
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTailoring the efficiency of porphyrin molecular frameworks for the electroactivation of molecular N2†
María
Romero-Angel‡
 a,
Roumayssa
Amrine‡
b,
Beatriz
Ávila-Bolívar
b,
Neyvis
Almora-Barrios
a,
Carolina R.
Ganivet
a,
Natalia M.
Padial
a,
Vicente
Montiel
b,
José
Solla-Gullón
*b,
Sergio
Tatay
*a and
Carlos
Martí-Gastaldo
*a
a,
Roumayssa
Amrine‡
b,
Beatriz
Ávila-Bolívar
b,
Neyvis
Almora-Barrios
a,
Carolina R.
Ganivet
a,
Natalia M.
Padial
a,
Vicente
Montiel
b,
José
Solla-Gullón
*b,
Sergio
Tatay
*a and
Carlos
Martí-Gastaldo
*a
aInstituto de Ciencia Molecular, Universidad de València, c/Catedrático José Beltrán, 2, 46980, Paterna, Spain. E-mail: sergio.tatay@uv.es; carlos.marti@uv.es
bInstitute of Electrochemistry, University of Alicante, Apdo. 99, E-03080 Alicante, Spain. E-mail: jose.solla@ua.es
First published on 25th March 2024
Abstract
The combination of compositional versatility and topological diversity for the integration of electroactive species into high-porosity molecular architectures is perhaps one of the main appeals of metal–organic frameworks (MOFs) in the field of electrocatalysis. This premise has attracted much interest in recent years, and the results generated have also revealed one of the main limitations of molecular materials in this context: low stability under electrocatalytic conditions. Using zirconium MOFs as a starting point, in this work, we use this stability as a variable to discriminate between the most suitable electrocatalytic reaction and specific topologies within this family. Our results revealed that the PCN-224 family is particularly suitable for the electroreduction of molecular nitrogen for the formation of ammonia with faradaic efficiencies above 30% in the presence of Ni2+ sites, an activity that improves most of the catalysts described. We also introduce the fluorination of porphyrin at the meso position as a good alternative to improve both the activity and stability of this material under electrocatalytic conditions.
Introduction
The structural and compositional versatility intrinsic to metal–organic frameworks (MOFs) and their ultrahigh porosity make them interesting platforms for catalysis. Compared to metal complexes, metal oxides, and zeolites, to name a few, the molecular nature of MOFs enables an exquisite level of control over their structural, chemical, and physical properties. Their large internal surface areas and high density of active sites can facilitate substrate accessibility and mass transport for better catalytic performance.1 Also, the isolation of catalytic sites in the framework backbone can contribute to the stabilization of highly reactive intermediates whereas the ordered arrangement of molecular components can help delineate structure–activity relationships to support the design of improved catalysts. Besides, for a given geometry and connectivity of the linker and metal node, MOFs can exist in different topologies, that will result in different pore sizes, dimensionalities (i.e. mass transport), coordination geometries and variable densities of the active sites.2–5 On top of this, the pore environment around the active site can be further modified to regulate redox properties and binding energies of the metal centers, stabilize intermediates, promote proton transfer, or locally modify proton concentration.6Provided the samples have sufficient chemical stability, their use in photo and electrocatalytic processes is of great importance to demonstrate the potential of reticular design in catalysis.7 MOFs have been used as catalysts with promising results in reactions as the hydrogen evolution reaction (HER),8 oxygen reduction/evolution reaction (ORR/OER),9 CO2 reduction reaction (CO2RR),10 and nitrogen reduction reaction (NRR).11 Porphyrin molecules are particularly appealing in this context. Besides their intrinsic electrocatalytic activity in solution,12–14 their rigid and robust backbones allow for the assembly of reticular frameworks with fine control over their intermolecular distance, their density, the chemical environment, and greater/lower accessibility to substrates depending on the net porosity of the material. This ease to direct framework assembly is supported by the huge number of porphyrin-MOFs reported with different metal nodes and secondary building units (SBUs).15–17 Among them, Zr/carboxylate-based SBUs, in particular, those based on the Zr6O4(OH)4 cluster (Zr6), seem to be one of the most synthesized, at least in terms of electrocatalytic efficiency, with at least one member of this subfamily standing out in the HER, OER, CO2RR or NRR. This is arguably due to the superior thermal and water/moisture stability of Zr-MOFs,18,19 that are expected to prevent their degradation under the conditions imposed by electrocatalytic reactions. The Zr6 SBU also combines a high versatility in its connectivity index, from 12 to 6 connection points, with a high persistence in dissolution, thus facilitating the synthesis of different topologies with variable porosities for a large pool of potential catalysts (Fig. 1).
 | ||
| Fig. 1 Some reported topologies for common Zr-oxo clusters and TCPP organic linkers. Representative MOF examples are given for each corresponding net connectivity and topology. | ||
Most porphyrin-based molecular frameworks have been built from tetratopic meso-tetra(carboxyphenyl)porphyrin (TCPP) linkers, or some other related porphyrins with reduced/increased topicity and/or extended backbones.20,21 Here we use several representative TCPP–MOFs to systematically study the electrocatalytic activity of such frameworks for ammonia generation as a function of their chemical stability, metal substitution (MTCPP), or the functionalization of the porphyrin ring in the metal position with fluorine atoms (TCPP–F) for tailorable pore environments.
Results and discussion
Framework selection and synthesis
As mentioned above, there are multiple Zr–TCPP MOFs reported, many of which are built from the ubiquitous Zr6 cluster. However, this rich variability also imposes some difficulties in controlling their synthesis as phase-pure frameworks due to the formation of defective phases,22 the concurrent formation of different polymorphs23 or, different degrees of sensitivity to experimental conditions between different crystallographic phases.24In the search for a synthetically robust platform compatible with the iterative variation of metals and linkers, we chose to test in our laboratory some of the most representative members of the Zr–TCPP MOF family (Fig. 1) including: MOF-525,24 PCN-223,25 MOF-545![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 24 (also referred to as PCN-22226 and MMPF-627), PCN-224,28 and PCN-226.29 The reader should note that in this list we included not only the classical Zr6 clusters, but also the closely related Zr8 node and the rod-shape Zr-oxo SBU in PCN-226 for the shake of completeness. We attempted the synthesis of all these MOFs based on the original reported procedures,24–26,29 except for the case of PCN-224, for which we chose to follow the revisited synthetic protocol recently reported by Lotsch's group.22 According to our powder X-ray diffraction (PXRD) data (Fig. 2), we straightforwardly obtained phase pure crystalline phases of MOF-525, PCN-224 and PCN-226, while our attempts for the synthesis of MOF-545 and PCN-223 were unsuccessful and yielded amorphous phases. These results highlight the difficulty in controlling the synthesis of Zr–TCPP MOF, where different members of the family share very similar or even equal compositions. Our results are in line with recent studies that demonstrate the acute influence of water content on controlling phase formation in Zr–porphyrin-based MOFs30 and the problems for their synthesis reproducibility due to their sensitivity to slight changes in the experimental conditions.23,31,32
24 (also referred to as PCN-22226 and MMPF-627), PCN-224,28 and PCN-226.29 The reader should note that in this list we included not only the classical Zr6 clusters, but also the closely related Zr8 node and the rod-shape Zr-oxo SBU in PCN-226 for the shake of completeness. We attempted the synthesis of all these MOFs based on the original reported procedures,24–26,29 except for the case of PCN-224, for which we chose to follow the revisited synthetic protocol recently reported by Lotsch's group.22 According to our powder X-ray diffraction (PXRD) data (Fig. 2), we straightforwardly obtained phase pure crystalline phases of MOF-525, PCN-224 and PCN-226, while our attempts for the synthesis of MOF-545 and PCN-223 were unsuccessful and yielded amorphous phases. These results highlight the difficulty in controlling the synthesis of Zr–TCPP MOF, where different members of the family share very similar or even equal compositions. Our results are in line with recent studies that demonstrate the acute influence of water content on controlling phase formation in Zr–porphyrin-based MOFs30 and the problems for their synthesis reproducibility due to their sensitivity to slight changes in the experimental conditions.23,31,32
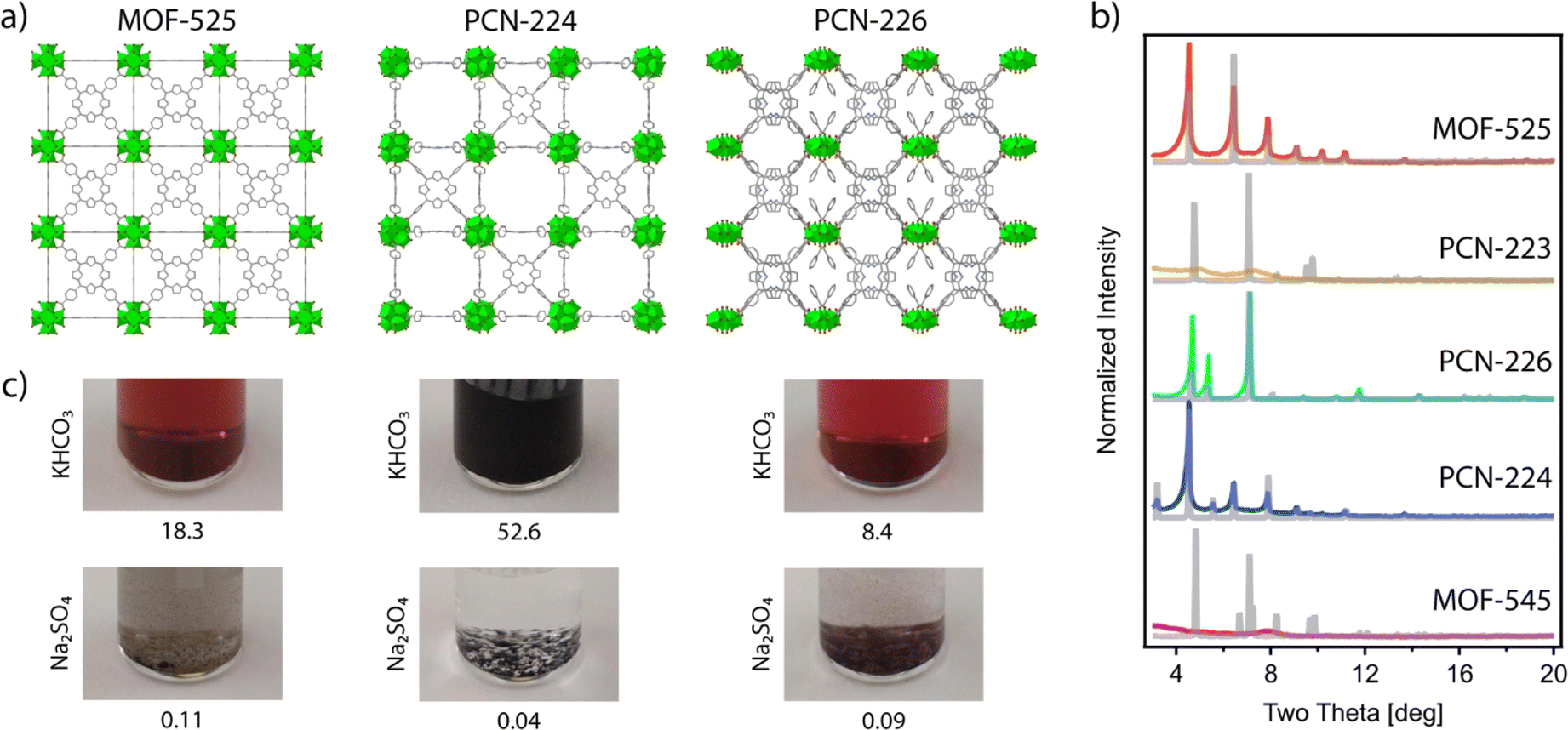 | ||
| Fig. 2 (a) From left to right, crystal structures of the frameworks: MOF-525, PCN-224 and PCN-226. (b) Experimental (colored lines) and simulated (gray lines) PXRD patterns for different Zr–TCCP MOFs referred to in the text. (c) Images showing suspensions of MOF-525, PCN-224, and PCN-226 after immersion at room temperature in KHCO3 and Na2SO4 solutions for 24 h. The numbers below each image show the percentage of Zr leached into the corresponding solution, as determined by ICP. | ||
As a result of our initial screening, we selected MOF-525, PCN-224, whose crystal structure is represented in Fig. 2c, and PCN-226 as potential platforms to implement our meta-substituted TCPP roadmap.
Stability under electrochemical conditions
Among possible applications of Zr–TCPP MOF in electrocatalysis, first we focused on the CO2RR, not only because of the need to find alternatives that contribute to the mitigation of the climate crisis,33 but also because, in the past decade, porphyrins have attracted remarkable interest for the electrochemical reduction of CO2.34 These experiments are usually carried out in the presence of basic bicarbonate buffer. Although, Zr-MOFs are often considered relatively stable in alkali media,18 the presence of bicarbonate makes these reaction conditions somewhat more demanding.35 In order to examine the chemical stability of the synthesized MOF-525, PCN-224, and PCN-226 frameworks in CO2RR media, the samples were immersed in 0.5 M KHCO3 aqueous solutions at room temperature. After 24 h, solutions had turned dark brown in all cases, and inductively coupled plasma (ICP) analysis confirmed the presence of significant amounts of Zr in the solution that oscillated between 52.6 and 18.3% of the total metal content in the frameworks. This is indicative of a severe degradation associated with the decomposition of Zr-oxo clusters in this medium, which would be a serious concern for their implementation under electrocatalytic conditions (Fig. 2c). We reasoned that the problem was the coordinating ability of bicarbonate and decided to try some other related catalytic reaction conditions that imposed a less coordinating medium. The key role of ammonia today is as the basic feedstock for inorganic fertilizers that currently support food production for around half of the world's population. Industrial ammonia production is currently derived from fossil fuels and is responsible for the emission of more CO2 than any other chemical-making reaction. Lately, ammonia is also being considered as a renewable energy carrier. For these reasons, the use of the NRR as a sustainable approach to NH3 production is currently being intensely investigated. Compared to other energy related applications, MOFs have been much less studied in this context.36,37 To confirm the stability of these frameworks under the conditions required for NRR electrochemical tests, we soaked the as-made materials in 0.1 M Na2SO4 aqueous solutions at room temperature. Compared to the use of bicarbonate, all the solutions remained transparent after 24 hours with negligible metal leaching determined by ICP, which remained below 0.11 Zr% in all cases (Fig. 2c).Our chemical stability tests in conventional aqueous electrolytic solutions suggest that MOF-525, PCN-226 and PCN-224 do not display sufficient chemical stability to be used for the electrochemical CO2RR but they show comparatively higher stability in the medium used for the NRR. Among them, PCN-224 showed the highest chemical stability, displaying a minimum percentage of metal leaching after 24 hours (0.04%). Consequently, PCN-224 is the ideal candidate to investigate the electrochemical activity of the pristine framework or other derivatives belonging to the same family towards the production of ammonia from nitrogen.
PCN-224–M series for electrochemical nitrogen reduction
The nature of the metal centre on the MTCPP unit has been reported to have a marked effect on the catalytic performance.38–40 Consequently, we synthesized a series of metalated analogues of PCN-224 that we refer to as PCN-224–M (M = Co2+, Ni2+, Cu2+), by reacting ZrOCl2 in the presence of the metalated TCPP porphyrin linkers under the same conditions used for the synthesis of PCN-224. See ESI S2† for the experimental details. According to their PXRD profiles (Fig. 3a), the PCN-224–M samples isolated are isostructural to the non-metalated framework confirming the formation of phase pure solids. It is worth noting that we also attempted the synthesis of the PCN-224–Fe analogue. Iron is essential for the biological fixation of molecular nitrogen, and Fe-based compounds have been widely applied industrially in this context.41 Unfortunately, all our attempts led to the formation of either amorphous solids or the PCN-222–Fe framework (Fig. S10†).26 Energy dispersive X-ray spectroscopy (EDX) single-point mapping measurements of the PCN-224–M solids confirmed values close to the expected 1:4 M/Zr ratio consistent with the corresponding unit formula. Crystal size and morphology were studied by using scanning electron microscopy (SEM). Fig. 3b shows little variation in morphology across the series. All solids display cube-like crystals; however, two different size ranges are observed depending on the sample. While PCN-224–Cu shows crystals of about 1 μm very similar to those of PCN-224, the Co and Ni analogues are comparatively bigger. Leaving aside this small divergence in size, the properties of the PCN-224–M series agree well with the characteristics reported for this MOF and rule out the formation of contaminant oxide phases. Permanent porosity was also analysed with N2 isotherms at 77 K (Fig. S12†).28 All samples exhibit a reversible type-I isotherm. While PCN-224 shows the maximum N2 uptake with a value consistent with that originally reported,15 the metalated analogues display comparatively lower uptakes as previously reported by others.42,43
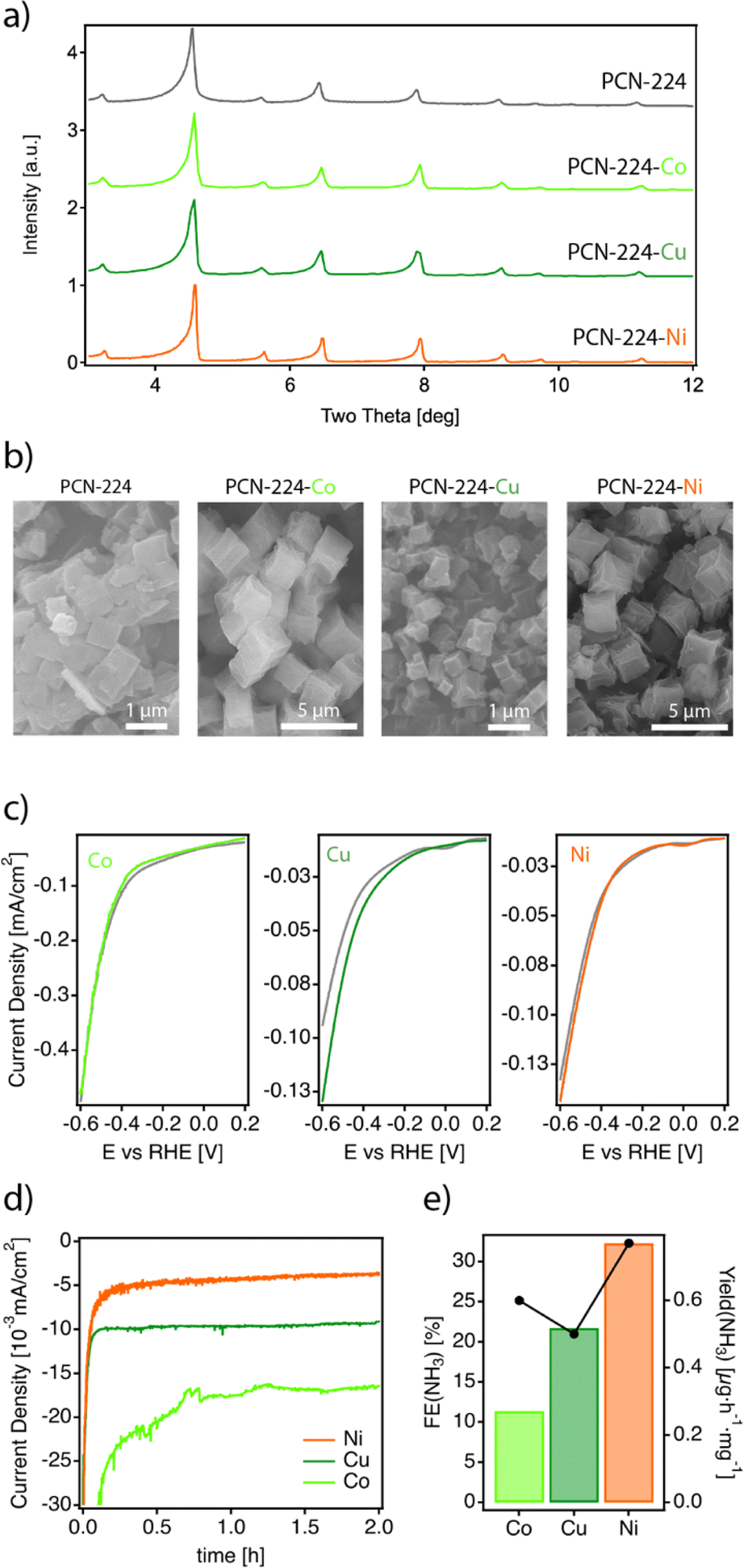 | ||
| Fig. 3 (a) Powder diffraction of PCN-224 and the metalated PCN-224–M derivatives. (b) SEM images of the isolated solids showing the formation of cube-like crystals with varying sizes. (c) CV at 50 mV s−1 for each PCN-224–M framework under an Ar (gray) and N2 atmosphere (color). (d) CA at −0.04 V vs. RHE for the same materials and (e) comparison of faradaic efficiencies and yields for ammonia generation. | ||
Next, the electrocatalytic activity of the materials toward the NRR was systematically investigated. For this purpose, electrodes of the different samples were prepared, by mixing the corresponding solids with the binder agent, and sprayed over a Toray carbon paper using an air brushing technique for a final electrode loading of about 0.7 mg cm−2. A platinum wire and a leakless AgCl/Ag (saturated KCl) were used as the counter and reference electrodes, respectively. All potentials were referred to the reversible hydrogen electrode (RHE). Cyclic voltammetry (CV) curves were recorded at 50 mV s−1 in Ar and N2 saturated 0.1 M Na2SO4 solution. Chronoamperometry (CA) measurements (electrolysis) were subsequently carried out at −0.04 V, −0.14 V and −0.34 V vs. RHE for 2 h. The experimental details are included in ESI Sections S2 and S7†. It is worth highlighting that these experiments were performed in the same Na2SO4 aqueous electrolyte for which we confirmed the stability of PCN-224 under open circuit potential conditions with negligible metal leaching after 24 hours. Faradaic efficiencies (FEs) and ammonia yields were determined using the amount of ammonia produced as determined by the indophenol blue method.42 Before each electrolysis, five voltammetry cycles between 0.56 V and −0.94 V vs. RHE were recorded to verify the status of the electrodes. Fig. 3c displays the voltammetric profiles corresponding to PCN-224–Co, PCN-224–Ni and PCN-224–Cu in the potential range from 0.2 V to −0.6 V vs. RHE. The voltammetric responses of the electrodes in Ar (gray line) and N2 (color line) are very similar, with a slight increase of the negative currents in the N2 saturated solution electrolyte which could be related to an enhanced activity for the electrochemical reduction of N2 to ammonia. To better evaluate the electrocatalytic properties of the samples towards N2 electroreduction, CAs were performed at different potentials of interest. Fig. 3d shows the variation of current density versus time at a constant potential of −0.04 V (the potential at which ammonia production reached its maximum) for all catalysts. The results demonstrate that the behaviour of the samples is strongly determined by the type of metal incorporated. In agreement with the voltametric curves (Fig. 3c), Ni and Cu samples exhibited lower current density compared to Co. In addition, all samples showed a relatively stable current during the electrolysis time. Regarding the FE (Table S3†), the results indicate that the FE towards ammonia ranges from 4 to 32% depending on the nature of the metal. Interestingly, in all cases, the highest FE value is achieved at −0.04 V; the Co, Ni, and Cu samples show FE values of about 11.4, 32.3 and 21.7%, respectively. In all cases, the FEs systematically decrease at more negative potentials due to the increased effect of the HER. Thus, at −0.34 V vs. RHE, the FE values are the lowest independent of the nature of the electrode. The FE values discussed previously clearly indicate that at a relatively positive potential value of −0.04 V vs. RHE, the incorporation of a metal into the PCN-224 framework significantly enhances the electrocatalytic activity of the electrocatalyst, with the Ni metalated being the most efficient one with an FE of about 32%. To rule out the possible contribution of the Toray paper, control experiments were performed with the bare Toray paper electrode. The results are shown in Fig. S27 and S28.† The results indicate the absence of detectable ammonia during these control experiments. To better understand our finding, Fig. 4 compares this 32% FE value with the FEs reported with other electrocatalysts, measured in a comparable range of potentials of ± 0.2 V and obtained in alkaline electrolyte which is expected to suppress the hydrogen evolution reaction for improved NRR activities (Table S4†). The FE of 32.29% registered for PCN-224–Ni is quite higher than the 16.2% reported for the related 2D microporous MOF Fe–TCPP, built from Zn(II) paddlewheel clusters and the metalated TCPP linker.44,45 Other electrocatalysts prepared by the calcination/pyrolysis of MOFs to generate the corresponding Bi nanoparticles,46 CeO2 nanorods47 Y-stabilized ZrO2,48 Co3O4 nanopolyhedra,49 iron doped nanocarbons,50 or NiO/Ni microtubes51 all supported on carbon also display comparatively lower activities below a FE of 13%. Higher activities have been reported for other MOF composites reliant on the use of MXenes as ZIF-67@Ti3C2,51 but they are comparatively lower with efficiencies near 20%. The activity associated with Zr6 clusters in defective UiO-66 frameworks has been also shown to be very high for this type of reaction with efficiencies up to 48%.52 This comparison clearly points out the relevance of our results as it implicates the MOF exclusively as a crystalline support of porphyrin complexes acting as electrocatalytic centers.51 Despite the excellent results in terms of FE, it is important to mention that the electrodes cannot be reused, and the FE values decrease significantly during their second use. This suggests a possible degradation of the electrodes under electrochemical working conditions, which is not obvious for the Cu and Ni derivatives (Fig. S26a†), but is more evident for the cobalt derivative (Fig. S26b†). In the last case, we observed a strong colouration of the solution that agrees well with the characteristic MTCPP UV-vis signals, which was concomitant with the loss of performance. In addition, ICP analysis of the isolated electrolyte solution after electrocatalytic experiments with PCN-224–Ni confirmed an increase in the nickel concentration in the electrolyte buffer of 29.2 ppb compared to the 0.1 ppb present before the reaction, which might be indicative of framework degradation. These results highlight the need to improve the stability of the samples under electrochemical working conditions.
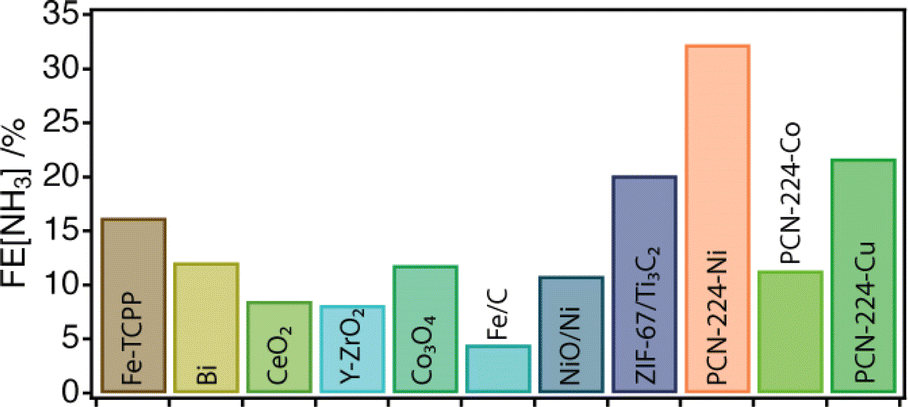 | ||
| Fig. 4 Overview of the FE of the PCN-224–M family compared to that of other NRR electrocatalysts measured in alkaline electrolytes and under comparable voltage conditions. See ESI Table S1† for more details. | ||
Enhancement of NRR performance by porphyrin functionalization: PCN-224–Ni(F)
The high activity of PCN-224–Ni for electrochemical nitrogen reduction encouraged us to target further improvements, particularly in terms of stability, based on the functionalization of the porphyrin ring. Inspired by similar modifications on molecular catalysts for the electroreduction of CO2,12 we prepared the meta-fluorinated analogue of TCPP starting from the commercially available linker. As shown in Fig. 5a TCPP–F was obtained from the corresponding methyl esters after hydrolysis in basic media. Meta-substituted TCPP–F methyl esters were synthesized in moderate yields by using Lindsey's reaction between pyrrole and commercially available 3-fluoromethyl 4-formylbenzoate to obtain the corresponding TCPP–F methyl esters. In turn, NiTCPP–F was obtained by reacting NiCl2 in DMF at reflux overnight. The NiTCPP–F linker was then reacted with the zirconium salt to produce the corresponding PCN-224–Ni(F) framework according to the same protocol used for the non-fluorinated family. PCN-224–Ni(F) was isolated as micrometric cubes (Fig. 5b). As summarised in ESI Section S4†, the PXRD pattern is consistent with the formation of an isostructural PCN-224 framework with an experimental surface area of 1.400 m2 g−1. To the best of our knowledge, this is the first example of this family of frameworks prepared from functionalized porphyrin units.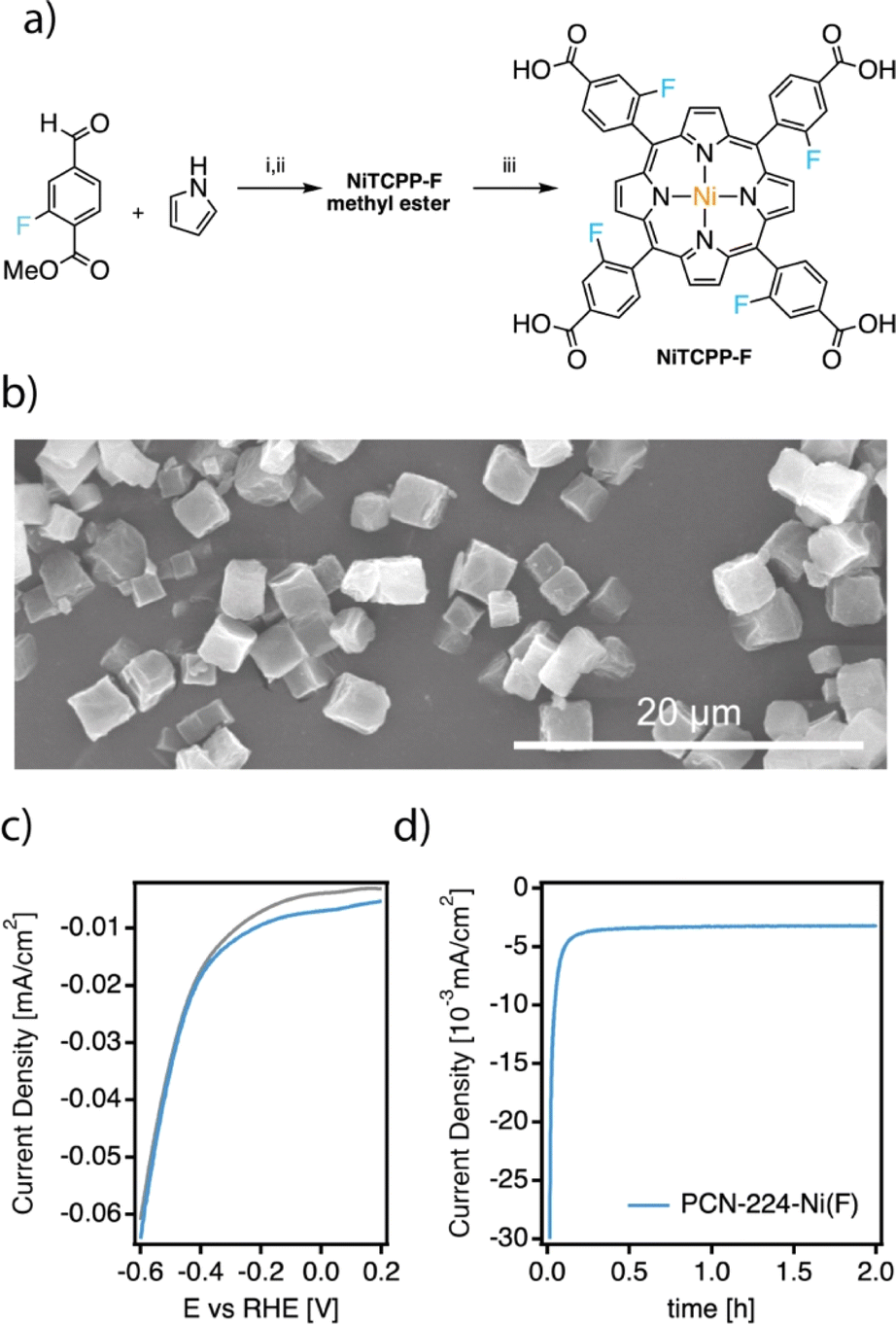 | ||
| Fig. 5 (a) Experimental procedure for the synthesis of the NiTCCP–F linker: (i) DCM, pyrrole, BF3·OEt2, stirring 2 h, then add p-chloranil, reflux for 2 h, (ii) Ni2+, DMF, reflux overnight, and (iii) MeOH, THF and KOH, reflux overnight and wash with acetic acid. (b) SEM micrograph of PCN-224–Ni(F) crystals. (c) CV at 50 mV s−1 under an Ar (gray) and N2 atmosphere (color) and (d) CA at −0.04 V vs. RHE of the fluorinated framework. | ||
To evaluate the impact of the electron withdrawing fluorine groups on the electrocatalytic NRR we prepared the corresponding electrodes and tested the system under the same conditions used for the PCN-224–M family. The CV of the sample in Ar and N2-saturated solution was essentially similar to those shown by PCN-224–Ni in Fig. 3c, although for the PCN-224–Ni(F) sample (Fig. 5c), the voltammetric responses revealed an increase in current in the N2-saturated electrolyte which could be related to an enhanced activity for the electrochemical reduction of N2 to ammonia. With respect to the FE values (Table S4†), the incorporation of F also improved the FE at −0.04 V, from 32.3% to about 34.5%. As for the unfluorinated samples, for more negative potentials than −0.04 V, the FEs systematically decrease due to the increased effect of the HER.37 Regarding the ammonia yield rates, a maximum value of 0.9 μgNH3 h−1 mgcat−1 was obtained at −0.04 V vs. RHE. Remarkably, compared to PCN-224–Ni, PCN-224–Ni(F) showed constant FE values up to the third electrode use. This implies an increased stability of the samples under electrochemical working conditions. Hydrophobic fluorination has been used before to enhance MOF stability,53–55 and in the particular case of the NRR, to increase local concentration of N2 around the catalytic centre, thus limiting proton transportation and favouring the NRR over the HER.56–58 To confirm the improved stability associated with the incorporation of fluorinated substituents into the porphyrin ring, we analysed the electrolyte solutions after the first electroreduction reaction by ICP. Compared to the near 30 ppb leached by PCN-224–Ni, its fluorinated equivalent is only 4.3 ppb confirming a significant improvement in the stability of this MOF under electrocatalytic conditions. We also incubated both MOFs in the electrolyte used in the electrocatalytic experiment for 24 h. Fig. S30† shows that while PCN-224–Ni amorphizes considerably under these conditions, the crystallinity of the fluorinated derivative is retained without apparent amorphization. The water isotherms of PCN-224–Ni and PCN-224–Ni(F) at 298 K (Fig. S29†) provide additional evidence of the enhanced stability towards hydrolysis of the fluorinated framework, which is less prone to collapse and displays a much higher water uptake than PCN-224–Ni despite the increase in hydrophobicity.
Although all these experiments support the enhanced stability and reusability of the fluorinated framework, they do not provide a clear answer for the origin of the experimentally observed enhancement of FE. To clarify which was the most likely cause for the observed change in performance, we next carried out DFT calculations.
Computational rationalization of performance
We first performed DFT calculations by using the cluster models representative of the catalytic centers in the corresponding frameworks to obtain the density of states of the MTCPP–X (M = Ni, Co, Cu and X = H, F) units and the adsorption energies of the reactants by using the VASP code and the hybrid functional HSE06. For more details on the computational methodology see ESI Section S9.† Our results only show a small effect of the metal on the electronic structure of the MTCPP units (Table S5 and Fig. S31†), suggesting that the effect of the metal might have a more important role in determining the reaction mechanism in line with previous reports.59Based on these preliminary results we analyzed separately the two generally accepted mechanisms for the NRR. First, we considered an associative Heyrovsky pathway60 that relies on the adsorption of N2 molecules to the active site followed by hydrogenation, from the combination of protons in solution and electrons from the electrode. However, our simulations reveal that this mechanism is quite unlikely in our case. The binding of N2 with the MTCPP–X units is very weak or metastable with N2–M distances near 3.3 Å in all cases, compared to the 2.0 (Co), 2.5 (Cu), and 2.7 Å (Ni) that would be required for nitrogen adsorption and activation (Fig. S32†).
We next considered the Tafel–Volmer mechanism,61 which involves hydrogen dissociation (1), electron transfer to the catalyst (2), the adsorption of solvated protons from solution (3), and the adsorption of N2 for its reduction (4). We calculated the adsorption energy for all systems by combining steps (2)–(4) correlating the trends in catalytic activity with the transition metal and fluorination of the porphyrin ring (Fig. 6a). As anticipated by our experimental results, the N2 molecule was preferentially adsorbed in NiTCPP–H and NiTCPP–F, compared to the Co and Cu analogues. According to our calculations, the N2 molecule not only interacts preferably with the Ni site, but also forms a hydrogen bonding with the proton adsorbed by the porphyrin for enhancing adsorption and reducing the energy barrier in the rate determining step (Fig. 6b). This effect is even more acute for a fluorinated framework, for which the electron transfer and nitrogen adsorption are comparatively more favourable making it the best electrocatalyst of the family (Table S6†). The addition of the proton to the porphyrin core in CuTCPP–H is stabilized by 0.01 eV, although it elongates the Cu–N interactions with the pyrrole nitrogens by 0.5 Å, suggesting that the N2 is weakly bound as suggested before by the Heyrovsky mechanism. Finally, the electrons in CoTCPP–H are located at the lowest chemical potentials (2.65 eV) across the series (Table S6†). This suggests a poorer ability for accepting electrons in the first step of the NRR for the lowest electrocatalytic activity of the family.
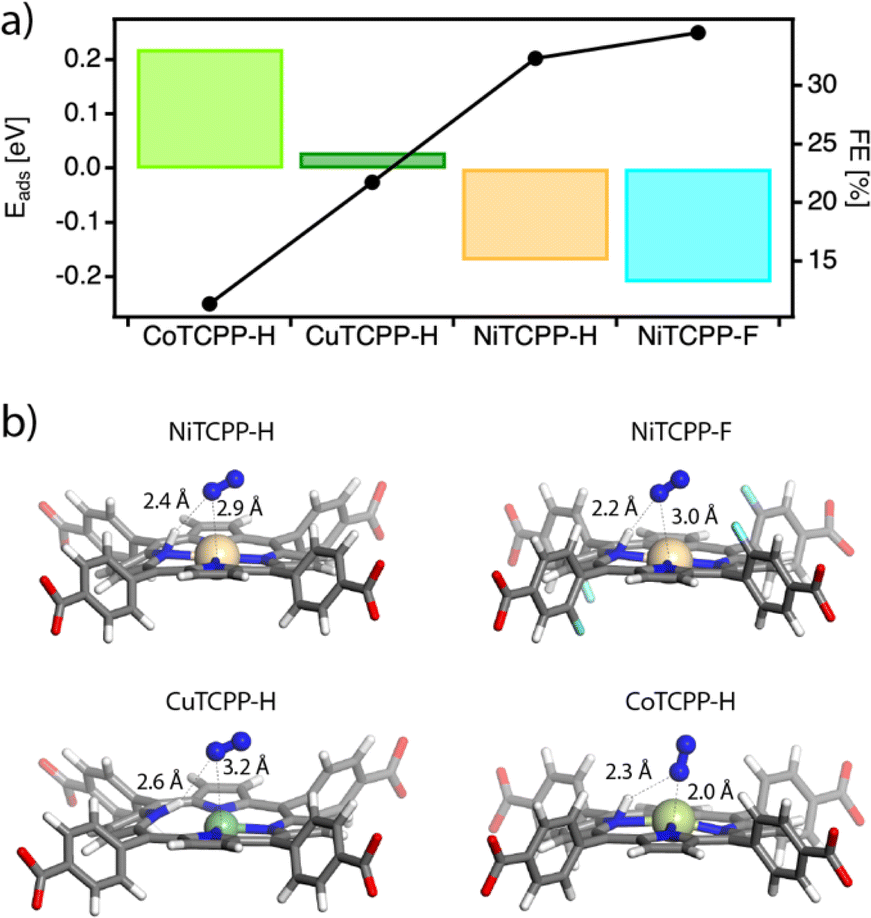 | ||
| Fig. 6 (a) DFT adsorption energy (Eads) calculated for MTCPP–X (M = Ni, Co, Cu and X = H, F) cluster models according to the Tafel–Volmer mechanism (left axis) and correlation with the experimental FE values (right axis). (b) Calculated models showing the adsorption of N2 molecules (in blue) to the catalytic site and distances with the protonated porphyrin in all cases studied. | ||
Conclusions
Based on the topological diversity of Zr MOFs as a starting point, we have identified the PCN-224 family as one of the best candidates for a systematic analysis of their electrocatalytic activity based on their superior chemical stability in the presence of basic aqueous electrolytes. The results obtained in the electroreduction of molecular nitrogen into ammonium reveal a direct impact of the nature of the metal on the electrocatalytic activity, with faradaic efficiency values above 30% for PCN-224–Ni. While this activity is well above that of other reference electrocatalysts in this reaction, our study also reveals important limitations to the reuse of electrodes resulting from a low stability of the MOF under the experimental conditions. The functionalization of porphyrin in the meso position has allowed us to synthesize the fluorinated analogue of this framework. PCN-224–Ni(F) displays a significant improvement in the stability of the electrode under equivalent conditions to reach efficiencies close to 35% as a result of its intrinsic hydrophobicity.Author contributions
All authors contributed to the discussion, participated in the writing of the original draft, and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the H2020 Program (ERC-2021-COG-101043428), the Generalitat Valenciana (PROMETEU/2021/054, IDIFEDER/2021/075, MFA/2022/026 and SEJIGENT/2021/059), the Spanish Government (RTI2018-098568-A-I00, CEX2019-000919-M, PID2020-118117RB-I00, EUR2021-121999 & CNS2022-135677), and VLC-Biomed AP2022-5. M. R. A. acknowledges the Spanish Government for a predoctoral grant (PRE-C-2018-0109) and S. T. for a RyC contract (RYC-2016-19817). N. M. P. thanks the La Caixa Foundation for a Postdoctoral Junior Leader–Retaining Fellowship (ID 100010434, fellowship code LCF/BQ/PR20/11770014). We also thank the University of Valencia for research facilities (NANBIOSIS). R. A. acknowledges the Ministry of Higher Education and Scientific Research of the People's Democratic Republic of Algeria for the financial support to carry out her PhD study at the University of Alicante, Spain.Notes and references
- A. Bavykina, N. Kolobov, I. S. Khan, J. A. Bau, A. Ramirez and J. Gascon, Chem. Rev., 2020, 120, 8468–8535 CrossRef CAS PubMed.
- M. Li, D. Li, M. O’Keeffe and O. M. Yaghi, Chem. Rev., 2013, 116, 12466–12535 Search PubMed.
- J. Lyu, X. Zhang, K. Otake, X. Wang, P. Li, Z. Li, Z. Chen, Y. Zhang, M. C. Wasson, Y. Yang, P. Bai, X. Guo, T. Islamoglu and O. K. Farha, Chem. Sci., 2018, 10, 1186–1192 RSC.
- X. Gong, Y. Shu, Z. Jiang, L. Lu, X. Xu, C. Wang and H. Deng, Angew. Chem., Int. Ed., 2020, 59, 5326–5331 CrossRef CAS PubMed.
- V. Guillerm and M. Eddaoudi, Ind. Eng. Chem. Res., 2022, 61, 12641–12648 CrossRef CAS.
- Z. Ji, H. Wang, S. Canossa, S. Wuttke and O. M. Yaghi, Adv. Funct. Mater., 2020, 30, 2000238 CrossRef CAS.
- Y. Peng, S. Sanati, A. Morsali and H. García, Angew. Chem., Int. Ed., 2023, 62, e202214707 CrossRef CAS PubMed.
- S. Navalón, A. Dhakshinamoorthy, M. Álvaro, B. Ferrer and H. García, Chem. Rev., 2023, 123, 445–490 CrossRef PubMed.
- (a) X. F. Lu, B. Y. Xia, S. Zang and X. W. D. Lou, Angew. Chem., Int. Ed., 2020, 59, 4634–4650 CrossRef CAS PubMed; (b) X. Sun, S. Wang, Y. Hou, X. F. Lu, J. Zhang and X. Wang, J. Mater. Chem. A, 2023, 11, 13089–13106 RSC.
- C. Wang, Z. Lv, W. Yang, X. Feng and B. Wang, Chem. Soc. Rev., 2023, 52, 1382–1427 RSC.
- (a) H. Zhong, M. Wang, M. Ghorbani-Asl, J. Zhang, K. H. Ly, Z. Liao, G. Chen, Y. Wei, B. P. Biswal, E. Zschech, I. M. Weidinger, A. V. Krasheninnikov, R. Dong and X. Feng, J. Am. Chem. Soc., 2021, 143, 19992–20000 CrossRef CAS PubMed; (b) Y. Wang, Q. Li, W. Shi and P. Cheng, Chin. Chem. Lett., 2020, 31, 1768–1772 CrossRef CAS; (c) X. Sun, S. Wang, Y. Hou, X. F. Lu, J. Zhang and X. Wang, J. Mater. Chem. A, 2023, 11, 13089–13106 RSC.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, Science, 2012, 338, 90–94 CrossRef CAS PubMed.
- Y. Ishimizu, Z. Ma, M. Hada and H. Fujii, Inorg. Chem., 2021, 60, 17687–17698 CrossRef CAS PubMed.
- A. Zhang, D. Si, H. Huang, L. Xie, Z. Fang, T. Liu and R. Cao, Angew. Chem., Int. Ed., 2022, 61, e202203955 CrossRef CAS PubMed.
- W.-Y. Gao, M. Chrzanowski and S. Ma, Chem. Soc. Rev., 2014, 43, 5841–5866 RSC.
- J. Chen, Y. Zhu and S. Kaskel, Angew. Chem., Int. Ed., 2021, 60, 5010–5035 CrossRef CAS PubMed.
- Z. Liang, H.-Y. Wang, H. Zheng, W. Zhang and R. Cao, Chem. Soc. Rev., 2021, 50, 2540–2581 RSC.
- A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Nat. Rev. Mater., 2016, 1, 15018 CrossRef CAS.
- S. Yuan, L. Feng, K. Wang, J. Pang, M. Bosch, C. Lollar, Y. Sun, J. Qin, X. Yang, P. Zhang, Q. Wang, L. Zou, Y. Zhang, L. Zhang, Y. Fang, J. Li and H. Zhou, Adv. Mater., 2018, 30, e1704303 CrossRef PubMed.
- B. F. Abrahams, B. F. Hoskins, D. M. Michail and R. Robson, Nature, 1994, 369, 727–729 CrossRef CAS.
- X. Zhang, M. C. Wasson, M. Shayan, E. K. Berdichevsky, J. Ricardo-Noordberg, Z. Singh, E. K. Papazyan, A. J. Castro, P. Marino, Z. Ajoyan, Z. Chen, T. Islamoglu, A. J. Howarth, Y. Liu, M. B. Majewski, M. J. Katz, J. E. Mondloch and O. K. Farha, Coord. Chem. Rev., 2021, 429, 213615 CrossRef CAS PubMed.
- C. Koschnick, R. Stäglich, T. Scholz, M. W. Terban, A. von Mankowski, G. Savasci, F. Binder, A. Schökel, M. Etter, J. Nuss, R. Siegel, L. S. Germann, C. Ochsenfeld, R. E. Dinnebier, J. Senker and B. V. Lotsch, Nat. Commun., 2021, 12, 3099 CrossRef CAS PubMed.
- S. M. Shaikh, P. M. Usov, J. Zhu, M. Cai, J. Alatis and A. J. Morris, Inorg. Chem., 2019, 58, 5145–5153 CrossRef CAS PubMed.
- W. Morris, B. Volosskiy, S. Demir, F. Gándara, P. L. McGrier, H. Furukawa, D. Cascio, J. F. Stoddart and O. M. Yaghi, Inorg. Chem., 2012, 51, 6443–6445 CrossRef CAS PubMed.
- D. Feng, Z.-Y. Gu, Y.-P. Chen, J. Park, Z. Wei, Y. Sun, M. Bosch, S. Yuan and H.-C. Zhou, J. Am. Chem. Soc., 2014, 136, 17714–17717 CrossRef CAS PubMed.
- D. Feng, Z.-Y. Gu, J.-R. Li, H.-L. Jiang, Z. Wei and H.-C. Zhou, Angew. Chem., Int. Ed., 2012, 124, 10453–10456 CrossRef.
- Y. Chen, T. Hoang and S. Ma, Inorg. Chem., 2008, 51, 12600–12602 CrossRef PubMed.
- D. Feng, W.-C. Chung, Z. Wei, Z.-Y. Gu, H.-L. Jiang, Y.-P. Chen, D. J. Darensbourg and H.-C. Zhou, J. Am. Chem. Soc., 2013, 135, 17105–17110 CrossRef CAS PubMed.
- M. O. Cichocka, Z. Liang, D. Feng, S. Back, S. Siahrostami, X. Wang, L. Samperisi, Y. Sun, H. Xu, N. Hedin, H. Zheng, X. Zou, H.-C. Zhou and Z. Huang, J. Am. Chem. Soc., 2020, 142, 15386–15395 CrossRef CAS PubMed.
- C. Koschnick, M. W. Terban, S. Canossa, M. Etter, R. E. Dinnebier and B. V. Lotsch, Adv. Mater., 2023, 2210613 Search PubMed.
- J. Lyu, X. Gong, S.-J. Lee, K. Gnanasekaran, X. Zhang, M. C. Wasson, X. Wang, P. Bai, X. Guo, N. C. Gianneschi and O. K. Farha, J. Am. Chem. Soc., 2020, 142, 4609–4615 CrossRef CAS PubMed.
- H. L. B. Boström, S. Emmerling, F. Heck, C. Koschnick, A. J. Jones, M. J. Cliffe, R. A. Natour, M. Bonneau, V. Guillerm, O. Shekhah, M. Eddaoudi, J. Lopez-Cabrelles, S. Furukawa, M. Romero-Angel, C. Martí-Gastaldo, M. Yan, A. J. Morris, I. Romero-Muñiz, Y. Xiong, A. E. Platero-Prats, J. Roth, W. L. Queen, K. S. Mertin, D. E. Schier, N. R. Champness, H. H. -M. Yeung and B. V. Lotsch, Adv. Mater., 2023, e2304832 Search PubMed.
- P. Gilding, Nature, 2019, 573, 311 CrossRef CAS PubMed.
- P. Gotico, Z. Halime and A. Aukauloo, Dalton Trans., 2020, 49, 2381–2396 RSC.
- J. Chu, F.-S. Ke, Y. Wang, X. Feng, W. Chen, X. Ai, H. Yang and Y. Cao, Commun. Chem., 2020, 3, 5 CrossRef CAS PubMed.
- Y. Wang, Q. Li, W. Shi and P. Cheng, Chin. Chem. Lett., 2020, 31, 1768–1772 CrossRef CAS.
- M. A. Mushtaq, M. Arif, G. Yasin, M. Tabish, A. Kumar, S. Ibraheem, W. Ye, S. Ajmal, J. Zhao, P. Li, J. Liu, A. Saad, X. Fang, X. Cai, S. Ji and D. Yan, Renewable Sustainable Energy Rev., 2023, 176, 113197 CrossRef CAS.
- I. Bhugun, D. Lexa and J.-M. Savéant, J. Am. Chem. Soc., 1996, 118, 3982–3983 CrossRef CAS.
- J. Bonin, A. Maurin and M. Robert, Coord. Chem. Rev., 2017, 334, 184–198 CrossRef CAS.
- C.-X. Zhao, B.-Q. Li, J.-N. Liu, J.-Q. Huang and Q. Zhang, Chin. Chem. Lett., 2019, 30, 911–914 CrossRef CAS.
- T. Wang, Z. Guo, X. Zhang, Q. Li, A. Yu, C. Wu and C. Sun, J. Mater. Sci. Technol., 2023, 140, 121–134 CrossRef CAS.
- S.-C. Qi, Z.-H. Yang, R.-R. Zhu, X.-J. Lu, D.-M. Xue, X.-Q. Liu and L.-B. Sun, J. Mater. Chem. A, 2021, 9, 24510–24516 RSC.
- P. M. Stanley, K. Hemmer, M. Hegelmann, A. Schulz, M. Park, M. Elsner, M. Cokoja and J. Warnan, Chem. Sci., 2022, 13, 12164–12174 RSC.
- M. Zhao, Y. Wang, Q. Ma, Y. Huang, X. Zhang, J. Ping, Z. Zhang, Q. Lu, Y. Yu, H. Xu, Y. Zhao and H. Zhang, Adv. Mater., 2015, 27, 7372–7378 CrossRef CAS PubMed.
- M. Cong, X. Chen, K. Xia, X. Ding, L. Zhang, Y. Jin, Y. Gao and L. Zhang, J. Mater. Chem. A, 2021, 9, 4673–4678 RSC.
- D. Yao, C. Tang, L. Li, B. Xia, A. Vasileff, H. Jin, Y. Zhang and S. Qiao, Adv. Energy Mater., 2020, 10, 2001289 CrossRef CAS.
- Y. Liu, C. Li, L. Guan, K. Li and Y. Lin, J. Phys. Chem. C, 2020, 124, 18003–18009 CrossRef CAS.
- S. Luo, X. Li, M. Wang, X. Zhang, W. Gao, S. Su, G. Liu and M. Luo, J. Mater. Chem. A, 2020, 8, 5647–5654 RSC.
- L. Wen, X. Li, R. Zhang, H. Liang, Q. Zhang, C. Su and Y.-J. Zeng, ACS Appl. Mater. Interfaces, 2021, 13, 14181–14188 CrossRef CAS PubMed.
- R. Zhang, L. Jiao, W. Yang, G. Wan and H.-L. Jiang, J. Mater. Chem. A, 2019, 7, 26371–26377 RSC.
- (a) S. Luo, X. Li, W. Gao, H. Zhang and M. Luo, Sustainable Energy Fuels, 2019, 4, 164–170 RSC; (b) X. Liang, X. Ren, Q. Yang, L. Gao, M. Gao, Y. Yang, H. Zhu, G. Li, T. Ma and A. Liu, Nanoscale, 2021, 13, 2843–2848 RSC; (c) X. He, Y. Liao, J. Tan, G. Li and F. Yin, Electrochim. Acta, 2022, 409, 139988 CrossRef CAS.
- G. Li and F. Yin, Electrochim. Acta, 2022, 139988 Search PubMed.
- J. B. Decoste, G. W. Peterson, M. W. Smith, C. A. Stone and C. R. Willis, J. Am. Chem. Soc., 2012, 134, 1486–1489 CrossRef CAS PubMed.
- T.-H. Chen, I. Popov, O. Zenasni, O. Daugulis and O. Š. Miljanić, Chem. Commun., 2013, 49, 6846–6848 RSC.
- J. Castells-Gil, F. Novio, N. M. Padial, S. Tatay, D. Ruíz-Molina and C. Martí-Gastaldo, ACS Appl. Mater. Interfaces, 2017, 9, 44641–44648 CrossRef CAS PubMed.
- Y. Lv, Y. Wang, M. Yang, Z. Mu, S. Liu, W. Ding and M. Ding, J. Mater. Chem. A, 2020, 9, 1480–1486 RSC.
- H. He, H.-K. Li, Q.-Q. Zhu, C.-P. Li, Z. Zhang and M. Du, Appl. Catal., B, 2022, 316, 121673 CrossRef CAS.
- L. Wen, K. Sun, X. Liu, W. Yang, L. Li and H. Jiang, Adv. Mater., 2023, 35, 2210669 CrossRef CAS PubMed.
- S. Hamad, N. C. Hernandez, A. Aziz, A. R. Ruiz-Salvador, S. Calero and R. Grau-Crespo, J. Mater. Chem. A, 2015, 3, 23458–23465 RSC.
- J. Heyrovský, Recl. Trav. Chim. Pays-Bas, 1927, 46, 582–585 CrossRef.
- A. R. Kucernak and C. Zalitis, J. Phys. Chem. C, 2016, 120, 10721–10745 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta07004b |
| ‡ These authors contributed to the work equally. |
| This journal is © The Royal Society of Chemistry 2024 |