
Decoupling Li+ conductivity and mechanical stability in a thermally reversible concentrated sulfone-based gel electrolyte for lithium metal batteries†
Xueao
Jiang
a,
Jiayin
Chen
a,
Junfeng
Zeng
a,
Weijian
Liu
a,
Xuansheng
Wu
a,
Yang
Lv
a,
Fangyan
Liu
b,
Ce
Zhang
c,
Zhiyong
Li
 d,
Xiwen
Wang
*a and
Shiguo
Zhang
*a
d,
Xiwen
Wang
*a and
Shiguo
Zhang
*a
aState Key Laboratory of Advanced Design and Manufacturing Technology for Vehicle, College of Materials Science and Engineering, Hunan University, Changsha 410082, P. R. China. E-mail: wangxw@hnu.edu.cn; zhangsg@hnu.edu.cn
bDepartment of Physics, City University of Hong Kong, Hong Kong 999077, Hong Kong
cNanophotonics and Optoelectronics Research Center, Qian Xuesen Laboratory of Space Technology, China Academy of Space Technology, Beijing 100094, China
dSchool of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang, Henan 453007, P. R. China
First published on 18th July 2024
Abstract
Increasing the liquid content has been recognized as an effective strategy to enhance the Li+ conductivity and electrode compatibility of gel electrolytes for lithium-metal batteries (LMBs). However, a low gelator content reduces the mechanical strength of gels. Herein, a mechanically stable gel electrolyte comprising 7 wt% methylcellulose (MC), 3 wt% 12-hydroxyoctadecanoic acid (12-HOA), and 90 wt% concentrated sulfone electrolyte (CSE) was developed. Because of its high electrolyte uptake, the MC/12-HOA gel electrolyte exhibits an ionic conductivity of 0.25 mS cm−1, which is similar to that of the neat CSE, and a Li+ transference number of 0.57, which is even higher than that of the CSE. Abundant ether groups on MC and hydroxyl groups on 12-HOA can strongly immobilize TFSI−via a hydrogen bond, endowing the MC/12-HOA gel with an elastic modulus of 10 MPa enough to form an 80 μm – thick self-supporting film and suppress Li dendrite growth. Additionally, a reversible gel–sol transition occurs in the MC/12-HOA gel at 329–331 K, which can be utilized to promote electrode wetting and seal the damages of electrolytes. As a result, the MC/12-HOA gel electrolyte displays reliable safety, practical flexibility, and suitable electrochemical performance in both the Li‖LiFePO4 and Li‖LiNi0.8Co0.1Mn0.1O2 cells, suggesting the great promise of the MC/12-HOA scaffold in gel electrolytes with different solvents for a wide range of applications in flexible devices.
Introduction
Rechargeable lithium-ion batteries (LIBs) have already overwhelmed the current market of portable electronics and the electric market with its flagship layered-structured metal oxide cathode and graphite anode. As an indispensable component of LIBs, their electrolytes always rely on flammable, toxic, and volatile organic solvents, which pose safety concerns such as leakage, fire, and explosion.1,2 To pursue higher energy density in batteries, Li metal batteries (LMBs) have become the hot spot in both the academic and industrial communities owing to the ultrahigh theoretical capacity (3860 mA h g−1) and lowest redox potential (−3.04 V versus the standard hydrogen electrode) of Li metal anodes.3 However, the Li dendrite growth in the liquid electrolyte aggravates safety issues.4,5 Thus, to fundamentally address these safety problems, it is crucial to develop nonflammable solid-state electrolytes (SSEs) to replace liquid electrolytes. Unfortunately, inorganic and polymer SSEs suffer from a poor electrode/electrolyte contact.6One of the most promising approaches to concurrently realize high safety and sufficient interface contact in LMBs is using flexible gel electrolytes (GEs).7 It features a relatively high volume fraction of a liquid electrolyte supported by a polymer scaffold (gelator). To meet the high safety requirements of LMBs, the liquid components of GEs are usually ionic liquid (IL) electrolytes or concentrated electrolytes, which possess nonflammability, low volatility, high thermal stability, and a wide electrochemical window.8–11 Furthermore, the high-energy density LMBs call for GEs with high room-temperature ionic conductivity, although the ionic conductivity of electrolytes is much higher than that of SSEs. Because of low gelator content, Li+ motion in GEs bears a close resemblance to that in the corresponding liquid organic electrolyte.1 Thus, there are two strategies to achieve appropriately high ionic conductivity and lithium-ion transference number of GEs: one is to improve the Li+ transportation properties of liquid electrolytes in GEs, and the other is to increase the liquid electrolyte content in GEs. However, excess electrolyte uptake, namely the higher concentration of mobile counterions, will result in the swelling of the polymer scaffold, which dramatically weakens the mechanical strength and finally destroys the polymer gel network.12–14 To balance the Li+ conductivity and mechanical strength of GEs with IL or concentrated electrolytes, their gelator contents have been controlled in the range of 20–40 wt%.15–17 Thus, it is urgent to solve the dilemma that requires the sacrifice of ionic conductivity in exchange for the strength of GEs.
As a component that is equally important to liquid electrolytes in GEs, a gelator should have a high intrinsic mechanical strength, high porosity, and a high liquid absorption rate to absorb as much electrolytes as possible. As inexhaustible renewable organic polymers in nature, cellulose fibrils have abundant functional groups, which interact with the electrolyte components to enhance the dissociation of Li salts, promote the electrolyte uptake, and reinforce the mechanical properties of GEs.18–20 In addition, the very high aspect ratio (>100) and high modulus (88–110 GPa) of cellulose fibrils endow the GEs with mechanically strong interwoven networks at a low concentration of gelator.21,22 Because of these merits, the cellulose fibril scaffold could uptake over 300 wt% conventional LIB electrolytes (1 M LiPF6 in carbonates), without the swelling phenomenon.23–25 Although the ionic liquid or concentrated electrolytes have physicochemical properties differing from the dilute carbonate electrolytes or aqueous solutions, it is promising that a trace amount of cellulose could “solidify” abundant quantities of liquids. However, a polymer gel containing cellulose and IL/concentrated electrolytes has yet to be explored for advanced LMBs.
In this work, we designed highly Li+-conductive, mechanically reliable, and thermally reversible polymer GEs using a lithium bis(trifluoromethanesulfonyl)imide (LiTFSI)/sulfone (SL) concentrated electrolyte, MC, and a 12-hydroxy octadecanoic acid (12-HOA) gelator, as illustrated in Fig. 1. Compared to other aprotic solvents, the sulfone shows nonflammability and high anodic stability (>5.0 V vs. Li+/Li).26 Meanwhile, dissolving a high concentration (>3 M) of Li salts in the SL significantly suppresses the reduction of SL on the Li metal.27 Thus, concentrated SL electrolytes have been widely studied in the high-voltage LMBs.28 Through the hydrogen bonds and van der Waals forces, the gelation of concentrated LiTFSI/SL electrolytes (CSEs) could be achieved with a low concentration of 7 wt% MC and 3 wt% 12-HOA. The as-prepared concentrated SL-based GE (M/H-GE) showed a very similar ionic conductivity, Li+ transference number (tLi+), and electrochemical window to the neat CSE due to its ultrahigh liquid electrolyte content. Meanwhile, the elastic modulus of M/H-GE is high enough to support itself to produce an 80 μm – thick membrane, which shows no swelling and no liquid leakage during the mechanical tests. Moreover, the M/H-GE undergoes a fast and reversible gel–sol transition at a relatively low temperature, which induces sufficient wetting of electrodes and self-healing of GEs. Because of these desirable properties, a high capacity, high coulombic efficiency, excellent cycling stability, and good reliability against abuse can be realized in the quasi-solid-state Li‖Li symmetric and LiFePO4 (LFP)‖Li LMBs with the M/H-GE.
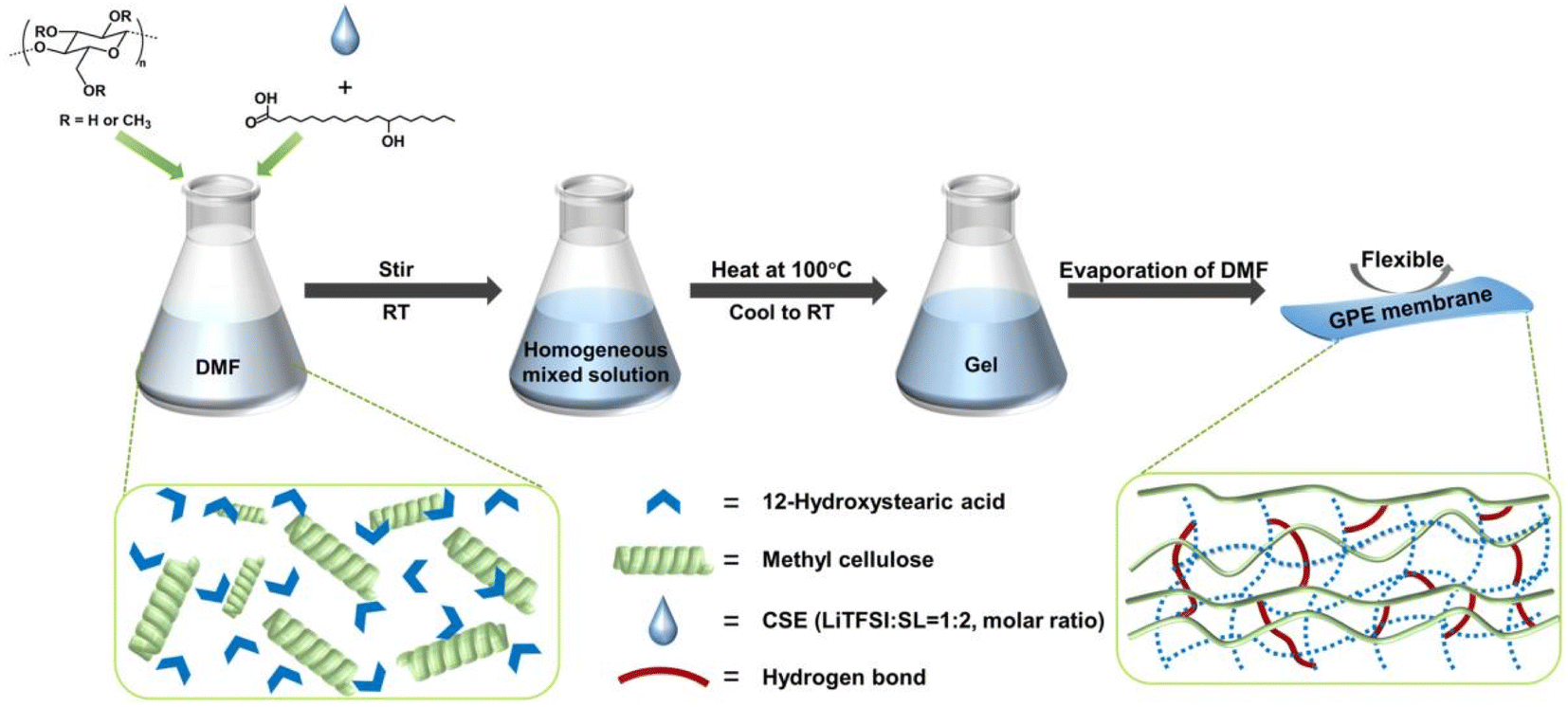 | ||
Fig. 1 Schematic illustration of the preparation of the M/H-GE membrane using MC, 12-HOA, and CSE (LiTFSI![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :SL = 1:2). :SL = 1:2). | ||
Results and discussion
Before the preparation of GEs, the concentrated sulfone electrolyte was synthesized by dissolving a LiTFSI salt in the sulfone in a molar ratio of 1/2. Since the oxygen coordination number in the first ion solvation shell of Li+ was 4.0, all the sulfone solvents were solvated by Li+ to generate a well-known tetrahedral solvation arrangement.29 The elimination of free sulfone solvents will greatly enhance the cathodic and anodic stability of electrolytes. In this CSE, Li+ exchanges the ligands (sulfone and TFSI−) rapidly to cause fast Li-ion hopping conduction, which could provide high tLi+ under anion-blocking conditions.30 Then, the CSE, MC, and 12-HOA were dissolved in dimethylformamide (DMF), and the thermogelation was finished after several heating/cooling processes. After the evaporation of DMF, a M/H-GE membrane can be obtained (Fig. 1).As shown in Fig. S1,† the GE membranes using the MC/12-HOA (M/H) gelator are self-supporting, transparent, and smooth. The SEM images further confirm the surface morphology and thickness of GE membranes (Fig. 2a–d and S2†). All the GE membranes are flat, which ensures sufficient contact of electrolytes with the Li foil. To achieve an areal conductance close to that of a liquid electrolyte using a 25 μm separator, the reduction in the thickness is equally important to the increase in the ionic conductivity of GEs. Moreover, the cell-level energy density of solid-state LMBs is inversely related to the thickness of GEs.31 Notably, the thickness of the M/H-GE membrane is about 80 μm, while that of the GE membrane using 10 wt% MC gelator alone (M-GE) increases to 205 μm. Even if the MC gelator content increases to 15 wt%, the thickness of the GE membrane remains 151 μm. Thus, the introduction of 12-HOA in the MC gelator leads to a significant decrease in the thickness of the electrolyte, without inducing the risk of membrane fracture.
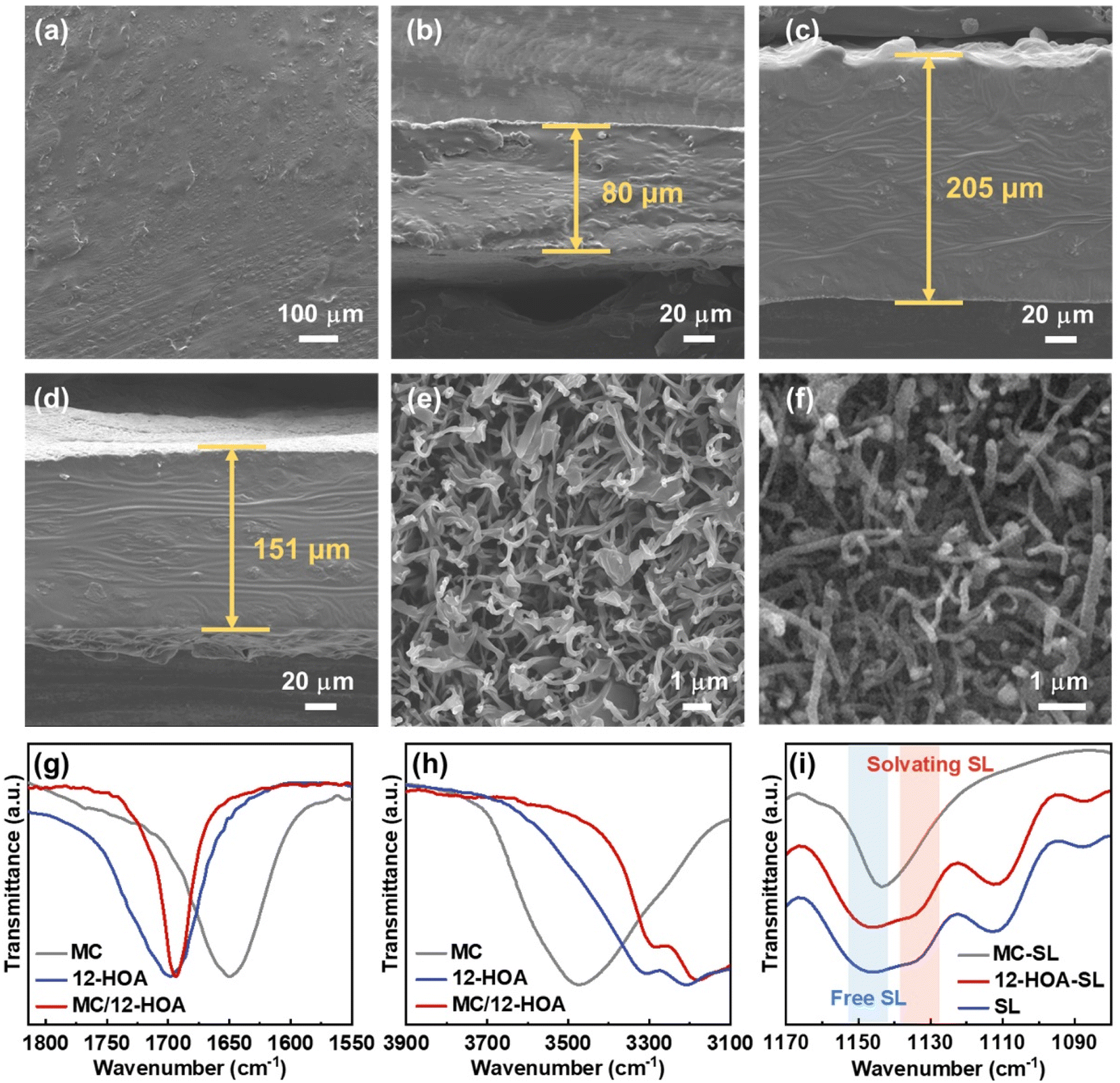 | ||
| Fig. 2 Morphology and chemical structure of GE membranes: (a) top-view SEM image of the M/H-GE membrane. Cross-sectional SEM images of (b) the M/H-GE membrane, (c) GE membrane using a 10 wt% MC gelator and (d) GE membrane using a 15 wt% MC gelator. Top-view SEM images of (e) the M/H polymer scaffold and (f) pure MC polymer scaffold. FTIR spectra of the MC, 12-HOA, and M/H polymer scaffold at wavenumbers of (g) 1550–1825 cm−1 and (h) 3100–3900 cm−1. (i) FTIR spectra of the M-GE, H-GE, and M/H-GE at wavenumbers of 1080–1170 cm−1. | ||
To detect the effect of the 12-HOA gelator on the thickness of GE membranes, the microstructure of dried gels derived from GEs was investigated. After the freeze-drying of the CSE, we obtained the MC and M/H polymer scaffold. Both the polymer scaffolds show tightly interwoven network structures with fibrils in the diameters of 100–150 nm in their corresponding SEM images (Fig. 2e and f). This interconnected network provides abundant large pores formed by the space between fibrils, which encapsulate the CSE and function as the Li+ motion pathway in GEs. Based on the results from the weighting method, the porosity of the MC and M/H polymer scaffold was 93.7% and 96.1%, respectively. Thus, there is almost no difference between the microstructures of the MC and M/H polymer scaffold. Therefore, intermolecular interaction between the MC and 12-HOA may play a more vital role in sustaining the structure of thin M/H-GE membranes.
The chemical structures of the MC, 12-HOA, and M/H polymer scaffold were further evaluated by Fourier transform infrared (FTIR) spectra. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration band at about 1700 cm−1 in the FTIR spectra of the 12-HOA and M/H sample results from the formation of the COOH dimer via a hydrogen bond, similar to the supramolecular ionogel with it (Fig. 2g).16,32 Considering that the symmetric stretching vibration of the anhydride is located at 1780 cm−1, no reaction occurs between the –COOH group of 12-HOA and the –OH group of MC in the M/H polymer scaffold.33 Meanwhile, the intensity of the O–H stretching vibration bands (3350 cm−1) decreased when the MC was incorporated with 12-HOA, indicating the intermolecular hydrogen bonds between MC and 12-HOA (Fig. 2h).24 Together with the hydrogen bond of the COOH dimer and the van der Waals forces between the elongated alkyl chains of 12-HOA, these intermolecular hydrogen bonds endow the M/H polymer scaffold with a three-dimensional network structure and a relatively strong mechanical strength. Additionally, the peaks at 1144 and 1130 cm−1 are assigned to the symmetric SO2 stretching vibration of the free and solvating SL (Fig. 2i).34 The relative intensity of these two peaks reflects the solvation degree of SL with Li+. It is apparent that the gelation of the CSE with MC will destroy the tetrahedral solvation complex of [Li(SL)2]+, as evidenced by the disappearance of the solvating SL peak. However, the [Li(SL)2]+ solvation structure could be preserved upon the addition of 12-HOA, which guarantees the pathway for fast Li-ion hopping conduction.
O stretching vibration band at about 1700 cm−1 in the FTIR spectra of the 12-HOA and M/H sample results from the formation of the COOH dimer via a hydrogen bond, similar to the supramolecular ionogel with it (Fig. 2g).16,32 Considering that the symmetric stretching vibration of the anhydride is located at 1780 cm−1, no reaction occurs between the –COOH group of 12-HOA and the –OH group of MC in the M/H polymer scaffold.33 Meanwhile, the intensity of the O–H stretching vibration bands (3350 cm−1) decreased when the MC was incorporated with 12-HOA, indicating the intermolecular hydrogen bonds between MC and 12-HOA (Fig. 2h).24 Together with the hydrogen bond of the COOH dimer and the van der Waals forces between the elongated alkyl chains of 12-HOA, these intermolecular hydrogen bonds endow the M/H polymer scaffold with a three-dimensional network structure and a relatively strong mechanical strength. Additionally, the peaks at 1144 and 1130 cm−1 are assigned to the symmetric SO2 stretching vibration of the free and solvating SL (Fig. 2i).34 The relative intensity of these two peaks reflects the solvation degree of SL with Li+. It is apparent that the gelation of the CSE with MC will destroy the tetrahedral solvation complex of [Li(SL)2]+, as evidenced by the disappearance of the solvating SL peak. However, the [Li(SL)2]+ solvation structure could be preserved upon the addition of 12-HOA, which guarantees the pathway for fast Li-ion hopping conduction.
Compared to the M/H gelator, it seems that other common polymer gelators such as polyvinylidene fluoride (PVDF), polyethylene oxide (PEO), and polymethyl methacrylate (PMMA) cannot encapsulate 90 wt% CSE, solidify, and maintain acceptable mechanical stability (Fig. S3†). In sharp contrast, the M/H-GE membrane shows excellent flexibility, compression toughness, and elasticity. It does not break after folding for 180° or even twice, and can undergo twisting deformation with a good recovery (Fig. S1† and 3a). According to the tensile stress–strain curves (Fig. 3b), the tensile strengths of the M-GE and M/H-GE membrane are 0.06 and 0.78 MPa, respectively. The value of the M/H-GE membrane is higher than that of the reported polymer ionogels (0.2–0.5 MPa) with IL contents ≥80 wt%.35–38 The M/H-GE could withstand a compression stress of 0.2 MPa at 90% stain (Fig. S4†). In the rheological results at room temperature (30 °C) (Fig. 3c–e), both the M-GE and M/H-GE samples have a storage modulus (G′) higher than the loss modulus (G′′) at room temperature, implying their elastic nature.16 Note that the G′ value of M/H-GE samples is about 10 MPa, an order of magnitude higher than that of the M-GE sample. For the GE membrane with 10 wt% 12-HOA (H-GE), it possesses G′ values similar to those of the M/H-GE sample in the low-strain region (<0.1%), but the decrease in G′ at high strains, termed as the Payne effect, originates from the inhomogeneous dispersion of fillers and cross-linking in the gel.39 Thus, the cross-linking between 12-HOA and MC not only reinforces the polymer scaffold to enhance the mechanical strength of the GE membrane but also facilitates the distribution of 12-HOA and MC gelators in the M/H-GE membrane. Moreover, with the increase in temperature, the G′ values of M/H-GE are always greater than the G′′ values (Fig. 3f). This phenomenon suggests that the M/H-GE keeps mechanical robustness even at high temperatures, which holds great promise in the application of high-temperature LMBs.
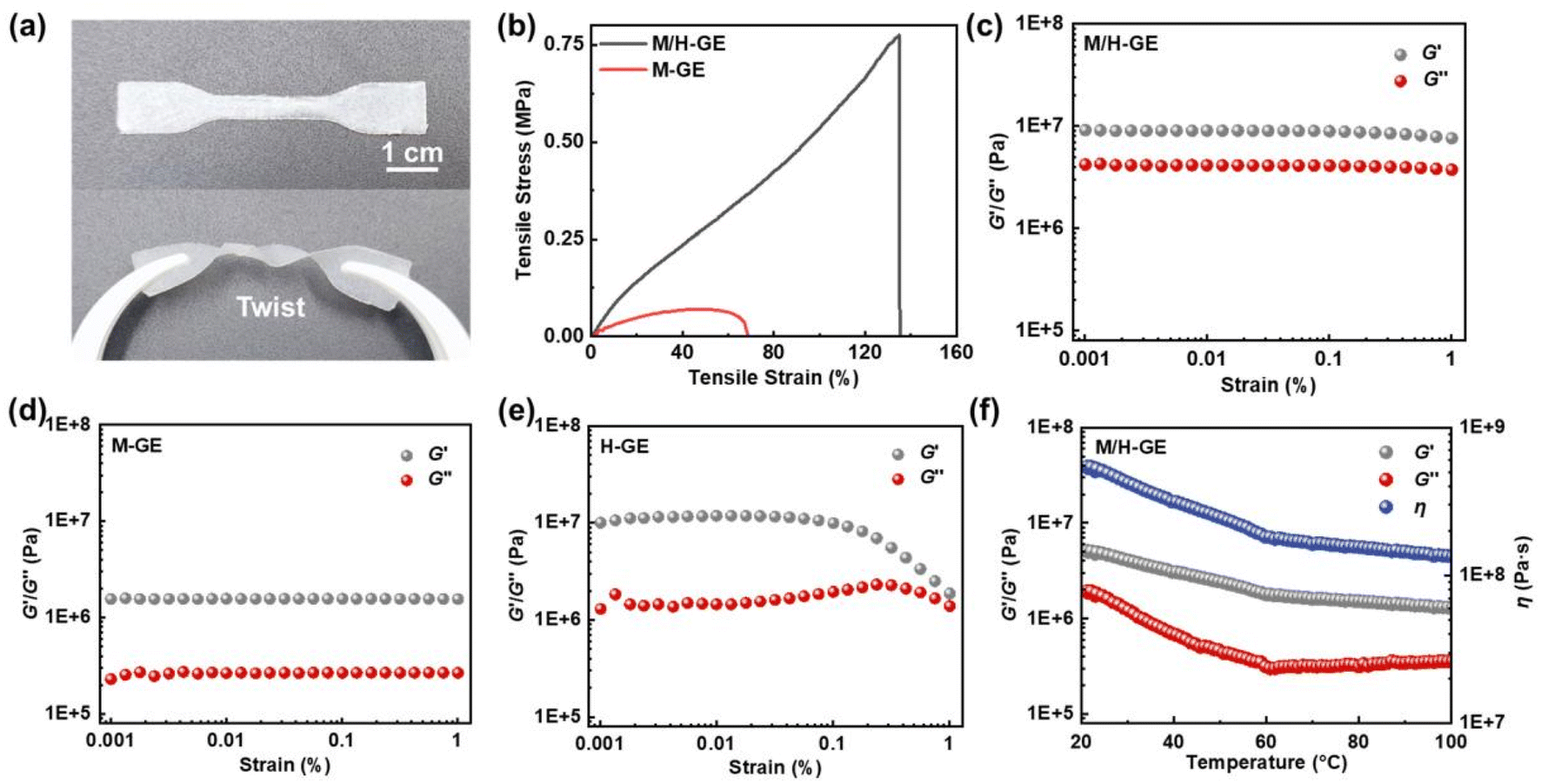 | ||
| Fig. 3 Mechanical properties of GEs: (a) digital photographs of the twisted M/H-GE membrane. (b) Tensile stress–strain curves of the M-GE and M/H-GE membranes. Strain-dependent storage modulus (G′) and loss modulus (G′′) curve of (c) M/H-GE, (d) M-GE, and (e) H-GE membranes. (f) Temperature-dependent rheological curves of the M/H-GE membrane (angular frequency = 10 rad s−1, strain = 1%). | ||
Since both the MC-based hydrogel and the 12-HOA-based supramolecular ionogel show a gel-to-sol transition with the increase in temperatures,16,40–42 the M/H-GE may also be thermo-sensitive. Before the investigation of the thermo-responsiveness of the M/H-GE, its thermal stability was measured. At 150 °C, the polypropylene separator shows obvious shrinkage but almost no degradation occurs for the M/H-GE membrane. The thermogravimetric (TGA) analysis of the M/H-GE membrane further confirms its high thermal stability with no significant weight loss observed up to 150 °C (Fig. S5†). The TGA curve of the M/H-GE membrane is analogous to that of the CSE, demonstrating that the solvation structure of [Li(SL)2]+ is well preserved in the M/H-GE.9,43 Then, the differential scanning calorimetry (DSC) plots of the M-GE, H-GE, and M/H-GE samples are compared in Fig. 4a and S6.† The endothermic peaks at 56–58 °C during the heating stage emerge reversibly in the DSC plots of the M/H-GE samples, but disappear in the M-GE. The thermo-responsive feature of the M/H-GE membrane emerges from 12-HOA (Fig. S6b†),16 while the relatively strong interaction between MC and Li+ will eliminate this property in the MC-based gel with Li salt – concentrated electrolytes.15,44 To reveal the mechanism of thermally induced phase transition in the M/H-GE, temperature-dependent FTIR analysis was performed. When the temperature increased from 40 to 100 °C, the peak at 1670 cm−1 eventually shifted to higher wavenumbers (Fig. S7†), indicating the dissociation of the COOH dimers in 12-HOA.16,32 Meanwhile, the symmetric stretching vibrations of CH2 at 2850 cm−1 become weaker at elevated temperatures (Fig. S8†), indicating diminished van der Waals forces between the alkyl chains of 12-HOA.45,46 These FTIR spectroscopy results prove that the breakdown of 12-HOA bridges in the M/H polymer scaffold at high temperatures contributes to the gel–sol transformation of the M/H-GE membrane.
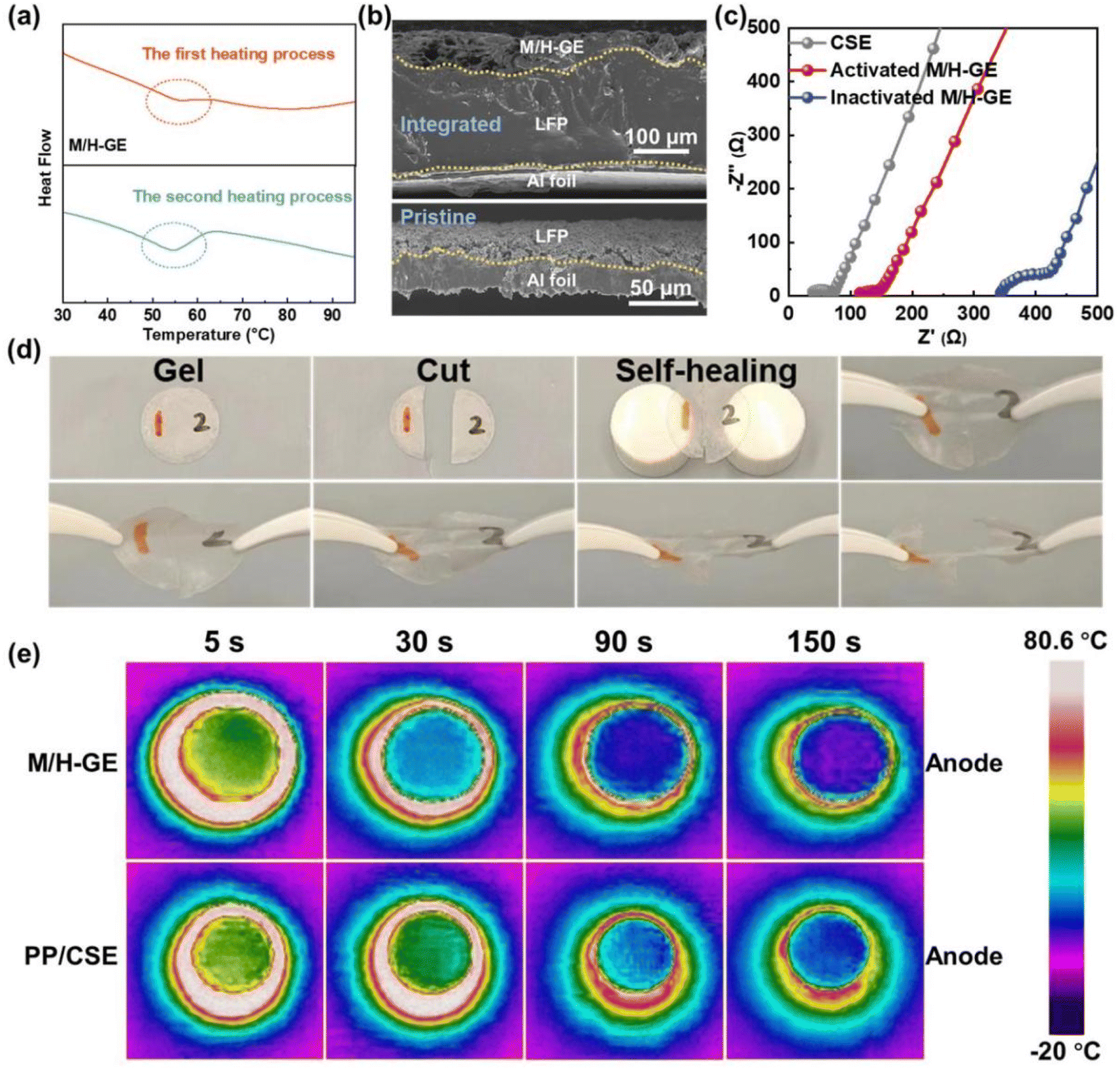 | ||
| Fig. 4 Thermal responsiveness of the M/H-GE membrane: (a) DSC curves of the M/H-GE membrane during the 1st and 2nd heating processes. (b) Cross-sectional SEM images of the pristine LFP electrode and M/H-GE-incorporated LFP electrode. (c) EIS spectra of the inactivated FP‖M/H-GE‖LFP cell, thermally activated FP‖M/H-GE‖LFP cell, and FP‖CSE‖LFP cell. (d) Photos of the M/H-GE membranes that were cut into two pieces and then healed at 70 °C. (e) Thermal images of the Li‖M/H-GE‖LFP cell and Li‖CSE‖LFP cell at different times during natural cooling after heating to 80 °C. | ||
The reversible and dynamic gel-to-sol transition endows the M/H-GE membrane with some special characteristics such as thermally induced electrode wetting, thermally induced healing, and fast cooling rate. The poor solid–solid contact between GEs and electrodes is one of the obstacles to the commercialization of gel-based solid-state LMBs, which leads to inferior ion migration and Li dendrite growth on Li metal anodes.47 Upon heating and converting the M/H-GE from gel to sol, the CSE/12-HOA sol infiltrates into the porous cathode prepared by the particles with irregular size and shape. After cooling to room temperature, a conformal electrolyte/electrode interface was constructed. As shown in the cross-sectional SEM images and corresponding EDX mappings of the LFP@M/H-GE-integrated electrode, the pores of the LFP electrode are effectively filled with gels (Fig. 4b and S9†). The sufficient contact between the electrode and the GE will reduce the interfacial resistance (Rint), as indicated by the electrochemical impedance (EIS) spectra of the asymmetric FePO4 (FP)‖LFP cells (Fig. 4c). The FP‖M/H-GE‖LFP cell was heated to 70 °C and then cooled to room temperature, denoted as the thermally activated FP‖M/H-GE‖LFP cell. Note that the activated cell shows an Rint value of 25.1 Ω, greatly lower than that of the inactivated FP‖M/H-GE‖LFP cell (78.1 Ω) and even close to that of the FP‖LFP cell using the liquid CSE (31.2 Ω). This facile in situ interface wetting strategy further avoids the use and potential leakage of wetting reagents.48 The self-healing ability of the GE was measured by cut-recover and tensile tests. As shown in Fig. 4d, the cut M/H-GE membrane can quickly recombine by putting them together at 70 °C for 1 h. The repaired M/H-GE membrane can still withstand a great deal of stretching and its tensile strength reaches 76.4% of the value of the intact M/H-GE membrane (Fig. S10†). While the cut M-GE membrane did not recombine after being put together at 70 °C for 1 h, indicating that the thermally induced healing property of the M/H-GE membrane may originate from the ability of 12-HOA to form new hydrogen bonds with the broken MC and 12-HOA chains (Fig. S11†). Moreover, the Li‖M/H-GE‖LFP cell and common Li‖LFP cell with the liquid CSE were heated to 80 °C and then allowed to cool naturally to record the cooling process thermography. A more even temperature distribution and a faster cooling rate were observed in the infrared thermal graphs of the Li‖M/H-GE‖LFP cell (Fig. 4e). Combined with the healing capability, the rapid cooling rate of the M/H-GE membrane can significantly enhance the reliability and safety of cells when it is used in LMBs.49
Because of the 90 wt% liquid electrolyte content in the M/H-GE membrane, it should show a Li+ transportation property and electrochemical stability analogous to the neat CSE. The bulk ionic conductivity of GEs was tested by the symmetric steel cells using EIS. As exhibited in Fig. 5a, the gelation of the CSE with MC/12-HOA minimally decreases the ionic conductivity from 0.27 to 0.25 mS cm−1 at 30 °C. When the temperature increases to 40 °C or higher values, the ionic conductivity of the M/H-GE is even higher than that of the neat CSE. In sharp contrast, the M-GE membrane has ionic conductivities much lower than those of the CSE at a temperature ranging from 30 to 70 °C (Fig. S12†), resulting from the destruction of the fast Li-ion hopping conduction pathway.50 By fitting the relationship between the conductivity and temperature, the activation energy (Ea) of GEs was calculated using the Arrhenius equation (Fig. S13†). The Ea value of the M/H-GE (12.9 kJ mol−1) is also similar to that of the neat CSE (10.2 kJ mol−1). Apart from the ionic conductivity and activation energy, the Li+ transference number (tLi+) is one of the important requirements for the electrolytes because it directly reflects the fraction of charge carried by Li+ ions. By an AC impedance/DC polarization combined technique using the Li symmetric cell,51 the tLi+ value of M/H-GE was determined to be 0.57, higher than that of the SL electrolyte (0.51) (Fig. 5b and S14†). As a result, the Li+ conductivity of the M/H-GE membrane (0.143 mS cm−1) is even higher than that of the liquid CSE (0.137 mS cm−1).
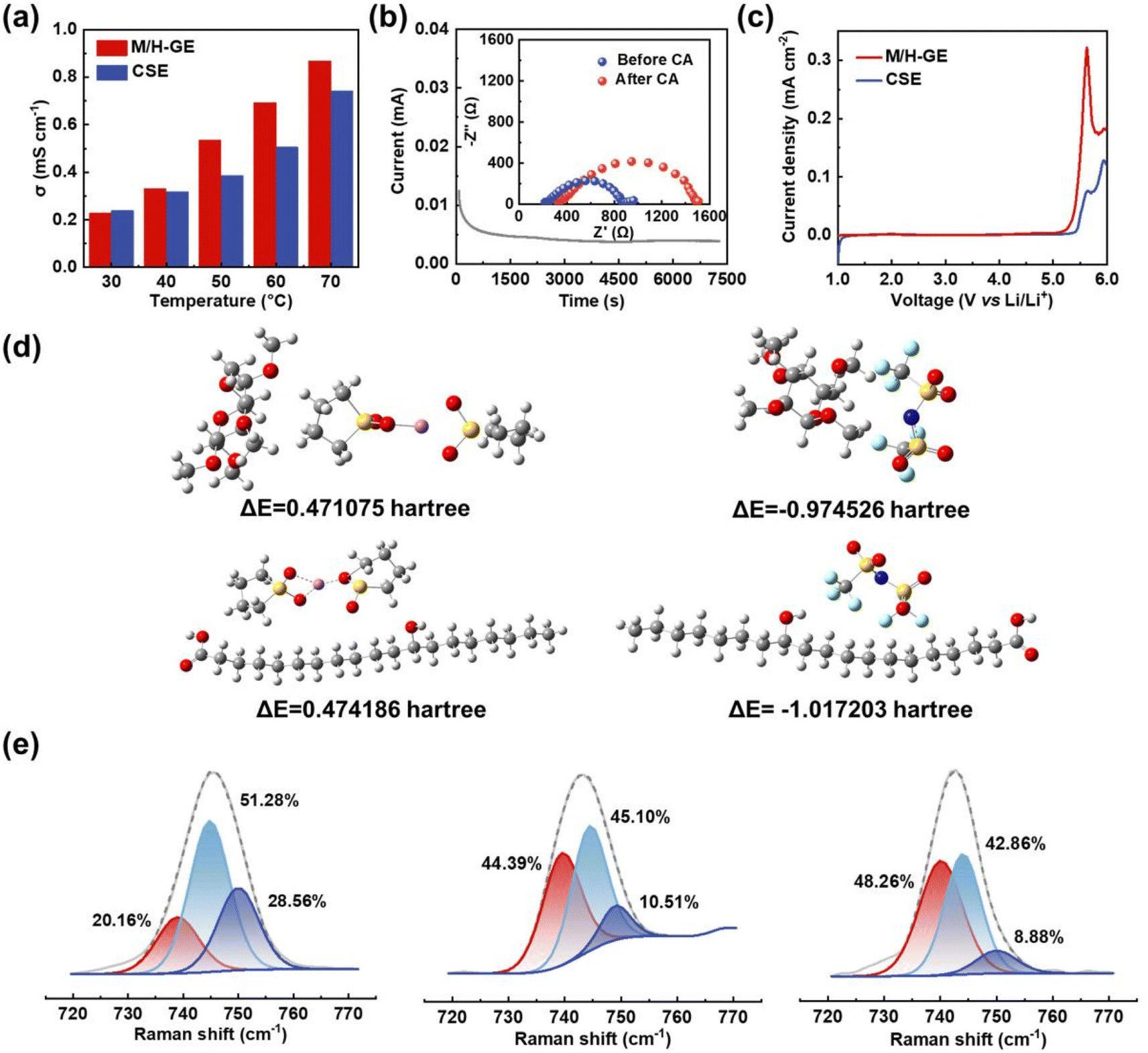 | ||
| Fig. 5 Li+ conduction properties and mechanism of GEs: (a) EIS results of stainless steel symmetric cells using the M/H-GE membrane tested at 30–70 °C. (b) Direct current (DC) polarization of the Li‖M/H-GE‖Li cell at 10 mV (inset: EIS spectra before and after DC polarization). (c) LSV curves of the Li‖M/H-GE‖stainless steel cell and Li‖CSE‖stainless steel cell. (d) Simulated gelator–[Li(SL)2]+ complex cation interactions and gelator–TFSI− interactions. (e) Raman spectra of CSE, H-GE, M/H-GE membrane. | ||
Additionally, the linear scan voltammetry and differential pulse voltammetry tests show only a slight decline in the oxidation onset potential from 5.45 to 5.25 V vs. Li+/Li after the gelation of CSE by M/H (Fig. 5c and S15†). Moreover, the electrochemical floating test, a more stringent method to evaluate the oxidation stability of electrolytes, was conducted in the CSE and M/H-GE-based Li‖NCM811 half-cells. As shown in Fig. S16,† the cell potential is allowed to “float” at different potentials. The leakage currents of two cells are lower than 10 μA before 4.8 V, which indicates that both electrolytes can operate normally at 4.8 V vs. Li+/Li. Moreover, the leakage current of the M/H-GE-based half-cell is smaller than that of the CSE-based one, demonstrating a higher electrochemical oxidation stability of M/H-GE than that of the CSE.52–54 Thus, the M/H-GE membrane is also compatible with high-voltage layered oxide cathodes, which maximize the energy density advantages of LMBs.
Density functional theory (DFT) calculations were further performed to investigate the interactions between the CSE and polymer scaffolds, as well as their effect on the Li+ conduction properties of GEs (Fig. 5d). First, the deformation of the tetrahedral [Li(SL)2]+ complex by the ether groups of MC was verified (Fig. S17†). Then, the possible interactions between the gelators and [Li(SL)2]+ complex cation were simulated. It seems that [Li(SL)2]+ is not prone to form hydrogen bonds with MC or 12-HOA, as the ΔE values of all polymer–cation interactions are positive. In contrast, the interactions between the polymer scaffolds and TFSI− anion are spontaneous due to the high electronegativity of the F atoms of TFSI−. These interactions and van der Waals forces contribute to the gelation of CSE. Moreover, there are two advantages brought by the polymer-TFSI− anion interactions. On one hand, the TFSI− anions are tethered, allowing a high tLi+ to reduce the buildup of ion concentration gradients and suppress the lithium dendrites growth in LMBs.55 On the other hand, the dissociation of Li+–TFSI− contact ion pairs (CIPs) and aggregates (AGGs) is promoted, which releases more free TFSI− anions that can decompose on the Li surface and thus enables the formation of an anion-derived solid electrolyte interphase (SEI) film.56–58 To validate this, the Raman spectra of electrolytes were recorded and analyzed (Fig. 5e). The Raman band in the range of 720–770 cm−1 corresponds to the C–N–C bending vibration of the TFSI− anion and reflects the surrounding environment of the TFSI− anion.59 After fitting, it was found that the proportion of free TFSI− anions in the M/H-GE (48.26 at%) is higher than that in the M-GE (44.39 at%) and neat CSE (20.16 at%).
Benefiting from the high tLi+ value, the M/H-GE membrane shows excellent compatibility with the Li metal anode. According to the galvanostatic cycling tests (Fig. 6a), the symmetric Li‖M/H-GE‖Li cell exhibits stable overpotentials of <100 mV during the Li stripping/plating process. More importantly, no failures can be observed for the Li‖M/H-GE‖Li cell after 1500 h cycling at 0.1 mA cm−2. In sharp contrast, the Li‖CSE‖Li cell has a lower overpotential (∼85 mV) but experienced a sudden short circuit after 890 h, which originates from the penetration of Li dendrites. As shown in Fig. 6b and S18,† the surface of the Li anode cycled in the CSE is rough and covered with Li mossy, while the Li anode cycled in the M/H-GE remains flat without noticeable Li dendrites. As discussed above, the high content of free TFSI− anions in the gel or solid-state polymer electrolytes will lead to the formation of a robust and conductive anion-derive SEI film rather than a solvent-derived SEI film that consists of fragile organic components. Thus, X-ray photoelectron spectra (XPS) of cycled Li anodes were conducted to unveil the difference in the chemical composition of SEI films formed in the M/H-GE and CSE. As indicated by the F 1s and N 1s XPS high-resolution and full-survey XPS spectra (Fig. 6c, d and S19†), the LiF (685.1 eV) content and Li3N (397.2 eV) content of the M/H-GE-engineered SEI film are much higher than those in the CSE-engineered SEI film. The previous reports demonstrated that LiF plays a crucial role in enhancing the mechanical stability of SEI and regulating the Li+ diffusion across the SEI,60 while Li3N is a fast Li+ conductor (>1 mS cm−1) that increases the overall conductivity of the SEI.61 In addition, the gelation of the CSE with MC and 12-HOA will not promote the decomposition of sulfone molecules on the Li metal, as implied by the similar Li2CO3 contents of the M/H-GE-engineered SEI and CSE-engineered SEI films (Fig. S20†). These XPS results confirm that the M/H-GE facilitates the formation of a robust and conductive SEI film, which is another reason for its improved compatibility with the Li anode.
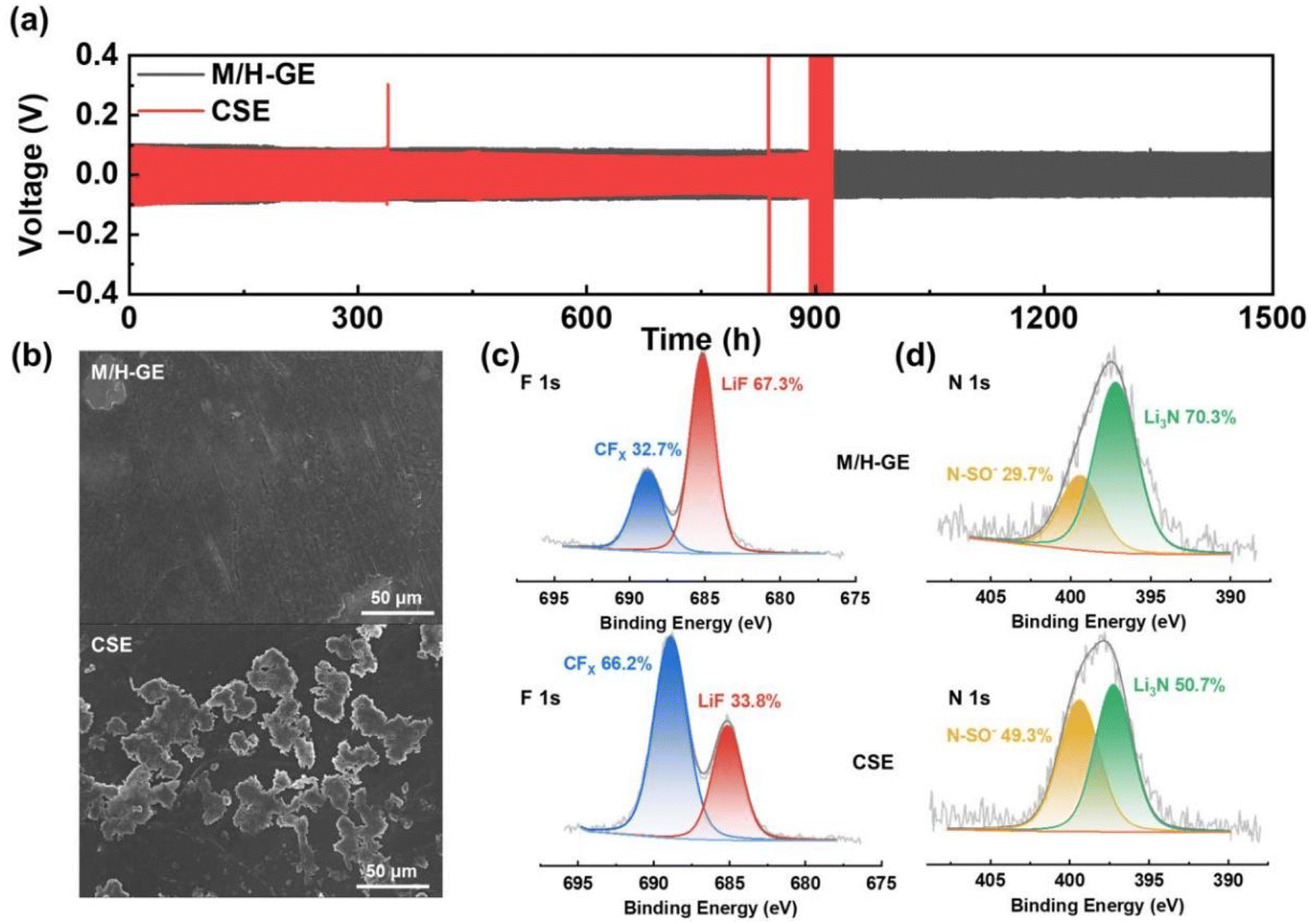 | ||
| Fig. 6 Li compatibility of the M/H-GE: (a) long-term cycling of the Li‖M/H-GE‖Li cell and Li‖CSE‖Li cell at 0.1 mA cm−2, and 0.1 mA h cm−2 Li plated and stripped per cycle. (b) SEM image of the deposited lithium surface after 25 cycles using different electrolytes. XPS (c) F 1s and (d) N 1s spectra of the cycled Li electrode taken from the Li‖M/H-GE‖Li cell and Li‖CSE‖Li cell. | ||
Because of the thermally induced electrode wetting and good compatibility with the Li anode of the M/H-GE, it is expected that the corresponding LMB full cells exhibit a favorable performance. As depicted in Fig. 7a, the Li‖M/H-GE‖LFP cell working at 0.1C and room temperature delivers an initial discharge capacity of 142.5 mA h g−1 and an average coulombic efficiency over 99.5%. After 100 cycles, it sustained a capacity of 128.9 mA h g−1. In contrast, the Li‖CSE‖LFP cell displays a lower initial capacity of 133.2 mA h g−1 and suffers from quick capacity fading after 80 cycles, which may result from the inferior stability of the electrolyte/Li interface. The C-rate capability of the Li‖M/H-GE‖LFP cell was also evaluated. At a rate of 1C, the capacity of about 100 mA h g−1 can be obtained (Fig. 7b). As the electrochemical stability window of M/H-GE is up to 5.25 V vs. Li+/Li, the feasibility of the M/H-GE membrane in the rigid state-of-the-art Ni-rich cathode-based LMB system was examined. When the cathode was altered to LiNi0.8Co0.1Mn0.1O2 (NCM811) and the corresponding areal loading surged to 8.67 mg cm−2, the Li‖M/H-GE‖NCM811 cell still possesses an initial capacity of 153.5 mA h g−1 and a capacity retention of 77.1% after 400 cycles at 1C (Fig. 7c and d). In contrast, the Li‖CSE‖NCM811 cell at 1C suffers from severe polarization and its specific capacity decreases drastically to < 50 mA h g−1 (Fig. S21†). The areal capacity (1.33 mA h cm−2) of this Li‖M/H-GE‖NCM811 cell at such a high rate of 1C is considerably higher than most previously reported Li‖NCM cells using the ionogels or gels prepared with concentrated electrolytes, as summarized in Table S1.†9,14,16,58,62–66 Furthermore, the M/H-GE membrane well inherited the superior flexibility of the MC/12-HOA polymer scaffold and the high safety of the CSE electrolyte, as demonstrated in Fig. 7e. The Li‖M/H-GE‖LFP pouch cell could provide a stable output voltage of 3.44 V in the folding condition or rolling up condition. Even if the pouch cell was completely deteriorated, it could light up a red light-emitting diode (LED) bulb. After 5 h, the cutting-open cell can still work as a power source. By employing an N/P ratio of 1.1, the graphite‖M/H-GE‖NCM811 and graphite‖CSE‖NCM811 full cells were also prepared. Except for the slightly higher initial capacity, both the coulombic efficiency and the cycling stability of the graphite‖M/H-GE‖NCM811 cell are similar to those of the graphite‖CSE‖NCM811 cell (Fig. S22†). All these excellent battery and safety performances indeed promise the practical application of the M/H-GE in solid-state LMBs and commercial Li-ion batteries.
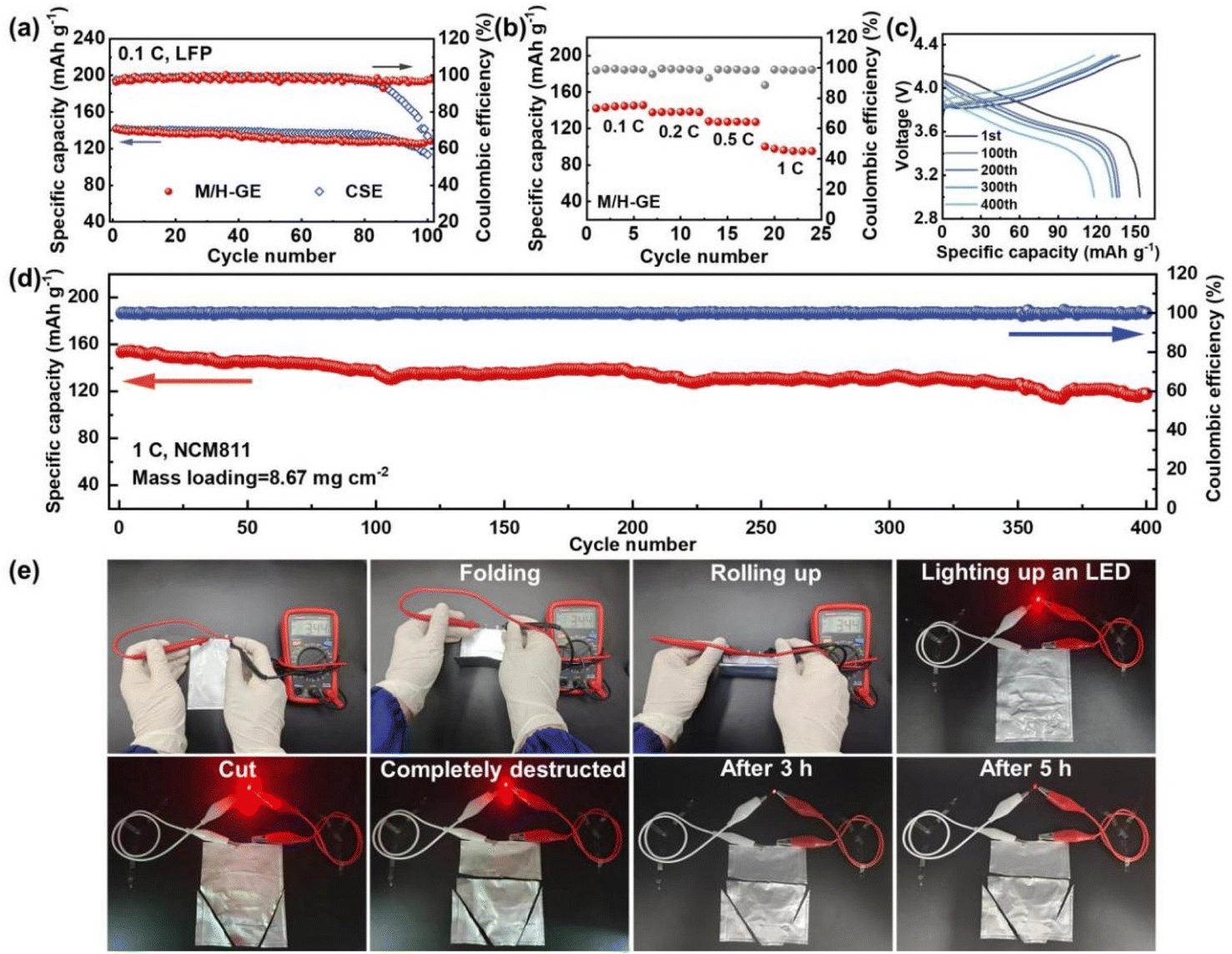 | ||
| Fig. 7 Room-temperature electrochemical and safety performance of M/H-GE-based LMBs: (a) cycling performances of the Li‖M/H-GE‖LFP cell and Li‖CSE‖LFP cell at 0.1C. (b) C-rate capability of the Li‖M/H-GE‖LFP cell. (c) Charge–discharge curves and (d) cycling performances of the Li‖M/H-GE‖NCM811 cell at 1C. (e) Flexibility tests and abuse tolerance tests of Li‖M/H-GE‖LFP pouch cells. | ||
Conclusion
In summary, we have designed a mechanically stable gel polymer electrolyte with an ultrahigh liquid electrolyte or concentrated sulfone electrolyte content of 90 wt% by employing a hybrid gelator of MC and 12-HOA. Owing to the 90 wt% CSE content, the M/H-GE exhibits a room-temperature ionic conductivity (0.25 mS cm−1) similar to the liquid CSE. Meanwhile, the strong hydrogen bonds between MC/12-HOA and the gelator/TFSI− anion effectively immobilize the CSE to provide a storage modulus of gel as high as 10 MPa enough to sustain an 80 μm – thick self-supporting film. DFT calculations and spectroscopic characterizations revealed the evidence of favorable gelator/TFSI− interactions that increase the tLi+ value of GE to 0.57 and promote the dissociation of Li+–TFSI− contact ion pairs (CIPs) and aggregates (AGGs) to improve the compatibility with the Li metal anode, as proved by the stable Li stripping/plating behavior of the Li‖M/H-GE‖Li cell with small polarization over 1500 h. Moreover, the M/H-GE features a thermally reversible sol-to-gel phase transition, which endows it with thermally induced electrode wetting, thermally induced healing, and a rapid cooling rate. Benefiting from these properties, the Li‖M/H-GE‖NCM811 cell shows an initial capacity of 153.5 mA h g−1 and a capacity retention of 77.1% after 400 cycles at 1C and room temperature. The MC/12-HOA-supported gel electrolyte present here not only offers a novel strategy to construct multifunctional and high-performance gel electrolytes for practical LMBs but also holds great promise in the application of other flexible and wearable devices.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.† The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
X. J., X. W., and S. Z. conceived the idea and designed the experiments. X. J. and J. C. synthesized the materials. J. Z. provided theoretical calculations. X. J. fabricated the cells and analyzed the data from the electrochemical measurements with the help of W. L., X. W., and F. L. X. J., X. W., and S. Z. co-wrote the manuscript. J. C., J. Z., W. L., and X. W. discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
X. J., J. C., and J. Z. contributed equally to this work. We thank the Analytical Instrumentation Center of Hunan University for SEM and rheological measurements. We acknowledge the support of Open Foundation of State Key Laboratory of Advanced Design and Manufacturing Technology for Vehicle (72275002), National Natural Science Foundation of China (52072118, 52373206, and 51904098), Research Fund of YueLu Mountain Industrial Innovation Center (2023YCII0137), Natural Science Foundation of Hunan Province (2019JJ50050), Key Laboratory Research Fund of Guangxi (EMFM20212205), and Open Research Fund of School of Chemistry and Chemical Engineering, Henan Normal University (No. 2024Z04).References
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- X. Feng, M. Ouyang, X. Liu, L. Lu, Y. Xia and X. He, Energy Storage Mater., 2018, 10, 246–267 CrossRef.
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS PubMed.
- X. B. Cheng, R. Zhang, C. Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef CAS PubMed.
- A. Kushima, K. P. So, C. Su, P. Bai, N. Kuriyama, T. Maebashi, Y. Fujiwara, M. Z. Bazant and J. Li, Nano Energy, 2017, 32, 271–279 CrossRef CAS.
- Y. Tian, T. Shi, W. D. Richards, J. Li, J. C. Kim, S. H. Bo and G. Ceder, Energy Environ. Sci., 2017, 10, 1150–1166 RSC.
- W. Ren, C. Ding, X. Fu and Y. Huang, Energy Storage Mater., 2021, 34, 515–535 CrossRef.
- N. Chen, H. Zhang, L. Li, R. Chen and S. Guo, Adv. Energy Mater., 2018, 8, 1702675 CrossRef.
- M. Li, Y. Gao, D. Yu, Z. Hu, Z. Liu, X. Wang, Q. Weng, Y. Chen, Y. Zhang and S. Zhang, Energy Storage Mater., 2023, 59, 102753 CrossRef.
- D. E. Ciurduc, N. Boaretto, J. P. Fernández-Blázquez and R. Marcilla, J. Power Sources, 2021, 506, 230220 CrossRef CAS.
- S. Wang, Y. Jiang and X. Hu, Adv. Mater., 2022, 34, 2200945 CrossRef CAS PubMed.
- E. Vasheghani-Farahani, J. H. Vera, D. G. Cooper and M. E. Weber, Ind. Eng. Chem. Res., 1990, 29, 554–560 CrossRef CAS.
- Y. Liu, L. Hou, Y. Jiao and P. Wu, ACS Appl. Mater. Interfaces, 2021, 13, 13319–13327 CrossRef CAS PubMed.
- A. J. D'Angelo and M. J. Panzer, Adv. Energy Mater., 2018, 8, 1801646 CrossRef.
- S. Chereddy, J. Aguirre, D. Dikin, S. L. Wunder and P. R. Chinnam, ACS Appl. Energy Mater., 2020, 3, 279–289 CrossRef CAS.
- X. Jiang, Z. Liu, W. Liu, D. Yu, J. Zhang, X. Wang, Y. Zhang and S. Zhang, Matter, 2024, 7, 1558–1574 CrossRef CAS.
- C. Capiglia, Y. Saito, H. Yamamoto, H. Kageyama and P. Mustarelli, Electrochim. Acta, 2000, 45, 1341–1345 CrossRef CAS.
- D. Klemm, B. Heublein, H. P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS PubMed.
- T. Xu, K. Liu, N. Sheng, M. Zhang, W. Liu, H. Liu, L. Dai, X. Zhang, C. Si, H. Du and K. Zhang, Energy Storage Mater., 2022, 48, 244–262 CrossRef.
- Z. Wang, P. Heasman, J. Rostami, T. Benselfelt, M. Linares, H. Li, A. Iakunkov, F. Sellman, R. Östmans, M. M. Hamedi, I. Zozoulenko and L. Wågberg, Adv. Funct. Mater., 2023, 33, 2212806 CrossRef CAS.
- D. Klemm, F. Kramer, S. Moritz, T. Lindström, M. Ankerfors, D. Gray and A. Dorris, Angew. Chem., Int. Ed., 2011, 50, 5438–5466 CrossRef CAS PubMed.
- H. Chan, K. Jung, W. Kim, L. Zhai and J. Kim, Cellulose, 2019, 26, 5821–5829 CrossRef.
- N. Mittal, S. Tien, E. Lizundia and M. Niederberger, Small, 2022, 18, 2107183 CrossRef CAS PubMed.
- J. Wang, C. Wang, W. Wang, W. Li and J. Lou, Chem. Eng. J., 2022, 428, 132604 CrossRef CAS.
- J. Wan, J. Zhang, J. Yu and J. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 24591–24599 CrossRef CAS PubMed.
- K. Xu and C. A. Angell, J. Electrochem. Soc., 2002, 149, A920 CrossRef CAS.
- X. Ren, S. Chen, H. Lee, D. Mei, M. H. Engelhard, S. D. Burton, W. Zhao, J. Zheng, Q. Li, M. S. Ding, M. Schroeder, J. Alvarado, K. Xu, Y. S. Meng, J. Liu, J. G. Zhang and W. Xu, Chem, 2018, 4, 1877–1892 CAS.
- W. Wu, Y. Bai, X. Wang and C. Wu, Chin. Chem. Lett., 2021, 32, 1309–1315 CrossRef CAS.
- T. A. Pham, K. E. Kweon, A. Samanta, V. Lordi and J. E. Pask, J. Phys. Chem. C, 2017, 121, 21913–21920 CrossRef CAS.
- Y. Ugata, S. Sasagawa, R. Tatara, K. Ueno, M. Watanabe and K. Dokko, J. Phys. Chem. B, 2021, 125, 6600–6608 CrossRef CAS PubMed.
- J. Wu, L. Yuan, W. Zhang, Z. Li, X. Xie and Y. Huang, Energy Environ. Sci., 2021, 14, 12–36 RSC.
- D. A. S. Grahame, C. Olauson, R. S. H. Lam, T. Pedersen, F. Borondics, S. Abraham, R. G. Weiss and M. A. Rogers, Soft Matter, 2011, 7, 7359–7365 RSC.
- L. Hou and P. Wu, Cellulose, 2019, 26, 2759–2769 CrossRef CAS.
- J. Fu, X. Ji, J. Chen, L. Chen, X. Fan, D. Mu and C. Wang, Angew. Chem., 2020, 132, 22378–22385 CrossRef.
- Z. Luo, W. Li, J. Yan and J. Sun, Adv. Funct. Mater., 2022, 32, 2203988 CrossRef CAS.
- L. M. Zhang, Y. He, S. Cheng, H. Sheng, K. Dai, W. J. Zheng, M. X. Wang, Z. S. Chen, Y. M. Chen and Z. Suo, Small, 2019, 15, 1804651 CrossRef PubMed.
- Z. Cao, H. Liu and L. Jiang, Mater. Horiz., 2020, 7, 912–918 RSC.
- Q. Xia, W. Li, X. Zou, S. Zheng, Z. Liu, L. Li and F. Yan, Mater. Horiz., 2022, 9, 2881–2892 RSC.
- A. R. Payne, J. Appl. Polym. Sci., 1964, 8, 2661–2686 CrossRef.
- H. Zhang, S. Ma, Q. Zhang, M. Cao, Y. Wang, Y. Gu and X. Xu, ACS Appl. Mater. Interfaces, 2020, 12, 41819–41831 CrossRef CAS PubMed.
- Y. Li, N. Khuu, A. Gevorkian, S. Sarjinsky, H. Therien-Aubin, Y. Wang, S. Cho and E. Kumacheva, Angew. Chem., Int. Ed., 2017, 56, 6083–6087 CrossRef CAS PubMed.
- Y. J. Kim and Y. T. Matsunaga, J. Mater. Chem. B, 2017, 5, 4307–4321 RSC.
- J. Kawaji, M. Morishima, M. Hirooka and T. Okumura, Electrochim. Acta, 2023, 447, 142115 CrossRef CAS.
- R. Mantravadi, P. R. Chinnam, D. A. Dikin and S. L. Wunder, ACS Appl. Mater. Interfaces, 2016, 8, 13426–13436 CrossRef CAS PubMed.
- V. Kanakaiah, M. Latha, B. Sravan, A. Palanisamy and J. V. Rani, J. Electrochem. Soc., 2014, 161, A1586–A1592 CrossRef CAS.
- S. Chen, B. Zhang, N. Zhang, F. Ge, B. Zhang, X. Wang and J. Song, ACS Appl. Mater. Interfaces, 2018, 10, 5871–5879 CrossRef CAS PubMed.
- S. Xia, X. Wu, Z. Zhang, Y. Cui and W. Liu, Chem, 2019, 5, 753–785 CAS.
- C. Z. Zhao, B. C. Zhao, C. Yan, X. Q. Zhang, J. Q. Huang, Y. Mo, X. Xu, H. Li and Q. Zhang, Energy Storage Mater., 2020, 24, 75–84 CrossRef.
- J. He, A. Bhargav, L. Su, J. Lamb, J. Okasinski, W. Shin and A. Manthiram, Nat. Energy, 2024, 9, 446–456 CrossRef CAS.
- K. Dokko, D. Watanabe, Y. Ugata, M. L. Thomas, S. Tsuzuki, W. Shinoda, K. Hashimoto, K. Ueno, Y. Umebayashi and M. Watanabe, J. Phys. Chem. B, 2018, 122, 10736–10745 CrossRef CAS PubMed.
- J. Evans, C. A. Vincent and P. G. Bruce, Polymer, 1987, 28, 2324–2328 CrossRef CAS.
- D. Yu, Z. Ma, Z. Liu, X. Jiang, H. A. Younus, X. Wang and S. Zhang, Chem. Eng. J., 2023, 457, 141043 CrossRef CAS.
- Q. Zhao, P. Chen, S. Li, X. Liu and L. A. Archer, J. Mater. Chem. A, 2019, 7, 7823–7830 RSC.
- M. He, L. Hu, Z. Xue, C. C. Su, P. Redfern, L. A. Curtiss, B. Polzin, A. von Cresce, K. Xu and Z. Zhang, J. Electrochem. Soc., 2015, 162, A1725–A1729 CrossRef CAS.
- J. Zhu, Z. Zhang, S. Zhao, A. S. Westover, I. Belharouak and P. F. Cao, Adv. Energy Mater., 2021, 11, 1–18 Search PubMed.
- H. Wang, J. Song, K. Zhang, Q. Fang, Y. Zuo, T. Yang, Y. Yang, C. Gao, X. Wang, Q. Pang and D. Xia, Energy Environ. Sci., 2022, 15, 5149–5158 RSC.
- M. Yao, Q. Ruan, Y. Wang, L. Du, Q. Li, L. Xu, R. Wang and H. Zhang, Adv. Funct. Mater., 2023, 33, 2213702 CrossRef CAS.
- J. Sun, C. He, Y. Li, Q. Zhang, C. Hou, M. De Volder, K. Li and H. Wang, Energy Storage Mater., 2023, 54, 40–50 CrossRef.
- D. M. Seo, P. D. Boyle, R. D. Sommer, J. S. Daubert, O. Borodin and W. A. Henderson, J. Phys. Chem. B, 2014, 118, 13601–13608 CrossRef CAS PubMed.
- J. Tan, J. Matz, P. Dong, J. Shen and M. Ye, Adv. Energy Mater., 2021, 11, 2100046 CrossRef CAS.
- M. S. Kim, Z. Zhang, J. Wang, S. T. Oyakhire, S. C. Kim, Z. Yu, Y. Chen, D. T. Boyle, Y. Ye, Z. Huang, W. Zhang, R. Xu, P. Sayavong, S. F. Bent, J. Qin, Z. Bao and Y. Cui, ACS Nano, 2023, 17, 3168–3180 CrossRef CAS PubMed.
- Q. Ruan, M. Yao, S. Luo, W. Zhang, C. J. Bae, Z. Wei and H. Zhang, Nano Energy, 2023, 113, 108571 CrossRef CAS.
- G. Tan, F. Wu, C. Zhan, J. Wang, D. Mu, J. Lu and K. Amine, Nano Lett., 2016, 16, 1960–1968 CrossRef CAS PubMed.
- Q. Sun, S. Wang, Y. Ma, D. Song, H. Zhang, X. Shi, N. Zhang and L. Zhang, Adv. Mater., 2023, 35, 2300998 CrossRef CAS PubMed.
- X. Yu, Z. Wang, X. Deng, K. Hu, Q. Liu, Y. Shen, W. Yu and X. Hu, ACS Energy Lett., 2024, 9, 1826–1834 CrossRef CAS.
- Y. Zhai, W. Hou, M. Tao, Z. Wang, Z. Chen, Z. Zeng, X. Liang, P. Paoprasert, Y. Yang, N. Hu and S. Song, Adv. Mater., 2022, 34, 2205560 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta02561j |
| This journal is © The Royal Society of Chemistry 2024 |