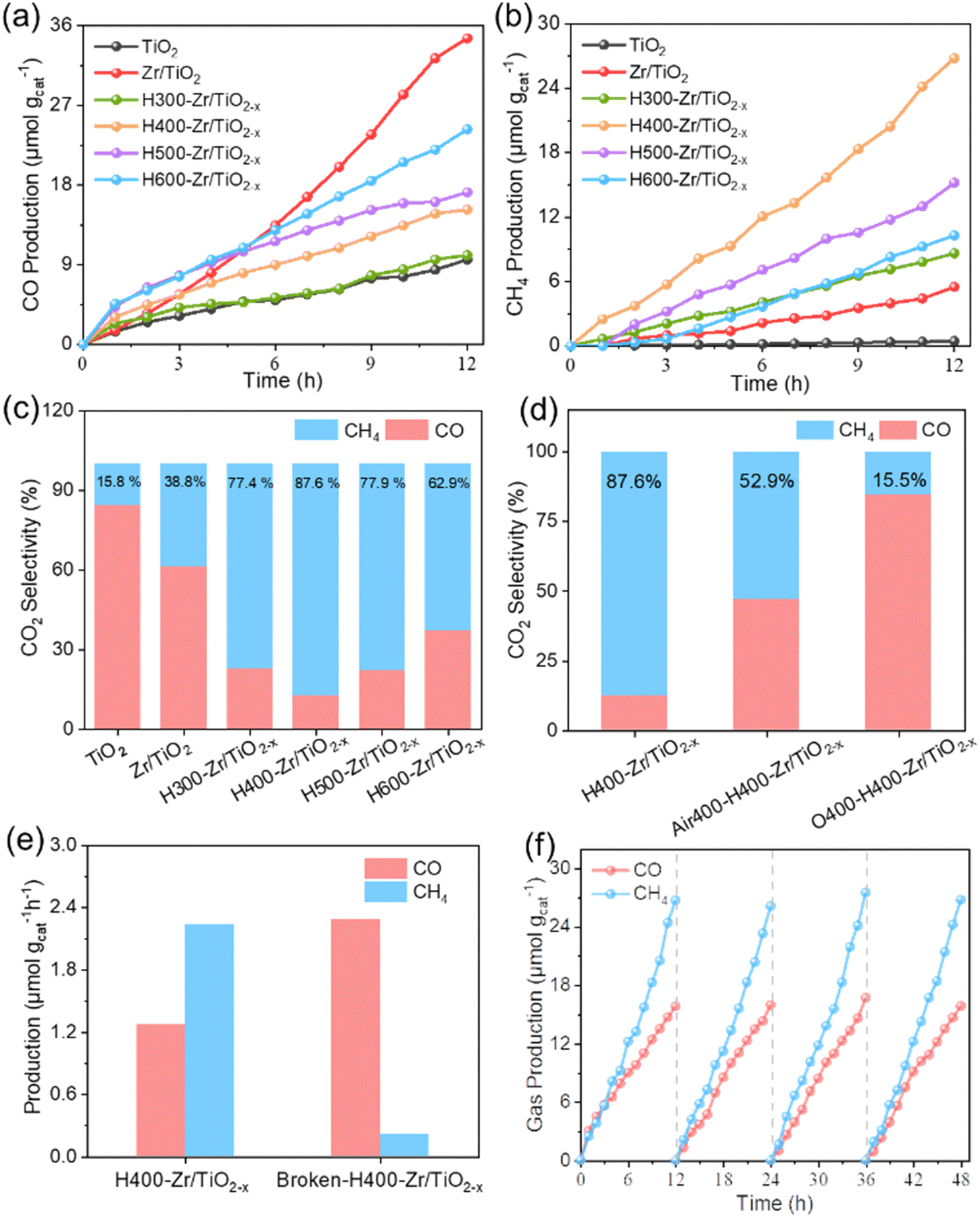 | ||
| Fig. 4 The CO2RR performance of CN and Cux-CN catalysts. (a) and (b) Time-dependent yield of CO and CH4 products under light irradiation over various Zr/TiO2−x NFs materials. (c) Product selectivity of photocatalytic CO2 reduction over various Zr/TiO2−x NFs materials. (d) Product selectivity of H400-Zr/TiO2−x and H400-Zr/TiO2−x post-treated with different contents of O2 to remove OVs. (e) CO2 photoreduction performance of H400-Zr/TiO2−x NFs before and after crushing. (f) Cycling stability tests over H400-Zr/TiO2−x NFs. | ||
The production of CO from the photoreduction of CO2 needs to consume two electrons and protons while the production of CH4 requires eight electrons and protons.12 By calculating the number of consumed electrons, the product selectivity of CO and CH4 can be calculated.
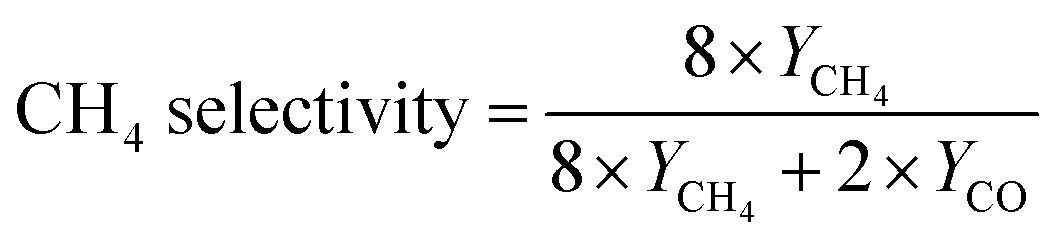
where YCH4 and YCO represent the yield (production rate) of CH4 and CO, respectively.
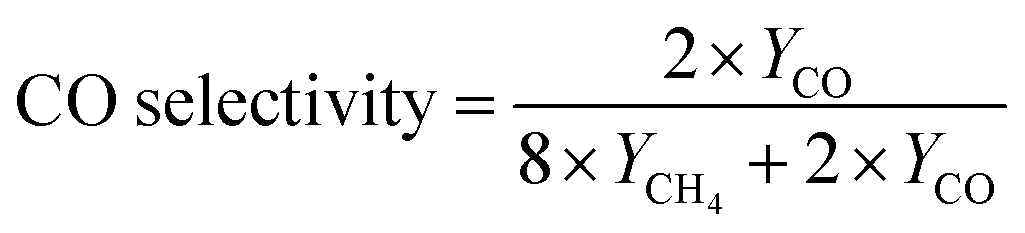
The results in Fig. 4c show that H400-Zr/TiO2−x NFs achieved 87.6% selectivity for CO2 reduction into CH4, which was better than most of the reported results (Fig. S11, ESI†). To in-depth investigate the effect of OVs on the product selectivity of the CO2RR, H400-Zr/TiO2−x NFs were further calcinated in atmospheres with different content of oxygen to remove OVs. H400-Zr/TiO2−x NFs calcined under an air atmosphere with low oxygen content are considered to have OVs partially filled and are denoted as air-H400-Zr/TiO2−x NFs. In contrast, H400-Zr/TiO2−x NFs calcined under an oxygen atmosphere with a high oxygen content are considered to obtain high-vacancy filled O-H400-Zr/TiO2−x NFs. The performance of the above obtained materials was determined, and it was found that both their selectivity and performance were greatly reduced as the amount of OVs decreased (Fig. S9b, ESI†). The selectivity of CH4 was reduced from 87.6% over H400-Zr/TiO2−x NFs to 52.9% over air-H400-Zr/TiO2−x NFs and was further reduced to 15.5% over O-H400-Zr/TiO2−x NFs (Fig. 4d). The nanofiber structure could also affect the product selectivity of the CO2RR. To verify this opinion, the CO2RR performance was characterized and evaluated over the crushed NFs with a destroyed fiber structure. Fig. S12 (ESI†) shows the SEM image and XRD pattern of the crushed NFs with their fiber structure destroyed. It can be observed that the compositions of NFs before and after the structural damage are both indexed to the TiO2 anatase phase. The CO2 photoreduction performance in Fig. 4e illustrates that Broken-H400-Zr/TiO2−x NFs has a rather low yield rate of CH4 yield, reflecting the superiority of fiber structure in photoconverting CO2 into CH4. Thus, it can be concluded that the synergistic effect between OVs and the nanofiber structure of H400-Zr/TiO2−x produces a high selectivity of CH4 in the CO2RR process.
The photocatalytic CO2RR performance characterized without CO2, without H2O, without catalyst and without light were also investigated (Fig. S13, ESI†). In these four experimental conditions, the performance of CO2 photoreduction was negligible, suggesting that the CO2 photoreduction process requires the involvement of CO2, H2O, photocatalyst and light. For the as-prepared flexible nanofibers, it can be simply collected from the reactor and then reused without a further centrifugation and redispersion process, which could avoid many disadvantages such as difficult recovery of powder and secondary pollution to the environment. Taking the 12 h test as a cycle, the production rate of CH4 was almost unchanged during the overall testing period of four cycles (Fig. 4f), and the selectivity for CO2 reduction remains stable over four cycles (Fig. S14, ESI†). The XRD pattern and SEM image of H400-Zr/TiO2−x NFs after the measurement of the CO2RR in Fig. S15 (ESI†) show that there was no significant change in the composition and morphology structure of H400-Zr/TiO2−x NFs. These results demonstrate that H400-Zr/TiO2−x NFs possess excellent cycling stability. The XRD patterns and SEM images of other Zr/TiO2−x material photocatalytic measurements were also characterized, showing the strong stability of the fibers (Fig. S16, ESI†). Fig. S17a and b (ESI†) display the optical images of the folded and opened H400-Zr/TiO2−x NFs and the corresponding photocatalytic CO2RR properties were demonstrated in Fig. S17c (ESI†). It is obvious that there was no significant decrease in the performance of the fibers before and after folding. Thus, when the flexible nanofiber photocatalyst was not in use, it can be simply folded to reduce the occupied space, and when it needs to be used, it is opened without much change in the photocatalytic performance, suggesting great potential for practical application.
DFT simulation was further adopted to investigate the selectivity mechanism of NFs. The structural models of Gibbs free energy calculations were constructed as shown in Fig. S18 (ESI†). Fig. 5 demonstrates the calculated Gibbs free energies of the reaction pathway of the H400-Zr/TiO2−x NFs catalyst for CO2 reduction to CH4 and CO with the computation details presented in Table S1 (ESI†). It can be found that the Gibbs free energy of the adsorbed activation (*CO2) of H400-Zr/TiO2−x NFs is less than 0, indicating that the catalyst facilitates the activation of CO2. The next step of the reaction is to generate COOH* by hydrogenation and COOH* is subsequently converted to OH* and CO*. OH* can easily be converted to water while the transformation of CO* involves two competing reactions. One is the desorption of *CO from the catalyst surface to form CO, and the other one is the transformation of CO* to CHO* by hydrogenation which is a prerequisite for the formation of CH4. As shown in Fig. 5a, the desorption of CO* requires more energy than the hydrogenation of *CO, demonstrating that CO2 is more likely to generate CH4 on the surface of H400-Zr/TiO2−x NFs, furthermore, from the situ infrared absorption spectra of the H400-Zr/TiO2−x NFs in Fig. 5b, peaks at 1100 cm−1 and1225 cm−1 are attributed to the CHO* and CH3O* groups, which are intermediate products promoting the formation of CH4.12 In conclusion, the introduction of OVs provides an effective strategy for the conversion of active CO2 to CH4.49,50 The valence band positions of Zr/TiO2 and H400-Zr/TiO2−x NFs were calculated through the XPS valence band spectrum which are 2.46 eV and 2.56 eV, respectively (Fig. S19, ESI†). The band gap width can be obtained on the UV-vis diffuse reflectance spectra of Fig. 2f. Based on the above results, the band structure of Zr/TiO2 and H400-Zr/TiO2−x NFs can be drawn, which shows the thermodynamically feasibility for CO2 reduction (Fig. 5c). A schematic illustration of the mechanism for the photocatalytic CO2RR is shown in Fig. 5d. It is proposed that the OVs act as traps to capture the photogenerated electrons, and thus reduce the recombination of electron–hole pairs.51 Simultaneously, the unsaturated Ti could interact with C and O, resulting in a high catalytic performance and selectivity of CH4.
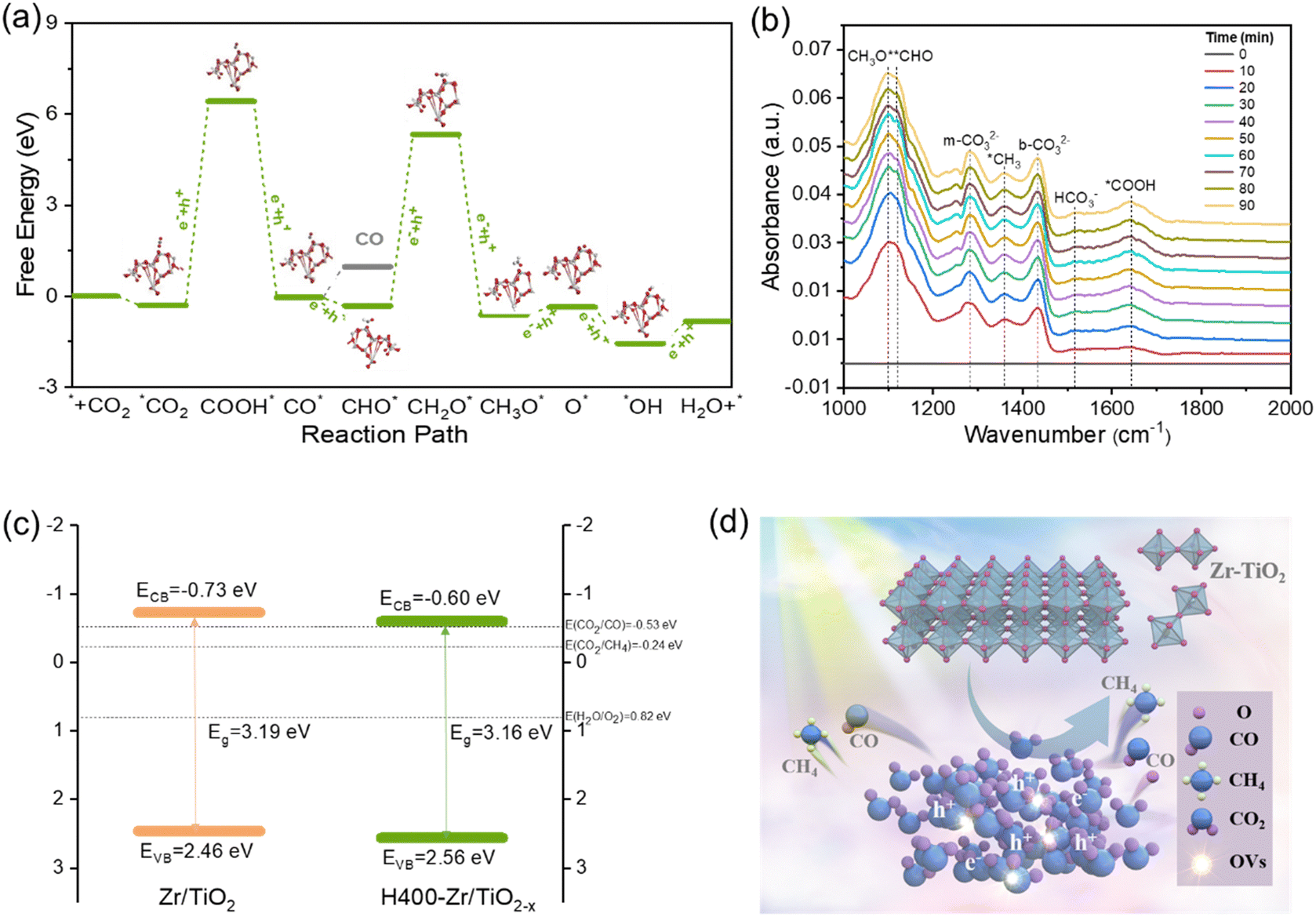 | ||
| Fig. 5 (a) Calculated Gibbs free energy diagrams of CO2 photoreduction to CO/CH4 for the H400-Zr/TiO2−x NFs. (b) In situ FTIR characterization for the adsorption and activation of CO2 on the H400-Zr/TiO2−x NFs catalyst. (c) Evaluated band structures of Zr/TiO2 and H400-Zr/TiO2−x NFs. (d) Schematic illustration for the photocatalytic mechanism over Zr/TiO2 and H400-Zr/TiO2−x NFs. | ||
Conclusions
H400-Zr/TiO2−x NFs with abundant OVs were fabricated and employed in the photocatalytic CO2RR process. It was found that H400-Zr/TiO2−x NFs have a large surface area of 158.6 m2 g−1, widened absorption spectrum with the absorption tail extended to 800 nm, and promoted separation and migration properties of charge carriers. DFT simulation confirms that the existence of OVs could reduce the surface free energy barrier for the transformation from the intermediate CO* to CHO*, which is conductive to producing CH4. Additionally, the nanofiber structure is verified to facilitate the evolution of CH4. Benefiting from the synergetic effect between OVs and nanofiber structure, H400-Zr/TiO2−x NFs present a rather high selectivity of CH4 with the value about 87.6% in the photoreduction process of CO2. Furthermore, the folding process of H400-Zr/TiO2−x NFs have negligible influence on the efficiency and selectivity of the CO2RR process, indicating their great potential for practical applications.Author contributions
Shan Jiang and Haoze Li contributed equally to this work.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Fundamental Research Funds for the Central Universities (no. 2232023D-02, and 2232021A-02), and the National Natural Science Foundation of China (no. 52202361 and 52122312).Notes and references
- M. Meinshausen, N. Meinshausen, W. Hare, S. B. Raper, K. Frieler, R. Knutti, D. J. Frame and M. R. Allen, Nature, 2009, 458, 1158 CrossRef CAS PubMed.
- H. Lin, S. Luo, H. Zhang and J. Ye, Joule, 2022, 6, 294 CrossRef CAS.
- T. Yang, M. Kuang and J. Yang, Nano Res., 2023, 16, 8670 CrossRef CAS.
- L. Wang, S. Jiang, W. Gui, H. Li, J. Wu, H. Wang and J. Yang, Small Struct., 2023, 4, 2300142 CrossRef CAS.
- E. C. Ra, K. Y. Kim, E. H. Kim, H. Lee, K. An and J. S. Lee, ACS Catal., 2020, 10, 11318 CrossRef CAS.
- L. Wang, D. Lv, Z. Yue, H. Zhu, L. Wang, D. Wang, X. Xu, W. Hao, S. X. Dou and Y. Du, Nano Energy, 2019, 57, 398–404 CrossRef CAS.
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962 CrossRef CAS PubMed.
- M. Li, F. Zhang, M. Kuang, Y. Ma, T. Liao, Z. Sun, W. Luo, W. Jiang and J. Yang, Nano-Micro Lett., 2023, 15, 238 CrossRef CAS PubMed.
- J. Fu, K. Jiang, X. Qiu, J. Yu and M. Liu, Mater. Today, 2020, 32, 222 CrossRef CAS.
- L. Wang, X. Zhao, D. Lv, C. Liu, W. Lai, C. Sun, Z. Su, X. Xu, W. Hao, S. X. Dou and Y. Du, Adv. Mater., 2020, 32, 2004311 CrossRef CAS PubMed.
- S. Si, H. Shou, Y. Mao, X. Bao, G. Zhai, K. Song, Z. Wang, P. Wang, Y. Liu, Z. Zheng, Y. Dai, L. Song, B. Huang and H. Cheng, Angew. Chem., Int. Ed., 2022, 61, e202209446 CrossRef CAS PubMed.
- E. Gong, S. Ali, C. B. Hiragond, H. S. Kim, N. S. Powar, D. Kim, H. Kim and S. In, Energy Environ. Sci., 2022, 15, 880 RSC.
- J. Jiang, X. Wang, Q. Xu, Z. Mei, L. Duan and H. Guo, Appl. Catal., 2022, 316, 121679 CrossRef CAS.
- Y. Wu, J. Wu, C. Zhu, L. Zhang and J. Yan, Chem. Eng. J., 2023, 465, 142798 CrossRef CAS.
- X. Sun, H. Huang, Q. Zhao, T. Ma and L. Wang, Adv. Funct. Mater., 2020, 30, 1910005 CrossRef CAS.
- X. Xiong, Y. Zhao, R. Shi, W. Yin, Y. Zhao, G. I. N. Waterhouse and T. Zhang, Sci. Bull., 2020, 65, 987 CrossRef CAS PubMed.
- X. Li, J. Yu and M. Jaroniec, Chem. Soc. Rev., 2016, 45, 2603–2636 RSC.
- J. Jeon, D. H. Kweon, B. J. Jang, M. J. Ju and J. B. Baek, Adv. Sustainable Syst., 2020, 4, 2000197 CrossRef CAS.
- Z. Wang, W. Zhou, X. Wang, X. Zhang, H. Chen, H. Hu, L. Liu, J. Ye and D. Wang, Catal., 2020, 10, 654 CAS.
- X. Wang, Z. Jiang, H. Chen, K. Wang and X. Wang, J. Alloys Compd., 2022, 896, 163030 CrossRef CAS.
- Y. Zhao, G. Chen, T. Bian, C. Zhou, G. I. N. Waterhouse, L. Z. Wu, C. H. Tung, L. J. Smith, D. O'Hare and T. Zhang, Adv. Mater., 2015, 27, 7824 CrossRef CAS PubMed.
- K. Wang, Y. Du, Y. Li, X. Wu, H. Hu, G. Wang, Y. Xiao, S. Chou and G. Zhang, Carbon Energy, 2022, 5, 1 Search PubMed.
- A. Nada, W. M. El Rouby, M. Bekheet, M. Antuch, M. Weber, P. Miele, R. Viter, S. Roualdes, P. Millet and M. Bechelany, Appl. Surf. Sci., 2020, 505, 144419 CrossRef CAS.
- F. Xu, J. Zhang, B. Zhu, J. Yu and J. Xu, Appl. Catal., B, 2018, 230, 194 CrossRef CAS.
- Q. Ren, Y. He, H. Wang, Y. Sun and F. Dong, ACS Catal., 2022, 12, 14015–14025 CrossRef CAS.
- J. Low, B. Cheng and J. Yu, Appl. Surf. Sci., 2017, 392, 658 CrossRef CAS.
- X. Xiong, C. Mao, Z. Yang, Q. Zhang, G. I. N. Waterhouse, L. Gu and T. Zhang, Adv. Energy Mater., 2020, 10, 2002928 CrossRef CAS.
- Z. Li, L. Zhang, M. Nishiura, G. Luo, Y. Luo and Z. Hou, J. Am. Chem. Soc., 2020, 142, 1966 CrossRef CAS PubMed.
- Z. Deng, J. Ji, M. Xing and J. Zhang, Nanoscale Adv., 2020, 2, 4986 RSC.
- C. Jia, Z. Xu, D. Luo, H. Xiang and M. Zhu, Adv. Fiber Mater., 2022, 4, 573 CrossRef CAS PubMed.
- Y. Wu, J. Wu, C. Zhu, L. Zhang and J. Yan, Chem. Eng. J., 2023, 465, 142798 CrossRef CAS.
- Y. Wu, J. Wu, C. Zhu, L. Zhang and J. Yan, Chem. Eng. J., 2023, 465, 142798 CrossRef CAS.
- F. Zhang, J. Chen, G. G. Wallace and J. Yang, Prog. Mater. Sci., 2023, 133, 101069 CrossRef CAS.
- F. Zhang, J. Chen and J. Yang, Adv. Fiber Mater., 2022, 4, 720 CrossRef CAS.
- X. Lin, S. Xia, L. Zhang, Y. Zhang, S. Sun, Y. Chen, S. Chen, B. Ding, J. Yu and J. Yan, Adv. Mater., 2022, 34, 2200756 CrossRef CAS PubMed.
- J. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- G. Cheng, M. S. Akhtar, O. B. Yang and F. J. Stadler, ACS Appl. Mater. Interfaces, 2013, 5, 6635 CrossRef CAS PubMed.
- S. M. Wu, X. L. Liu, X. L. Lian, G. Tian, C. Janiak, Y. X. Zhang, Y. Lu, H. Z. Yu, J. Hu, H. Wei, H. Zhao, G. G. Chang, G. Van Tendeloo, L. Y. Wang, X. Y. Yang and B. L. Su, Adv. Mater., 2018, 30, 1802173 CrossRef PubMed.
- X. Wei, J. Cao and F. Fang, RSC Adv., 2018, 8, 31822 RSC.
- S. Wang, Z. Zhang, W. Huo, X. Zhang, F. Fang, Z. Xie and J. Jiang, Chem. Eng. J., 2021, 403, 126331 CrossRef CAS.
- F. Xu, Z. Tang, S. Huang, L. Chen, Y. Liang, W. Mai, H. Zhong, R. Fu and D. Wu, Nat. Commun., 2015, 6, 1 Search PubMed.
- X. Y. Kong, Y. Y. Choo, S. P. Chai, A. K. Soh and A. R. Mohamed, Chem. Commun., 2016, 52, 14242 RSC.
- V. Etacheri, C. D. Valentin, J. Schneider, D. Bahnemann and S. Pillai, J. Photochem. Photobiol., C, 2015, 25, 1 CrossRef CAS.
- L. Jiang, Y. Li, X. Wu and G. Zhang, Sci. China Mater., 2021, 64, 2230 CrossRef CAS.
- J. Y. Zhu, Y. P. Li, X. J. Wang, J. Zhao, Y. S. Wu and F. T. Li, ACS Sustainable Chem. Eng., 2019, 7, 14953 CrossRef CAS.
- X. Xiong, C. Mao, Z. Yang, Q. Zhang, G. I. N. Waterhouse, L. Gu and T. Zhang, Adv. Energy Mater., 2020, 10, 025102 Search PubMed.
- H. Li, A. Li, Z. Zhao, M. Li and Y. Song, Small Struct., 2020, 1, 2000028 CrossRef.
- S. Cao, T.-S. Chan, Y.-R. Lu, X. Shi, B. Fu, Z. Wu, H. Li, K. Liu, S. Alzuabi, P. Cheng, M. Liu, T. Li, X. Chen and L. Piao, Nano Energy, 2020, 67, 104287 CrossRef CAS.
- J. Di, C. Chen, C. Zhu, P. Song, J. Xiong, M. Ji, J. Zhou, Q. Fu, M. Xu, W. Hao, J. Xia, S. Li, H. Li and Z. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 30786 CrossRef CAS PubMed.
- X. Yang, S. Wang, N. Yang, W. Zhou, P. Wang, K. Jiang, S. Li, H. Song, X. Ding, H. Chen and J. Ye, Appl. Catal., 2019, 259, 118088 CrossRef CAS.
- A. Naldoni, M. Altomare, G. Zoppellaro, N. Liu, Š. Kment, R. Zbořil and P. Schmuki, ACS Catal., 2018, 9, 345 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc00098f |
| ‡ Shan Jiang and Haoze Li contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |