
Proximity labeling expansion microscopy (PL-ExM) evaluates interactome labeling techniques†
Sohyeon
Park
a,
Xiaorong
Wang
b,
Yajin
Mo
a,
Sicheng
Zhang
 c,
Xiangpeng
Li
d,
Katie C.
Fong‡
c,
Clinton
Yu
b,
Arthur A.
Tran
e,
Lorenzo
Scipioni
fg,
Zhipeng
Dai
d,
Xiao
Huang
h,
Lan
Huang
b and
Xiaoyu
Shi
*acgi
c,
Xiangpeng
Li
d,
Katie C.
Fong‡
c,
Clinton
Yu
b,
Arthur A.
Tran
e,
Lorenzo
Scipioni
fg,
Zhipeng
Dai
d,
Xiao
Huang
h,
Lan
Huang
b and
Xiaoyu
Shi
*acgi
aCenter for Complex Biological Systems, University of California, Irvine, Irvine, CA 92697, USA. E-mail: xiaoyu.shi@uci.edu
bPhysiology and Biophysics, University of California, Irvine, Irvine, CA 92697, USA
cDepartment of Developmental and Cell Biology, University of California, Irvine, Irvine, CA 92697, USA
dDepartment of Bioengineering and Therapeutic Sciences, University of California, San Francisco, San Francisco, CA 94143, USA
eCardiovascular Research Institute, School of Medicine, University of California, San Francisco, San Francisco 94143, USA
fLaboratory for Fluorescence Dynamics, University of California, Irvine, Irvine, CA 92697, USA
gDepartment of Biomedical Engineering, University of California, Irvine, Irvine, CA 92697, USA
hSchool of Biomedical Engineering, Science and Health Systems, Drexel University, Philadelphia, PA 19104, USA
iDepartment of Chemistry, University of California, Irvine, Irvine, CA 92697, USA
First published on 30th July 2024
Abstract
Understanding protein–protein interactions (PPIs) through proximity labeling has revolutionized our comprehension of cellular mechanisms and pathology. Various proximity labeling techniques, such as HRP, APEX, BioID, TurboID, and μMap, have been widely used to biotinylate PPIs or organelles for proteomic profiling. However, the variability in labeling precision and efficiency of these techniques often results in limited reproducibility in proteomic detection. We address this persistent challenge by introducing proximity labeling expansion microscopy (PL-ExM), a super-resolution imaging technique that combines expansion microscopy with proximity labeling techniques. PL-ExM enabled up to 17 nm resolution with microscopes widely available, providing visual comparison of the labeling precision, efficiency, and false positives of different proximity labeling methods. Our mass spectrometry proteomic results confirmed that PL-ExM imaging is reliable in guiding the selection of proximity labeling techniques and interpreting the proteomic results with new spatial information.
Introduction
Most cellular functions are realized by a set of protein–protein interactions (PPIs) called the protein interactome. Studies on the interactome of a hub protein have transformed our understanding of health and diseases.1–4 Proximity labeling (PL) is a powerful technique used to label interacting proteins for further proteomic identification. In this method, a protein of interest is fused to or labeled by an enzyme. When activated, this enzyme modifies nearby molecules by attaching a small probe like biotin. This spatial labeling allows for subsequent enrichment and identification of these neighboring molecules with mass spectrometry (MS), shedding light on potential interaction partners or local cellular environments of the protein of interest. Several PL methods, such as HRP,5–7 APEX,8–10 BioID,11–13 TurboID,14,15 and μMap1 have been widely used to reveal the organellar proteome9,16 and network of interactions in cells,13,17–19 aiding in understanding diseases and discovering therapeutic targets.1–4Despite its advantages, the variability in PL methods often leads to limited overlap in MS results, even when analyzing the same protein of interest.20 For example, a comparison of proximity labeling mass spectrometry (PL-MS) methods showed less than 25% overlap in interactomes detected by APEX2 and BioID for the same bait valosin-containing protein (VCP).18 This is because labeling precision, labeling efficiency, and false positives of each PL technique and experiment can differ significantly. The direct causes include the choice of enzymes, probes, labeling duration, reaction conditions, and macromolecular crowding in the biological samples.21,22 These variations in the labeling step are further complicated by the nonspecific pulldown that happens during the enrichment process. Consequently, the MS results from different PL experiments for the same bait protein show only a small overlap. Therefore, careful assessment of the labeling quality is essential for selecting the optimal PL method and for interpreting the PL-MS results.
However, evaluating the labeling precision and efficiency of PL is a persistent challenge because the labeling radius ranging from 10 nm to 100 nm is blow the diffraction limit of light microscopy. Commonly used microscopy techniques, such as confocal, Airyscan, and structured illumination microscopy (SIM), offer resolutions from 250 nm to 120 nm, which is insufficient to evaluate the precision of PL. Although electron microscopy and super-resolution microscopy, such as stochastic optical reconstruction microscopy (STORM) and stimulated emission depletion microscopy (STED), offer improved resolutions, the accessibility to these advanced microscopes is limited in most proteomics laboratories.8,10,21,23 This scenario underscores the pressing requirement for more accessible super-resolution imaging techniques, which can both evaluate PL techniques and interpret the accuracy of resultant proteomes.
An emerging super-resolution approach called expansion microscopy (ExM) offers a new way to obtain super-resolution on regular microscopes by physically enlarging the cells.24 Combining ExM with PL, we developed a super-resolution imaging method called proximity labeling expansion microscopy (PL-ExM). This method can qualitatively visualize the labeling radius, efficiency, and false-positive of PL experiments on microscopes widely available, such as confocal and Airyscan microscopes. For example, PL-ExM can theoretically provide a resolution of 12 nm on an Airyscan microscope by expanding a cell by 10 times in each dimension. PL-ExM is compatible with any PL method that biotinylates proteins, for instance, APEX- and HRP-catalyzed PL. Notably, HRP-catalyzed tyramide signal amplification (TSA) was recently employed to enhance signals for ExM,25 albeit not for PL assessment or proteome characterization. In our work, PL-ExM has been specifically designed and optimized for these goals.
Using PL-ExM, we compared the labeling precision, efficiency, and false positives for different PL methods, including APEX2- and HRP-catalyzed PL with various labeling durations. The imaging results matched the proteome results detected by MS. The agreement confirms that PL-ExM is a reliable method to assess the equality of PL as an accurate guidance for optimization of interactome labeling.
Results
Principle and workflow
PL-ExM provides super resolution to visualize the proximity-labeled proteins by physically expanding the cells and tissues in a swellable hydrogel. The effective imaging resolution of an expanded sample is the microscope's resolution divided by the sample's expansion factor. PL-ExM is compatible with a wide range of light microscopes, including confocal, Airyscan, light sheet, SIM, STORM, and STED, as well as with most ExM protocols that yield various expansion factors. For example, if a proximity-labeled sample is expanded fourfold and imaged with a confocal microscope that has a resolution of 280 nm, the effective imaging resolution will be 70 nm. The sample expansion allows for the visualization of a wealth of structural details previously unresolvable by diffraction-limited microscopes alone (Fig. 1A).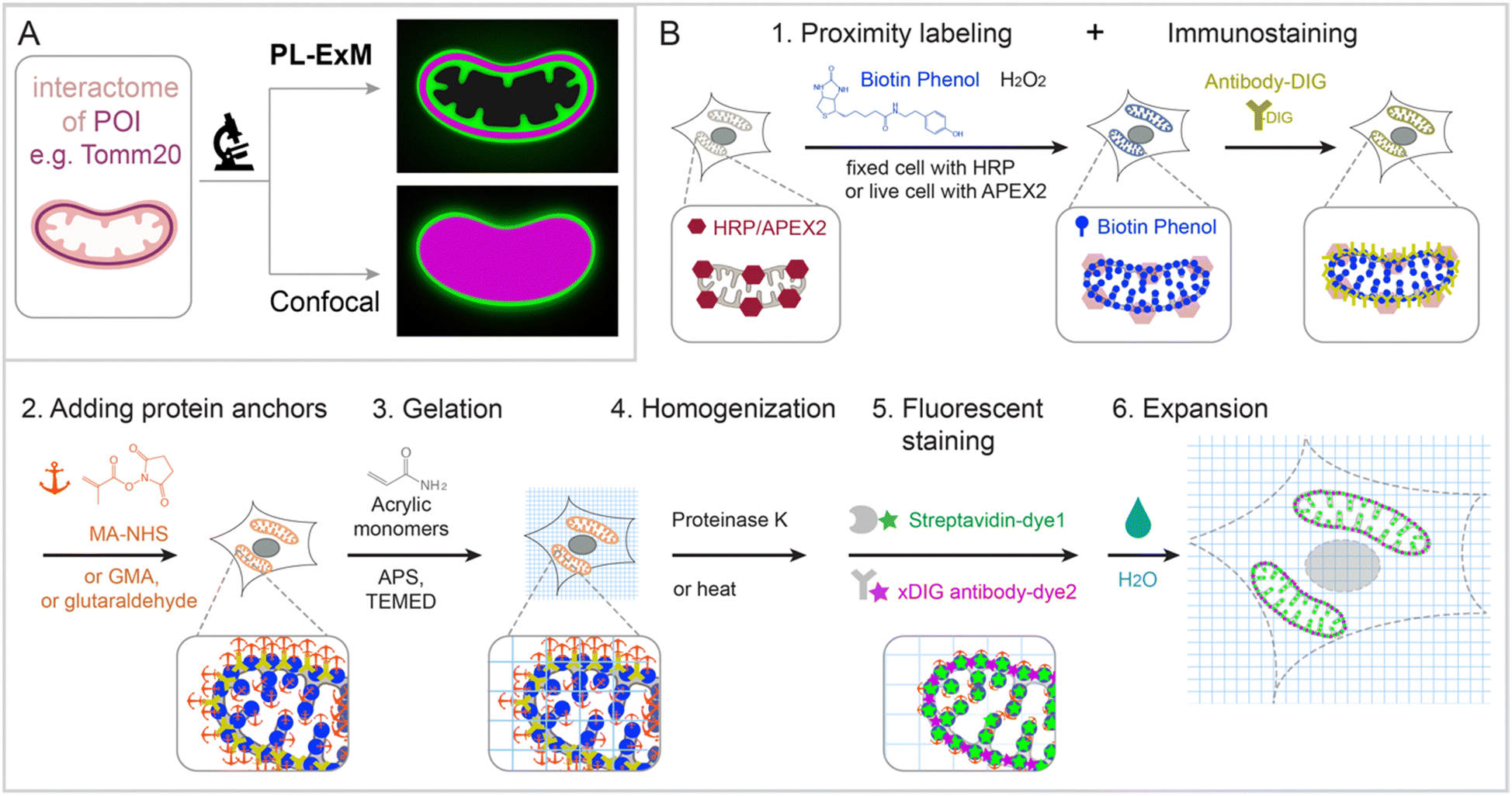 | ||
| Fig. 1 Graphic abstract and workflow of PL-ExM. In the showcase, Tomm20 is the bait for the PL and the target for the immunostaining. (A) Graphic abstract of PL-ExM method. PL-ExM offers super resolution to visualize small interactome structures that present the ground truth. Diffraction-limited microscopy, such as confocal microscopy, misses structural details in the ground truth. (B) The PL-ExM workflow comprises six steps. (1) PL catalyzed by enzymes (HRP, APEX, TurboID etc.) and delivered by biotin phenol. Following PL, a protein of interest is labeled with antibodies conjugated with digoxigenin (antibody-DIG). (2) Adding protein anchors, such as MA-NHS, GMA or glutaraldehyde. (3) Gelation with acrylic and acrylate monomers. (4) Denaturation using proteinase K or heat denaturation. (5) Fluorescent staining: stain the biotin and DIG with fluorescently conjugated streptavidin and anti-DIG antibodies. (6) Expansion: expand hydrogel through immersion in pure water. | ||
Swellable hydrogels, composed of various recipes and subjected to different expansion procedures, can expand from 3 to 20 times in each dimension.24,26–31 The most widely used gel formula for ExM includes acrylamide, sodium acrylate, N,N′-methylenebisacrylamide, ammonium persulfate (APS), and N,N,N′,N′-tetramethylethylenediamine (TEMED).32–34 This particular hydrogel typically expands about fourfold in each dimension when emersed in pure water. Modifying the crosslinkers or the duration of hydrolysis can induce the hydrogel to expand up to 13-fold in a single round.26–30 Through multiple rounds of expansion, a length expansion factor of approximately 15 to ∼20 times can be achieved.31 By varying the combination of microscope type and expansion protocol, PL-ExM can attain resolutions theoretically from 12 nm to 70 nm (Fig. S1, ESI†).
The PL-ExM workflow consists of six steps (Fig. 1B): (1) PL and immunostaining, (2) adding protein anchors, (3) gelation, (4) homogenization, (5) fluorescent staining, and (6) expansion. The workflow can start with any PL method that biotinylates proteins including HRP,5–7 APEX,8–10 BioID,11–13 TurboID,14,15 and μMap.1 We showcase peroxidase-based PL of mitochondria in our workflow, given its widespread use.9,22 Initially, peroxidase HRP or APEX2 is introduced to the bait protein of the interactome. In the presence of hydrogen peroxide (H2O2) and biotin–phenol, proteins within the labeling radius of the peroxidase are biotinylated. Simultaneously, a protein of interest is immunostained with antibodies conjugated with digoxigenin (DIG). The expansion procedure, encompassing steps 2 through 6, follows the PL and immunostaining. In step 2, proteins are chemically modified with anchoring molecules, such as glutaraldehyde (GA), methacrylic acid N-hydroxysuccinimide ester (MA-NHS), or glycidyl methacrylate (GMA). These anchors all aim to covalently crosslinking proteins to polyacrylic chains during hydrogel formation within and around the cells in step 3. Subsequently, cells embedded in the hydrogel (step 3) undergo homogenization, facilitated by proteinase K digestion or heat denaturation (step 4). This homogenization disrupts the protein interactions, enabling isotropic expansion of the sample in the final step (step 6). Prior to expansion, the biotinylated interactome and DIG-labeled proteins of interest are stained with fluorescently conjugated streptavidin and anti-DIG antibodies, respectively (step 5). Introducing fluorescent dyes after gelation avoids the quenching effects of free radical polymerization reactions.27,33–37 Our label-retention expansion microscopy (LR-ExM) technique demonstrates that post-digestion fluorescent staining of biotin or DIG probes significantly enhances the signal-to-noise ratio in ExM images.33 Thus, PL and ExM are integrated seamlessly into the PL-ExM workflow through these six steps. Detailed chemical reactions underlining each step in the workflow are described in Fig. S2 (ESI†).
PL-ExM provides the super resolution to visualize the proximity-labeled structures
We demonstrated the super resolution of PL-ExM by comparing images of proximity-labeled mitochondria in U2OS cells without expansion (Fig. 2A–F) and with expansion (Fig. 2G–R). The bait protein, TOMM20, located on the outer mitochondrial membrane (OMM), was co-stained with HRP-conjugated antibodies and DIG-conjugated antibodies. HRP catalyzed the biotinylation of proteins within its labeling radius using biotin–phenol. The duration of the PL was 30 seconds. The DIG-conjugated antibodies marked the location of TOMM20 in the second channel. Both the expanded and non-expanded samples were imaged using the same Airyscan microscope, which has a measured resolution of 139 nm (Fig. S1A, ESI†). Given that this resolution was too low to detect the PL radius of HRP, the cross-sectional fluorescence intensity plots of non-expanded mitochondria showed single broad bands (Fig. 2F). Conversely, the images of samples expanded by a factor of 4.2 with PL-ExM revealed the mitochondria's hollow structure (Fig. 2J). The measured effective resolution of PL-ExM was 35 nm. The visualization of a hollow structure with strong peripheral signals and a weaker internal signal (Fig. 2J) suggested that the proximity labeled interactome not only includes the OMM proteins, such as translocases of the outer membrane proteins (TOMs), but also those inside mitochondria, such as the translocases of the inner membrane proteins (TIMs). The identities of these proteins were confirmed by our PL-MS analyses of samples prepared alongside those used for imaging (Fig. 3).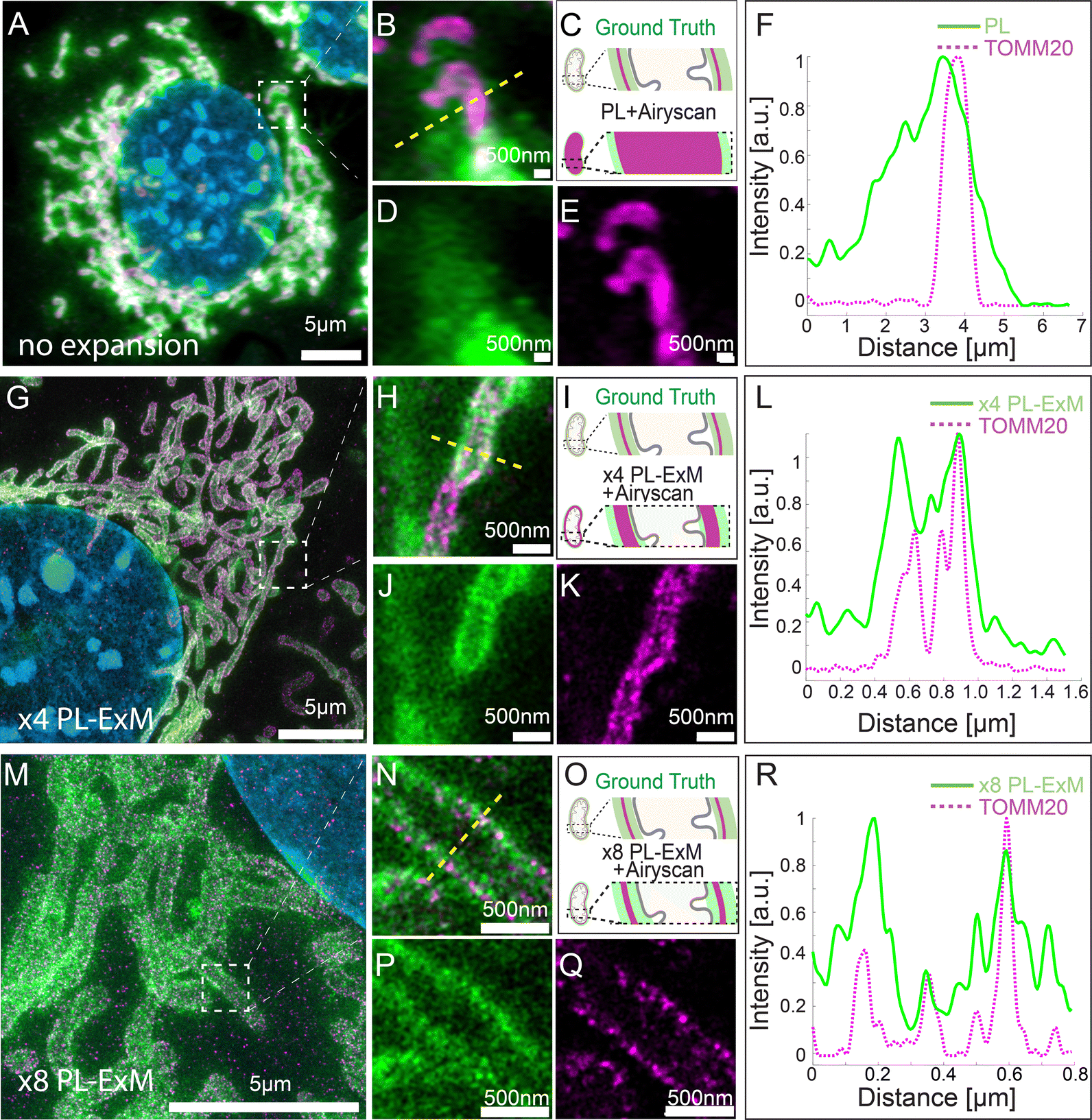 | ||
| Fig. 2 PL-ExM enables super-resolution visualization of the proximity-labeled interactome structure. All images were captured from MEF cells processed as follows: TOMM20 was proximity-labeled to visualize its interactome (green) and simultaneously immunostained (magenta). The nucleus was counterstained with DAPI (blue). All images were acquired using an Airyscan microscope. (A) displays a representative image of a non-expanded sample. (B) shows an enlarged view of the region indicated in (A). (C) provides schematics of the ground-truth structure of proximity-labeled TOMM20 (green) alongside immunostained TOMM20 (magenta), and the anticipated image without expansion. (D) represents the PL channel of (B). (E) illustrates the TOMM20 immunostaining channel of (B). (F) presents a representative histogram of the fluorescence intensity across a mitochondrion section from image (B) of the non-expanded sample. (G) depicts a representative PL-ExM image of a sample expanded 4.2 times, termed ×4 PL-ExM. (H) is a magnified view of the boxed area marked in (G). (I) Schematics showing the same ground truth structure as in (C), and the expected image after 4.2 times expansion. (J) is the PL-ExM channel of (H). (K) is the TOMM20 immunostaining channel of (H). (L) shows a representative histogram of the fluorescence intensity across a mitochondrion section from the ×4 PL-ExM image. (M) features a representative PL-ExM image of a sample expanded 8.2 times, named ×8 PL-ExM. (N) is a magnified view of the boxed area in (M). (O) Schematics of the same ground truth structure as in (C), and the expected image after 8 times expansion. (P) is the PL-ExM channel of (N). (Q) is the TOMM20 immunostaining channel of (N). (R) is a representative histogram of the fluorescence intensity across a mitochondrion section from the ×8 PL-ExM image. In all histograms (F), (L) and (R), the fluorescence intensity data were normalized for each channel. (A), (G), (M), (N), (P) and (Q) are maximum intensity projections of 3D z-stacks. (B), (D), (E), (H), (J) and (K) are single-slice images of 3D z-stacks. The length expansion factors were 4.2 for images (G), (H), (J) and (K), and 8.2 for (M), (N), (P) and (Q). All scale bars are in pre-expansion units. | ||
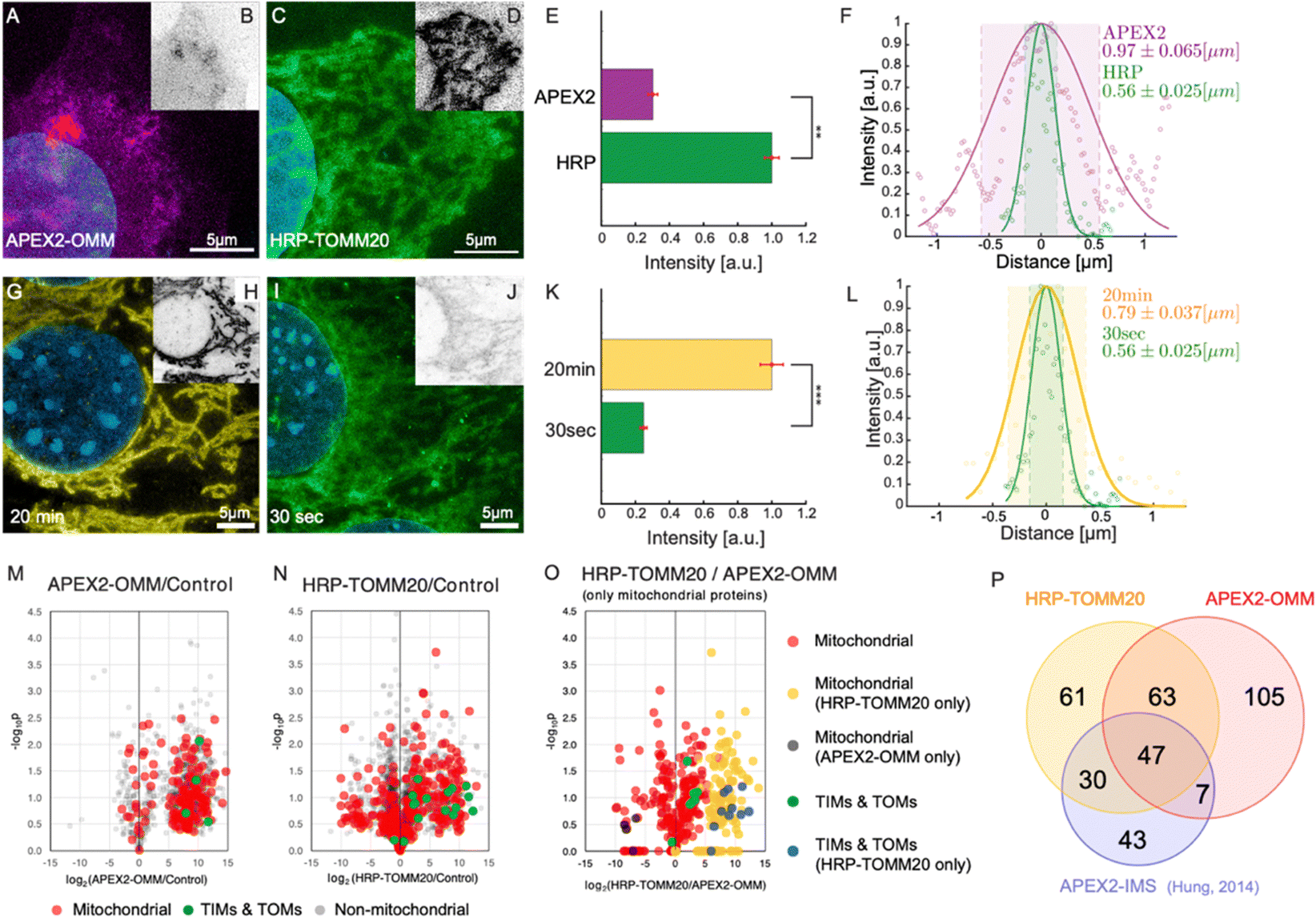 | ||
| Fig. 3 PL-ExM assesses the labeling resolution and efficiency of PL catalyzed by APEX2 and HRP. For APEX2, U2OS cells overexpressing APEX2-OMM were used, while for HRP, U2OS cells were immunostained with HRP-conjugated anti-TOMM20 antibodies. All images were captured using a confocal microscope under identical imaging conditions. (A) Displays a representative PL-ExM image following APEX2-catalyzed PL. (B) Presents grayscale versions of (A), with matched contrast settings with (D) for quantitative analysis. (C) Shows a representative PL-ExM image following HRP-catalyzed PL. (D) Presents grayscale versions of (C), with matched contrast settings with (B) for quantitative analysis. (A)–(D) Represent maximum intensity projections of 3D z-stacks. (E) Summarizes the fluorescence intensity from PL-ExM images of APEX2- and HRP-labeled samples with a sample size of n ≥ 3 per condition. The statistical significance is denoted by a p-value of less than 0.01. (F) Displays a histogram illustrating the fluorescence intensity across a mitochondrion's cross-section from a PL-ExM image of an APEX2 and HRP samples, with a mitochondrial diameter measured at 0.97 ± 0.065 μm and 0.56 ± 0.030 μm respectively. These statistics are derived from 90 measurements across three independent samples. In the comparison of 20-minute versus 30-second reaction durations (G)–(J), HRP-catalyzed PL was used on MEF cells with TOMM20 immunostained with HRP-conjugated antibodies. (G) Presents a PL-ExM image of HRP-catalyzed PL with a 20-minute H2O2 treatment. (I) Depicts a PL-ExM image following a 30-second H2O2 treatment. (H) and (J) Are grayscale versions of G and I, respectively, with the same contrast setting for quantitative analysis. (G)–(J) Are maximum intensity projections from 3D z-stacks. (K) Compares labeling efficiency of samples proximity labeled for 20 min and 30 seconds, demonstrating that 20 min labeling duration achieved approximately four times greater labeling efficiency than the 30-second labeling, with a p-value less than 0.001. This bar chart encapsulates the fluorescence intensity from PL-ExM images for both durations, n ≥ 3 per condition. (L) Illustrates a histogram of the fluorescence intensity from a mitochondrion's cross-section in a 20-minute- and 30 second-samples, with a mitochondrial diameter of 0.79 ± 0.037 μm and 0.56 ± 0.025 μm respectively. These values are averaged from 90 measurements from three independent samples. (M) Volcano plot demonstrates mitochondrial proteins enriched by APEX2-OMM. (N) Volcano plot demonstrates mitochondrial proteins enriched by HRP-TOMM20. The log2 fold-change is plotted on the x-axis, calculated from the relative normalized abundances of proteins in labeled versus control samples. Subunits of the TIM/TOM complex are highlighted in green, other mitochondrial proteins identified by MitoCarta are in red, and non-mitochondrial proteins are in gray. (O) Compares mitochondrial protein enrichment by APEX2-OMM against HRP-TOMM20, with log2 fold-change on the x-axis, representing the relative normalized abundances from HRP-TOMM20 versus APEX2-OMM. TIM/TOM complex subunits quantified by both APEX and HRP are in green; those exclusively quantified by HRP are in blue. Other mitochondrial proteins are in red unless only quantified by APEX (black) or HRP labeling (blue). (P) Depicts the overlap of enriched mitochondrial proteins identified by APEX2-OMM, TOMM20-HRP, and APEX2-IMS.9 The expansion factors for images (A)–(D) and (G)–(J) range from 4.1 to 4.2. All scale bars are 5 μm in pre-expansion units. | ||
The resolution achievable with PL-ExM can be enhanced using hydrogels with larger expansion factors. Using the hydrogel formula from TREx protocol,29 we achieved 8.2-fold expansion of proximity-labeled cells. Consequently, ×8 PL-ExM attained a measured effective resolution of 17 nm after expanding the gel 8.2 times in each dimension (Fig. 2M and N), which is two times better than the resolution achieved by PL-ExM using 4× expandable hydrogel. The higher resolution enabled further and clearer distinction of two narrow, well-separated peaks corresponding to proximity-labeled proteins at the cross-section of a mitochondrion (Fig. 2P and R), compared with those in ×4 PL-ExM images (Fig. 2J and L). The separation of these peaks indicated a mitochondrial diameter of approximately 500 nm (Fig. 2R).
To make sure the expansion procedure does not cause local distortions to mitochondria, we measured and compared mitochondrial diameters with and without expansion with an Airyscan microscope. The results confirmed the expansion didn’t cause mitochondria distortion. Previous works on ExM methods also reported that the expansion of mitochondria is isotropic and faithful.35
PL-ExM assesses the precision and efficiency of proximity labeling methods
In this section, we demonstrate how PL-ExM evaluates the labeling quality of different PL methods. We compared the labeling quality of two enzymes (APEX2 versus HRP) over two reaction durations (30 seconds versus 20 minutes) for the same bait protein. We analyzed the labeling resolution and efficiency in each set of conditions. The labeling resolution is critical for determining the spatial selectivity of the interactome and the rate of false positives, whereas the labeling efficiency reflects the interactome coverage. We measured the average mitochondrial diameter (n ≥ 90) from PL-ExM images, and we compared total fluorescence intensity from the streptavidin–dye to evaluate labeling efficiency among different PL conditions. To guarantee a fair comparison, all samples were labeled concurrently in the same batches (n > 3) and imaged with consistent microscope settings on the same days. It is worth noting that the fluorescent signal is determined by both the enzyme's labeling capability and the number of enzymes per target. Because an antibody may recognize multiple epitopes in the same targeted protein, the measured labeling efficiency of HRP-conjugated antibodies includes signal amplification.We assessed two widely used enzymes, APEX2 and HRP using PL-ExM. Mitochondrial outer membrane proteins were selected as the bait proteins, due to their well-documented interactomes via PL-MS9,38 which provide references for validating our PL-ExM assessments. In our experiments, APEX2-catalyzed PL was performed on U2OS cells overexpressing APEX2-OMM (Fig. 3A and Fig. S4A, ESI†), where OMM is a peptide targeting the outer mitochondrial membrane. HRP-catalyzed PL was performed on U2OS cells, which were immunostained with anti-TOMM20 antibodies conjugated with HRP (Fig. 3C and Fig. S4B, ESI†). The same biotin–phenol and reaction duration were used in APEX2 and HRP experiments. PL-ExM imaging revealed that HRP-catalyzed PL achieved approximately fourfold greater labeling efficiency than that of APEX2-catalyzed PL (Fig. 3B, D and E). Moreover, HRP-catalyzed PL demonstrated a higher labeling precision (Fig. S4, ESI†). The labeling precision was reflected in the measured mitochondrial diameters. A smaller average mitochondrial diameter of 0.56 μm ± 0.030 μm was obtained from the HRP (Fig. 3F, green). In contrast, APEX2-catalyzed PL resulted in a more diffusive signal surrounding the mitochondria (Fig. 3A and Fig. S4A, ESI†), resulting in a significantly larger average mitochondrial diameter of 0.97 μm ± 0.065 μm (Fig. 3F, purple). The lower labeling efficiency and precision of APEX-catalyzed PL might be due to the suboptimal permeability of biotin–phenol in live cells and the relatively lower enzymatic activity of APEX2 than that of HRP.
Experimental conditions, such as labeling duration, buffer, and temperature, can also significantly affect PL precision and efficiency. Compared with buffer and temperature, it is more difficult to keep the labeling duration consistent between experiments due to operator inconsistencies. Therefore, it is essential to measure how much labeling duration could affect the PL. We compared two H2O2 treatment durations, 30 seconds and 20 minutes (Fig. 3G–L). The PL-ExM results showed a quadrupling of labeling efficiency when the duration was extended to 20 minutes, as opposed to the 30-second condition (Fig. 3K). Surprisingly, the labeling precision reflected by measured mitochondrial diameters did not differ significantly between the two durations (Fig. 3L). The 20-minute labeling exhibited a slightly larger mitochondrial diameter, on average 0.79 μm (Fig. 3L, yellow), compared with 0.56 μm with the 30-second labeling (Fig. 3L, green). These results suggest that the efficiency of HRP-catalyzed PL increases significantly over time, while the labeling precision only decreases slightly.
We conducted PL-MS proteomic profiling (Fig. 3M–P) on samples prepared alongside those used for imaging (Fig. 3A–L). Cells biotinylated by APEX2 and HRP were lysed, and the biotinylated proteins were affinity purified, digested, and subsequently analyzed by MS. Relative to non-PL controls, quantitative MS analyses confirmed that both APEX2 and HRP methods could effectively enrich mitochondrial proteins (Fig. 3M and N). These results are in line with previous findings using APEX2 for mitochondrial intermembrane space (IMS) labeling9 (Fig. 3P). Notably, the HRP-labeled samples exhibited more robust labeling of TIMs and TOMs proteins compared to APEX2 samples (Fig. 3O), which corroborated the labeling efficiency assessment results (Fig. 3A–E).
This suggests that HRP-catalyzed PL is more effective at labeling proteins in close proximity to the bait, TOMM20. Such proteomic findings corroborate the PL-ExM imaging results. Collectively, PL-ExM proves to be a sensitive and reliable method for assessing and optimizing PL experimental conditions.
PL-ExM is compatible with cultured cells
PL-ExM is compatible with organelles, cell lines and tissues. In the preceding sections, we demonstrated PL-ExM on mitochondria. In this section, we will apply the method on various organelles including microtubules and cilia in U2OS and MEF cells (Fig. 4). We will also demonstrate PL-ExM in mouse brain tissues (Fig. 5).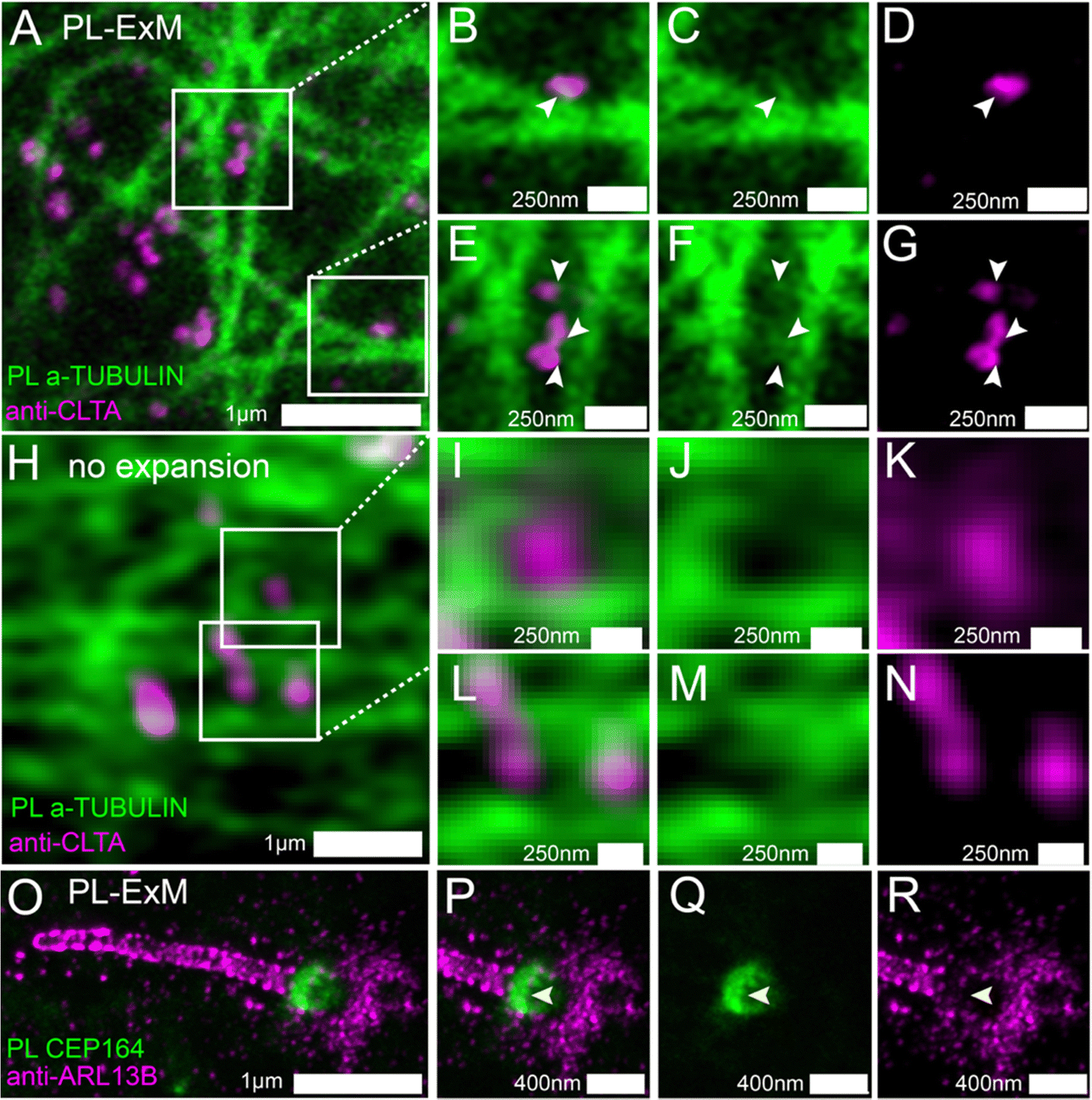 | ||
| Fig. 4 Two-color PL-ExM imaging to elucidate spatial relationships between interacting proteins. (A)–(G) PL-ExM images of proximity-labeled α-TUBULIN (green) and immunostained CLTA (magenta) in U2OS cells. (B)–(G) A magnified view of the areas boxed in (A), where arrows point out the co-localization of clathrin-coated pits (immunostained with anti-CLTA antibodies) and bud-like structures protruding from microtubules. (H)–(N) Airyscan images of non-expansion U2OS cells with proximity-labeled α-TUBULIN (green) and immunostained CLTA (magenta). (I)–(N) Magnified views of the areas indicated in (H). (O)–(R) PL-ExM images of a primary cilium of a MEF cell with proximity-labeled CEP 164 (green) and immunostained ARL13B (magenta). (P)–(R) Malignified views of the ciliary base in (O), where arrows point to an area with little co-localization between the PL and immunostaining channels. Images (A)–(N) are single-slice images, whereas (O)–(R) are maximum intensity projections of z-stacks. The length expansion factors are 4.1 for (A)–(G) and 8.4 for (O)–(R). All images were captured using an Airyscan microscope, and scale bars are in pre-expansion units. | ||
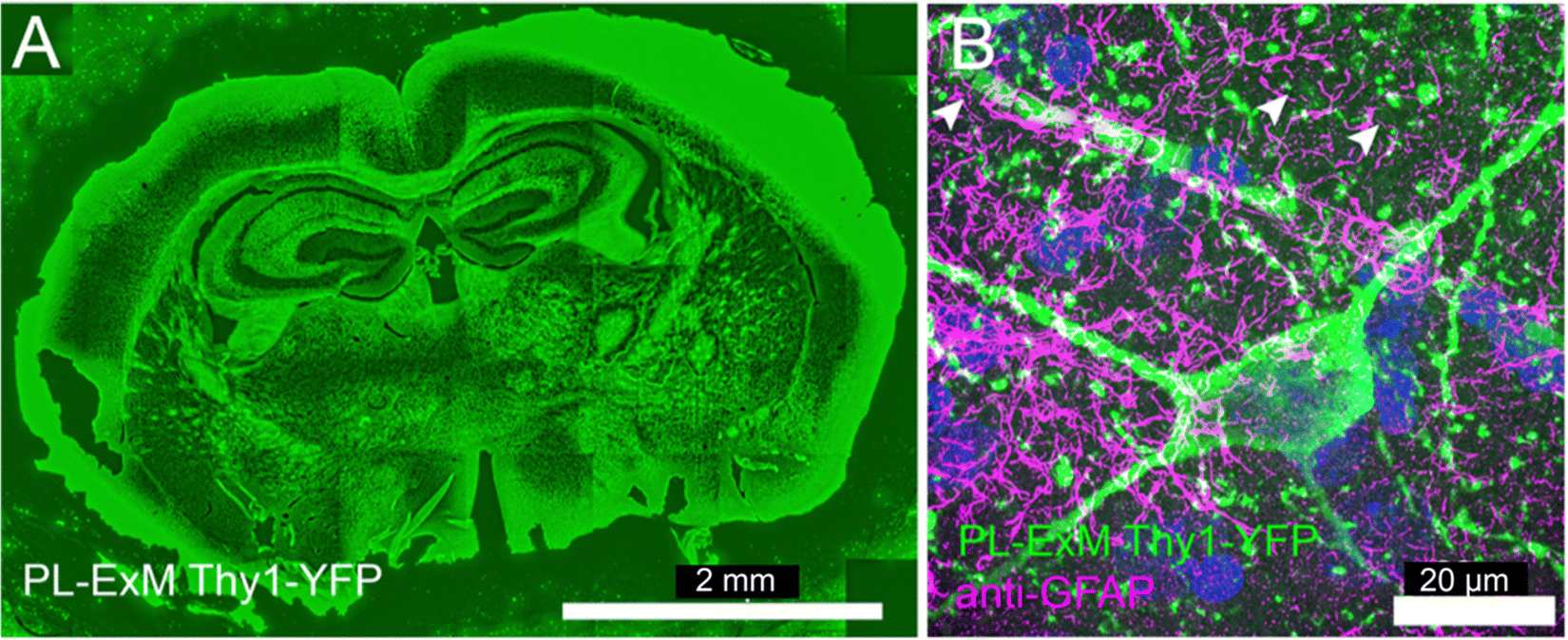 | ||
| Fig. 5 Two-color PL-ExM images of mouse brain sections. The PL-ExM images, captured using an Airyscan microscope, depict 20-μm sections of a mouse brain expressing Thy1-YFP. PL was applied to Thy1-YFP (green) and GFAP was immunostained (magenta). (A) Shows the proximity-labeled Thy1-YFP channel across an entire mouse brain slice. (B) Provides a magnified view of a region from (A), displaying both the proximity-labeled Thy1-YFP (green) and immunostained GFAP (magenta). Both images are maximum intensity projections of z-stack. The orthogonal views of the z-stack can be found in Fig. S7 (ESI†). The length expansion factor is 4.0. Scale bars are 2 mm for (A) and 20 μm for (B) in pre-expansion units. | ||
We used two-color PL-ExM to visualize the proximity-labeled interactome in one channel and a nearby non-bait protein in the other channel. Fig. 4A–G show the proximity-labeled α-TUBULIN and their spatial relationship with immunostained Clathrin A (CLTA). With the super resolution provided by PL-ExM, the PL channel revealed small clusters budding from the microtubules, as indicated by arrows in Fig. 4C and F. Notably, many of these clusters partially overlapped with the clathrin-coated pits (Fig. 4B and E), suggesting that clathrin-coated pits are components of the interactome of the microtubule. Such spatial information about interactomes were not detectable without expansion, owing to the limited resolution of conventional imaging techniques (Fig. 4H–N).
We further applied PL-ExM on the primary cilium, an organelle challenging to image due to its tiny size and composition of low-abundance proteins (Fig. 4O–R). The primary cilium functions as a sensory organelle that orchestrates signaling pathways, including the sonic hedgehog signaling, through regulatory GTPases, such as ADP-ribosylation factor-like protein 13B (ARL13B). Mick et al. developed an innovative approach known as cilia-APEX, which biotinylated ciliary interactome for MS analysis.39 Here, we present PL-ExM as an adjunct to cilia-APEX, offering spatial information about PPIs. We used two-color PL-ExM to concurrently image proximity-labeled distal appendage (DA) component CEP164 at the base of the cilium and immunostained ARL13B in MEF cells. With an 8.4-fold expansion, we successfully resolved the donut-shaped DA disk and the distribution of AL13B along the cilium (Fig. 4O). The images revealed a subtle overlap between the proximity-labeled DAs and the immunostained ARL13B (Fig. 4P–R), suggesting that ARL13B either interacts transiently with DAs or does not interact with DAs.
PL-ExM is compatible with tissues
Deep tissue imaging presents inherent challenges due to light scattering caused by the layers of cells and extracellular matrix. The expansion process integral to PL-ExM converts intact tissue into a hydrogel that is optically clear, aligning with the same principles of tissue clearing as the CLARITY method.40 Consequently, PL-ExM not only enhances resolution but also offers tissue clearing, enabling clearer and deeper visualization of interactomes within tissue samples.We recommend using HRP-catalyzed PL-ExM for tissues. HRP-conjugated antibodies can be tagged to the proteins of interest in fixed tissue samples. On the other hand, live-cell PL methods that require gene editing, such as APEX and BioID, may not be suitable for tissues, especially human tissues. We used PL-ExM to visualize proximity-labeled neurons in mouse brain sections. We performed PL of the neuron marker Thy1 using HRP. In 20-μm tissue sections, antibodies conjugated with HRP permeated tissue thoroughly for PL to occur robustly along the z-axis of the tissue (Fig. S7, ESI†). The ×4 PL-ExM images revealed the distribution of the proximity-labeled Thy1 throughout the brain section (Fig. 5A). While the noise level in tissues was higher than that in cultured cells, the dendrites and axons of neurons were distinctly visible in the PL channel (Fig. 5B). Additionally, we co-immunostained for the astrocyte marker Glial fibrillary acidic protein (GFAP). The two-color PL-ExM images captured the intricate spatial relationship between astrocytes and neurons, underscoring their interactive nature (Fig. 5B).
Discussion
Throughout this work, we found that the variabilities in PL quality between experiments were often underestimated. The smallest labeling radius of the peroxidase-catalyzed PL measured by PL-ExM is 19 nm, and the largest is more than 100 nm. The labeling radius varied greatly depending on the enzyme, labeling duration, biological targets, and other experimental conditions. Our results are within the range of labeling radii of HRP and APEX reported by electron microscopy studies, spanning from a few nanometers to 300 nm.5,8,20 A recent STORM study reported a labeling radius of 269 nm for HRP PL and emphasized that the quality of PL can differ not just between various PL techniques but is also subjected to sample conditions and operator errors.21 Factors such as the high concentration of radical quenchers present in the cytosol and mitochondrial matrix41 and the effects of macromolecular crowding42 can impact both the PL precision and efficiency. Biological systems, with their inherent variability and dynamic nature – shaped by genetics, environmental factors, or the physiological state of the samples – can introduce additional variability into PL outcomes. Therefore, having a reliable visualization method to evaluate the quality of PL experiments is necessary.PL-ExM offers the necessary resolution and sensitivity to directly visualize and evaluate the labeling resolution and efficiency of PL in both cells and tissues. While we focused on the evaluation of peroxidase-based PL techniques, we also demonstrated the compatibility of PL-ExM with PL based on biotin ligase, such as TurboID (Fig. S5, ESI†). Given these capabilities, we advocate for PL method developers to employ super-resolution imaging tools like PL-ExM to characterize and refine new PL techniques. Similarly, for users of PL-MS, we recommend the assessment of sample preparation with PL-ExM to corroborate the spatial context of their proteomic results. The PL-ExM images provide new spatial information, not only for validating proteomic data but also for identifying potential false positives.
During the expansion procedure of PL-ExM, the homogenization step is designed to disentangle neighboring proteins, facilitating isotropic expansion. A relevant question may arise: Does this homogenization step result in the loss of interactome detection in the images due to the disruption of PPIs? The answer is no. This is because the interactome is captured by the PL process, which occurs when the cells are still intact, before the expansion procedure begins. Proteins within the PL radii are tagged with biotin while the cell architecture is preserved. Thus, provided that the biotin-marked proteins are imaged successfully at the end of the process, any disruption of PPIs during the expansion will not lead to incomplete interactome detection. The effective imaging of biotin post-expansion has been validated by the LR-ExM technique that we recently developed.33
A crucial consideration in PL-ExM is the fidelity of expansion. Anisotropic expansion could distort the interactome structure, leading to unreliable observation. To prevent this, our team, and other developers of ExM have meticulously optimized the process to ensure isotropic expansion. This involves refining fixation methods, enhancing protein anchoring efficiency, perfecting sample homogenization techniques, and developing precise hydrogel recipes.37,43–45 We have extensively addressed strategies to guarantee isotropic expansion across various biological samples in a recent review.37 The PL-ExM technique has been fine-tuned to faithfully represent the proximity-labeled interactome, accommodating different enzymes and labeling conditions. Anchoring agents such as MA-NHS, glutaraldehyde, and glycidyl methacrylate have all proven effective for anchoring biotinylated proteins. Consistent with other ExM methods, proteinase K digestion remains the go-to approach for sample homogenization in PL-ExM.
The next question is about the resolution limit achievable by PL-ExM. In this study, we attained an effective resolution of 17 nm by expanding cells 8.2-fold using the TREx protocol29 and utilizing an Airyscan microscope for imaging (Fig. 2M–R). The resolution of PL-ExM can be further enhanced by employing a larger expansion factor26–31 and by imaging with more advanced microscopes, such as STORM, photoactivated localization microscopy (PALM), and STED. However, there is an intrinsic resolution limit in PL-ExM, dictated by the pore size of the hydrogel prior to expansion. The capacity of the hydrogel to precisely anchor biomolecules is contingent upon these pore sizes. Consequently, any structural features finer than the pores will likely be distorted during the expansion process.
Conclusions and future directions
PL-ExM provides superior imaging resolution and detection sensitivity to assess the labeling precision and efficiency of PL techniques. By integrating the PL with ExM, PL-ExM facilitates resolutions as fine as 17 nm on widely accessible microscopes, such as confocal and Airyscan systems. The detection sensitivity of single fluorophores allows us to compare labeling efficiency between different PL techniques at the single-cell level. Through our analysis of APEX2- and HRP-catalyzed PL, we demonstrated that PL-ExM possesses the necessary resolving power to accurately measure labeling radii and has the sensitivity to discern labeling efficiency among diverse PL methods. The consistency between our imaging results and proteomic data from PL-MS corroborates PL-ExM's efficacy as a reliable method for quality control in PL-based research.Looking ahead, it is promising to use PL-ExM to elucidate the three-dimensional spatial relationships within the interactome. Our two-color PL-ExM images of microtubules and cilia serve as a prelude for this application. In these images, one channel delineates the proximity-labeled interactome, whereas the second channel pinpoints a specific protein. This specific protein can be selected from the interacting proteins identified via PL-MS. Envision a scenario where we superimpose all proteins identified onto the interactome structure using PL-ExM. This would reveal the spatial organization of proteins that underpin the function of the bait protein. However, the challenge of spatially dissecting the whole interactome stems from the need to enhance PL-ExM's multiplexity. Integration with highly multiplexed immunostaining methods, such as Immuno-SABER,46 could enable PL-ExM to map each protein within the interactome comprehensively.
Methods
Cell line generation
APEX2-OMM gene fragment (from a plasmid Addgene #238450) was cloned into a second generation 5′ self-inactivating lentiviral backbone (pHR) downstream of a SFFV promoter, using InFusion cloning (Takara Bio #638910). A pantropic VSV-G pseudotyped lentivirus was produced via transfection of Lenti-X 293T cells with the pHR transgene expression vector and viral packaging plasmids pCMVdR8.91 and pMD2.G using Fugene HD (Promega #E2312). At 48 hours, the viral supernatant was harvested, filtered through a 0.45 μm filter (Millipore #HAWP04700), and added onto the U2OS cells for transduction. APEX2-OMM cell lines are generated from Single-cell cloning of the transduced U2OS cells.The OMM-V5-LOV-Turbo gene fragment (from plasmid Addgene #199665) was cloned into a mammalian expression lentiviral vector with the TRE3G promoter (pCW, Addgene plasmid #41393). Lentivirus was produced by transfecting Lenti-X 293T cells with the transgene expression vector and viral packaging plasmids pCMVdR8.91 and pMD2.G using PEI (DNA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PEI = 1:3). After 48 hours, the viral supernatant was harvested, filtered through a 0.45 μm filter (Millipore #HAWP04700), and used to transduce U2OS cells. Following 48 hours of lentivirus infection, the U2OS cells were selected with U2OS culture medium containing 4 μg mL−1 puromycin for 48 hours until all untransduced cells were dead. The cell culture medium continued to contain 4 μg mL−1 puromycin until the cells were prepared for imaging setting.
:PEI = 1:3). After 48 hours, the viral supernatant was harvested, filtered through a 0.45 μm filter (Millipore #HAWP04700), and used to transduce U2OS cells. Following 48 hours of lentivirus infection, the U2OS cells were selected with U2OS culture medium containing 4 μg mL−1 puromycin for 48 hours until all untransduced cells were dead. The cell culture medium continued to contain 4 μg mL−1 puromycin until the cells were prepared for imaging setting.
Cell culture
MEF cells were cultured in DMEM, Glutamax (Thermofisher; 10566-016) supplemented with 15% Fetal Bovine Serum (FBS) and 1% antibiotics-antimycotic solution (Sigma Aldrich; A5955). The culture conditions were set at 37 °C and 5% CO2. U2OS (ATCC; HTB-96) and U2OS-APEX2-OMM cells were cultured in McCoy's 5a (ATCC; 30-2007) also supplemented with 10% FBS and 1% antibiotics-antimycotic solution under identical temperature and CO2 conditions. For PL-ExM experiments, cells were plated at a density of 104 cells per cm2 in 16-well chambers (Grace Bio-Labs; 112358) and cultivated until they reached 80% confluency. For MEF cell adhesion, chambers were pre-coated with a gelatin solution (Sigma-Aldrich; G1393-100ML) for one hour at 37 °C. MEF cells were seeded in 16-well chambers at the same density. Following a 16-hour incubation period, the cells underwent a starvation period of 24 hours in Opti-MEM reduced serum medium to induce ciliation.Animal sacrifice and brain slice preparation
Thy1-YFP mice were euthanized via CO2 inhalation and transcardially perfused with ice-cold 1× PBS buffer. Brains were removed carefully and fixed in freshly made 4% paraformaldehyde solution for 24 hours at 4 °C. Brains were then cryoprotected in 30% sucrose solution at 4 °C before embedding in OCT and storage at −80 °C. Frozen brains were sectioned at 20 μm on a Leica SM2000 R sliding microtome for subsequent immunohistochemical analyses. All animal protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of California, Irvine.HRP antibody-catalyzed PL for cultured cells, including the specific steps for fixation, blocking, and immunostaining
Cultured cells were fixed in different ways for the immunostaining of different organelles. For mitochondria, cells were fixed with a solution of 3% paraformaldehyde (PFA) and 0.1% Glutaraldehyde (GA) for 10 minutes at room temperature. This was followed by a reduction step with 0.1% sodium borohydride in PBS for 5 minutes. For microtubules, cells underwent a 30-second treatment with PEM buffer (comprising 100 mM Pipes, 1 mM EGTA, and 1 mM MgCl2, pH 6.9) followed by fixation with 3.2% PFA in PEM buffer for 10 minutes at room temperature, and again reduction with 0.1% sodium borohydride in PBS for 5 minutes. For the cilia, fixation was achieved using 4% PFA for 15 minutes at room temperature.Post-fixation, cells were washed three times with PBS, with a 5-minute interval between each wash. The cells were then treated with 3% hydrogen peroxide (H2O2, Sigma Aldrich; H1009) for 5 minutes at room temperature to inhibit endogenous peroxidase activity before the addition of any HRP to the system. The H2O2 reaction was terminated by the application of 2 mM L-ascorbic acid sodium (Alfa Aesar; A17759) for 5 minutes, followed by three further PBS washes. Subsequently, the fixed cells were permeabilized and blocked in a buffer containing 3% BSA, and 0.1% Triton X-100 in PBS for 30 minutes at room temperature, preparing them for the immunostaining process.
Primary antibodies were added to the fixed cells at a concentration of 2 μg mL−1 in blocking buffer (3% BSA in PBS) and incubated for 16 hours at 4 °C. The primary antibodies utilized in this study include Rabbit anti-TOMM20 (1:250 dilution, santa cruz; sc-11415), Rat anti-α-TUBULIN, tyrosinated, clone YL1/2 (1:5000 dilution, Millipore Sigma; MAB1864-I), Rabbit anti-clathrin heavy-chain (1:100 dilution, Abcam; ab21679), Rabbit anti-ARL 13B (1:100 dilution, Proteintech; 17711-1-AP), Mouse anti-CEP164 (1:100 dilution, Santa Cruz; sc-515403), Chicken anti-GFAP (1:1000 dilution, AbCam; ab4674), Rabbit anti-GFP (D5.1,1:200, Cell Signaling; 2956). Following incubation, cells were washed three times with blocking buffer, each followed by a 5-minute interval. The cells were then incubated with 3 μg mL−1 AffiniPure Goat anti-Rabbit (1:100, Jackson ImmunoResearch; 111-005-144), Goat anti-Mouse (1:100, Jackson ImmunoResearch; 115-005-146), or Goat anti-Rat (1:100, Jackson ImmunoResearch; 112-005-167) secondary antibodies in blocking buffer for 1 hour at room temperature. After secondary staining, cells were washed with blocking buffer, with a 5-minute interval between washes. Following the secondary antibody staining and subsequent washing steps, cells were incubated with ImmPRESS HRP Horse anti-Goat IgG Polymer Detection Kit (no dilution, Vector Laboratories; MP-7405) for 1 hour at room temperature. After incubation, the cells were washed three times with PBS to remove any unbound antibodies.
For PL, the cells were then treated with 0.5 mM solution of biotin phenol (Biotin tyramide, Sigma Aldrich; SML-2135) for 15 minutes at room temperature to allow for adequate labeling. Immediately prior to initiating the labeling reaction, a fresh solution of 2 mM H2O2 in PBS was prepared. This H2O2 solution was then promptly added to the cells, which were already in the biotin–phenol solution for 30 seconds, unless a different time was specified in the experimental conditions. To stop the reaction and prevent over-labeling, the cells were then treated with a quenching solution of 2 mM of L-ascorbic acid sodium salt for 5 minutes at room temperature.
APEX2-catalyzed PL for cultured cells
For APEX2-catalyzed PL in cultured cells, the permeability of biotin phenol is a critical factor that influences labeling efficacy, especially when labeling is performed on live cells. Our optimization experiments showed that incubating cells with 1 mM biotin–phenol for 2 hours at 37 °C yields the most effective labeling. Just before starting the labeling reaction, a fresh 2 mM H2O2 solution in PBS was prepared. This H2O2 solution was then immediately added to the biotin–phenol solution with the cells for a duration of 1 minute. To terminate the reaction and prevent over-labeling, a quenching step was conducted using a 2 mM of L-ascorbic acid sodium solution for 5 minutes, followed by three subsequent PBS washes to thoroughly remove any unreacted compounds.Subsequent to the PL, U2OS-APEX2-OMM cells were fixed with 4% PFA solution for 15 minutes at room temperature. After fixation, cells were washed three times with PBS to ensure the removal of excess fixative.
TurboID-catalyzed PL for cultured cells
After seeding and culturing U2OS cells with proper density overnight, McCoy's 5a medium was changed with fresh medium supplemented with 2 mg mL−1 puromycin for stably expressing the corresponding TurboID fusion construct. TurboID-PL labeling was initiated by changing the medium to fresh medium containing 100 μM biotin for 2 h in dark, and then expose to a visible LED light source for 30 min. During this process, the cells were incubated at 37 °C under 5%CO2. Note, the cells were kept in complete dark after adding puromycin. The labeling reaction was stopped by washing cells three times with ice-cold PBS and followed by immediate fixation.HRP antibody catalyzed PL for mouse brain tissues
We initiated the process by air-drying a tissue slide for 30 minutes, followed by rehydration in PBS for 10 minutes. After two additional PBS washes, the tissue was treated with 3% H2O2 for 5 minutes to quench endogenous peroxidase activity. This was stopped by adding a 2 mM solution of L-ascorbic acid sodium and incubating for 5 minutes, followed by three PBS washes. The tissue was then permeabilized and blocked using a buffer containing 3% BSA, and 0.1% Triton X-100 in PBS for one hour at room temperature.Overnight primary antibody staining was conducted at 4 °C using Rabbit anti-GFP (a dilution of 1:200, Cell Signaling; 2956). This was followed by a 2.5-hour incubation with Goat anti-Rabbit secondary antibody (a dilution of 1:100, Jackson ImmunoResearch; 111-005-144), and a 2.5-hour tertiary staining with ImmPRESS HRP Horse x Goat Polymer Detection Kit (no dilution, Vector Laboratories; MP-7405). After the antibody staining, the tissue was incubated in a 0.5 mM biotin–phenol solution (Biotin tyramide, Sigma Aldrich; SML-2135) for 15 minutes. Immediately before the PL reaction, a fresh 2 mM H2O2 solution in PBS was prepared and added to the tissue sample for 30 seconds. The PL reaction was then halted using a 2 mM of L-ascorbic acid sodium solution for 5 minutes.
Subsequent to PL, additional immunostaining was performed for GFAP using Chicken anti-GFAP primary antibody (1:1000 dilution, AbCam; ab4674) for 2.5 hours, followed by secondary antibody staining with Donkey anti-Chicken Dig-MA-NHS (prepared in our laboratory) for another 2.5 hours. Post-immunostaining, the tissue was anchored for 10 minutes using 0.25% glutaraldehyde solution. The tissue sample then underwent gelation, staining, and expansion following the procedures outlined in the label-retention expansion microscopy33,36 protocols. All reactions were carried out at room temperature, with three PBS washes after each step, unless otherwise specified.
Protein anchoring, gelation, denaturation, post-digestion fluorescent staining, and expansion steps of the ×4 PL-ExM
After completing PL and immunostaining, samples underwent a protein anchoring step using one of three possible reagents: 0.25% glutaraldehyde (GA; Electron Microscopy Sciences; 16120) solution in PBS incubated at room temperature for 10 minutes, a 25 mM Methacrylic acid N-hydroxysuccinimide ester (MA-NHS; Simga-Aldrich; 730300) solution in PBS for 1 hour, or a 0.04% glycidyl methacrylate (GMA; Sigma-Aldrich; 151238) solution in 100 mM sodium bicarbonate (pH 8.5) for 4 hours. These anchoring agents demonstrated comparable efficiencies for HRP- and APEX2-catalyzed PL. But GA and GMA are preferred for TurboID.The subsequent steps, including gelation, denaturation, fluorescent staining, and expansion, were carried out similarly to those described in label-retention expansion microscopy (LR-ExM)33,36 protocols. Briefly, for gelation, samples were incubated with a monomer solution (8.6 g sodium acrylate, 2.5 g acrylamide, 0.15 g N,N′-methylenebisacrylamide, 11.7 g sodium chloride in 100 mL PBS buffer) on ice for 5 minutes. Following this, the gelation solution was prepared by mixing the monomer solution with a 10% (w/v) N,N,N′,N′-tetramethylethylenediamine (TEMED) stock solution, a 10% (w/v) ammonium persulfate (APS) stock solution, and water in a volume ratio of 47:1:1:1. This gelation mixture was then added to the samples and incubated on ice for an additional 5 minutes. Once the gelation mixture had been applied, the samples were transferred to a humidity controlled chamber set at 37 °C to facilitate the gelation process. The samples remained in this environment for 2 hours to ensure complete gelation.
After one hour of gelation, the gelated samples were submerged in a proteinase K buffer composed of 8 units per mL proteinase K in a digestion buffer containing 50 mM Tris (pH 8.0), 1 mM EDTA, 0.5% Triton X-100, 1 M NaCl. Following digestion, the samples were thoroughly washed with an excess of DNase/RNase-free water. The duration of the proteinase K incubation was 16 hours at room temperature for cultured cells, and 1.5 hours at 78 °C for tissue samples.
After denaturation, the gelated samples were incubated in a staining solution with 3 uM fluorescently labeled streptavidin, such as streptavidin-Alexa Fluor 488, and fluorescently labeled anti-DIG antibodies, like anti-DIG-DyLight 594, for 24 hours at room temperature. The buffer for staining was composed of 10 mM HEPES and 150 mM NaCl in water, adjusted to pH 7.5.
The gelated and stained samples underwent expansion in DNase/RNase-free water for over 4 hours at room temperature. Once fully expanded, the gelated samples were carefully trimmed and placed onto poly-lysine-coated glass bottom multiwell plates or dishes, preparing them for subsequent imaging.
Protein anchoring, gelation, denaturation, post-digestion fluorescent staining, and expansion steps of the ×8 PL-ExM
The anchoring, digestion, and post-digestion fluorescent staining steps of the ×8 PL-ExM were identical to those of the ×4 PL-ExM. We modified the gel monomer recipe and expansion steps for the 8× PL-ExM based on the TREx protocol.29 Briefly, we incubated the samples with a monomer solution designed for ×8 expansion (1.1 M sodium acrylate, 2.0 M acrylamide, 50 ppm N,N′-methylenebisacrylamide in PBS) on ice for 5 minutes. Then we quickly added a gelation solution, a mixture of the monomer solution,1.5 ppt APS, and 1.5 ppt TEMED, to the samples and incubated them on ice for an additional 5 minutes. After this, we transferred the samples with the gelation solution to a 37 °C humidity-controlled chamber to allow gelation to occur for 2 hours. The expansion step was conducted similarly to that of the ×4 PL-ExM, with the main difference being an overnight expansion duration at room temperature.Image acquisition and analysis
Image acquisition and analysis for the PL-ExM data were carried out using a Zeiss LSM 980 and Zeiss LSM 900, both equipped with a 63× water immersion objective (Zeiss Plan Apo 63× NA 1.15). The non-expanded samples were imaged with Airyscan mode using Zeiss LSM 980 using the same 63× water immersion objective (Zeiss Plan Apo 63× NA 1.15). Confocal imaging was conducted on the Zeiss LSM 980 with the 63× water immersion objective (Zeiss Plan Apo 63× NA 1.15) or on a spinning-disk confocal microscope (Nikon CSU-W1 Sora) with a 40× water-immersion objective (Nikon CFI Apo 40× WI NA 1.15). We analyzed the fluorescence intensity of both Airyscan and confocal images using the open-source software Fiji (ImageJ). No deconvolution was applied to any images in this study.Image intensity quantitative analysis and statistics
For the quantitative analysis of image intensity, images were first denoised by defining noise as| Noise = 0.1 × (Intensitymax − Intensitymin) |
Protein purification and digestion for MS
The cell pellets were lysed in lysis buffer [50 mM Tris–HCl, 500 mM NaCl, 0.2% SDS, 1% Triton, 1 mM Tris(2-carboxyethyl) phosphine hydrochloride (TCEP), 10 mM sodium azide, 10 mM sodium ascorbate, 5 mM TROLOX, protease inhibitor cocktail (pH 7.5)] with sonication on ice. The lysates were centrifuged at 13000 rpm for 15 minutes to remove cell debris, and the supernatant was incubated with streptavidin Mag Sepharose resin (Cytiva) for overnight at 4 °C with rotation. The streptavidin beads were then washed twice with four buffers containing: (A) 2% SDS at room temperature; (B) 50 mM Tris–HCl, 500 mM NaCl, 2% Triton-X; (C) 50 mM Tris–HCl, 250 mM NaCl, 0.5% SDS, 0.5% Triton-X and (D) 2 M urea, 50 mM Tris–HCl at 4 °C. The bound proteins were then reduced, alkylated, and digested on-bead by LysC in 8 M urea/25 mM NH4HCO3 for 4 hours, followed by trypsin in 1.5 M urea/25 NH4HCO3 overnight at 37 °C. The peptide digests were extracted and desalted with C18 tip (Agilent) prior to liquid chromatography tandem mass spectrometry (LC MS/MS).47
Mass spectrometry analysis
The peptide digests were subjected to LC MS/MS analysis using an UltiMate 3000 RSLC system (Thermo Fisher Scientific) coupled in-line to an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific). Reverse-phase separation was performed on a 50 cm × 75 μm I.D. Acclaim® PepMap RSLC column. Peptides were eluted using a gradient of 4% to 22% B over 87 minutes at a flow rate of 300 nL min−1 (solvent A: 100% H2O, 0.1% formic acid; solvent B: 100% acetonitrile, 0.1% formic acid). Each cycle consisted of one full Fourier transform scan mass spectrum (375–1500 m/z, resolution of 120000 at m/z 400) followed by data-dependent MS/MS scans acquired in the Orbitrap with HCD NCE 30% at top speed for 3 seconds. Target ions already selected for MS/MS were dynamically excluded for 30 s. Protein identification and label-free quantitation was carried out using MaxQuant as described.48 Raw spectrometric files were searched using MaxQuant (v. 2.0.3.0) against a FASTA of the complete human proteome obtained from SwissProt (version from April 2023). The first search peptide tolerance was set to 15 ppm, with main search peptide tolerance set to 4.5 ppm. Trypsin was set as the digestive enzyme with max 2 missed cleavages. Methionine oxidation and protein N-terminal acetylation were set as variable modifications, while cysteine carbamidomethylation was set as a fixed modification. Peptide spectra match and protein FDRs were both set as 0.01. For quantitation, intensities were determined as the full peak volume over the retention time profile. “Unique plus razor peptides” was selected as the degree of uniqueness required for peptides to be included in quantification. The resulting iBAQ values for each identified protein by MaxQuant were used for comparing protein relative abundances.
For the experiments presented in Fig. 3M–P, we conducted two sets of quantitative mass spectrometry analyses to compare the APEX2- and HRP-catalyzed PL. For each group, negative controls were also included. Initially, we cultured U2OS-APEX2-OMM (experimental, and negative control) and wild-type U2OS cells (experimental, and negative control) in multiple 150 mm dishes. After trypsinization, we collected the cells by centrifugation at 1800 rpm for 3 minutes and used approximately 2 × 108 cells per condition. In Fig. 3M, O and Q, U2OS-APEX2-OMM cells were used. We treated the experimental and control groups with 500 μL of 1 mM Bitoin Phenol (BP) solution at 37 °C for 2 hours. Without removing the BP solution, we then treated the experimental group with an equal volume of 2 mM freshly prepared H2O2 solution for 1 minute. This was followed by quenching the reaction with 750 μL of 15 mM sodium ascorbate solution. The samples were washed twice with PBS, with a 3-minute interval between washes. After the PL step, each sample was fixed with 1% paraformaldehyde (PFA) solution; the control group was fixed immediately after BP incubation without H2O2 treatment. We homogenized the samples thoroughly after each step and centrifuged at 500G for 3 minutes to pellet the cells before proceeding to the next treatment. For Fig. 3N–P, where wild-type U2OS cells were used, we first fixed the cells with 0.1% glutaraldehyde (GA) for 10 minutes at room temperature and then washed them thrice with PBS for 3 minutes each. The cells were incubated with blocking buffer (3% BSA in PBS) for 30 minutes and then stained overnight at 4 °C with a primary antibody using Rabbit anti-TOMM20 (1:250 dilution, Santa Cruz; sc-11415). After three washes with blocking buffer, we stained the samples with 3 μg mL−1 AffiniPure Goat anti-Rabbit (1:100, Jackson ImmunoResearch; 111-005-144) for one hour at room temperature, followed by three additional washes. The samples were then stained with Goat anti-Horse HRP (no dilution, Vector Laboratories; MP-7405) for 1 hour at room temperature, washed, and incubated in 500 μL of 0.5 mM BP solution for 15 minutes at room temperature. The negative control was not treated further, whereas the experimental condition was treated with 500 μL of 2 mM H2O2 solution for 30 seconds at room temperature, followed by quenching with 750 μL sodium ascorbate solution. After a 5-minute incubation, we washed the samples thoroughly with PBS three times.
Image resolution measurement
For resolution measurement, we utilized 0.1 μm size fluorescent beads (TetraSpeck Microspheres, Invitrogen; T7279) to determine the resolving power of the Airyscan LSM980 microscope equipped with a 63× water immersion objective (NA1.15). We sampled 30 different beads to obtain the average full width half maximum (FWHM) along with the standard error. The effective resolution of PL-ExM was assessed by dividing the measured FWHM by the physical expansion factor of the hydrogel.Author contributions
S. P. and X. S. conceived and led the research. S. P. performed PL-ExM, imaging, and prepared samples for MS analysis. X. W. performed MS experiments and analysis. X. L. and X. S. initialized the concept and performed preliminary experiments. X. H. made plasmids, and generated cell lines with X. S. K. F. performed cell experiments under S. P. supervision. A. A. T. synthesized early versions of LR-ExM probes. Z. D. synthesized LR-ExM probes. L. S. assisted Airyscan imaging. L. H. led and supervised MS work. X. W. performed MS experiments. X. W., C. Y. and L. H analyzed MS data. K. F. assisted sample preparation. S. P, X. S., and L. H. drafted and edited the manuscript.Data availability
All data are available in the main text or the ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We acknowledge Professor Vivek Swarup and Dr Sudeshna Das for providing mouse brain tissues to this project. S. P. was supported by NIH Pathway to Independence Award (R00GM126136), an NSF grant (DMS1763272), and Simons Foundation grant (594598). X. S. was supported by the NIH Director's New Innovator Award (DP2GM150017) and the NSF Faculty Early Career Development (CAREER) Award (2341058). Z. D. was supported by the Chan Zuckerberg Initiative (CZI) Visual Proteomics Imaging Award. X. W., C. Y., and L. H. are supported by the NIH Maximizing Investigators’ Research Award (R35GM145249). Special thanks to the CZI Advancing Imaging through Collaborative Projects award for supporting our dissemination of PL-ExM and LR-ExM.References
- J. B. Geri, et al., Microenvironment mapping via Dexter energy transfer on immune cells, Science, 2020, 367, 1091–1097 CrossRef CAS PubMed.
- B. A. Killinger, et al., In situ proximity labeling identifies Lewy pathology molecular interactions in the human brain, Proc. Natl. Acad. Sci. U. S. A., 2022, 119(28), e2209742119 CrossRef CAS PubMed.
- A. Uezu, et al., Identification of an elaborate complex mediating postsynaptic inhibition, Science, 2016, 353, 1123–1129 CrossRef CAS PubMed.
- T. Takano, et al., Chemico-genetic discovery of astrocytic control of inhibition in vivo, Nature, 2020, 588, 296–302 CrossRef CAS PubMed.
- N. Kotani, et al., Biochemical visualization of cell surface molecular clustering in living cells, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 7405–7409 CrossRef CAS PubMed.
- K. Honke and N. Kotani, Identification of cell-surface molecular interactions under living conditions by using the enzyme-mediated activation of radical sources (EMARS) method, Sensors, 2012, 12, 16037–16045 CrossRef CAS PubMed.
- S. Jiang, et al., A proteomics approach to the cell-surface interactome using the enzyme-mediated activation of radical sources reaction, Proteomics, 2012, 12, 54–62 CrossRef CAS PubMed.
- J. D. Martell, et al., Engineered ascorbate peroxidase as a genetically encoded reporter for electron microscopy, Nat. Biotechnol., 2012, 30, 1143–1148 CrossRef CAS PubMed.
- V. Hung, et al., Proteomic Mapping of the Human Mitochondrial Intermembrane Space in Live Cells via Ratiometric APEX Tagging, Mol. Cell, 2014, 55, 332–341 CrossRef CAS PubMed.
- S. S. Lam, et al., Directed evolution of APEX2 for electron microscopy and proximity labeling, Nat. Methods, 2015, 12, 51–54 CrossRef CAS PubMed.
- K. J. Roux, D. I. Kim, M. Raida and B. Burke, A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells, J. Cell Biol., 2012, 196, 801–810 CrossRef CAS PubMed.
- D. I. Kim, et al., An improved smaller biotin ligase for BioID proximity labeling, Mol. Biol. Cell, 2016, 27, 1188–1196 CrossRef CAS PubMed.
- C. D. Go, et al., A proximity-dependent biotinylation map of a human cell, Nature, 2021, 595, 120–124 CrossRef CAS PubMed.
- A. Mair, S. L. Xu, T. C. Branon, A. Y. Ting and D. C. Bergmann, Proximity labeling of protein complexes and cell-type-specific organellar proteomes in Arabidopsis enabled by TurboID, eLife, 2019, 8, e47864 CrossRef CAS PubMed.
- K. F. Cho, et al., Proximity labeling in mammalian cells with TurboID and split-TurboID, Nat. Protoc., 2020, 15, 3971–3999 CrossRef CAS PubMed.
- J. Hwang, et al., A Golgi rhomboid protease Rbd2 recruits Cdc48 to cleave yeast SREBP, EMBO J., 2016, 35, 2332–2349 CrossRef CAS PubMed.
- A. C. Gingras, K. T. Abe and B. Raught, Getting to know the neighborhood: using proximity-dependent biotinylation to characterize protein complexes and map organelles, Curr. Opin. Chem. Biol., 2019, 48, 44–54 CrossRef CAS PubMed.
- N. Zuzow, et al., Mapping the mammalian ribosome quality control complex interactome using proximity labeling approaches, Mol. Biol. Cell, 2018, 29, 1258–1269 CrossRef CAS PubMed.
- B. T. Lobingier, et al., An Approach to Spatiotemporally Resolve Protein Interaction Networks in Living Cells, Cell, 2017, 169, 350–360 CrossRef CAS PubMed.
- W. Qin, K. F. Cho, P. E. Cavanagh and A. Y. Ting, Deciphering molecular interactions by proximity labeling, Nat. Methods, 2021, 18, 133–143 CrossRef CAS PubMed.
- J. V. Oakley, et al., Radius measurement via super-resolution microscopy enables the development of a variable radii proximity labeling platform, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2203027119 CrossRef CAS PubMed.
- M. G. Kang and H. W. Rhee, Molecular Spatiomics by Proximity Labeling, Acc. Chem. Res., 2022, 55, 1411–1422 CrossRef CAS PubMed.
- V. Dumrongprechachan, et al., Cell-type and subcellular compartment-specific APEX2 proximity labeling reveals activity-dependent nuclear proteome dynamics in the striatum, Nat. Commun., 2021, 12, 4855 CrossRef CAS PubMed.
- F. Chen, P. W. Tillberg and E. S. Boyden, Expansion microscopy, Science, 2015, 347, 543–548 CrossRef CAS PubMed.
- U. T. Wang, et al., Protein and lipid expansion microscopy with trypsin and tyramide signal amplification for 3D imaging, Sci. Rep., 2023, 13, 21922 CrossRef CAS PubMed.
- S. Truckenbrodt, et al., X10 expansion microscopy enables 25-nm resolution on conventional microscopes, EMBO Rep., 2018, 19, e45836 CrossRef PubMed.
- S. Truckenbrodt, C. Sommer, S. O. Rizzoli and J. G. Danzl, A practical guide to optimization in X10 expansion microscopy, Nat. Protoc., 2019, 14, 832–863 CrossRef CAS PubMed.
- H. E. Park, et al., Scalable and Isotropic Expansion of Tissues with Simply Tunable Expansion Ratio, Adv. Sci., 2019, 6, 1901673 CrossRef CAS PubMed.
- H. G. J. Damstra, et al., Visualizing cellular and tissue ultrastructure using Ten-fold Robust Expansion Microscopy (TREx), eLife, 2022, 11, e73775 CrossRef CAS PubMed.
- K. A. Saal, et al., Heat denaturation enables multicolor X10-STED microscopy, Sci. Rep., 2023, 13, 5366 CrossRef CAS PubMed.
- J. B. Chang, et al., Iterative expansion microscopy, Nat. Methods, 2017, 14, 593–599 CrossRef CAS PubMed.
- F. Chen, P. W. Tillberg and E. S. Boyden, Optical imaging. Expansion microscopy, Science, 2015, 347, 543–548 CrossRef CAS PubMed.
- X. Shi, et al., Label-retention expansion microscopy, J. Cell Biol., 2021, 220(9), e202105067 CrossRef CAS PubMed.
- P. W. Tillberg, et al., Protein-retention expansion microscopy of cells and tissues labeled using standard fluorescent proteins and antibodies, Nat. Biotechnol., 2016, 34, 987–992 CrossRef CAS PubMed.
- T. J. Chozinski, et al., Expansion microscopy with conventional antibodies and fluorescent proteins, Nat. Methods, 2016, 13, 485–488 CrossRef CAS PubMed.
- S. Park, Y. Zhuang and X. Shi, Label-Retention Expansion Microscopy (LR-ExM) Enables Super-Resolution Imaging and High-Efficiency Labeling, J. Vis. Exp., 2022, 188, e63793 Search PubMed.
- Y. Y. Zhuang and X. Y. Shi, Expansion microscopy: A chemical approach for super-resolution microscopy, Curr. Opin. Struct. Biol., 2023, 81, 102614 CrossRef CAS PubMed.
- V. Hung, et al., Spatially resolved proteomic mapping in living cells with the engineered peroxidase APEX2, Nat. Protoc., 2016, 11, 456–475 CrossRef CAS PubMed.
- D. U. Mick, et al., Proteomics of Primary Cilia by Proximity Labeling, Dev. Cell, 2015, 35, 497–512 CrossRef CAS PubMed.
- K. Chung, et al., Structural and molecular interrogation of intact biological systems, Nature, 2013, 497, 332–337 CrossRef CAS PubMed.
- L. Y. Bourguignon, Biochemical analysis of ligand-induced receptor patching and capping using a novel immunolactoperoxidase iodination technique, J. Cell Biol., 1979, 83, 649–656 CrossRef CAS PubMed.
- D. Szklarczyk, et al., STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets, Nucleic Acids Res., 2019, 47, D607–D613 CrossRef CAS PubMed.
- F. Chen, P. W. Tillberg and E. S. Boyden, Expansion microscopy, Science, 2015, 347, 543–548 CrossRef CAS PubMed.
- D. Gambarotto, et al., Imaging cellular ultrastructures using expansion microscopy (U-ExM), Nat. Methods, 2019, 16, 71–74 CrossRef CAS PubMed.
- I. Kubalová, et al., Prospects and limitations of expansion microscopy in chromatin ultrastructure determination, Chromosome Res., 2020, 28, 355–368 CrossRef PubMed.
- S. K. Saka, et al., Immuno-SABER enables highly multiplexed and amplified protein imaging in tissues, Nat. Biotechnol., 2019, 37, 1080–1090 CrossRef CAS PubMed.
- C. Yu, et al., Characterization of Dynamic UbR-Proteasome Subcomplexes by In vivo Cross-linking (X) Assisted Bimolecular Tandem Affinity Purification (XBAP) and Label-free Quantitation, Mol. Cell. Proteomics, 2016, 15, 2279–2292 CrossRef CAS PubMed.
- J. Cox and M. Mann, MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification, Nat. Biotechnol., 2008, 26, 1367–1372 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb00516c |
| ‡ Current Address: School of Criminal Justice and Criminalistics, California State University, Los Angeles; Los Angeles 90042, USA. |
| This journal is © The Royal Society of Chemistry 2024 |