
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDissolution of the Ti porous transport layer in proton exchange membrane water electrolyzers†
Junsic
Cho‡
 a,
Dong Hyun
Kim‡
a,
Min Wook
Noh
a,
Haesol
Kim
a,
Hong-Gyun
Oh
b,
Pilyoung
Lee
c,
Soobin
Yoon
c,
Wangyun
Won
d,
Young-June
Park
*c,
Ung
Lee
*efg and
Chang Hyuck
Choi
*ah
a,
Dong Hyun
Kim‡
a,
Min Wook
Noh
a,
Haesol
Kim
a,
Hong-Gyun
Oh
b,
Pilyoung
Lee
c,
Soobin
Yoon
c,
Wangyun
Won
d,
Young-June
Park
*c,
Ung
Lee
*efg and
Chang Hyuck
Choi
*ah
aDepartment of Chemistry, Pohang University of Science and Technology (POSTECH), Pohang 37673, Republic of Korea. E-mail: chchoi@postech.ac.kr
bGlobal Sales Department, Shinsung C&T, Suwon 16648, Republic of Korea
cHydrogen and Fuel Cell Development Center, Hyundai Motor Group, Yongin 16891, Republic of Korea. E-mail: yjpark2935@hyundai.com
dDepartment of Chemical and Biological Engineering, Korea University, 145, Seoul 02841, Republic of Korea
eClean Energy Research Center, Korea Institute of Science and Technology, Seoul 02792, Republic of Korea. E-mail: ulee@kist.re.kr
fGreen School, Korea University, 145 Anam-ro, Seongbuk-gu, Seoul 02841, Republic of Korea
gKIST Europe, Korea Institute of Science and Technology Europe, Campus E71, Saarbrücken 66123, Germany
hInstitute for Convergence Research and Education in Advanced Technology (I-CREATE), Yonsei University, Seoul 03722, Republic of Korea
First published on 30th July 2024
Abstract
The titanium porous transport layer (PTL) is a key component in proton exchange membrane water electrolyzers (PEMWEs), facilitating efficient water supply to the catalyst layer while rapidly removing oxygen bubbles. However, in the highly anodic operating environment of PEMWEs, the Ti PTL suffers from degradation, limiting the lifetime of the device. To gain deeper insights into Ti PTL degradation, here we monitor the potential/time-resolved Ti dissolution rates by coupling a PEMWE with an online inductively coupled plasma-mass spectrometer (ICP-MS). The results show that the dissolution of the Ti PTL is a complex and dynamic (electro)chemical event. Initiated by the decreased interfacial pH (even at pH < 1) due to proton accumulation during PEMWE operation, Ti dissolution intensifies with increasing bias potential. However, the dissolved Ti ions are simultaneously hydrolyzed, forming surface Ti oxides that slow down the dissolution rate. Coating the Ti PTL surface with Pt and IrO2 effectively reduces Ti dissolution, albeit at a higher cost, but they are also susceptible to dissolution during operation. Interestingly, the dissolution profiles of Pt and IrO2 deposited on the Ti PTL differ significantly from their conventional behavior, which requires further investigation for reliable prediction and optimization of new PTL designs for practical implementation in PEMWEs.
Introduction
In the quest for a sustainable, carbon-neutral future, there is a growing energy paradigm shift away from polluting fossil fuels toward a cleaner and more sustainable society.1 At the heart of this new energy economy is green hydrogen.2 A critical technology driving this transformation is the proton exchange membrane water electrolyzer (PEMWE),1 which produces hydrogen gas from abundant water with minimal, or ideally zero, greenhouse gas emissions. The PEMWE has demonstrated promising performance and rapid response to fluctuations in power supply, making it well-suited for integration with renewable energy sources. However, its harsh operating conditions (>2 V, temperature of 60–80 °C, and acidic environment) limit the choice of materials library that can be used in the PEMWE, as they must have high corrosion resistance.3,4Titanium offers relatively high corrosion resistance under such conditions, with high electrical conductivity and mechanical strength.5 Therefore, in addition to the bipolar plates, the porous transport layer (PTL), an essential component of the PEMWE for efficiently supplying the water flow to the catalyst layer while facilitating the removal of oxygen bubbles, is typically made of metallic Ti.6–10 Its remarkable corrosion resistance is due to the formation of a stable protective oxide layer (generally TiO2) on its surface when exposed to air or water. This layer is extremely adherent and has the ability to repair itself if damaged, providing robust protection against various corrosive environments. However, the poor electrical conductivity of the protective layer leads to an increase in the ohmic resistance between the PTL and other components of the PEMWE, which can significantly deteriorate the energy efficiency of the PEMWE.11,12 This drawback has prompted several strategies to ensure durable operation, including optimizing the PTL morphology and coating the metallic Ti surface with thin layers of noble metals (e.g., Pt, Ir, etc.).13
It should be noted, however, that titanium and even noble metals used to protect Ti are not completely immune to corrosion. Using a scanning flow cell (SFC) coupled to an inductively coupled plasma-mass spectrometer (ICP-MS), several research groups have investigated the potential-resolved electrochemical stability of Ti-containing mixed oxide catalysts (e.g., Ir–TiOx,14–16 Ir/CuTiONx,17 and RuIr–TiOx18) in 0.1 M HClO4 electrolyte. While these studies focused primarily on the dissolution of noble metal catalysts and the subsequent decline in their oxygen evolution reaction (OER) activity — and thus the results may not fully represent the behavior of the Ti PTL in the PEMWE — they showed appreciable Ti dissolution, following a trend similar to that of noble metal dissolution. Meanwhile, Rakousky et al. observed the presence of Tin+ species at the cathode in an aged catalyst-coated membrane (CCM) after >1000 h of conventional PEMWE operation (2 A cm−2 and 80 °C).12 Since the cathode in a pristine CCM was free of Ti and this element was only present in the anode catalyst (a mixture of IrO2 and TiO2) and Ti PTL, Ti dissolution under PEMWE operation was clearly identified. In addition, Ti crossover from the anode PTL to cathode was also observed in the PEMWE for H2O2 synthesis, where Ti felt was used as the PTL but the anode catalyst was only IrO2.19 Thus, the previous findings highlight the need for a deeper understanding of Ti PTL dissolution under PEMWE conditions in order to develop rational PTL designs and operation strategies to meet industrial requirements.
In this study, Ti PTL dissolution in the PEMWE was studied using an online ICP-MS, which allowed potential/time-resolved and quantitative monitoring of the Ti dissolution rate during electrochemical operation. We clearly identified non-negligible Ti dissolution, the rate of which is strongly influenced by the feedstock (or interfacial) pH and applied potential. On the other hand, dissolved Ti ions undergo hydrolysis and form a Ti oxide passivation layer, which suppresses the Ti dissolution rate. We investigated this complex event, during which dissolution and redeposition processes dynamically compete, under various potential profiles on commercialized Ti PTLs with and without Pt and IrO2 coating layers. Our findings highlight the necessity of similar or further modified diagnostics for better design and evaluation of PTLs to ensure their high durability during PEMWE operation.
Results and discussion
For the online monitoring of Ti dissolution from the PTL, we coupled a PEMWE cell with an ICP-MS (Fig. 1a). At the anode, the Ti PTL without an OER catalyst, typically Ir(Ox), was used as the electrode. On the other hand, at the cathode, a Pd/C catalyst rather than conventional Pt/C, which was coated on the gas-diffusion layer (GDL), was employed. It is of note that these distinct configurations of electrodes were due to the elimination of any interferences of Ir or Pt dissolution from the conventional catalytic substances for accurate investigation on Pt/IrO2-coated Ti PTLs that will be discussed later, and due to minimization of bubble formation that can disrupt stable operation of the online monitoring. The anode and cathode were separated by a Nafion membrane, which was extended out of the electrolyzer cell and connected with a saturated Ag/AgCl reference electrode. This unique cell configuration,20 beyond the conventional two-electrode system, provided excellent experimental reproducibility and allowed us to accurately control and monitor the anode (or cathode) potential in the electrochemical device (Fig. S1†). Heating rods were inserted into the back plates of the cell to heat the cell. During the measurement, feedstock was supplied at a constant flow rate of 5 mL min−1 on each electrode side, of which 400 μL min−1 was introduced into the ICP-MS (see details in the Experimental methods).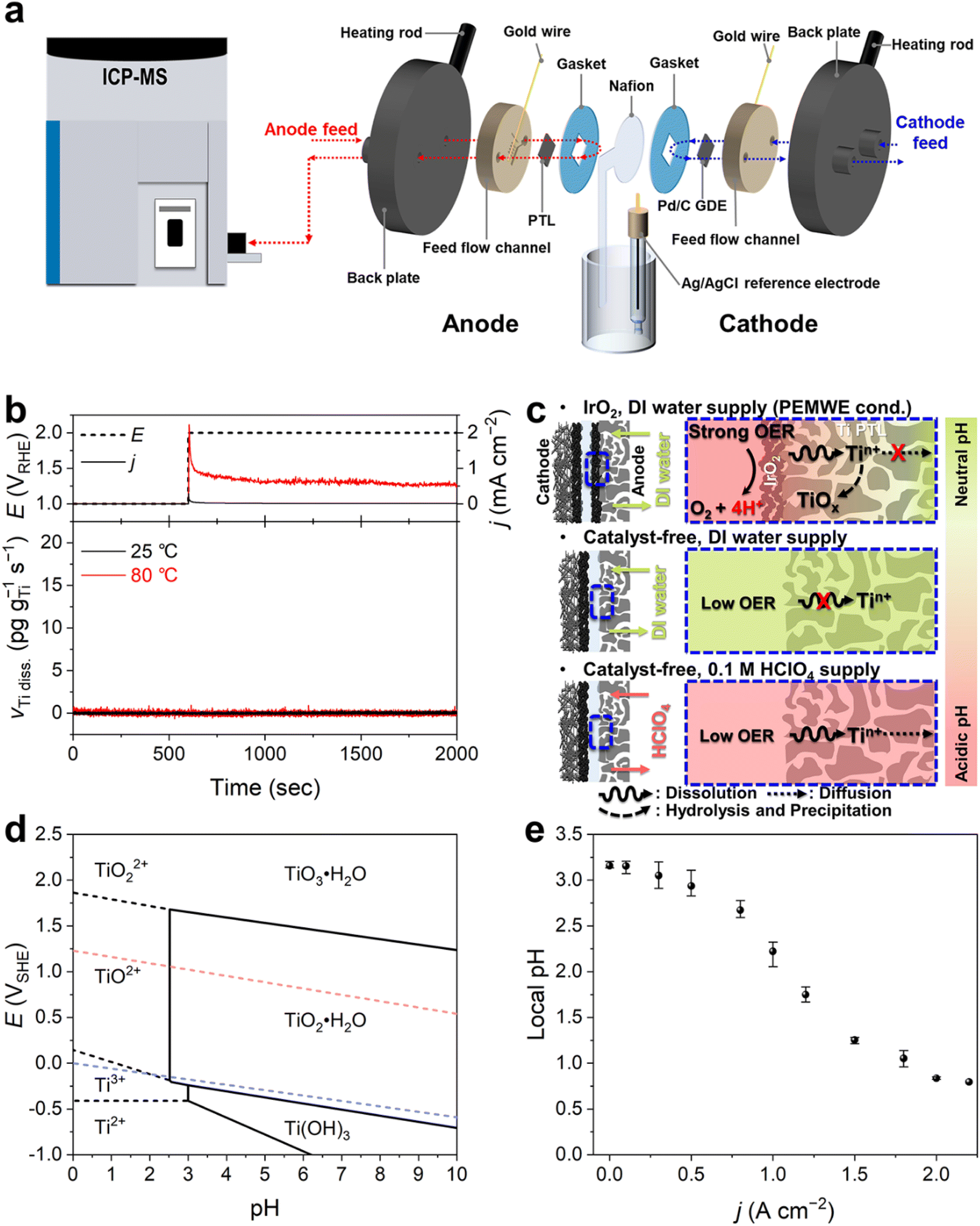 | ||
| Fig. 1 (a) Schematic representation of the online PEMWE/ICP-MS setup. (b) Real-time Ti dissolution measured at a static potential of 2 VRHE with a supply of O2-saturated DI water at 25 and 80 °C. (c) Schematic representation of Ti dissolution behavior under three different operating conditions: PEMWE operation (i.e., Ti PTL with a catalyst layer and DI water supply) and our model systems of catalyst-free Ti PTLs with DI water or 0.1 M HClO4 supply. (d) Pourbaix diagram of Ti. (e) Estimated local pHs at the electrode–membrane interface measured at different current densities. | ||
Ti dissolution was first investigated at 25 and 80 °C with a continuous flow of deionized (DI) water, a typical feedstock for commercialized PEMWEs, during the anode polarizations at 1 and 2 V vs. the reversible hydrogen electrode (RHE; Fig. 1b), which are in the relevant potential range for rest and OER operating potentials, respectively. However, at both temperatures, we found no discernible Ti dissolution during the operation. This result does not agree with previous findings that showed non-negligible Ti dissolution after PEMWE operation,12,21 thereby implying that our experimental conditions insufficiently reflect those in practical PEMWEs.
We envisioned that this discrepancy primarily originated from different H2O consumption and H+ production rates, leading to consequent differences in local pH at the PTL–electrolyte interface (Fig. 1c). In practical PEMWEs, ampere-order current density (j) is typically achieved for efficient evolution of O2 gas. We also confirmed that our system also reached j >2 A cm−2 when using the IrO2 catalyst layer (Fig. S2†). This leads to considerable H+ production at the anode and its possible accumulation at the interface, lowering the interfacial pH.22,23 However, in our system, the Ti PTL free from the anode catalyst resulted in a marginal j less than 1 mA cm−2 even at 2 VRHE (Fig. 1b), and therefore no considerable changes in the interfacial pH can be reasonably deduced. The importance of feedstock pH on the Ti dissolution can also be understood with the Pourbaix diagram of Ti (Fig. 1d), which shows that pH lower than approximately 2.5 is thermodynamically needed for the exergonic dissolution process of Ti to TiO2+ or TiO22+ at potential above 1 VRHE. It is of note that, at a given pH 7 of DI water, the most stabilized phase of Ti is solid TiO2·H2O or TiO3·H2O depending on the potential applied.24
To confirm this hypothesis, i.e., greater accumulation of H+ at the interface with increasing j, we estimated the current-dependent local pH changes using the method recently developed by Sauvé et al.,25 which follows a quasi-equilibrium potential between hydrogen oxidation and reduction reactions on Pt and estimates the interfacial pH (Fig. S3†). The result showed that even at zero current, the interface exhibited a slightly acidic nature (pH 3.2, Fig. 1e), due to the sulfonic acid group-terminated Nafion membrane.26,27 However, as the current increased, the pH decreased significantly, and it even reached below pH 1 when the j exceeded 1.8 A cm−2. Therefore, for subsequent online ICP-MS experiments, we opted to use a 0.1 M HClO4 solution instead of DI water as the feedstock. This choice was made to more accurately simulate the interfacial conditions of the real PEMWE without an anode catalyst and to avoid any chemical decomposition and precipitation of dissolved Ti ions into solid TiOx as they moved away from the diffusion layer before being analyzed by ICP-MS (Fig. 1c).
In this measurement, we monitored the dissolution rate of Ti in O2-saturated 0.1 M HClO4 during four consecutive potential protocols (Fig. 2). Protocol 1 entailed the potentiostatic operation of the PEMWE, with a potential hold of the Ti PTL at 2 VRHE for 30 min. Conversely, Protocols 2 and 3 involved potentiodynamic conditions, comprising three cyclic voltammograms (CVs) at a scan rate of 5 mV s−1 and 60 cycles of potential pulses of 10 s each within a potential range of 1–2 VRHE, respectively. Typically, the measurement was performed in the order of Protocols 1 → 2 → 3 (Fig. 2a), although the reverse order, Protocols 3 → 2 → 1 (Fig. 2b), was also evaluated. Protocol 4 was then implemented, which corresponded to potential rest, i.e., open circuit potential (OCP), for 40 min. Before and after each protocol, the Ti PTL was polarized at 1 VRHE to stabilize the ICP-MS signals.
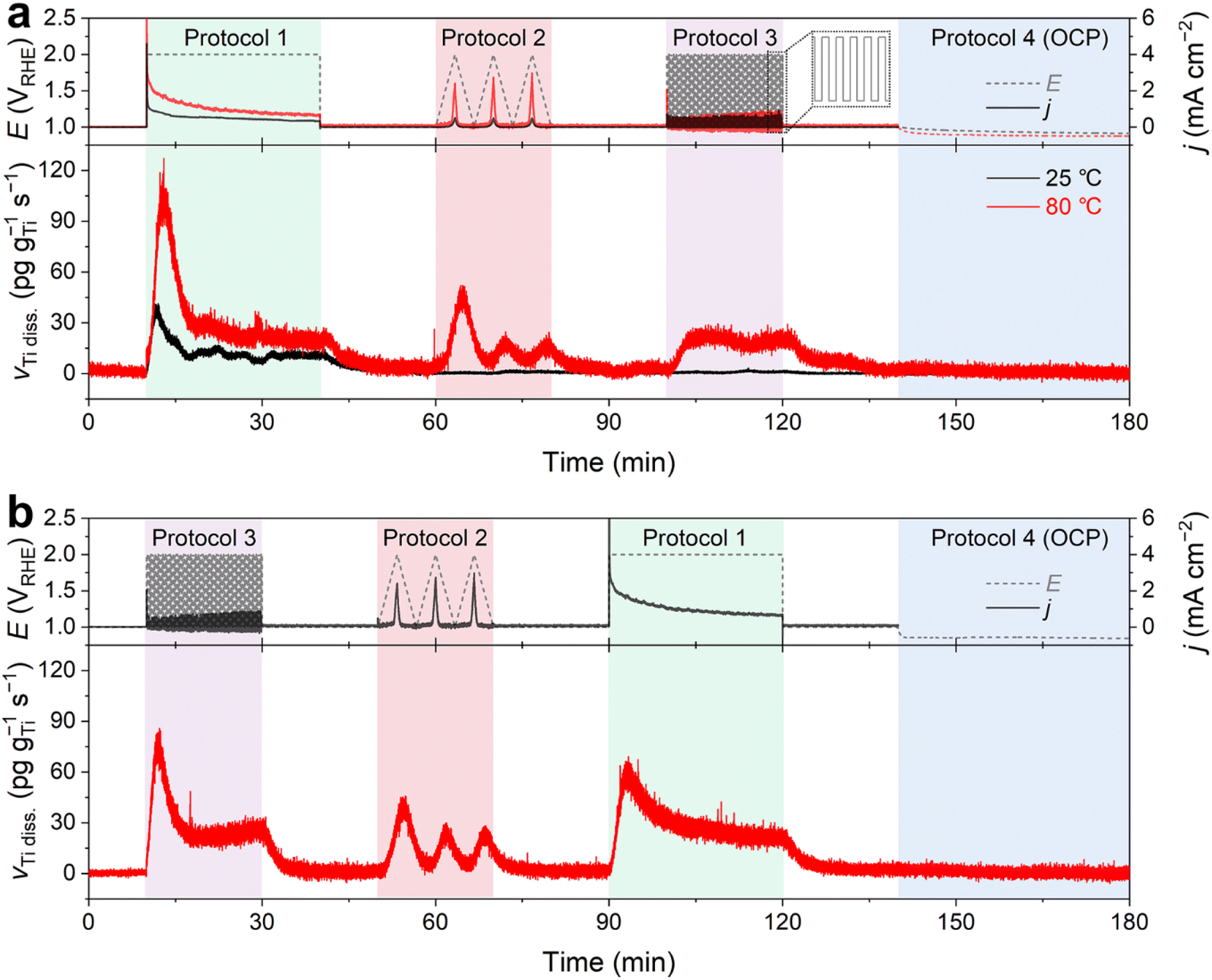 | ||
| Fig. 2 Real-time dissolution profiles of the Ti PTL measured during potential hold (Protocol 1), 3 cycles of CV (Protocol 2), and potential pulses (Protocol 3). For comparison, Ti dissolution was monitored in the order of (a) Protocols 1 → 2 → 3 at 25 and 80 °C and (b) Protocols 3 → 2 → 1 at 80 °C with a supply of O2-saturated 0.1 M HClO4 feedstock. At the end of the measurements, the Ti PTL was not externally biased but rested at OCP (Protocol 4). | ||
In contrast to the results observed with DI water (Fig. 1b and S4†), we observed significant Ti dissolution in the HClO4 feedstock. At 25 °C, significant Ti dissolution rate was found in Protocol 1, but it was almost negligible in the following Protocols 2–4. On the other hand, at 80 °C, a practically more relevant temperature, much greater dissolution of Ti was observed in Protocols 1–3 (Fig. 2a). In addition, it is noteworthy that the Ti dissolution signals were observed in both the anolyte and catholyte (Fig. S5†). This result indicates the crossover of Ti ions from the anode to the cathode, which is consistent with previous results measured after long-term operation of the PEMWE.12 However, the Ti signal measured in the anolyte was approximately 6 times higher than that in the catholyte, suggesting that the diffusion of dissolved Ti ions from the anode to the cathode was relatively marginal under our experimental conditions. Therefore, the following investigations focused solely on the dissolution of the Ti PTL in the anolyte at 80 °C.
In the standard protocol sequence (Fig. 2a), no perceptible Ti dissolution was detected at 1 VRHE. However, at the initial potential jump to 2 VRHE in Protocol 1, the Ti PTL exhibited substantial Ti dissolution of approximately 100 pg gTi−1 s−1. However, the intense Ti dissolution rapidly decreased within 500 seconds and stabilized at approximately 25 pg gTi−1 s−1 during the potential hold. Afterward, in Protocol 3, a consistent Ti dissolution rate was observed, quantitatively similar to the plateau observed in Protocol 1. However, the initial intense Ti dissolution, seen in Protocol 1, was not found in Protocol 3, while the Ti PTL underwent the same potential shift from 1 to 2 VRHE and experienced this drastic potential change repeatedly. On the other hand, in the reverse protocol sequence (Fig. 2b), the initially intense but subsequently attenuated Ti dissolution was also recorded in Protocol 3, when it preceded all other protocols. The same conclusion was reached when the potential excursion started with Protocol 2 (Fig. S6†). Therefore, these results implied that the most intense Ti dissolution occurs at the initial potential transition from 1 to 2 VRHE, but thereafter its dissolution rate becomes relatively mitigated regardless of either potentiostatic or pulsed operation at 2 VRHE.
It is generally accepted that the Ti surface can be protected against corrosion by forming stable passivation films, e.g., Ti oxide or hydroxide, even in acidic environments.28,29 However, the intense Ti dissolution at the first potential transition from 1 to 2 VRHE allowed us to consider either that the Ti PTL surface is not fully protected by the passivation film, but contains some pits or defective sites, or that the native passivation layer is only quasi-stable, resulting in considerable Ti dissolution from an exposed metallic Ti and/or quasi-stable Ti passivation layer under the highly anodic conditions of 2 VRHE (Fig. 3a). Nevertheless, the subsequent exponential decay of the Ti dissolution rate implies the hydrolysis of dissolved Ti ions at the interface and the formation of a relatively stable or thicker passivation layer on the Ti PTL, as also explained in the literature.28–30 Scanning electron microscopy (SEM) and energy-dispersive spectroscopy (EDS) of the Ti PTL revealed the increase in oxygen content after its polarization at 2 VRHE, corroborating the formation of an additional passivation layer (Fig. 3b and S7†). However, the marginal but consistent Ti dissolution during potential hold at 2 VRHE and potential pulses between 1 and 2 VRHE suggests that even the newly formed passivation layer may not be completely immune to corrosion, possibly because the thermodynamically most stable phase under such conditions is Ti ions (TiO2+ and TiO22+) rather than solid Ti oxides. Computational modeling based upon the above simple mechanistic scenario toward Ti dissolution also successfully describes the trend of experimental results (Fig. 3c and S8†), i.e., initially intensive but subsequently marginal Ti dissolution, while more complex reactions may need to be further considered for more elaborate prediction. We note that Ti dissolution likely occurs by electrochemical reactions,31 not chemical reactions29 since (1) no significant dissolution of Ti was observed at potential hold at 1 VRHE before and after the protocols and during potential rest at OCP and (2) the Ti dissolution rate increased with increasing potential as shown in Protocol 2.
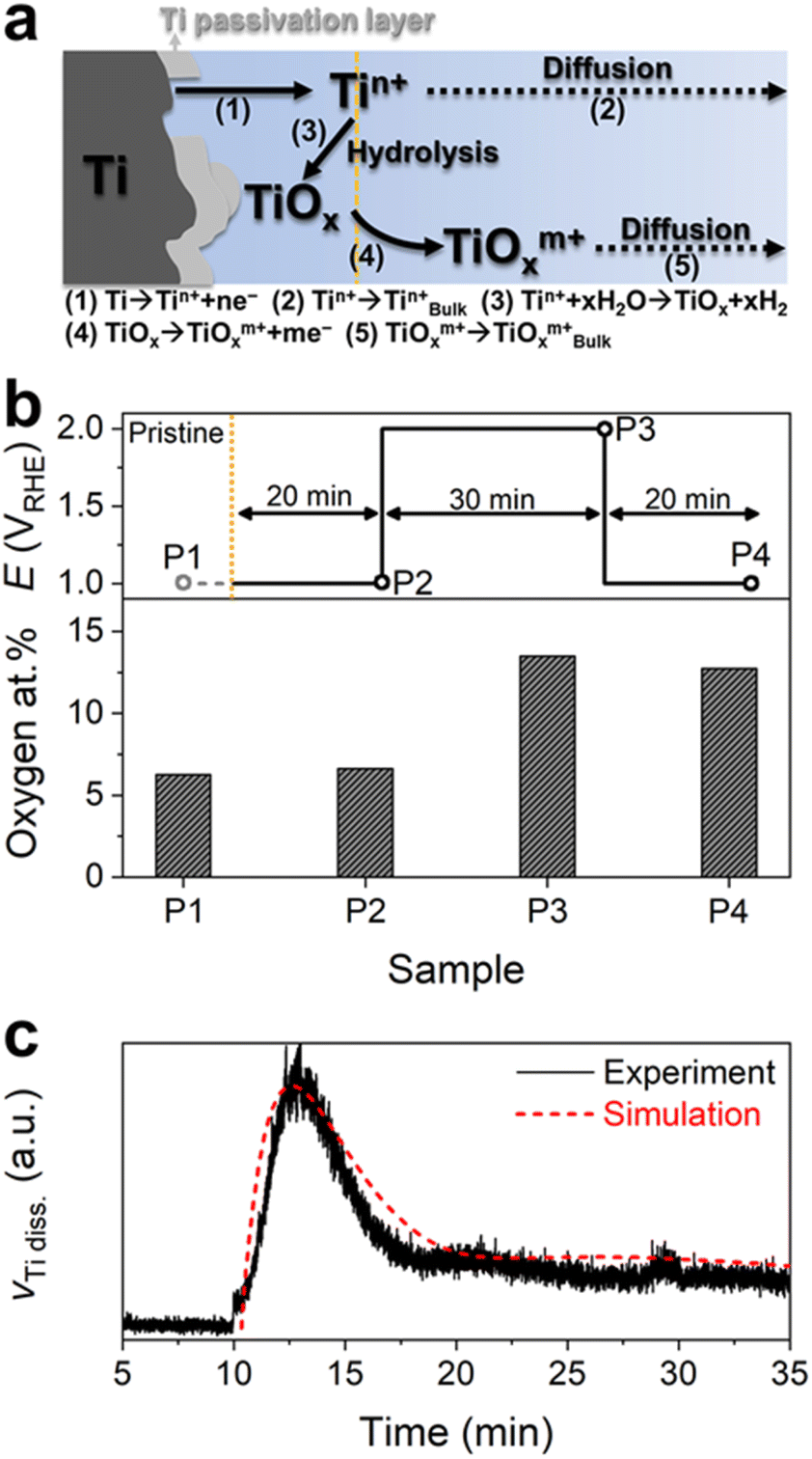 | ||
| Fig. 3 (a) Schematic representation of the expected Ti PTL dissolution mechanism under highly anodic conditions. The reaction equation of each step is also shown. (b) Oxygen atomic percentages on the Ti PTL before (P1; gray circle) and after specific potential application (P2, P3, and P4; black circles) in O2-saturated 0.1 M HClO4. Oxygen content was estimated by EDS analysis. (c) Comparison of experimental and simulated Ti dissolution profiles at 2 VRHE. | ||
After confirmation of possible Ti dissolution from the bare Ti PTL under PEMWE-relevant operating conditions, we investigated the Ti dissolution behavior of the Ti PTL onto which thin protecting layers of Pt and IrO2 were coated. These modified Ti PTLs are commonly utilized in the PEMWE to mitigate the formation of Ti oxide layers and consequently suppress the increase in contact resistance between the catalyst layer and the PTL.12,32–35 In this study, we employed commercially available Pt- and IrO2-coated Ti PTLs, fabricated on identical Ti PTL substrates, as those investigated in Fig. 2. SEM and EDS studies confirmed the homogeneous dispersion of Pt and IrO2 over the Ti PTLs (Fig. 4a and b), thickness of which was approximately 100 and 350 nm (Fig. S9†), respectively.
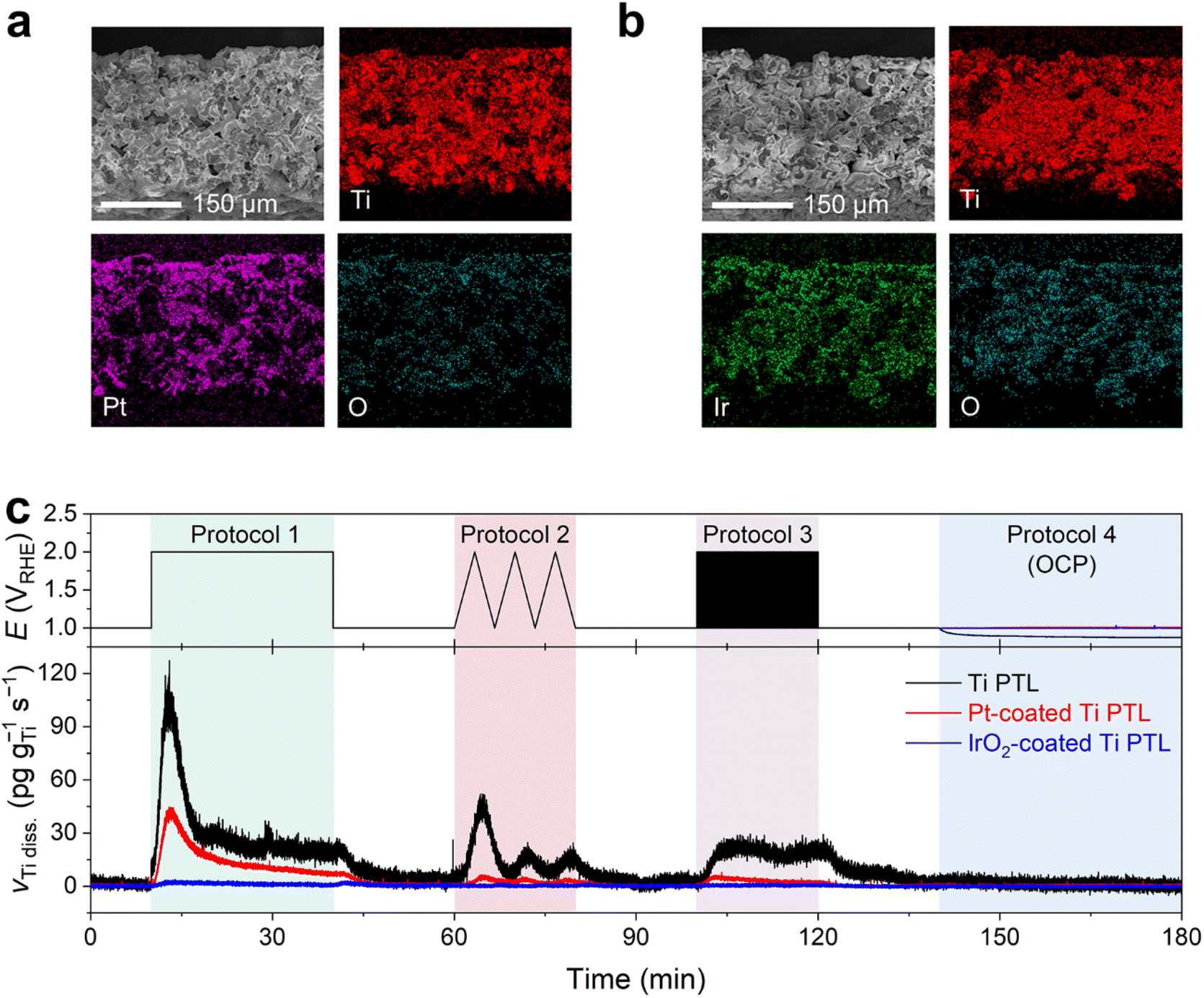 | ||
| Fig. 4 Cross-sectional SEM and EDS results of (a) Pt- and (b) IrO2-coated Ti PTLs. (c) Real-time Ti dissolution profiles of Pt/IrO2-coated Ti PTLs measured during potential hold (Protocol 1), 3 cycles of CV (Protocol 2), potential pulses (Protocol 3), and resting at OCP (Protocol 4) with a supply of O2-saturated 0.1 M HClO4 feedstock. | ||
Online ICP-MS results on Pt- and IrO2-coated Ti PTLs revealed a considerably reduced Ti dissolution rate compared to that of the bare Ti PTL (Fig. 4c; see S10† for the corresponding current densities). The total Ti dissolution amounts during the protocols were 125 ng gTi−1 for the bare Ti PTL, while Pt- and IrO2-coated Ti PTLs recorded only 39 and 4 ng gTi−1, respectively. Despite the effective protection provided by the IrO2 layer against Ti dissolution in our case, a direct comparison between Pt- and IrO2-coated Ti PTLs and the subsequent discussion on identifying a better protective layer may be less informative, as these modified Ti PTLs exhibit considerable differences in surface morphology (e.g., thickness of the protective layer; Fig. S9†) and noble metal content (Table S1†). More case studies with Pt/IrO2/or other protecting layer-coated Ti PTLs will be required to figure out the optimal composition and morphology of the protecting layer. However, at the very least, our results clearly demonstrate that the surface coating strategy is beneficial in mitigating undesirable Ti dissolution from Ti PTLs during PEMWE operation, in addition to the well-known electrical conductivity enhancement.
Our next question is then regarding the stability of the protecting layers during the operation, since even noble Pt and IrO2 are not immune to corrosion in such highly anodic environments.14–18,35–39
Fig. 5 displays Pt and Ir dissolution profiles measured upon the modified Ti PTLs during Protocols 1–4 (see Fig. S10† for the corresponding current densities). The results showed non-negligible dissolution of both protecting layers during the potential protocols. For the Pt-coated Ti PTL, Pt dissolution was found during potentiodynamic operation between 1 and 2 VRHE, but interestingly a significant Pt dissolution rate was also recorded during the potential hold at 1 VRHE that followed the end of each protocol. It is noteworthy that the Pt dissolution profile was different from that of conventional Pt particles deposited on a glassy carbon (GC) electrode, for which no considerable Pt dissolution was identified during the potential hold at 1 VRHE. Indeed, for the IrO2-coated Ti PTL, Ir dissolution occurred mainly during potentiodynamic operation, i.e., at the beginning and end of Protocol 1 and during Protocols 2 and 3, while conventional IrO2 showed much less Ir dissolution throughout the measurement except at the beginning of Protocol 1. Such fully different dissolution behaviors of Pt and IrO2 in Ti PTLs, compared to those of conventional Pt and IrO2, thus emphasize that the identity of the supporting electrode or substrate strongly influences the stability of Pt and IrO2, as also pointed out recently by Zlatar et al.40
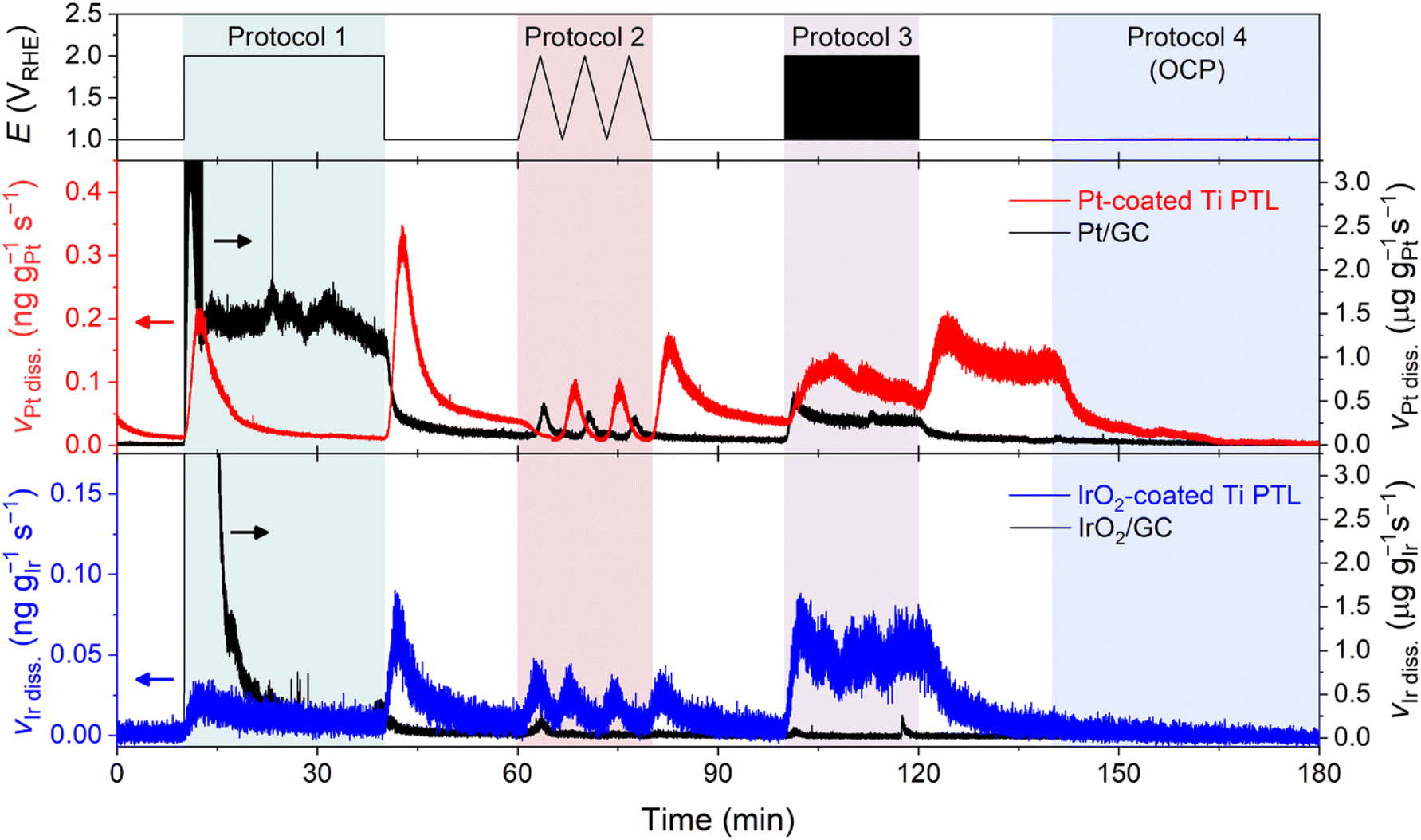 | ||
| Fig. 5 Real-time dissolution profiles of Pt and Ir from Pt/IrO2-coated Ti PTLs measured during potential hold (Protocol 1), 3 cycles of CV (Protocol 2), potential pulses (Protocol 3), and resting at OCP (Protocol 4) with a supply of O2-saturated 0.1 M HClO4 feedstock. For comparison, dissolution profiles of commercially available Pt and IrO2 nanoparticles deposited on a GC electrode were measured using an electrochemical flow cell at 25 °C. | ||
Conclusions
In summary, we have investigated the dissolution of Ti PTLs in a PEMWE by integrating it with an online ICP-MS. We have shown that Ti dissolution occurs when the potential exceeds 1 VRHE under both potentiostatic and potentiodynamic conditions. It is important to note, however, that our use of acidic 0.1 M HClO4 feedstock may exaggerate Ti dissolution rates compared to practical PEMWE operations, which typically use neutral DI water. In practical settings, neutral DI water facilitates rapid hydrolysis of dissolved Ti ions, resulting in the precipitation of TiOx and preventing the escape of Ti ions into the bulk feedstock (Fig. 1c). On the other hand, the use of 0.1 M HClO4 also places the entire Ti PTL in an acidic environment, which can cause the entire PTL surface to corrode. Despite this limitation, even limited Ti dissolution at the electrode–membrane interface, a region that can become highly acidic during high current operations, could significantly degrade PEMWE performance. In more detail, the dissolved Ti ions can precipitate on the anode and cathode as TiOx, blocking the active surface of catalyst layers and reducing the electrochemically active surface area.41 Since TiOx is an insulator, its accumulation can also lead to loss of electrical contact with active catalytic entities.11,12 Furthermore, the possible retention of dissolved Ti cations on the membrane through strong electrostatic interaction with anionic functional groups (e.g., –SO3− in Nafion) can hinder efficient proton transport and reduce cell performance.42,43 While protective layers on Ti PTLs, i.e., Pt and IrO2, can mitigate Ti dissolution to some extent, they can also dissolve under highly anodic conditions, and their dissolution behavior is significantly different from that of conventional Pt and IrO2. This discrepancy thus highlights the critical importance of stability measurements directly on practically relevant electrode materials, rather than on well-understood and relatively simple model substances, for accurate evaluation of their stability. In addition, while we have investigated three commercialized Ti PTLs in this study, a similar approach can be extended to other PTL electrodes with different protective layer loadings, morphologies, fabrication methods, etc., which may affect the dissolution rates of Ti and protective layers.44,45 Therefore, more comprehensive studies are needed to deepen our understanding and ultimately establish a better design of new Ti PTLs. Our results and the analytical platform used in this study lay the groundwork for such future research.Experimental methods
Online ICP-MS measurements
The anode was a commercial Ti PTL or Pt-/IrO2-modified Ti PTL, all provided by Shinsung C&T. The PTLs were tailored to 0.36 cm2 (0.6 cm × 0.6 cm) and directly introduced into the PEMWE without any OER catalysts. On the other hand, the cathode was a GDL (0.36 cm2), on which Pd/C (20 wt% Pd, Sigma-Aldrich) was spray-coated with a target loading of 1 mgcat. cm−2. To prepare the GDL, a highly hydrophobic carbon mesoporous layer (MPL) was deposited on a carbon paper (MGL280, AvCarb©) by spraying an ink, prepared by dispersing 100 mg of Ketjen black EC-300J (KB) and 200 mg of polytetrafluoroethylene (PTFE; 60 wt%, Sigma-Aldrich) in 20 mL of isopropyl alcohol (99.5%, Sigma-Aldrich). The KB content in the MPL was adjusted to 2 mg cm−2. The carbon paper was then sequentially heat-treated at 240 and 340 °C under an Ar flow for 30 min each.Online metal dissolution was analyzed using an ICP-MS (iCAP RQ, Thermo Fisher Scientific) coupled to a homemade PEMWE (Fig. 1a), which verified the comparable performance to other commercial PEMWEs when using a commercial membrane electrode assembly (MEA; Boyaz energy; anode – 4 mg cm−2 IrO2 and cathode – 0.5 mgPt cm−2 Pt/C) (Fig. S2†). The anode and cathode were separated by a Nafion 115 membrane (DuPont), which was extended from the PEMWE and connected to a saturated Ag/AgCl reference electrode to control the bias potential using a potentiostat (VMP3, Bio-Logic Science Inc).20 To avoid unwanted Ti dissolution from a conventional Ti bipolar plate, feed flow channels were fabricated with polyether ether ketone (PEEK). Since the PEEK is an electrical insulator, an Au wire was connected to each electrode to provide electrical contact. All these compartments were assembled in the following order — back plate, flow channel, Au wire, Ti PTL, gasket, Nafion, gasket, Pd/C GDE, Au wire, flow channel, and back plate (Fig. 1a) — and then tightened with six bolts to a torque of 18 Nm. Heating rods were inserted into the back plates of the PEMWE to control the cell temperature, and ICP-MS studies were performed at 25 and 80 °C.
The feedstock was either O2-saturated DI water (>18.2 MΩ, Arium® Pro, Sartorius) or 0.1 M HClO4 (diluted from 70% HClO4, Sigma-Aldrich), which flowed through each PEEK flow channel at a flow rate of 5 mL min−1 using a peristaltic pump (LEPP-150L, Labscitech). Prior to introduction of the feedstock into the ICP-MS, 400 μL min−1 of the outflow was separated and mixed with 0.5 M HNO3 containing 5 ppb 187Re as an internal standard at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixing ratio using a Y-connector. Online metal dissolution was estimated from the ratio of metal ions (48Ti, 193Ir, and 195Pt) to 187Re signals during electrochemical measurements.
:1 mixing ratio using a Y-connector. Online metal dissolution was estimated from the ratio of metal ions (48Ti, 193Ir, and 195Pt) to 187Re signals during electrochemical measurements.
In control experiments, the dissolution of Pt and IrO2 nanoparticles was investigated at 25 °C using a homemade electrochemical flow cell, which was used in our previous work.46 Briefly, the cell consisted of a U-shaped channel (1 mm diameter) and two openings (3 mm diameter). On one opening side, a mirror-polished 3 mm GC electrode (002012, ALS) was introduced as a working electrode, on which 50 μg cm−2 of Pt black (HiSPEC 1000, Alfa Aesar) or IrO2 powder (Alfa Aesar) was deposited by drop-casting of an ink, prepared by dispersing 10 mg of Pt black or IrO2 powder in a mixed solution of DI water (5894 μL), isopropyl alcohol (1079 μL), and 5 wt% Nafion solution (100 μL). On the other opening side, a 3 mm Teflon tube sealed at one end with a PTFE membrane (WP-020-80, Sumitomo Electric Ind., Ltd) was brought up to the working electrode surface to extract the evolved gases by vacuum. The counter electrode was a graphite rod separated from the electrolyte by a Nafion 115 membrane. The reference electrode was a saturated Ag/AgCl electrode connected directly to the electrolyte outlet.
Local pH measurements
The local pH change at the anode was measured by a method recently developed by Sauvé et al.,25 which monitors the OCP decay transient (potential of the H2/H+ equilibrium) on Pt. Our homemade PEMWE was used as the electrochemical platform, but a commercial Pt/C MEA (Boyaz energy) was installed. The MEA consisted of a Nafion 115 membrane, and Pt/C was deposited on both sides of the membrane with a loading of 0.5 mgPt cm−2. A GDL and a Pt-coated Ti PTL were attached to the anode and cathode sides, respectively. H2 gas, humidified at 50 °C, was introduced into the anode side at a flow rate of 200 sccm using a mass flow controller (M3030VA with an LTI1000 readout box, LineTech), and Ar-saturated DI water was introduced into the cathode side at a flow rate of 5 mL min−1. After stabilization of the potential signals at the specified j, which ranged from 0.0 to 2.2 A cm−2, the OCP decay transient was recorded with a data collection interval of 0.2 ms for 1 s. The local pH was calculated using eqn (1) with the OCP value measured at 20 ms after the potential transient to OCP.| Local pH = EOCP@20 ms (VSHE)/−0.070 (@80 °C) | (1) |
Physical characterization
SEM and EDS analyses were performed at an accelerating voltage of 5 and 15 kV, respectively, using a HITACHI S-4800. For cross-sectional SEM and EDS analyses, PTL samples were prepared by immersion in liquid N2 followed by cutting. The noble metal contents on the modified Ti PTLs were measured using an X-ray fluorescence spectrometer (SIMULTIX12; RIGAKU).Computational modeling
A computational modeling framework was developed using MATLAB to analyse the dynamic behaviour of Ti PTL dissolution, described by a system of conditional ordinary differential equations. These equations model the concentration changes of ions and predict the real-time Ti dissolution profile, thereby introducing nonlinear dynamics. The “ode45” solver in MATLAB 2023a was employed for its robust handling of both stiff and non-stiff ordinary differential equation systems, ensuring accurate simulations across varied parameter sets. To refine the model and align it with experimental observations, genetic algorithm optimization was utilized to adjust the kinetic parameters, aiming to minimize the discrepancy between the predictions and experimental data.Data availability
Data for this article are available at the Zenodo repository at https://doi.org/10.5281/zenodo.13117968.Author contributions
Junsic Cho: formal analysis, investigation, visualization, writing – original draft; Dong Hyun Kim: formal analysis, investigation, visualization, writing – original draft; Min Wook Noh: validation; Haesol Kim: methodology; Hong-Gyun Oh: resources; Pilyoung Lee: resources; Soobin Yoon: investigation; Wangyun Won: investigation; Young-June Park: conceptualization, project administration, funding acquisition; Ung Lee: formal analysis, data curation, writing – original draft; Chang Hyuck Choi: writing – original draft, supervision, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2022M3H4A3A01083536 and 2022K1A4A7A04095890). This research was also supported by the Hyundai Motor Group and Shinsung C&T.References
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2017, 16, 16–22 CrossRef
.
- S. E. Hosseini and M. A. Wahid, Renewable Sustainable Energy Rev., 2016, 57, 850–866 CrossRef CAS
- H. Jin, B. Ruqia, Y. Park, H. J. Kim, H.-S. Oh, S.-I. Choi and K. Lee, Adv. Energy Mater., 2021, 11, 2003188 CrossRef CAS
- M. Bonanno, K. Müller, B. Bensmann, R. Hanke-Rauschenbach, D. Aili, T. Franken, A. Chromik, R. Peach, A. T. S. Freiberg and S. Thiele, Renewable Sustainable Energy Rev., 2024, 9, 2300281 CAS
- N. D. Tomashov, R. M. Altovsky and G. P. Chernova, J. Electrochem. Soc., 1961, 108, 113 CrossRef CAS
- O. Panchenko, E. Borgardt, W. Zwaygardt, F. J. Hackemüller, M. Bram, N. Kardjilov, T. Arlt, I. Manke, M. Müller, D. Stolten and W. Lehnert, J. Power Sources, 2018, 390, 108–115 CrossRef CAS
- J. K. Lee, C. H. Lee and A. Bazylak, J. Power Sources, 2019, 437, 226910 CrossRef CAS
- T. Schuler, J. M. Ciccone, B. Krentscher, F. Marone, C. Peter, T. J. Schmidt and F. N. Büchi, Adv. Energy Mater., 2020, 10, 1903216 CrossRef CAS
- T. L. Doan, H. E. Lee, S. S. H. Shah, M. Kim, C.-H. Kim, H.-S. Cho and T. Kim, Int. J. Energy Res., 2021, 45, 14207–14220 CrossRef CAS
- D. H. Jeon, S. Kim, M. Kim, C. Lee and H.-S. Cho, J. Power Sources, 2023, 553, 232322 CrossRef CAS
- T. Srour, K. Kumar, V. Martin, L. Dubau, F. Maillard, B. Gilles, J. Dillet, S. Didierjean, B. Amoury, T. D. Le and G. Maranzana, Int. J. Hydrogen Energy, 2024, 58, 351–361 CrossRef CAS
- C. Rakousky, U. Reimer, K. Wippermann, M. Carmo, W. Lueke and D. Stolten, J. Power Sources, 2016, 326, 120–128 CrossRef CAS
- A. Lim, H.-Y. Jeong, Y. Lim, J. Y. Kim, H. Y. Park, J. H. Jang, Y.-E. Sung, J. M. Kim and H. S. Park, Sci. Adv., 2021, 7, eabf7866 CrossRef CAS PubMed
- S. Cherevko, T. Reier, A. R. Zeradjanin, Z. Pawolek, P. Strasser and K. J. J. Mayrhofer, Electrochem. Commun., 2014, 48, 81–85 CrossRef CAS
- M. van der Merwe, R. Garcia-Diez, L. Lahn, R. E. Wibowo, J. Frisch, M. Gorgoi, W. Yang, S. Ueda, R. G. Wilks, O. Kasian and M. Bär, ACS Catal., 2023, 13, 15427–15438 CrossRef CAS
- O. Kasian, T. Li, A. M. Mingers, K. Schweinar, A. Savan, A. Ludwig and K. Mayrhofer, J. Phys.: Energy, 2021, 3, 034006 CAS
- A. Lončar, D. Escalera-López, F. Ruiz-Zepeda, A. Hrnjić, M. Šala, P. Jovanovič, M. Bele, S. Cherevko and N. Hodnik, ACS Catal., 2021, 11, 12510–12519 CrossRef
- L. Lahn, A. M. Mingers, A. Savan, A. Ludwig and O. Kasian, ChemElectroChem, 2024, 11, e202300399 CrossRef CAS
- J.-P. B. Haraldsted, Z. Révay, R. Frydendal, A. Verdaguer-Casadevall, J. Rossmeisl, J. Kibsgaard and I. Chorkendorff, Mater. Today Energy, 2019, 14, 100352 CrossRef
- Q. Xu, S. Z. Oener, G. Lindquist, H. Jiang, C. Li and S. W. Boettcher, ACS Energy Lett., 2021, 6, 305–312 CrossRef CAS
- C. Rakousky, U. Reimer, K. Wippermann, S. Kuhri, M. Carmo, W. Lueke and D. Stolten, J. Power Sources, 2017, 342, 38–47 CrossRef CAS
- J. C. Fornaciari, L.-C. Weng, S. M. Alia, C. Zhan, T. A. Pham, A. T. Bell, T. Ogitsu, N. Danilovic and A. Z. Weber, Electrochim. Acta, 2022, 405, 139810 CrossRef CAS
- S. Liu, I. Zaharieva, L. D'Amario, S. Mebs, P. Kubella, F. Yang, P. Beyer, M. Haumann and H. Dau, Adv. Energy Mater., 2022, 12, 2202914 CrossRef CAS
-
M. Pourbaix, Atlas of Electrochemical Equilibria in Aqueous Solutions, National Association of Corrosion Engineers, 1974 Search PubMed
- E. R. Sauvé, B. Y. Tang, N. K. Razdan, W. L. Toh, S. Weng and Y. Surendranath, Joule, 2024, 8, 728–745 CrossRef
- V. Kozlov, N. Bunkin, I. Ps, A. Shkirin, S. Zakharov and Z. Aa, Water, 2013, 4, 129–154 Search PubMed
- S. A. Berlinger, B. D. McCloskey and A. Z. Weber, J. Phys. Chem. B, 2018, 122, 7790–7796 CrossRef CAS PubMed
- Y. Fovet, J.-Y. Gal and F. Toumelin-Chemla, Talanta, 2001, 53, 1053–1063 CrossRef CAS PubMed
- J.-F. Vanhumbeeck and J. Proost, Corros. Rev., 2009, 27, 117–204 CAS
- L. Fiedler, T.-C. Ma, B. Fritsch, J. H. Risse, M. Lechner, D. Dworschak, M. Merklein, K. J. J. Mayrhofer and A. Hutzler, ChemElectroChem, 2023, 10, e202300373 CrossRef CAS
-
R. Srinivasan and F. Fasmin, An Introduction to Electrochemical Impedance Spectroscopy, CRC Press, 2021 Search PubMed
- C. Liu, M. Carmo, G. Bender, A. Everwand, T. Lickert, J. L. Young, T. Smolinka, D. Stolten and W. Lehnert, Electrochem. Commun., 2018, 97, 96–99 CrossRef CAS
- C. Liu, M. Shviro, A. S. Gago, S. F. Zaccarine, G. Bender, P. Gazdzicki, T. Morawietz, I. Biswas, M. Rasinski, A. Everwand, R. Schierholz, J. Pfeilsticker, M. Müller, P. P. Lopes, R.-A. Eichel, B. Pivovar, S. Pylypenko, K. A. Friedrich, W. Lehnert and M. Carmo, Adv. Energy Mater., 2021, 11, 2002926 CrossRef CAS
- T. L. Doan, T. N. Nguyen, Y. S. Jung, C. Lee, M. Kim, S. Lee, H.-S. Cho and T. Kim, Int. J. Hydrogen Energy, 2024, 55, 839–847 CrossRef CAS
- C. Rakousky, G. P. Keeley, K. Wippermann, M. Carmo and D. Stolten, Electrochim. Acta, 2018, 278, 324–331 CrossRef CAS
- S. Cherevko, S. Geiger, O. Kasian, A. Mingers and K. J. J. Mayrhofer, J. Electroanal. Chem., 2016, 774, 102–110 CrossRef CAS
- O. Kasian, J.-P. Grote, S. Geiger, S. Cherevko and K. J. J. Mayrhofer, Angew. Chem., Int. Ed., 2018, 57, 2488–2491 CrossRef CAS PubMed
- S. Cherevko, A. A. Topalov, A. R. Zeradjanin, G. P. Keeley and K. J. J. Mayrhofer, Electrocatalysis, 2014, 5, 235–240 CrossRef CAS
- A. A. Topalov, S. Cherevko, A. R. Zeradjanin, J. C. Meier, I. Katsounaros and K. J. J. Mayrhofer, Chem. Sci., 2014, 5, 631–638 RSC
- M. Zlatar, D. Escalera-López, M. G. Rodríguez, T. Hrbek, C. Götz, R. Mary Joy, A. Savan, H. P. Tran, H. N. Nong, P. Pobedinskas, V. Briega-Martos, A. Hutzler, T. Böhm, K. Haenen, A. Ludwig, I. Khalakhan, P. Strasser and S. Cherevko, ACS Catal., 2023, 13, 15375–15392 CrossRef CAS
- S. Cherevko, Curr. Opin. Electrochem., 2018, 8, 118–125 CrossRef CAS
- S. A. Grigoriev, D. G. Bessarabov and A. S. Glukhov, Russ. J. Electrochem., 2017, 53, 808–812 CrossRef CAS
- Q. Feng, X. Z. Yuan, G. Liu, B. Wei, Z. Zhang, H. Li and H. Wang, J. Power Sources, 2017, 366, 33–55 CrossRef CAS
- C. Liu, K. Wippermann, M. Rasinski, Y. Suo, M. Shviro, M. Carmo and W. Lehnert, ACS Appl. Mater. Interfaces, 2021, 13, 16182–16196 CrossRef CAS PubMed
- C. Liu, J. A. Wrubel, E. Padgett and G. Bender, Appl. Energy, 2024, 356, 122274 CrossRef CAS
- J. Cho, T. Lim, H. Kim, L. Meng, J. Kim, S. Lee, J. H. Lee, G. Y. Jung, K.-S. Lee, F. Viñes, F. Illas, K. S. Exner, S. H. Joo and C. H. Choi, Nat. Commun., 2023, 14, 3233 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta02755h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |