
Efficient narrowband bluish-green emitters derived from a double-carbazole-fused organoboron multiple resonance skeleton with internal-structure modification†
Tong-Yuan
Zhang‡
a,
Ying-Chun
Cheng‡
a,
Hui
Wang
a,
Feng
Huang
a,
Xin
Xiong
a,
Xiao-Chun
Fan
*a,
Jia
Yu
ab,
Kai
Wang
 *ac and
Xiao-Hong
Zhang
*ab
*ac and
Xiao-Hong
Zhang
*ab
aInstitute of Functional Nano & Soft Materials (FUNSOM), Soochow University, Suzhou, Jiangsu 215123, P. R. China. E-mail: xcfan@suda.edu.cn; wkai@suda.edu.cn; xiaohong_zhang@suda.edu.cn
bJiangsu Key Laboratory of Advanced Negative Carbon Technologies, Soochow University, Suzhou, 215123, Jiangsu, P. R. China
cJiangsu Key Laboratory for Carbon-Based Functional Materials & Devices, Soochow University, Suzhou, 215123, Jiangsu, P. R. China
First published on 18th March 2024
Abstract
The double-carbazole-fused organoboron framework CzBN is a widely used parent skeleton in designs of multiple resonance (MR) emitters. Currently, the majority of CzBN derivatives are derived from peripheral modification strategies. Here, we focus on internal-structure modification of the CzBN skeleton and thus develop two narrowband bluish-green MR emitters by directly embedding thiophene units into the CzBN skeleton. This incorporation of thiophene units not only extends the π-conjugated backbone towards red-shifted emission, but also improves the MR characteristics and structural rigidity for narrowband features. Both emitters achieve narrowband bluish-green emissions with extremely small full width at half maximum of ca. 23 nm in dilute toluene. Moreover, the optimized sensitized device based on BTPCzBN as the terminal emitter achieves a high maximum external quantum efficiency of up to 24% with suppressed efficiency roll-off. This work not only presents a distinct strategy for the development of CzBN derivatives via internal-structure modification of MR skeletons, but also provides valuable inspiration for the design of heteroatom-fused narrowband emitters.
 Kai Wang | Kai Wang received his BS degree from the Department of Materials Science and Engineering, Beihang University in 2012 and received his PhD degree from the Technical Institute of Physics and Chemistry of the Chinese Academy of Sciences in 2017. Then, he carried out postdoctoral research at the Institute of Functional Nano & Soft Materials (FUNSOM), Soochow University. Now, he is an associate professor at Soochow University. His research interests mainly focus on organic optoelectronic materials and devices. |
Introduction
Thermally activated delayed fluorescence (TADF) emitters have undergone rapid development over the past decade, due to their great potential to replace conventionally fluorescent and phosphorescent emitters in commercial organic light-emitting diodes (OLEDs).1–4 However, a traditional TADF emitter based on a donor–acceptor (D–A) structure generally exhibits a broad emission spectrum with a full width at half maximum (FWHM) of approximately 80–100 nm, which results in a disadvantage in terms of the colour purity.5–9 To address this challenge, Hatakeyama et al.10 proposed an innovative molecular design concept by using the multiple resonance (MR) effect. To be specific, they strategically introduced electron-rich atoms (i.e., nitrogen, N)/electron-deficient atoms (i.e., boron, B) at the ortho-position of polycyclic aromatic hydrocarbons (PAHs), forming alternately separated frontier molecular orbital (FMO) distributions that ensure TADF characteristics.11–14 More importantly, this type of point-to-point FMO distribution also effectively weakens bonding/anti-bonding characteristics and synergistically inhibits molecular relaxation in both the ground and excited states, resulting in narrowband emissions with exceptional colour purities comparable to those inorganic ones.15–17 Undoubtedly, the exploration of these new-type narrowband MR emitters is both appealing and imperative.Currently, the design of a new-structure MR emitter is typically derived from modification of a parent MR skeleton.18,19 For example, the renowned pure-blue MR emitter v-DABNA can be taken as a derivative of the very first MR emitter DBANA-1 employing the same MR skeleton.20 This suggests that some of the previously reported MR skeletons play an important role in the development of MR emitters. Among these MR skeletons, the double-carbazole-fused organoboron MR framework CzBN has emerged as one of the most representative and widely used parent skeletons, mainly due to its favourable photophysical properties and multiple modification sites located at its peripheries.10 To date, over 40 CzBN derivatives have been reported, some of which have achieved state-of-the-art performance in OLEDs.6,15,21,22 Their molecular design strategies mainly involve two categories, i.e., (i) π-conjugated extension23–26 and (ii) peripheral substitution,18,27–29 both of which well maintain the profile of the CzBN skeleton. Generally, their key synthesis procedures (e.g., borylation) can be realized using similar precursors and reaction conditions as those for CzBN.30,31 Moreover, these strategies could draw good lessons from conventional emitters with long-range charge-transfer (LRCT), thus facilely obtaining CzBN derivatives with a desirable and decent performance.32 Nevertheless, these approaches may also induce obvious LRCT contributions that can be detrimental to maintaining narrowband emissions.33,34 To the best of our knowledge, there have been few investigations focusing on directly modifying the internal structure of the parent CzBN, although such derivatives could be taken as new MR backbones while retaining most of the active sites of the parent CzBN skeleton, thus being able to not only serve as emitters but also having potential as skeletons to derive more MR emitters by suitable decorations. Possible challenges include: their chemical syntheses may require different precursors and reaction conditions, and the variability in the MR backbones may affect the internal MR features, thus creating more uncertainty in the final performance of target emitters.
Herein, we proposed a MR-parent modification approach by directly embedding an aromatic heterocycle thiophene into the CzBN skeleton (Fig. 1). Two homologous CzBN-based MR emitters, namely BTPCzBN and BTPBN, were thus designed and synthesized with asymmetric and symmetric molecular structures, respectively. The thiophene group was selected as the embedded unit between pyrrole and benzene rings, as it is an aromatic and rigid group with cyclic delocalized π-orbitals,2,35 which allows for further π-orbital extension of the CzBN skeleton.36,37 Theoretical calculations reveal that the embedded thiophene units in both emitters sufficiently contribute to their MR distributions, thereby narrowing their energy gaps without harming their narrowband features.38 As a result, BTPCzBN and BTPBN both exhibit similar bathochromic narrowband bluish-green emissions in dilute solution, with small FWHMs of 23 and 25 nm, respectively. These FWHM in the energy unit are even narrower than that of the original CzBN skeleton, indicating that the incorporation of thiophene units can effectively mitigate the spectral broadening resulting from structural modifications, thereby highlighting the efficacy of our strategy. Ultimately, an optimized hyperfluorescence (HF) OLED based on BTPCzBN exhibits superior performance with a maximum external quantum efficiency (EQE) of up to 24% and a narrow FWHM of 32 nm. This work presents two potential MR skeletons which expand the molecular library of MR emitters based on the CzBN skeleton, thus opening up a new horizon for the further development of narrowband MR emitters.
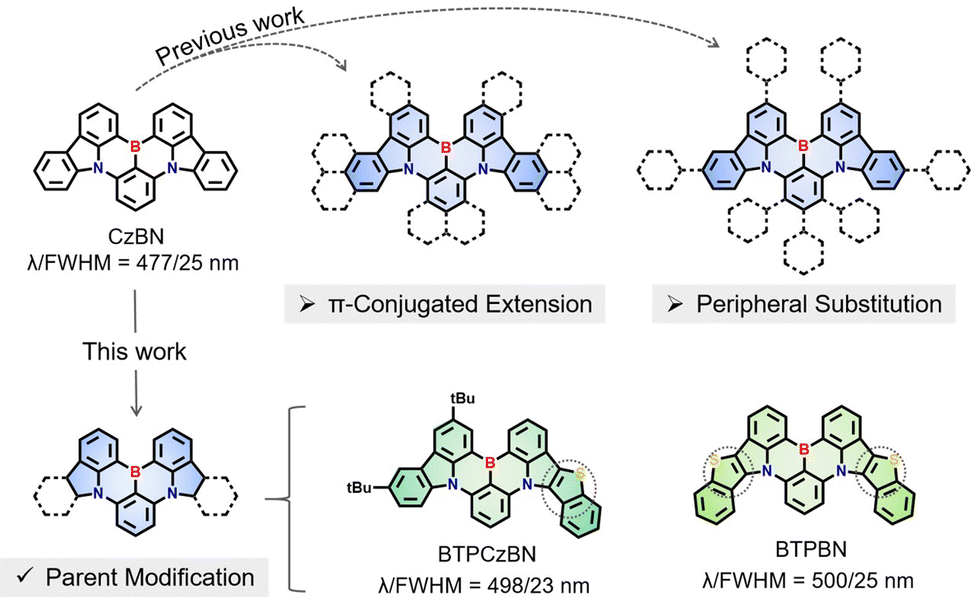 | ||
| Fig. 1 Previous works of CzBN derivatives and the design strategy of BTPCzBN and BTPBN in this work.15,18,21–29 | ||
Results and discussion
The synthesis routes of BTPCzBN and BTPBN are described in Scheme S1 (ESI†). The intermediate 9-(2-bromo-3-fluorophenyl)-3,6-di-tert-butyl-9H-carbazole (1a) was first obtained according to the literature. Then, the target compounds BTPCzBN and BTPBN were synthesized via two steps in decent total yields, including an electrophilic substitution reaction and a one-pot lithium–borylation–annulation reaction. All these synthesised compounds were fully characterized by nuclear magnetic resonance (NMR) and MALDI-TOF mass spectrometry analysis, as illustrated in the ESI.† Furthermore, the decomposition temperatures (Td, corresponding to 5% weight loss) of BTPCzBN and BTPBN were monitored to be around 380 °C and 449 °C, respectively, using thermogravimetric analysis. During the differential scanning calorimetry measurements, the glass transition temperature of BTPCzBN was determined at 130 °C, while no glass transition signal was detected for BTPBN (Fig. S1, ESI†). These results suggest that both emitters possess good thermal and morphological stability, which are favourable for OLED applications via the vacuum evaporation process.Single crystals of BTPCzBN and BTPBN were successfully obtained using the gas phase diffusion method with a mixed system of N,N-dimethylformamide and ethanol, and then characterized by the single-crystal X-ray diffraction (XRD) measurement (see Tables S1 and S2 for the crystal information, ESI†). As shown in Fig. 2, for two molecules, the lengths of the thiophene–pyrrole neighbouring bonds (i.e., C1–C2 and C1′–C2′) are both around 1.38 Å, even shorter than those of typical aromatic C–C bonds (e.g., 1.40 Å in benzene), indicating their higher bond energies and rigid structures. Although there are stronger hydrogen–hydrogen steric repulsions, which give rise to higher torsions of around 20° observed between the C1–E1, A1′–C1′, and C1′–E1′ planes than the original carbazole side without thiophene participation (i.e., A1–C1), due to the unique pentacyclic structures of thiophene units and the planar sp2 hybridization of the sulphur atoms inside, both molecules still display much more planar geometries than other reported sulphur-embedded MR emitters. Consequently, in both their molecular stacking modes, strong intermolecular interaction were observed with short plane-to-plane distances of nearly 3.50 Å.1
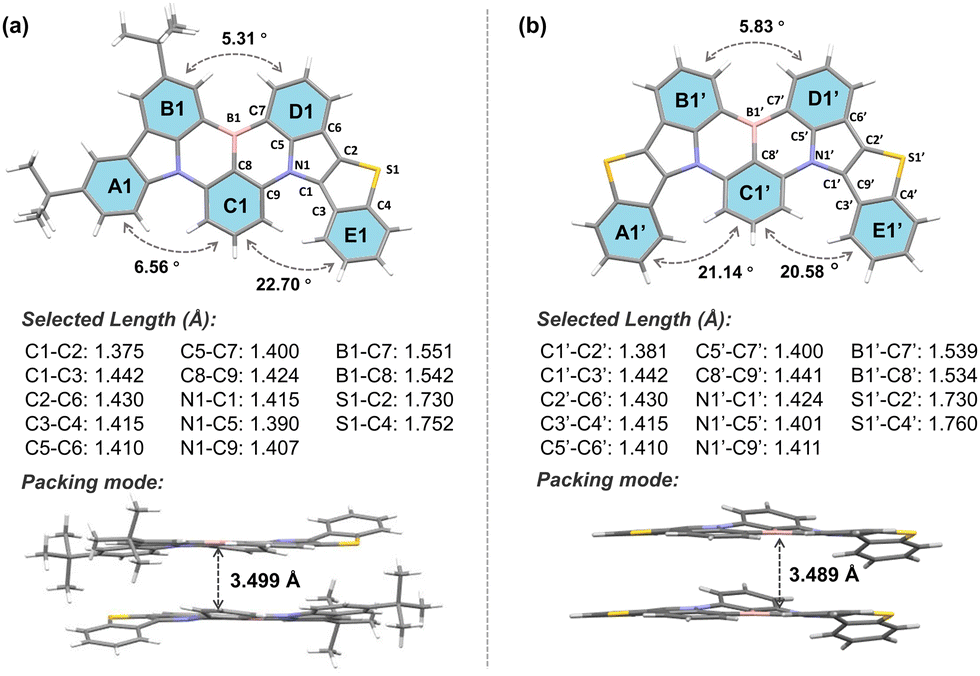 | ||
| Fig. 2 Molecular geometries of (a) BTPCzBN and (b) BTPBN in their single crystal samples with selected key bond lengths, dihedral angles, and its packing modes with highlighted intermolecular plane-to-plane distances (CDCC 2305070 and 2297252†). | ||
To explore the electronic features of BTPCzBN and BTPBN, we then performed density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations by using the Gaussian 16 package based on the PBE0/6-31g(d,p) level. As displayed in Fig. 3, in BTPCzBN and BTPBN molecules, the original parts from CzBN (i.e., the uncoloured parts in Fig. 3) still demonstrate typical MR features, i.e., the highest occupied molecular orbitals (HOMOs) are mainly delocalized on the nitrogen atoms (N) as well as the ortho- and para-positions of carbon atoms directly connected to them, while the lowest unoccupied molecular orbitals (LUMOs) are mainly located on the boron atoms as well as the meta-positions relative to the nitrogen atoms. Meanwhile, the small portion of FMOs delocalized on the thiophene segments present clear hybridized π-bonding/non-bonding characteristics, thereby slightly enhancing the overall MR featured π-conjugations without harming molecular rigidity, which is beneficial for narrowing the emission bandwidths. These results indicate that the introduction of thiophene units can significantly enhance π-conjugations without damaging the MR characteristics. As a result, the HOMO–LUMO energy gaps of BTPCzBN and BTPBN are predicted to be evidently reduced compared to that of the parent CzBN (see Table S1, ESI†), suggesting a potential bathochromic shift in their emission spectra. In previous works, introducing sulphur atoms in MR skeletons often induces serious structural relaxation, thus broadening emission spectra. To check this issue of our molecules, we further optimized the geometries at the first excited state (S1). For either molecule, the root mean square displacement (RMSD) values between the optimized ground state (S0) and S1 structures are estimated to be around 0.03 Å, suggesting negligible structural relaxation upon excitation. This can be attributed to the unique pentacyclic rigid structures of the thiophene units with the planar sp2 hybridized sulphur atoms, as mentioned above.
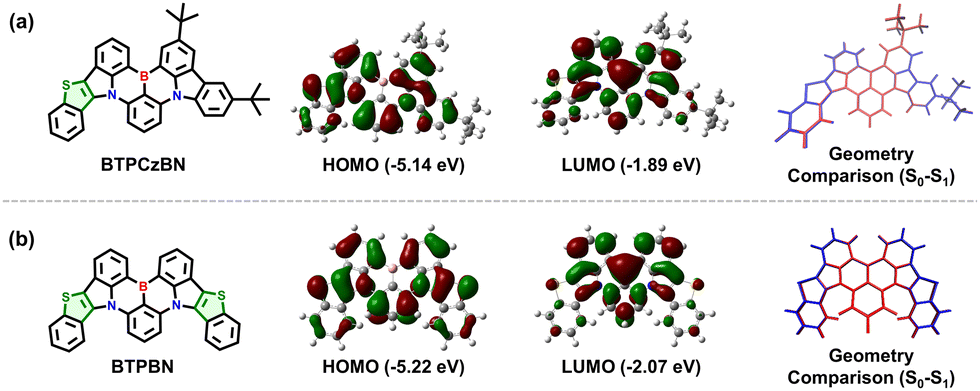 | ||
| Fig. 3 Theoretical calculation results of BTPCzBN and BTPBN. Molecular structure, the HOMO and LUMO distributions, geometry displacements between optimized S0 (blue) and S1 (red) states of (a) BTPCzBN and (b) BTPBN. | ||
Room-temperature ultraviolet-visible (UV-vis) absorption and photoluminescence (PL) spectra of BTPCzBN and BTPBN were then obtained in dilute toluene (1.0 × 10−5 M). As shown in Fig. 4, the absorption peaks ranging from 300 to 420 nm should be attributed to their high-energy n–π* and π–π* transitions; notably, for both BTPCzBN and BTPBN, similar sharp absorptions are observed with peaks at 473 and 481 nm, respectively, which are caused by the short-range charge-transfer (SRCT) transitions. In terms of the room-temperature PL spectra, BTPCzBN and BTPBN display similar bluish-green sharp emissions with peaks at 498 and 500 nm, accompanied by narrow FWHMs of 23 and 25 nm (energy unit: 97 and 101 meV), respectively. In previous studies, CzBN derivatives generally suffer from unavoidable spectral broadening caused by structural modifications. However, in our case, arising from the extended MR featured π-conjugated frameworks with incorporation of thiophene units, BTPCzBN and BTPBN successfully break the trade-off between the redshift and the spectral broadening, achieving redshifted emissions with FWHMs even narrower than that of CzBN in the energy unit (peak = 473 nm, FWHM = 25 nm/138 meV).15,19 This suggests the advantage of our strategy in developing CzBN-based MR derivatives. Moreover, given the bluish-green narrowband spectra of BTPCzBN and BTPBN, we expected these two emitters to have potential as skeletons in developing MR emitters at long-wavelengths with suitable decorations. The PL spectra of BTPCzBN and BTPBN diluted in other solvents were also recorded. As shown in Fig. S2 and Table S4 (ESI†), BTPCzBN and BTPBN both exhibited typical positive solvation behaviours, i.e., the PL spectra become slightly red-shifted and broadening as the solvent polarity increases, which can be attributed to the well-preserved MR features and rigid molecular frameworks.
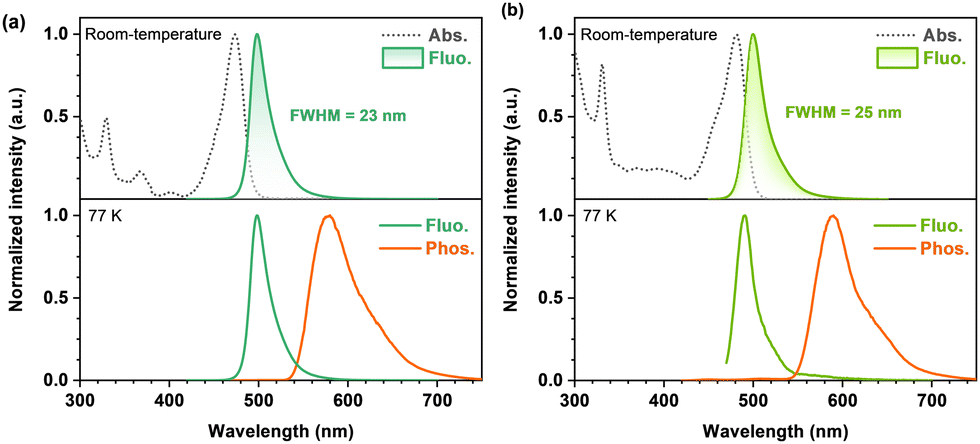 | ||
| Fig. 4 Photophysical properties of BTPCzBN and BTPBN. UV-vis absorption and photoluminescence spectra of (a) BTPCzBN and (b) BTPBN in dilute toluene (1 × 10−5 M). | ||
To determine the energy levels of the lowest excited states, fluorescence and phosphorescence spectra of BTPCzBN and BTPBN at low-temperature (77 K) were obtained in dilute toluene (Fig. 4). Based on the onsets of fluorescence and phosphorescence spectra, the S1 energy levels of BTPCzBN and BTPBN are estimated to be 2.14 and 2.15 eV, respectively, while the energy levels of their lowest triplet state (T1) are estimated to be 1.89 and 1.82 eV, respectively. Their corresponding S1-T1 energy differences (ΔESTs) were thus determined to be 0.25 and 0.33 eV, respectively (see Table S5, ESI†). These values appear excessively large to support an efficient reverse intersystem crossing (RISC) process. Therefore, to further assess their TADF characteristics, transient PL decays of 1 wt% BTPCzBN and BTPBN doped 9-(2-(9-phenyl-9H-carbazol-3-yl)phenyl)-9H-3,9′-bicarbazole (PhCzBCz) films were measured at room-temperature. As shown in Fig. S3 (ESI†), both transient decay curves exhibit similar dual-component decay trends on the nanosecond and microsecond scales, respectively. These suggest the existence of TADF contributions to both their overall fluorescence intensities. Meanwhile, the PL quantum yields (PLQYs) were measured to be 91% and 80% for BTPCzBN and BTPBN, respectively. Based on the fit lifetimes, and proportion of the prompt and delayed components, the corresponding dynamic parameters were further calculated. As summarized in Table S6 (ESI†), BTPCzBN and BTPBN exhibit close radiation decay rates (kr) as fast as 1.64 × 108 and 0.74 × 108 s−1, respectively, beneficial for singlet exciton utilizations. On the other hand, due to their large ΔEST values, both emitters exhibit very poor RISC rates (kRISCs) of 1.03 × 103 and 1.10 × 103 s−1, respectively. Such inefficient RISC processes will inevitably cause severe accumulation and serious loss of triplet excitons upon electrical excitations, thus badly impairing their performances in OLEDs. This is also a common issue encountered by most of the reported MR emitters. Fortunately, this issue can be well addressed by using the HF OLED structure with a suitable assistant TADF sensitizer.39–41
To evaluate the electroluminescence (EL) properties of BTPCzBN and BTPBN, we then employed them as the terminal emitters in the emitting layer (EML) and fabricated OLEDs with the HF device structure being as follows: indium tin oxide (ITO)/N,N-bis(p-tolyl)aniline (TAPC) (30 nm)/tris(4-carbazoyl-9-ylphenyl)amine (TCTA) (10 nm)/EML (20 nm)/3,3′-[5′-[3-(3-pyridinyl)phenyl][1,1′:3′,1′′-terphenyl]3,3′′diyl]bis-pyridine (TmPyPB) (40 nm)/lithium fluoride (LiF) (1 nm)/aluminium (Al) (150 nm). In the devices, TmPyPB was selected as the electron transporting layer (ETL); TATC and TAPC were utilized as the hole transporting layers (HTLs). In the EML, PhCzBCz with high triplet energy (ET1 = 2.96 eV) was applied as the host material due to its excellent capability of approaching carrier balance and thus effectively confining excitons within the EML; in addition, to facilitate triplet harvesting, a sky-blue TADF emitter 5CzBN showing a fast RISC process was selected as the assistant sensitizer since its PL spectrum highly overlaps with the absorption spectra of BTPCzBN and BTPBN (depicted in Fig. S4, ESI†), thus providing an efficient energy transfer channel from the sensitizer (5CzBN) to the two terminal emitters. Here, the doping concentration of the sensitizer was optimized to be 10 wt%. The corresponding energy diagram and chemical structures of the materials used are displayed in Fig. 5a and b. The detailed EL data of the corresponding devices are provided in Table S7 (ESI†).
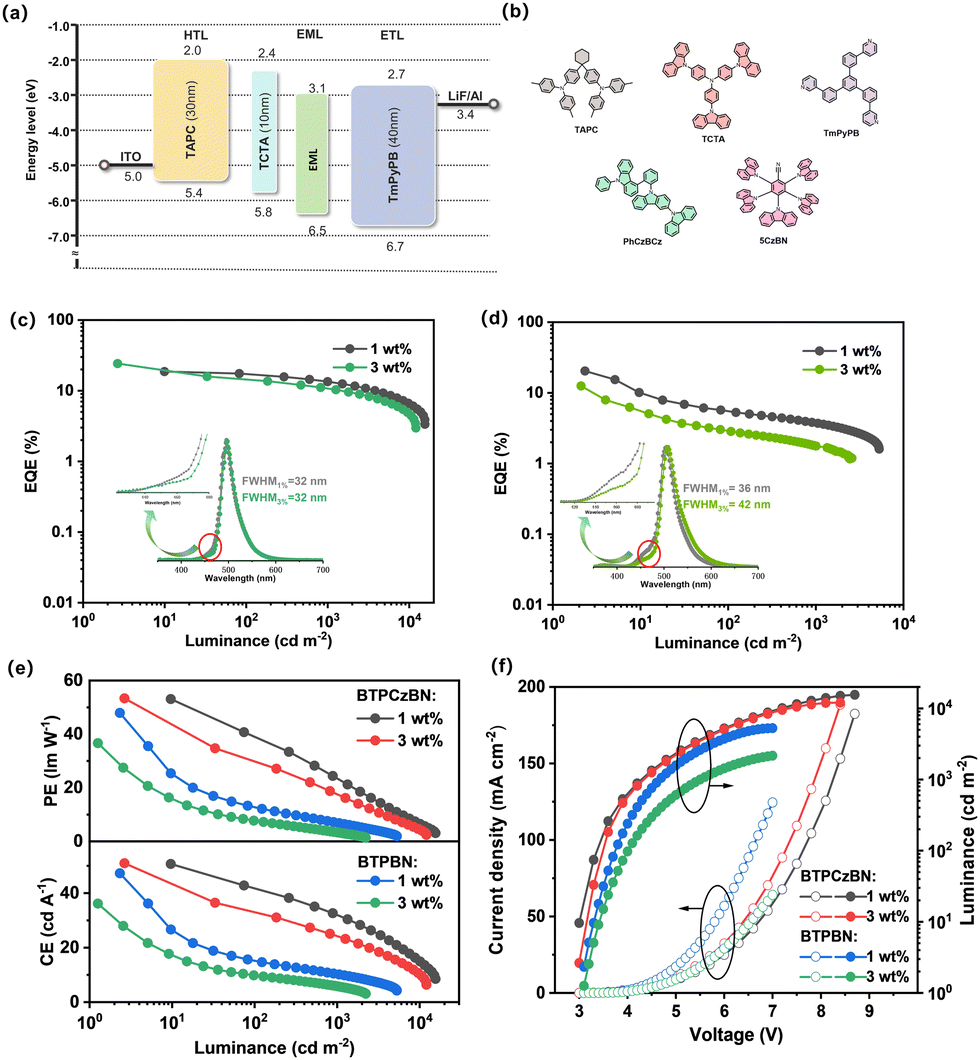 | ||
| Fig. 5 (a) Device architecture for the MR-TADF OLEDs with energy level alignment; (b) the chemical structures of the relevant materials used in the device, EQE-luminance curves with EL for (c) BTPCzBN and (d) BTPBN; (e) current efficiency (CE)- and power efficiency (PE)-luminance curves; (f) voltage–luminance–current density curves. | ||
For practical OLED fabrication, HF devices with most often employed doping concentrations of terminal emitters (i.e., 1 wt%) were first exploited.42 As depicted in Fig. 5, both devices achieve high maximum EQEs of over 20%, suggesting the efficacy of these HF systems. However, when comparing their overall efficiency performance trends, we found that the efficiency roll-off of the 1 wt% BTPBN-doped device (EQE of 5.2% at 100 cd cm−2) is much more serious than that of the 1 wt% BTPBN-doped device (EQE of 17.2% at 100 cd cm−2). This can be attributed to the close intersystem crossing (ISC) (kISC = 0.57 × 108 s−1) and kr (0.74 × 108 s−1) rates of BTPBN, resulting in the ISC process becoming a competitive exciton loss channel, especially at high luminance. In addition, both devices exhibit narrowband EL spectra with peaks at 496 and 504 nm and FWHMs of 32 and 36 nm, respectively, corresponding to CIE coordinates of (0.15, 0.52) and (0.25, 0.60). Notably, there are slight shoulders observed around the onset positions of the EL spectra, which can be attributed to the residual emissions from the TADF sensitizer 5CzBN, which arises from inadequate energy transfer between the sensitizer and terminal emitters due to the low doping concentration of the terminal emitters (Fig. S5, ESI†).42 Therefore, we further fabricated the devices by increasing the doping concentration of the terminal emitters to 3 wt%. As expected, for both 3 wt% terminal emitter-based devices, contributions of 5CzBN in the EL spectra are successfully mitigated due to the more sufficient energy transfer process. However, a dramatically decreased maximum EQE of 12.5% is also accompanied for the 3 wt% BTPBN-doped device. Its EL spectral bandwidth is also evidently broadened especially at the tail position. The much inferior performance is mainly due to the severe molecular packing of its nearly planar structure, leading to evident excimer formation with serious concentration quenching, as further evidenced by the results of their non-doped devices (Fig. S6, ESI†). Meanwhile for BTPCzBN, the molecular packing trend is not obvious due to the steric protection of the peripheral tert-butyl groups. As a result, the 3 wt% BTPBN-doped device achieves a better efficiency performance with a high maximum EQE of up to 24%. Moreover, compared with the 1 wt% BTPCzBN-doped device, the EL spectrum of the 3 wt% device shows negligible spectral broadening, with an identical FWHM of 32 nm. In addition, the EL spectra of the optimal HF devices at different luminance exhibit decent spectral stability (Fig. S7, ESI†). The results indicate that when optimizing the device performance of HF OLEDs based on MR type terminal emitters, it is important to consider both the competitive ISC process and aggregated trends of terminal emitters.
To further demonstrate the advantages of these HF systems, OLED devices using BTPCzBN, BTPBN, and 5CzBN as the sole dopants are fabricated as the controls. As depicted in Fig. S8–S10 (ESI†), both BTPCzBN- and BTPBN-based non-sensitized devices exhibit unsatisfactory performances (i.e., low EQEs and large efficiency roll-offs) due to their poor kRISC rates and thus inefficient TADF process; while the 5CzBN-based non-sensitized device shows better results due to its fast kRISC and much more efficient TADF properties. Fig. S11 (ESI†) further displays the EL decay characteristics of all the sensitized and non-sensitized devices. Delayed contribution is the most obvious for the 5CzBN based device, revealing its efficient RISC process for triplet harvesting. Meanwhile, the sensitized and non-sensitized device structures result in significant differences for both BTPCzBN and BTPBN based devices. For both emitters, the delayed components of the HF devices are substantially enhanced compared with the corresponding non-sensitized ones, suggesting the obvious contribution of the sensitizer 5CzBN to triplet harvesting. Fig. S5 (ESI†) shows the exciton harvesting mechanism of these HF devices.43 Exciton recombination mainly occurs on assistant sensitizer molecules 5CzBN because of its high doping concentration and “trapped” state energy level. The formed singlet excitons can directly transfer to the S1 states of terminal emitters via the Förster resonance energy transfer (FRET process), generating prompt fluorescence. Meanwhile, the formed triplet excitons are efficiently upconverted to the S1 state of 5CzBN due to its superior RISC process, and then undergo FRET process to the terminal emitter like the initial singlet excitons, eventually emitting delayed fluorescence. The FRET process can be evidenced by the faster exciton decay in HF devices than that in a non-sensitized device based on 5CzBN, as well as the nearly consistent PL spectra in non-sensitized and HF films (Fig. S12, ESI†).44 The above results further confirm the superiority of this HF system.
Conclusions
In summary, we have succeeded in developing two bluish-green MR emitters BTPCzBN and BTPBN, via direct internal-structure modification on the parent CzBN skeleton. The theorical calculation reveals that the embedded thiophene unit not only helps to efficiently expand the π-conjugation, but also shows a positive effect on enhancing the MR characteristic and suppressing the structural relaxation. As a result, both BTPCzBN and BTPBN exhibit narrowband bluish-green emissions with FHWMs of only 23 and 25 nm, respectively, which are even narrower than the CzBN skeleton in the energy unit, and thus present the potential as skeletons to further develop MR emitters emitting at long-wavelengths. To compensate for the lower RISC rates of both emitters, the well-established HF strategy is further employed in OLED fabrication. The corresponding HF device based on BTPCzBN achieves a maximum EQE of up to 24%, as well as a narrowband bluish-green EL with a small FWHM of 32 nm. We believe that our molecular design here greatly expands the diversity of MR emitters based on the CzBN framework.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 52130304, 51821002, 52373193), the National Key Research & Development Program of China (Grant No. 2020YFA0714601, 2020YFA0714604), the Science and Technology Project of Suzhou (No. ZXL2022490), the Natural Science Foundation of Jiangsu (BK20230507), the Jiangsu Funding Program for Excellent Postdoctoral Talent (2023ZB515), the China Postdoctoral Science Foundation (2023M742523), the China National Postdoctoral Program for Innovative Talents (BX20230254), Suzhou Key Laboratory of Functional Nano & Soft Materials, the Collaborative Innovation Center of Suzhou Nano Science & Technology, the 111 Project, Joint International Research Laboratory of Carbon-Based Functional Materials and Devices.References
- X. Song, D. Zhang, Y. Lu, C. Yin and L. Duan, Adv. Mater., 2019, 31, e1901923 CrossRef PubMed.
- R. K. Konidena, K. H. Lee, J. Y. Lee and W. P. Hong, Chem. – Asian J., 2019, 14, 2251–2258 CrossRef CAS PubMed.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed.
- C. Yin, D. Zhang, Y. Zhang, Y. Lu, R. Wang, G. Li and L. Duan, CCS. Chem., 2020, 2, 1268–1277 CrossRef CAS.
- Y. Wada, H. Nakagawa, S. Matsumoto, Y. Wakisaka and H. Kaji, Nat. Photonics, 2020, 14, 643–649 CrossRef CAS.
- N. Yadav, U. Deori, E. Ravindran, B. Sk and P. Rajamalli, J. Mater. Chem. C, 2023, 11, 16368–16376 RSC.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- Z. Bin, D. Shi, R. Su, W. Han, D. Zhang and L. Duan, Sci. Bull., 2020, 65, 153–160 CrossRef CAS PubMed.
- T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777–2781 CrossRef CAS PubMed.
- X.-C. Fan, K. Wang, Y.-Z. Shi, Y.-C. Cheng, Y.-T. Lee, J. Yu, X.-K. Chen, C. Adachi and X.-H. Zhang, Nat. Photonics, 2023, 17, 280–285 CrossRef CAS.
- F. Huang, X. C. Fan, Y. C. Cheng, H. Wu, X. Xiong, J. Yu, K. Wang and X. H. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202306413 CrossRef CAS PubMed.
- S. M. Suresh, E. Duda, D. Hall, Z. Yao, S. Bagnich, A. M. Z. Slawin, H. Bassler, D. Beljonne, M. Buck, Y. Olivier, A. Kohler and E. Zysman-Colman, J. Am. Chem. Soc., 2020, 142, 6588–6599 CrossRef CAS PubMed.
- M. Yang, I. S. Park and T. Yasuda, J. Am. Chem. Soc., 2020, 142, 19468–19472 CrossRef CAS PubMed.
- Y. T. Lee, C. Y. Chan, M. Tanaka, M. Mamada, K. Goushi, X. Tang, Y. Tsuchiya, H. Nakanotani and C. Adachi, Adv. Opt. Mater., 2022, 10, 202200682 Search PubMed.
- S. H. Jeong, J. S. Im, D. R. Lee, H. J. Ahn, J. Y. Kim, J.-H. Baek and J. Y. Lee, J. Mater. Chem. C, 2023, 11, 16288–16296 RSC.
- Y. Xu, J. Han, N. Li, Z. Huang, J. Miao and C. Yang, J. Mater. Chem. C, 2023, 11, 13733–13739 RSC.
- F. Huang, X. C. Fan, Y. C. Cheng, H. Wu, Y. Z. Shi, J. Yu, K. Wang, C. S. Lee and X. H. Zhang, Mater. Horiz., 2022, 9, 2226–2232 RSC.
- S. Xu, Q. Yang, Y. Zhang, H. Li, Q. Xue, G. Xie, M. Gu, J. Jin, L. Huang and R. Chen, Chin. Chem. Lett., 2021, 32, 1372–1376 CrossRef CAS.
- Y. Kondo, K. Yoshiura, S. Kitera, H. Nishi, S. Oda, H. Gotoh, Y. Sasada, M. Yanai and T. Hatakeyama, Nat. Photonics, 2019, 13, 678–682 CrossRef CAS.
- Y. Zhang, D. Zhang, J. Wei, Z. Liu, Y. Lu and L. Duan, Angew. Chem., Int. Ed., 2019, 58, 16912–16917 CrossRef CAS PubMed.
- Y. Zhang, D. Zhang, J. Wei, X. Hong, Y. Lu, D. Hu, G. Li, Z. Liu, Y. Chen and L. Duan, Angew. Chem., Int. Ed., 2020, 59, 17499–17503 CrossRef CAS PubMed.
- Y. Zhang, G. Li, L. Wang, T. Huang, J. Wei, G. Meng, X. Wang, X. Zeng, D. Zhang and L. Duan, Angew. Chem., Int. Ed., 2022, 61, e202202380 CrossRef CAS PubMed.
- X. F. Luo, H. X. Ni, H. L. Ma, Z. Z. Qu, J. Wang, Y. X. Zheng and J. L. Zuo, Adv. Opt. Mater., 2022, 10, 2102513 CrossRef CAS.
- X. F. Luo, S. Q. Song, H. X. Ni, H. Ma, D. Yang, D. Ma, Y. X. Zheng and J. L. Zuo, Angew. Chem., Int. Ed., 2022, 61, e202209984 CrossRef CAS PubMed.
- X. F. Luo, H. X. Ni, L. Shen, L. Wang, X. Xiao and Y. X. Zheng, Chem. Commun., 2023, 59, 2489–2492 RSC.
- Y. K. Qu, D. Y. Zhou, F. C. Kong, Q. Zheng, X. Tang, Y. H. Zhu, C. C. Huang, Z. Q. Feng, J. Fan, C. Adachi, L. S. Liao and Z. Q. Jiang, Angew. Chem., Int. Ed., 2022, 61, e202201886 CrossRef CAS PubMed.
- M. Yang, S. Shikita, H. Min, I. S. Park, H. Shibata, N. Amanokura and T. Yasuda, Angew. Chem., Int. Ed., 2021, 60, 23142–23147 CrossRef CAS PubMed.
- X. Cai, Y. Pan, X. Song, C. Li, Y. Pu, X. Zhuang, H. Bi and Y. Wang, Adv. Opt. Mater., 2023, 2302811 CrossRef.
- Y. C. Cheng, X. Tang, K. Wang, X. Xiong, X. C. Fan, S. Luo, R. Walia, Y. Xie, T. Zhang, D. Zhang, J. Yu, X. K. Chen, C. Adachi and X. H. Zhang, Nat. Commun., 2024, 15, 731 CrossRef CAS PubMed.
- X. Wu, J. W. Huang, B. K. Su, S. Wang, L. Yuan, W. Q. Zheng, H. Zhang, Y. X. Zheng, W. Zhu and P. T. Chou, Adv. Mater., 2022, 34, e2105080 CrossRef PubMed.
- C.-Z. Du, Y. Lv, H. Dai, X. Hong, J. Zhou, J.-K. Li, R.-R. Gao, D. Zhang, L. Duan and X.-Y. Wang, J. Mater. Chem. C, 2023, 11, 2469–2474 RSC.
- B. Du, K. Zhang, P. Wang, X. Wang, S. Wang, S. Shao and L. Wang, J. Mater. Chem. C, 2023, 11, 9578–9585 RSC.
- X. Xiong, Y.-C. Cheng, K. Wang, J. Yu and X.-H. Zhang, Mater. Chem. Front., 2023, 7, 929–936 RSC.
- H. Lee, R. Braveenth, J. D. Park, C. Y. Jeon, H. S. Lee and J. H. Kwon, ACS Appl. Mater. Interfaces, 2022, 14, 36927–36935 CrossRef CAS PubMed.
- X. Wang, Y. Zhang, H. Dai, G. Li, M. Liu, G. Meng, X. Zeng, T. Huang, L. Wang, Q. Peng, D. Yang, D. Ma, D. Zhang and L. Duan, Angew. Chem., Int. Ed., 2022, 61, e202206916 CrossRef CAS PubMed.
- D. Li, M. Li, D. Liu, J. Yang, W. Li, Z. Yang, H. Yuan, S. Jiang, X. Peng, G. X. Yang, W. Xie, W. Qiu, Y. Gan, K. Liu and S. J. Su, Adv. Opt. Mater., 2023, 11, 2301084 CrossRef CAS.
- J. Park, J. Lim, J. H. Lee, B. Jang, J. H. Han, S. S. Yoon and J. Y. Lee, ACS Appl. Mater. Interfaces, 2021, 13, 45798–45805 CrossRef CAS PubMed.
- P. Jiang, J. Miao, X. Cao, H. Xia, K. Pan, T. Hua, X. Lv, Z. Huang, Y. Zou and C. Yang, Adv. Mater., 2022, 34, e2106954 CrossRef PubMed.
- Y. Qiu, H. Xia, J. Miao, Z. Huang, N. Li, X. Cao, J. Han, C. Zhou, C. Zhong and C. Yang, ACS Appl. Mater. Interfaces, 2021, 13, 59035–59042 CrossRef CAS PubMed.
- T. Hua, L. Zhan, N. Li, Z. Huang, X. Cao, Z. Xiao, S. Gong, C. Zhou, C. Zhong and C. Yang, Chem. Eng. J., 2021, 426, 131169 CrossRef CAS.
- G. Liu, H. Sasabe, K. Kumada, H. Arai and J. Kido, Chem, 2022, 28, e202201605 CrossRef CAS PubMed.
- H. Nakanotani, T. Higuchi, T. Furukawa, K. Masui, K. Morimoto, M. Numata, H. Tanaka, Y. Sagara, T. Yasuda and C. Adachi, Nat. Commun., 2014, 5, 4016 CrossRef CAS PubMed.
- C. Yin, Y. Zhang, T. Huang, Z. Liu, L. Duan and D. Zhang, Sci. Adv., 2022, 8, eabp9203 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: General information, photophysical properties, device characterization. CCDC 2305070 and 2297252. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3tc04771g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |